

Background: Mosquito odorant-binding proteins constitute molecular targets for structure-based discovery of novel host-seeking disruptors.
Results: “Plus-C” AgamOBP48 exists as a three-dimensional domain-swapped dimer containing a combined binding site.
Conclusion: Domain swapping has important implications for AgamOBP48 binding specificity.
Significance: OBP dimerization should be considered in a successful OBP-based discovery strategy.
Keywords: Analytical Ultracentrifugation, Calorimetry, Chromatography, Crystal Structure, Fluorescence, Molecular Docking
Abstract
Much physiological and behavioral evidence has been provided suggesting that insect odorant-binding proteins (OBPs) are indispensable for odorant recognition and thus are appealing targets for structure-based discovery and design of novel host-seeking disruptors. Despite the fact that more than 60 putative OBP-encoding genes have been identified in the malaria vector Anopheles gambiae, the crystal structures of only six of them are known. It is therefore clear that OBP structure determination constitutes the bottleneck for structure-based approaches to mosquito repellent/attractant discovery. Here, we describe the three-dimensional structure of an A. gambiae “Plus-C” group OBP (AgamOBP48), which exhibits the second highest expression levels in female antennae. This structure represents the first example of a three-dimensional domain-swapped dimer in dipteran species. A combined binding site is formed at the dimer interface by equal contribution of each monomer. Structural comparisons with the monomeric AgamOBP47 revealed that the major structural difference between the two Plus-C proteins localizes in their N- and C-terminal regions, and their concerted conformational change may account for monomer-swapped dimer conversion and furthermore the formation of novel binding pockets. Using a combination of gel filtration chromatography, differential scanning calorimetry, and analytical ultracentrifugation, we demonstrate the AgamOBP48 dimerization in solution. Eventually, molecular modeling calculations were used to predict the binding mode of the most potent synthetic ligand of AgamOBP48 known so far, discovered by ligand- and structure-based virtual screening. The structure-aided identification of multiple OBP binders represents a powerful tool to be employed in the effort to control transmission of the vector-borne diseases.
Introduction
The olfactory system of mosquitoes plays a crucial role in their survival and reproductive success as it facilitates sugar feeding (1), mating (2), and oviposition (3) as well as host detection and blood feeding (4). Odorant-binding proteins (OBPs)2 are the first components of their odor detection unit. Hydrophobic odorant molecules entering the aqueous sensillar lymph of insect antennae are captured by OBPs that solubilize, carry, and deliver them to their cognate odorant receptors (5).
The fact that OBPs are so far the best characterized olfactory macromolecules has identified them as potential targets for the design of novel insect repellents or attractants. The AgamOBP1·DEET (N,N-diethyl-m-toluamide) complex structure has been the first example of a repellent recognized by an OBP (6).
More than 60 OBP-encoding sequences have been found in the A. gambiae genome (7–9). These proteins have been classified into three different groups based on primary protein sequence features as follows: “classic” OBPs with a typical six-cysteine signature; “atypical” OBPs, and Plus-C OBPs (8, 10, 11).
Several three-dimensional structures of insect OBPs have been determined to date, including those of the classic AgamOBPs 1 (6, 12), 4 (13), 7 (14), and 20 (15) as well as one member of the Plus-C group, AgamOBP47 (16). Their observed structural diversity results in a wide variety of cavities that differ in size, shape and position, solvent accessibility, and nature of amino acids involved in their wall formation and hence enable OBPs to selectively recognize and bind a wide range of organic molecules and naturally occurring odorants.
The Plus-C OBPs were first reported in Drosophila and Anopheles (9, 17) and recently to the pea aphid Acyrthosiphon pisum (Hemiptera) (18) indicating that protein evolution occurred before the divergence of diptera from other species; therefore, these proteins may also be present in other insect orders. The Anopheles gambiae Plus-C OBPs are distinguished from the other classes by their longer sequences and the presence of 12 cysteines in conserved positions, six of which correspond closely to the conserved cysteines residues of the classic OBPs (8, 19).
Previous studies have shown that two members of the A. gambiae Plus-C group, AgamOBP48 and AgamOBP47, are preferentially expressed in the antennae of female mosquitoes and belong to the subset of 10 OBPs that exhibit the highest transcript level in this olfactory organ (10, 19). Moreover, AgamOBP48, which displays the second highest expression level in antennae, was shown to be down-regulated after a blood meal, a finding suggesting that protein is an important determinant of the host-seeking behavior of female mosquitoes.
Dimerization of insect OBPs in solution has been reported several times (20–24) and has been also identified in the crystal state by x-ray crystallography since nearly all determined OBP crystal structures represent dimers (6, 14, 25–27). The dimerization of OBPs has been proposed to create new binding sites at the interface between the two subunits leading to the formation of dimers with binding features different from those of the monomeric proteins (6, 22, 28). In the case of Plus-C AgamOBP48, ex vivo yeast two-hybrid screens, in vitro co-immunoprecipitation experiments, and cross-linking studies have suggested that this protein is capable of forming readily detectable homodimers (28).
In this work, we describe the three-dimensional crystal structure and solution studies of the Plus-C group member, AgamOBP48. The AgamOBP48 structure constitutes a three-dimensional domain-swapped dimer thus far unique among dipteran OBPs. The three-dimensional domain swapping results in the creation of novel binding sites. Structural comparison with the monomeric AgamOBP47 reveals significant structural characteristics and differences that may play a role in dimer formation and consequently in odor detection. Finally, in silico and in solution binding studies with a synthetic compound, EN12, discovered by ligand- and structure-based virtual screening, reveal potential interactions and binding characteristics that promote its strong binding to AgamOBP48.
EXPERIMENTAL PROCEDURES
Bacterial Expression of AgamOBP48
AgamOBP48 cDNA (AF533512) (29) was PCR-amplified and subcloned into the NdeI and XhoI sites of the pET22b(+) expression vector (Novagen). The resultant pET22b(+)-AgamOBP48 plasmid was used to transform Escherichia coli Origami B(DE3) (Novagen) competent cells. Protein expression was performed as described previously (6). After centrifugation of the cell lysate at 15,000 × g for 30 min at 4 °C, the supernatant and pellet were analyzed by 12% SDS-PAGE. Nearly 80% of the expressed AgamOBP48 is aggregated in the form of inclusion bodies.
Purification of Recombinant Protein from Inclusion Bodies
Refolding of AgamOBP48 was performed according to Boix (30), with minor modifications. Following buffer exchange by dialysis to 10 mm Tris-HCl, pH 8.0 (Buffer A), the protein was applied on a HiTrap Q FF column (GE Healthcare) equilibrated with Buffer A and eluted with a 0–460 mm NaCl gradient. Fractions containing the target protein (judged by SDS-PAGE analysis) were loaded onto a Resource-Q column (GE Healthcare) and eluted with a linear gradient of 0–350 mm NaCl. The major peak of this step was then loaded onto a 16/73 Superdex75 gel filtration column (GE Healthcare) equilibrated with Buffer A containing 200 mm NaCl. Finally, the highly purified AgamOBP48 was desalted by dialysis and concentrated to 20 mg/ml in Buffer A.
Crystallization and Data Collection
AgamOBP48 single crystals were grown at 20 °C employing the sitting drop vapor diffusion technique. AgamOBP48 orthorhombic crystals belonging to space group P21212 were grown using a protein solution consisting of 7 mg/ml AgamOBP48/AgamOBP1 (1:1) with a reservoir solution of 0.1 m Tris-HCl, pH 8.5, and 25% (w/v) PEG3000.
Prior to data collection, crystals were transferred to an artificial mother liquor containing 10% (v/v) glycerol and flash-frozen in a nitrogen gas stream at 100 K. A total of 200° of data were collected.
Structure Determination
Data were integrated and scaled with the programs XDS (31) and SCALA of the CCP4 suite (32), and intensities were converted to amplitudes using TRUNCATE (33). Data analysis with phenix.xtriage from the Phenix suite (34) suggested strong anisotropy. Therefore, the scaled data were imported into the UCLA diffraction anisotropy server (35) for elipsoidal truncation and anisotropic scaling. Initial phases were obtained with PHASER (36) using the AgamOBP47 structure (Protein Data Bank code 3PM2) (16) as molecular replacement model. The molecular replacement solution yielded a single noncrystallographic symmetry dimer in the asymmetric unit corresponding to a solvent content of 37% (VM = 1.95 Å3 Da−1).
Maximum likelihood refinement of the molecular replacement solution (composed of positional minimization, B-factor optimization, and simulated annealing) was performed using the program phenix.refine (37), to give an initial model with R-factor of 0.343 (Rfree = 0.412). Alternate cycles of manual building with the program COOT (38) and phenix.refine employing positional and individual atomic displacement (B-factor) parameters resulted in a final model with R-factor of 0.198 (Rfree = 0.245). MolProbity (39) was used to assess the quality of the final structure. Details of data processing and refinement statistics are summarized in Table 1.
TABLE 1.
Statistics of data collection, processing and refinement of the AgamOBP48-PEG complex
Values in parentheses are for the outermost shell.
| Protein Data Bank code 4IJ7 | |
|---|---|
| SRS; λ | EMBL-DESY, X12; 1.07 Å |
| Space group | P21212 |
| Cell dimensions, a, b, c | 80.65, 89.11, 43.40 Å |
| α = β = γ = 90° | |
| Resolution | 38.22 to 2.25 Å |
| Outermost shell | 2.37 to 2.25 Å |
| Reflections measured | 123,630 (17,874) |
| Unique reflections (σ>0) | 15,437 (2,206) |
| Rsymma | 0.088 (0.53) |
| Completeness | 100% (100%) |
| 〈I/σI〉 | 18.6 (4.4) |
| Redundancy | 8.0 (8.1) |
| Wilson plot B-value | 24.8 Å2 |
| Final Rcrystb (Rfreec) | 19.8% (24.6%) |
| Error in coordinates by Luzzati plot | 0.26 Å |
| No. of water molecules in final cycle | 133 |
| Root mean square deviation from ideality | |
| Bond lengths | 0.011 Å |
| Bond angles | 1.15° |
| Average B-factor | |
| Protein atoms (chain A; chain B) | 32.0; 32.6 Å2 |
| Water molecules | 29.2 Å2 |
| PEG | 30.8 Å2 |
| Ramachandran favored/outliers | 98.2/0% |
a Rsymm = ΣhΣi|I(h) − Ii(h)/ΣhΣiIi(h), where Ii(h) and I(h) are the ith and the mean measurements of the intensity of reflection h.
b Rcryst = Σh|Fo − Fc|/ΣhFo, where Fo and Fc are the observed and calculated structure factors amplitudes of reflection h, respectively.
c Rfree is equal to Rcryst for a randomly selected 5% subset of reflections not used in the refinement.
Structural superimpositions were performed using DALI server (40). Possible hydrogen bonds were identified using the program HBPLUS (41). van der Waals contacts were determined by CONTACTS of the CCP4 suite for nonhydrogen atoms separated by less than 4 Å. Solvent-accessible areas were calculated with NACCESS (42). Protein cavities were visualized and analyzed by CAVER 2.0 (43). The dimerization interface of AgamOBP48 was analyzed by using the PISA (Protein Interfaces, Surfaces, and Assemblies) server (44). Protein domain motions were analyzed by using the DynDom server (45). All figures were created with PyMOL (46).
Gel Filtration Experiments
The GF column was calibrated for molecular weight estimations with bovine serum albumin (dimer, 132.8, and monomer, 66.4 kDa), carbonic anhydrase (29 kDa), and lysozyme (14.3 kDa) as molecular weight standards (Sigma). The plot of log(Mr) versus partition coefficients of the standards (Kav = Ve − Vo/Vt − Vo) yields a linear calibration curve log(Mr) = 5.2706 −2.5865 Kav, where Mr is the molecular weight in thousands; Ve is the elution volume of the standards; Vo (56.03 ml) is the void volume (elution volume for blue dextran 2000); and Vt (146.75 ml) is the total bed volume. The apparent molecular weights of AgamOBP48 monomer and dimer were estimated from their elution profiles, after storage of the concentrated ion exchange AgamOBP48 sample for 0, 48, and 96 h at 4 °C before GF chromatography.
Calorimetric Experiments
Differential scanning calorimetry (DSC) measurements were carried out on solutions of isolated AgamOBP48 monomer (“0-h” sample; see under “Gel Filtration Experiments”) and within 48 h after the gel filtration step. Protein concentration was 5 mg/ml in PBS buffer, pH 7. DSC experiments performed with a nano-DSC (CSC, TA Instruments Distributor) apparatus at 0.5 °C/min scan rate in the 10–110 °C range. This instrument uses capillary cells with 0.3 ml sensitive volume. The capillary cell design often prevents and/or mitigates aggregation phenomena. Data were analyzed by means of the software THESEUS (47). The excess molar heat capacity 〈ΔCp〉 or Cpex(T), i.e. the difference between the apparent molar heat capacity Cp(T) of the sample and the molar heat capacity of the “native state,” Cp,N(T), was recorded across the scanned temperature range. Details on the raw data treatment methods, (base-line scaling, etc.), are reported elsewhere (48, 49). The heat capacity drop, ΔdCp, across the signal was affected by a rather large error and was therefore not taken into account in this work. The value of the apparent denaturation enthalpy ΔdH was 230 ± 10 kJ mol−1 (monomer, mass of 19,200 Da).
Several thermodynamic models (see text) were tested to interpret the calorimetric data (the fitting functions of these models are described in the literature (48–51)). The theoretical models used to fit the experimental data were tested through the nonlinear Levenberg-Marquardt method (52). Best fitting parameters error was calculated in the 95% confidence limit.
Analytical Ultracentrifugation Experiments
All experiments were carried out using an XL-I analytical ultracentrifuge (Beckman-Coulter Inc.), with a UV-visible optics detection system, using an An60Ti rotor and 12-mm double sector centerpieces. Sedimentation equilibrium analyses were carried out at 20 °C, in PBS buffer, pH 7, at 15,000 rpm. Total protein concentration was 0.5 mg/ml. The “96-h” GF concentrated sample (see “Gel Filtration Experiments”) was stored for 2 months at 4 °C before dilution for the analytical ultracentrifugation experiments. The results of 18-h runs were analyzed using the SEDPHAT program (53). The density of the system: 1,005 g/ml and the Vbar 0.728 were calculated using the SEDNTERP program.
Fluorescent Assays
The affinity of AgamOBP48 for the EN12 was evaluated indirectly by determining the displacement of the fluorescent probe N-phenyl-1-naphthylamine (1-NPN) by the ligand, as described previously by Campanacci et al. (54) with minor modifications.
The emission spectra were recorded on a VarioScan Flash fluorimeter plate reader (Thermo Scientific) at 30 °C using black 96-well plates (Greiner, Bio-One). The probe was excited at 337 nm, and the emission spectra were recorded from 390 to 460 nm. The maximal fluorescence emission intensity in the presence of AgamOBP48 was observed between 408 and 410 nm, after subtraction of the background spectra (only ligand and/or 1-NPN in the buffer). Purified recombinant AgamOBP48 was assayed at a final volume of 200 μl. The buffer used for all measurements was PBS, pH 7.0. Fresh stock solutions of 1-NPN and EN12 were prepared in 100% DMSO. The final DMSO concentration of 2% was maintained in all dilutions.
The Kd1-NPN of the probe was determined in protein solutions containing 6 μm AgamOBP48 by plotting the fluorescence intensity as a function of 1-NPN concentration (0–100 μm). To determine the dissociation constant of EN12 (KdEN12), AgamOBP48 was assayed at a final concentration of 6 μm in the presence of varying concentrations of EN12 (0–50 μm) and a constant concentration of 1-NPN (60 μm).
Three independent experiments were run for each probe or ligand concentration. Data were analyzed by the use of the nonlinear regression program GraFit (55).
Molecular Docking
EN12 was studied for its binding to AgamOBP48 using the molecular docking program AutoDock 4.0 (56). The AutoDock empirical free energy model is able to predict binding free energies to within 2.5 kcal/mol. The Lamarckian genetic algorithm search method with 5,000,000 maximum number of energy evaluations was used, and the ligand was treated as flexible. Docking results were both visually inspected and quantitatively evaluated based on the estimated free energy of binding (FEB).
RESULTS
AgamOBP48 Crystallizes as a Three-dimensional Domain-swapped Dimer
The crystal structure of AgamOBP48 from A. gambiae, in complex with PEG, was determined at 2.25 Å resolution (Table 1). The three-dimensional structure revealed a compact three-dimensional domain-swapped dimer formed by exchange of the closely associated N- and C-terminal regions (Fig. 1, A and B and supplemental Video).
FIGURE 1.

Structure of the AgamOBP48. A and B, schematic representation of the three-dimensional domain-swapped dimer. Two views are shown, one from the face of the dimer formed by the swapped N- and C-terminal domain (A) and one from the face formed by the α5 and α5′ helices (B). Within each monomer, residues from the helices α1 to α5, α7, and the N-terminal stretch form the walls of a central cleft that is occupied by the C-terminal helix α8 of the adjacent subunit. A long tunnel with mouths on each side runs through the dimer interface. The center of the tunnel between α7 and α7′ helices is occupied by a PEG molecule. Chain A is colored red, and chain B is colored, green. Disulfide bonds are in yellow. The helices and residues in chain B are indicated with a prime (′). C, schematic representation of the AgamOBP48 monomer. Each monomeric subunit consists of three domains; the Core (indicated by red color) and the Cap and the NC-term domain (both presented by cyan color). D, comparison of AgamOBP48 with the Plus-C AgamOBP47. The AgamOBP48 adopts an open conformation as compared with the closed conformation of AgamOBP47 (orange). In AgamOBP47 the final C-terminal residue, Leu-173, is located in the center of the monomer and interacts with His-34. In contrast, the corresponding residue of AgamOBP48, Ile-172, interacts with Lys-21′ from the neighboring subunit (A). The root mean square deviation of Cα atoms between the overall structures of AgamOBP48 and AgamOBP47 is 2.35 Å, and between the two proteins excluding their NC-term domains is 0.85 Å.
Each 172-residue-long monomer of AgamOBP48 contains eight α-helices (α1 to α8) linked by flexible loops and stabilized by six disulfide bridges (Fig. 1C). The structure of the monomeric subunit can be divided into 3 main domains as follows: (i) the central “Core” domain that resembles the classic OBPs fold (helices α1–5, α7, and the 50-s loop); (ii) a flanking domain, called from now on as the “NC-term” domain (N-terminal residues 1–25 and C-terminal residues 148–172), and (iii) the “Cap” domain (helix α6 and the 120-s loop) (Fig. 1C). The two subunits orient themselves so that the NC-term domain of one monomer inserts into the center of the neighboring monomer, to form a compact homodimer (Fig. 1, A and B).
Inspection of the electron density maps showed a strong Fo − Fc positive peak accompanied by a 2Fo − Fc electron density, which can neatly accommodate one PEG molecule. The presence of PEG was subsequently confirmed by a simulated annealing omit map (Fig. 2). To further support this finding, the crystal structure of AgamOBP48 dimer was determined to a resolution of 3.3 Å using crystals formed in the absence of PEG (Protein Data Bank code 4KYN; data not shown). In this case, the electron density maps showed no electron density in this region, which gives additional confidence that the density we observed in the 2.2 Å crystal structure is derived from a PEG molecule.
FIGURE 2.
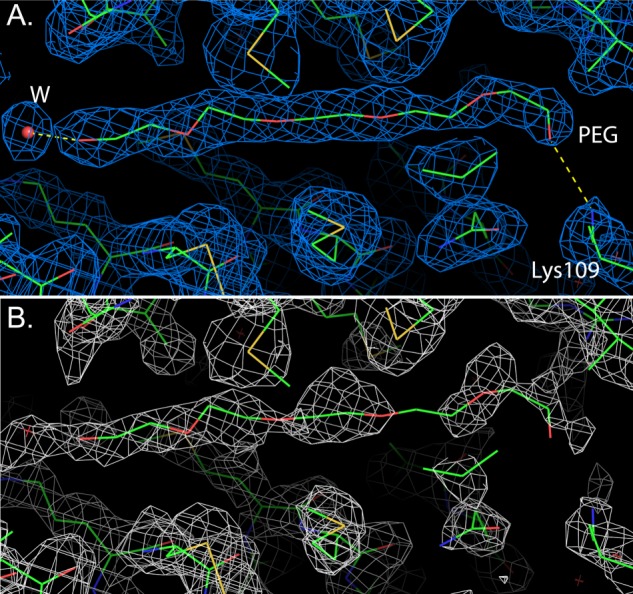
Electron density maps in the vicinity of the PEG-binding site. A, 2Fo − Fc electron density map of the final refined model. The polar contacts of PEG are depicted as yellow dashed lines. The water molecule that interacts with PEG is shown as a red sphere. PEG makes 15 van der Waals interactions mainly with nonpolar atoms of the binding side residues, as well as two putative hydrogen bonds with NZ of Lys-109 (OPEG-NZ; 2.84 Å) and a water molecule (OPEG–OW; 2.46 Å) (B). 2mFo − DFc simulated annealing omit electron density map obtained by omitting the PEG molecule and the surrounding protein residues. The contour levels corresponds to 1σ.
The PEG molecule is located at the interface between the α7 and α7′ helices, in the center of a double L-shaped, 51-Å long tunnel (Channel-1) with mouths on either side of the protein (Figs. 1, A and B, and 3A). The openings of this tunnel are created by the residues Ser-151, Glu-90, and Ser-93 of one chain and Ile-50′ and Glu-112′ of the other chain, whereas its walls are composed by residues from both chains.
FIGURE 3.
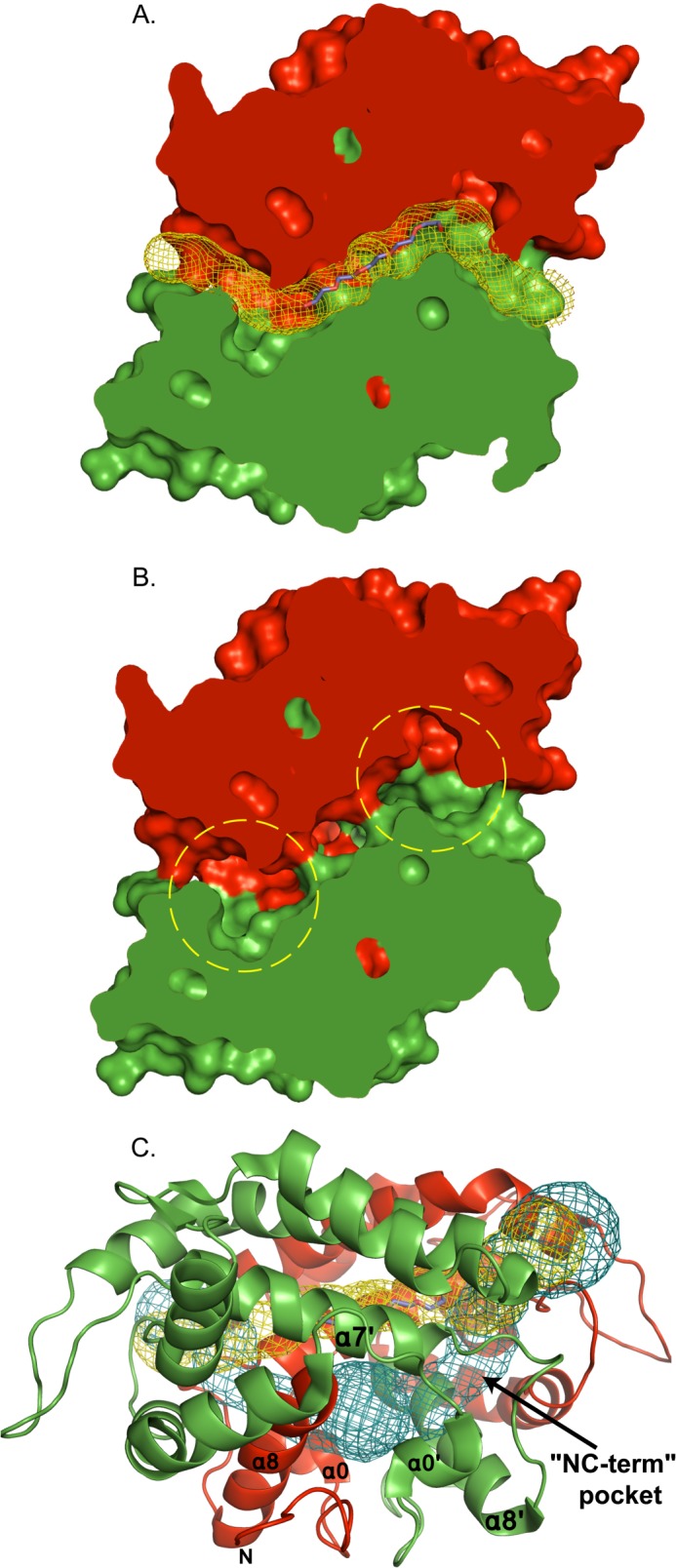
Channels of the AgamOBP48 three-dimensional swapped dimer. A and B, surface representation of the AgamOBP48 swapped dimer (slice). A, Channel-1 is represented by yellow mesh. This double L-shaped channel is 51 Å long with an average width of 2.5 Å. The PEG-binding site is located at the center of the tunnel. B, two deep pockets adjacent to the PEG-binding site are formed by residues of the NC-term domain (indicated by yellow dashed circles). C, Channel-2 is represented by blue mesh. Channel-2 is 56 Å long and shares the same mouth openings as Channel-1. The NC-term pockets are located in the regions where Channel-2 begins to deviate from Channel-1.
It is noteworthy that the shape and length of Channel-1 resembles the 56-Å long tunnel of the AgamOBP1 dimer, which was found to be occupied simultaneously by PEG and DEET (N,N-diethyl-m-toluamide) molecules (6). However, in the AgamOBP1 dimer, the full-length double L-shaped tunnel is generated by two individual 28-Å long L-shaped channels, one from each monomer, lying adjacently to each other. In contrast, the AgamOBP48 channel is formed at the dimer interface by the equal contribution of each monomer and therefore cannot exist in the monomeric form.
Two deep pockets lie underneath both ends of the PEG molecule, were also identified in the AgamOBP48 dimer (Fig. 3B). Each pocket is formed by residues of the NC-term domain of the corresponding subunit and is located between the α0 helix and the 150-s loop (Fig. 1C). The two pockets (NC-term pockets) communicate with each other and are joined into a central cavity positioned behind the PEG-binding site between helices α7, α7′, α8, α8′, and the two N-terminal stretches. This cavity is accessible to the bulk solvent through a second 56-Å long tunnel (Channel-2), which shares the same openings as Channel-1 (Fig. 3C). Similar to Channel-1, residues from both subunits participate in the formation of Channel-2. The residues that define Channel-1 and Channel-2 and the amino acids that make contact with the PEG molecule are listed in Table 2.
TABLE 2.
AgamOBP48 residues lining the central cleft and the two channels
EN12 is predicted to bind into the central cleft of the AgamOBP48 monomer. The combined PEG-binding site is part of Channel 1 and is found only in the dimer. A single copy of the NC-term binding pocket is present in the monomer. In the dimer, the NC-term pocket is duplicated and forms part of Channel 2. The residues that interact with the PEG molecule (P) in the crystal structure, and those that interact with the compound EN12 (L) in the modeled complexes are represented in bold.
| Central cleft, chain A | Channel 1 (PEG site),a chain A/B | Channel 2 (NC-term pockets), chain A/B |
|---|---|---|
| Lys-21 (L) | Gly-49 | Pro-15 (L) |
| Pro-22 (L) | Ile-50 | Cys-18 (L) |
| Met-23 (L) | Pro-51 | Cys-19 (L) |
| Leu-24 (L) | Pro-89 | Leu-45 (L)a |
| Val-25 (L) | Glu-90 | Ile-50 |
| Gly-27 (L) | Ser-93 | Pro-51 |
| Met-30 (L) | Leu-94 | Glu-90 |
| Met-31 (L) | Leu-105 (P/L) | Ser-93 |
| Leu-137 (L) | Lys-109 (P) | Leu-94 |
| Met-140 (L) | Glu-112 | Lys-109 |
| Gly-141 (L) | Ile-113 | Glu-112 |
| Met-144 (L) | Pro-131 | Met-142 |
| Gly-134 (L) | Phe-145 (L) | |
| Thr-135 (P/L) | Ala-146 (L) | |
| Arg-138 (P/L) | Gln-147 | |
| Cys-139 (P/L) | Cys-148 (L) | |
| Met-142 (P/L) | Pro-149 | |
| Met-143 (L) | Ala-150 (L) | |
| Ala-146 (P/L) | Ser-151 | |
| Gln-147 (L) | Phe-153 (L) | |
| Pro-149 | Leu-162 (L) | |
| Ala-150 | Arg-163 (L) | |
| Ser-151 | Gly-166 | |
| Ser-167 (L) |
a In the dimer only.
Dimer Interface Analysis
The interface present in the AgamOBP48 crystal dimer is hallmarked by hydrophobic interactions. Seven putative hydrogen bonds and six salt bridges between interface residues also contribute to the stability of the homodimer, including the interaction between the carbonyl oxygen of the last C-terminal amino acid Ile-172 and the amine nitrogen of Lys-21 from the other subunit (Fig. 4).
FIGURE 4.

Polar interactions at the dimer interface. The interface is stabilized by 13 polar interactions (depicted as yellow dashed lines). Disulfide bonds are in orange. Chain A is colored red, and chain B is in green. The helices and residues in chain B are indicated (′).
The PISA analysis suggested that the crystallographic dimer represents a very stable assembly in solution (ΔGdiss = 38.7 kcal/mol). Upon dimer formation a total surface area of 4,670 Å2 becomes buried, and the greatest contribution comes from the nonpolar atoms, which contribute 69.4% of the buried surface. Indeed, PISA reported a p value of the observed solvation free energy gain (ΔiG) on dimer formation equal to 0.103 (< 0.5), which indicates an interface with higher than average hydrophobicity.
Structural Analysis of AgamOBP48 Monomer
The individual domains of AgamOBP48 monomer are illustrated in Fig. 1C. Structurally, the Core domain shares a common architecture with classic OBPs and consists of four α-helices, α1 (aa 26–38), α3 (aa 54–64), α5 (88–108), and α7 (131–147) bearing six conserved cysteines. These Cys residues form three disulfide bridges linking helix α1 to α3 (Cys-33–Cys-59), α3 to the start of helix α7 (Cys-55–Cys-129), and the middle of α7 to α5 (Cys-139–Cys-102). A seventh cysteine (Cys-54) is located at the top of α3 helix and does not participate in a disulfide bond formation. Additionally, this domain also contains two nonbridged α-helices, α2 (aa 39–45) and α4 (aa 73–85), and a loop of 8 amino acids (50-s loop).
The Cap domain is composed of helix α6 (aa 109–118) and the 120-s loop (aa 119–130). The Cap and “α2–50-s loop” modules are oriented almost antiparallel to each other, and they interact through multiple hydrogen bonding that apparently serves to stabilize the local protein structure.
The most distinct protein region is the NC-term domain that is formed by residues of the long N-terminal stretch (aa 1–25), the C-terminal 150-s loop (aa 148–155), and the following C-terminal α8 helix (aa 156–172). The N-terminal component of this domain is characterized by an α-helical segment (α0; aa 14–19) that contains two additional cysteines located in adjacent positions. Cys-19 forms a disulfide bridge with Cys-148 that is positioned at the 150-s loop region, whereas Cys-18 makes a second disulfide bond with Cys-159 at the top of α8. The cysteine bridge Cys-19–Cys-148 can be considered the border between the Core and NC-term domains. In addition, the N- and C-terminal ends belonging to this domain are linked through a third disulfide bridge formed between Cys-5 and Cys-169 at the end of α8 helix.
Comparison with AgamOBP47 Revealed Putative Hinge Regions
The AgamOBP48 amino acid sequence exhibits 56% identity, 75% similarity, and one gap insertion (between aa 87 and 88) with the Plus-C AgamOBP47.
Overall, the tertiary structure of AgamOBP48 resembles the previously reported AgamOBP47 protein structure that has been crystallized as a monomer (16). Superimposition of AgamOBP48 and AgamOBP47 structures revealed that the major conformational difference between the two proteins occurs in the region of their NC-term domains, which form an angle of 27° (Fig. 1D). Specifically, the NC-term domain of AgamOBP48 structure is oriented away from the Core domain resulting in a monomer existing in an open conformation. In AgamOBP47, the corresponding region is in proximity to the Core domain, and its C-terminal extension enters the center of the monomer, where it participates in the formation of the binding site. Because three-dimensional domain swapping is often accompanied by the presence of hinge elements, the AgamOBP48 and AgamOBP47 structures were subjected to protein domain motion analysis using DynDom server (45) for the identification of putative hinge regions. In this analysis, AgamOBP48 and AgamOBP47 represented the open and closed conformers, respectively. The analysis proposed the existence of two hinge regions, the α0 helix and the 150-s loop, that could facilitate large conformational transition between the open and closed states. Amino acids Glu-17–Cys-18 of α0 helix and Val-152–Phe-153 of 150-s loop are potentially acting as hinge-bending residues. During the transition, the φ-dihedral angles of Glu-17 and Cys-18 change by 40.1 and −35.5°, respectively, whereas the corresponding values for Val-152 and Phe-153 are 21.0 and −55.3°, indicating that these residues contribute most to the intersubunit rotation. Therefore, a shift from a closed to an open conformation, controlled by the hinge regions, could be proposed as a possible mechanism required for the formation of a stable homodimer through three-dimensional domain swapping.
AgamOBP48 Is Self-assembled into Dimers in Solution
PISA analysis (see above) suggested that the AgamOBP48 dimer present in the crystal could also be stable in solution. This result prompted us to determine the oligomeric state of AgamOBP48 in solution by performing GF experiments on a calibrated Superdex75 column. Concentrated protein samples that applied to the GF column immediately after the ion exchange chromatography step were eluted as a single peak with apparent molecular mass of 24.4 kDa. However, a higher oligomeric form appeared as a left shoulder on the main peak when the protein sample was kept for 48 h at 4 °C before GF, although when the storage period was increased to 96 h at 4 °C, two distinct protein species with apparent molecular masses of 26.7 and 40.7 kDa were isolated (Fig. 5). Given that the estimated molecular weight of the AgamOBP48 monomer is 19.2 kDa, based on its amino acid composition, our analysis revealed that AgamOBP48 undergoes a time-dependent dimerization in solution. Moreover, the GF profiles showed that the dimer is the predominant form in solution. The higher apparent molecular weight of the eluted monomer indicates deviation from globular shape, which is in agreement with its elongated shape observed in the AgamOBP48 crystal structure. Additionally, the possible hinge movement of the NC-term domain in solution may lead to a significant increase of the hydrodynamic radius of the monomer unit.
FIGURE 5.
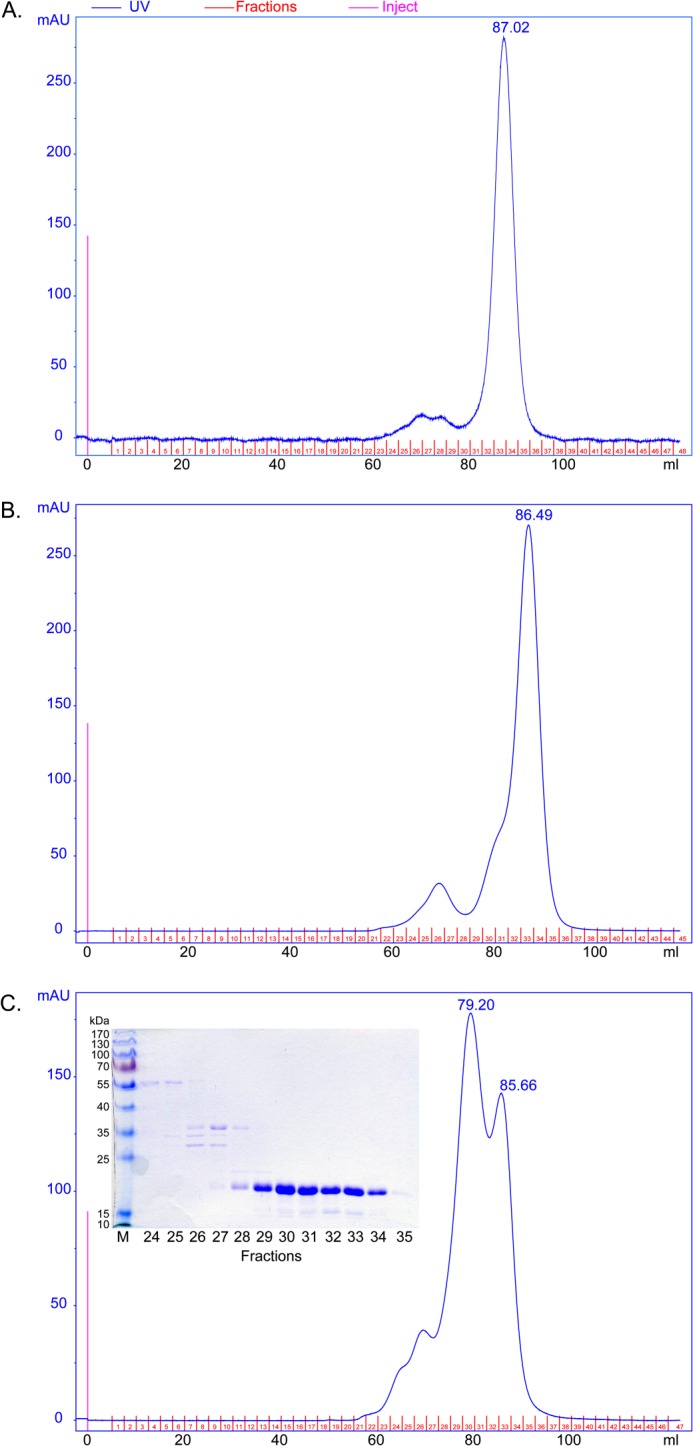
Gel filtration experiments of AgamOBP48. The concentrated pooled elution fraction from ion exchange chromatography was applied immediately (A), after 48 h (B), and after 96 h (C) of storage at 4 °C, to a calibrated Superdex 75 16/73 column. A, AgamOBP48 was eluted as a single peak corresponding to a monomer with apparent mass of 24.4 kDa. The monomer self-associates to form dimers as indicated by the appearance of a left shoulder (B) and a predominant peak (C), corresponding to a dimer with apparent mass of 40.7 kDa. Inset, SDS-PAGE analysis (12% stained with Coomassie) of the eluted fractions showed that both peaks contain AgamOBP48 protein with apparent mass of about 20 kDa. mAU, milliabsorbance units.
To further analyze the ability of AgamOBP48 to dimerize, the thermal stability of the isolated monomer (Fig. 5A), in 5 mg/ml solution, was investigated by means of DSC within 48 h after the gel filtration step. We observe a biphasic thermogram that covers an exceptionally large temperature range that indicates the presence of more than one species of the protein (Fig. 6A). Indeed, if we assume the presence of only a monomer in solution, the classical thermodynamic models, i.e. the assumption of the presence of two or more energetic domains in the monomer (taking also into account the case of the concomitant exothermic aggregation processes during denaturation), were tested and were not able to interpret these calorimetric data. However, the simplest hypothesis of the presence in solution of two species with different stability, i.e. a monomer and a dimer at a fixed ratio (as a first approximation and in line with the GF analysis suggestion), each of which undergoes in a simple one-step denaturation, was sufficient to produce an acceptable fit as a starting point (see Fig. 6A). The best fitting parameters corresponding to the theoretical curve calculated according to a single step denaturation model of a fixed ratio monomer/dimer system are as follows: monomer, composition 80%; denaturation temperature Td = 327.6 ± 0.1 K, ΔdH = 210 ± 10 kJ mol−1 (monomer); dimer, composition 20%, denaturation temperature Td = 340.6 ± 0.1 K, ΔdH = 330 ± 10 kJ mol−1 (monomer).
FIGURE 6.
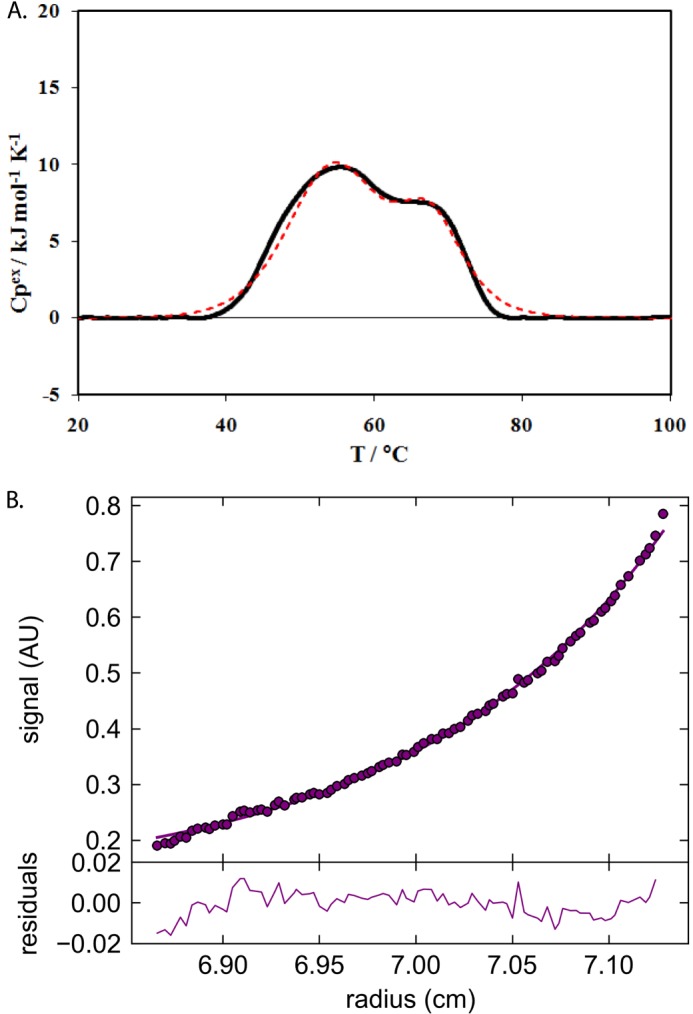
DSC and analytical ultracentrifugation experiments of AgamOBP48. A, differential scanning calorimetry on AgamOBP48 solution. Experimental (continuous line) and theoretical (dashed line) DSC thermal denaturation curves of AgamOBP48 (5 mg/ml in PBS buffer, pH 7, 0.5 °C/min scan rate). The theoretical curve was calculated according to a single step denaturation model of a fixed ratio monomer/dimer system (see text). B, analytical ultracentrifugation (AU) equilibrium experiment on AgamOBP48. The data were obtained at 280 nm and analyzed using SEDPHAT (circles represent the experimental data and lines fit the equivalent of 37,619 Da).
Τhe fixed monomer/dimer ratio assumed represents an average approximation in the temperature range investigated, and more refined models that include dissociation may produce a better fit. However, as observed with the GF, the initial monomer/dimer ratio seems time-dependent, and more refined models at this stage will be pure speculation without a better characterization of the kinetic features that are involved in the oligomer formation. Indeed, a further thermodynamic analysis is needed, i.e. the exploitation of the monomer-oligomer equilibrium and/or kinetics features in function of the concentration, temperature, presence of ligands, etc., that is beyond the purpose of this paper. Suffice it here to say that, independently of the denaturation mechanism peculiarities, these calorimetric data strongly support the evidence of the GF experiments, i.e. the presence of oligomers in solution.
Finally, to accurately determine the molecular weight of the oligomer observed in GF and DSC studies, we carried out sedimentation equilibrium experiments at 20 °C using analytical ultracentrifugation at 0.5 mg/ml AgamOBP48. The “96-h” GF-concentrated sample (Fig. 5C; fractions 29–34), stored for 2 months at 4 °C, was used for the preparation of the protein dilutions. A typical experiment is presented in Fig. 6B. The analysis showed that the molecule eventually forms dimers with an Mr of ∼37,619, in full agreement with the GF and the crystallographic results.
EN12 Fluorescence and in Silico Binding Studies
We have recently developed a ligand- and OBP structure-based screening protocol in an effort to identify multiple new and effective disruptors of the mosquito host seeking behavior.3 Several synthetic compounds targeting OBPs have been discovered. One of these compounds, (E)-3-[4-(dimethylamino)phenyl]-2-(2,2-dimethylpropanoyl)-2-propenenitrile (EN12) (Fig. 7, inset), was found to be the most potent binder of AgamOBP48 known to date. Fig. 7 shows the effect of EN12 on the fluorescence intensity of the AgamOBP48·1-NPN complex. Analysis of the data, as described in the figure legend, gave a KdEN12 value of 2.6 μm. The corresponding Kd values for the 1-NPN was 52.3 μm (data not shown).
FIGURE 7.

Fluorescence data analysis. Single site saturation curves were obtained by titration of AgamOBP48 (6 μm) with EN12. Replot of the fraction of 1-NPN displaced versus various concentrations of EN12. The fraction of the displaced fluorescent probe Y = 1 − ((FL − Fmin)/(Fmax − Fmin)) was plotted as a function of the EN12 concentration, where Fmax is the fluorescence intensity of the AgamOBP48·1-NPN complex in the absence of ligand; Fmin is the fluorescence intensity at saturating concentration of the ligand, and FL is the fluorescence intensity at any given EN12 concentration. Data were fit to the single binding site equation Y = Ymax·[L]/(Kd, app + [L]), where Ymax equals the fraction displaced at infinite ligand concentration, [L] is the concentration of ligand, and Kd, app is the concentration of ligand needed to displace half of the bound 1-NPN, which is assumed as the apparent dissociation constant of the respective ligand. EN12 binds to AgamOBP48 with a Kd, app (Kd apparent) value of 5.55 ± 0.49 μm. The dissociation constant, KdEN12 (2.58 ± 0.23 μm), was calculated using the equation KdEN12 = Kd, app/(1 + [1-NPN]/Kd1-NPN), where [1-NPN] is the probe concentration (60 μm), and Kd1-NPN is the dissociation constant of AgamOBP48·1-NPN complex (52.34 ± 4.80 μΜ; data not shown). Inset, chemical structure of EN12. Plot shows reduction of fluorescence intensity as a function of the EN12.
The resulting binding data fit the one binding site model, indicating the presence of a single binding site. However, given the symmetry of AgamOBP48 dimer, ligand binding to 2 equivalent binding sites, such as the NC-term pockets, cannot be excluded.
In the absence of direct crystallographic evidence for the binding mode of EN12 to AgamOBP48, we decided to perform in silico docking studies. EN12 is predicted to bind, with comparable affinities, either to the center of the PEG-binding site (Pose 1a, FEB = −7.83 kcal/mol, and Pose 1b, FEB = −7.67 kcal/mol) or to the two NC-term pockets of the dimer (FEB = −7.35 kcal/mol) (Fig. 8A). Poses 1a and 1b overlap with the five central ethylene glycol units of PEG molecule and make 69 van der Waals and 2 polar interactions via the carbonyl oxygen (Pose 1a) or the nitrile nitrogen (Pose 1b).
FIGURE 8.

EN12 docking to the AgamOBP48. A, EN12 is docked to the AgamOBP48 dimer. EN12 makes two polar contacts through its carbonyl oxygen (pose-1a in yellow; Kd, calc = 1.8 μm) or its nitrile nitrogen (pose-1b in orange; Kd, calc = 2.4 μm). The top-ranked EN12 pose docked to the NC-term pocket is shown in purple (Kd, calc = 4.1 μm). B, binding site predictions of AgamOBP48 monomer. Predicted EN12 poses in the vicinities of the NC-term pocket (Kd, calc = 9.3 μm) and the central cleft (Kd, calc = 15.2 μm) of AgamOBP48 monomer. C, each swapped α8 helix occupies the central cleft of its neighboring monomer reducing the volume available to ligands. The central cleft of AgamOBP48 dimer is predicted to accommodate less bulky molecules such as 1-NPN. The theoretical Kd values (Kd, calc) were calculated from the corresponding FEB values using the equation Kd, calc = exp(ΔG/(R·T)).
In each of the NC-term pockets, EN12 makes contacts mainly with residues of the one subunit and two additional van der Waals interactions with the backbone carbonyl oxygen and the side chain of Leu-45′ from the other subunit. The residues predicted by docking analysis, to interact with EN12, are listed in Table 2. Considering that AgamOBP48 may exist also as a monomer in solution (GF and DSC experiments), we subsequently carried out modeling studies on the monomeric form of the protein.
EN12 is predicted to bind to the NC-term pocket of the AgamOBP48 monomer (Fig. 8B) with an affinity similar to the one for the dimer (FEB = −6.86 kcal/mol). It has to be noted that the NC-term pocket is absent from the AgamOBP47 monomer. Therefore, this site constitutes a novel binding site of AgamOBP48.
In the AgamOBP47 monomer, two surface-binding cavities connected by a narrow channel have been predicted to accommodate the aromatic rings of 4-hydroxy-4′-isopropylazobenzene (AZO) molecule. Modeling of the AgamOBP48 monomer showed that EN12 can be docked into the bottom of this central cleft, with a calculated FEB value of −6.57 kcal/mol (Fig. 8B). In this position, EN12 makes contacts with several residues, including Met-30, Leu-137, and Arg-138. Their structurally equivalent amino acids in the AgamOBP47 structure, Phe-30, Leu-138, and Ala-139, have been suggested to interact with the ΑΖΟ compound (16).
However, in the AgamOBP48 dimer, this central cleft is partially occupied by the C-terminal part of helix α8′ of the neighboring subunit, leading to a volume reduction of the corresponding binding site (Fig. 8C). Although small hydrophobic ligands (e.g. 1-NPN) are predicted in silico to bind to this region of the AgamOBP48 dimer, this pocket is barely large enough to accommodate bulkier compounds like EN12, due to space restrictions between the ligand and the α8′ helix residues according to the analysis.
Based on the calculated FEB values, an undoubted conclusion in favor of the PEG-binding site over the NC-term pockets and vice versa is difficult. Nevertheless, our docking calculations are in close agreement with the results from the fluorescence binding assays. EN12 is suggested to bind either to a single site (PEG-binding site) or to 2 equivalent sites (NC-term pockets) of AgamOBP48 dimer.
In contrast, binding to the central cleft of the dimer can be more safely excluded due to the energetic cost of α8 helix displacement, and probably of dimer disruption, which would be required for EN12 binding to this site of the dimer. In support of this, dimer dissociation upon EN12 binding would result in sigmoidal rather than the hyperbolic binding curves that have been observed in this study.
DISCUSSION
The AgamOBP48 and AgamOBP47 proteins are to date the only two insect Plus-C OBPs of known three-dimensional structure. A comparison of their crystal structures revealed that although the two proteins share common secondary structural elements, they exist in two distinct tertiary conformations indicating distinguishable physiological function. The AgamOBP48 monomer adopts an open conformation, which has a profound influence on the tertiary and quaternary structure of the protein and also on the type and number of ligand-binding sites. In contrast to all the other AgamOBPs of known structure, the C-terminal extension of AgamOBP48 does not interact with the interior of the protein but orients toward the bulk solvent. This unique structural feature of AgamOBP48 monomer has two major consequences. First, it results in the creation of a novel binding site (NC-term pocket). Second, and more importantly, the open conformation promotes the formation of a stable three-dimensional domain-swapped homodimer with novel functionality. The dimerization results in the duplication of the NC-term pocket and the creation of an additional combined binding site where a PEG molecule was found to bind.
Three-dimensional domain swapping has been previously observed in the case of a bovine OBP (57) as well as in the honeybee (hymenoptera Apis mellifera) pheromone-binding protein ASP1 (58), but the AgamOBP48 swapped dimer represents the first example for a mosquito (dipteran) OBP. Notably, as in AgamOBP48, bovine OBP dimerization results in the generation of a novel, combined binding site.
Combining the experimental evidence from our crystal and solution studies, we were able to explain the regulatory properties of AgamOBP48 according to the Eisenberg's model for three-dimensional domain-swapped proteins (59). This model includes a transition from a closed to an open conformer, through hinge motions, followed by the interaction of two open monomers that leads to three-dimensional domain-swapped dimer formation.
Our gel filtration experiments show that monomer to dimer conversion occurs slowly in vitro. However, the calculated high free energy of dissociation suggests that, in all likelihood, the rate-determining step of this process is the interconversion between the closed and open states. Although we have observed the shelf assembly of monomers in the absence of ligand, it is tempting to speculate that ligand binding at the hinge region (NC-term pocket) may lock the monomer in its open conformation, thus accelerating dimer formation. However, because we have not yet been able to obtain the AgamOBP48·EN12 crystal complex, this hypothesis remains to be proven or not.
Crystallographic dimers have also been previously reported for AgamOBP1, AaegOBP1, and CquiOBP1. OBP1s were crystallized as dimers and found to exhibit an unusual binding pocket consisting of a long tunnel running through both monomers and occupied by a PEG molecule (12). AgamOBP48 dimer possesses a similarly sized and shaped tunnel, which is also occupied by a PEG molecule. Intriguingly, a comparative binding study of five A. gambiae OBPs against 20 virtual screening hits, in solution, revealed that EN12 has significant higher affinity for both AgamOBP1 and AgamOBP48 than for the other OBPs tested (data not shown). Previous studies using yeast two-hybrid screens, co-immunoprecipitation, and cross-linking techniques have suggested that AgamOBP48 is capable of generating not only homodimers but also heterodimers with AgamOBP1 (28). Moreover, in situ hybridization experiments have shown that the support cells producing AgamOBP48 and AgamOBP1 are located in close proximity to each other and to olfactory receptor neurons that express the odorant receptor AgamOR1 and thus co-exist in the lymph of the same sensilla (60). The accumulative outcome of these studies indicates a degree of common specificity for the two proteins and their cooperative action. The possible interplay of these two proteins needs to be explored in future research, as it will provide a great asset to the OBP-structure-based discovery of new repellents/attractants.
We have recently proposed for the first time OBPs as valuable molecular targets for the structure-based discovery and design of disruptors of normal olfactory and host-seeking mosquito behavior (6). Significantly, we have now developed and successfully validated a ligand- and structure-based discovery protocol that led to identification of novel bioactive leads such as EN12, attesting the applicability of the OBP structure-aided discovery method.3 In the effort to discover and design multiple disruptors of host-seeking behavior, several factors such as cooperation and oligomerization must be taken into account. Therefore, the study on OBPs of A. gambiae and the determination of their three-dimensional structures and binding specificities could help us understand the molecular basis of odorant detection in this and other anthropophilic species and develop safe, effective, and environmentally friendly strategies for mosquito control.
Acknowledgments
We acknowledge the late Dr. Harald Biessmann and Dr. Marika F. Walter (Developmental Biology Center, University of California) for kindly providing the AgamOBP48 gene (GenBankTM accession number AF533512). Work at the Synchrotron Radiation Sources, MAX-lab, Lund, Sweden, and EMBL Hamburg Outstation, Germany, was supported by funding provided by the European Community's Seventh Framework Programme Grant FP7/2007-2013 under BioStruct-X Grant 283570.
This work was supported by European Commission for the FP7-HEALTH-2007-2.3.2.9 Project “ENAROMaTIC” Grant GA-222927, FP7-REGPOT-2008-1 Project “EUROSTRUCT” Grant GA-230146, and FP7-REGPOT-2009-1 Project ARCADE Grant GA-245866.

This article contains a supplemental video.
The atomic coordinates and structure factors (codes 4IJ7 and 4KYN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
E. Eliopoulos, P. M. Guerin, K. Iatrou, and S. E. Zographos, manuscript in preparation.
- OBP
- odorant-binding protein
- AgamOBP48
- odorant-binding protein 48 from A. gambiae
- EN12
- (E)-3-[4-(dimethylamino)phenyl]-2-(2,2-dimethylpropanoyl)-2-propenenitrile
- GF
- gel filtration chromatography
- DSC
- differential scanning calorimetry
- FEB
- free energy of binding
- aa
- amino acid
- 1-NPN
- N-phenyl-1-naphthylamine.
REFERENCES
- 1. Foster W. A., Hancock R. G. (1994) Nectar-related olfactory and visual attractants for mosquitoes. J. Am. Mosq. Control Assoc. 10, 288–296 [PubMed] [Google Scholar]
- 2. Cabrera M., Jaffe K. (2007) An aggregation pheromone modulates lekking behavior in the vector mosquito Aedes aegypti (Diptera: Culicidae). J. Am. Mosq. Control Assoc. 23, 1–10 [DOI] [PubMed] [Google Scholar]
- 3. Bentley M. D., Day J. F. (1989) Chemical ecology and behavioral aspects of mosquito oviposition. Annu. Rev. Entomol. 34, 401–421 [DOI] [PubMed] [Google Scholar]
- 4. Takken W. (1991) The role of olfaction in host-seeking of mosquitoes: A review. Insect Science and Its Application 12, 287–295 [Google Scholar]
- 5. Leal W. S. (2003) in Insect Pheromone Biochemistry and Molecular Biology, The Biosynthesis and Detection of Pheromone and Plant Volatiles (Blomquist G. J., Vogt R. G., eds) pp. 447–476, Elsevier Academic Press, London [Google Scholar]
- 6. Tsitsanou K. E., Thireou T., Drakou C. E., Koussis K., Keramioti M. V., Leonidas D. D., Eliopoulos E., Iatrou K., Zographos S. E. (2012) Anopheles gambiae odorant-binding protein crystal complex with the synthetic repellent DEET: implications for structure-based design of novel mosquito repellents. Cell. Mol. Life Sci. 69, 283–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vogt R. G. (2002) Odorant-binding protein homologues of the malaria mosquito Anopheles gambiae; possible orthologues of the OS-E and OS-F OBPs of Drosophila melanogaster. J. Chem. Ecol. 28, 2371–2376 [DOI] [PubMed] [Google Scholar]
- 8. Xu P. X., Zwiebel L. J., Smith D. P. (2003) Identification of a distinct family of genes encoding atypical odorant-binding proteins in the malaria vector mosquito, Anopheles gambiae. Insect Mol. Biol. 12, 549–560 [DOI] [PubMed] [Google Scholar]
- 9. Zhou J. J., Huang W., Zhang G. A., Pickett J. A., Field L. M. (2004) “Plus-C” odorant-binding protein genes in two Drosophila species and the malaria mosquito Anopheles gambiae. Gene 327, 117–129 [DOI] [PubMed] [Google Scholar]
- 10. Biessmann H., Nguyen Q. K., Le D., Walter M. F. (2005) Microarray-based survey of a subset of putative olfactory genes in the mosquito Anopheles gambiae. Insect Mol. Biol. 14, 575–589 [DOI] [PubMed] [Google Scholar]
- 11. Pelosi P., Zhou J. J., Ban L. P., Calvello M. (2006) Soluble proteins in insect chemical communication. Cell. Mol. Life Sci. 63, 1658–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wogulis M., Morgan T., Ishida Y., Leal W. S., Wilson D. K. (2006) The crystal structure of an odorant-binding protein from Anopheles gambiae: evidence for a common ligand release mechanism. Biochem. Biophys. Res. Commun. 339, 157–164 [DOI] [PubMed] [Google Scholar]
- 13. Davrazou F., Dong E., Murphy E. J., Johnson H. T., Jones D. N. (2011) New insights into the mechanism of odorant detection by the malaria-transmitting mosquito Anopheles gambiae. J. Biol. Chem. 286, 34175–34183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lagarde A., Spinelli S., Tegoni M., He X., Field L., Zhou J. J., Cambillau C. (2011) The crystal structure of odorant-binding protein 7 from Anopheles gambiae exhibits an outstanding adaptability of its binding site. J. Mol. Biol. 414, 401–412 [DOI] [PubMed] [Google Scholar]
- 15. Ziemba B. P., Murphy E. J., Edlin H. T., Jones D. N. (2013) A novel mechanism of ligand binding and release in the odorant-binding protein 20 from the malaria mosquito Anopheles gambiae. Protein Sci. 22, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lagarde A., Spinelli S., Qiao H., Tegoni M., Pelosi P., Cambillau C. (2011) Crystal structure of a novel type of odorant-binding protein from Anopheles gambiae, belonging to the C-plus class. Biochem. J. 437, 423–430 [DOI] [PubMed] [Google Scholar]
- 17. Hekmat-Scafe D. S., Scafe C. R., McKinney A. J., Tanouye M. A. (2002) Genome-wide analysis of the odorant-binding protein gene family in Drosophila melanogaster. Genome Res. 12, 1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou J. J., Vieira F. G., He X. L., Smadja C., Liu R., Rozas J., Field L. M. (2010) Genome annotation and comparative analyses of the odorant-binding proteins and chemosensory proteins in the pea aphid Acyrthosiphon pisum. Insect Mol. Biol. 19, 113–122 [DOI] [PubMed] [Google Scholar]
- 19. Justice R. W., Dimitratos S., Walter M. F., Woods D. F., Biessmann H. (2003) Sexual dimorphic expression of putative antennal carrier protein genes in the malaria vector Anopheles gambiae. Insect Mol. Biol. 12, 581–594 [DOI] [PubMed] [Google Scholar]
- 20. Danty E., Briand L., Michard-Vanhée C., Perez V., Arnold G., Gaudemer O., Huet D., Huet J. C., Ouali C., Masson C., Pernollet J. C. (1999) Cloning and expression of a queen pheromone-binding protein in the honeybee: an olfactory-specific, developmentally regulated protein. J. Neurosci. 19, 7468–7475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campanacci V., Longhi S., Nagnan-Le Meillour P., Cambillau C., Tegoni M. (1999) Recombinant pheromone binding protein 1 from Mamestra brassicae (MbraPBP1). Functional and structural characterization. Eur. J. Biochem. 264, 707–716 [DOI] [PubMed] [Google Scholar]
- 22. Plettner E., Lazar J., Prestwich E. G., Prestwich G. D. (2000) Discrimination of pheromone enantiomers by two pheromone binding proteins from the gypsy moth Lymantria dispar. Biochemistry 39, 8953–8962 [DOI] [PubMed] [Google Scholar]
- 23. Honson N., Johnson M. A., Oliver J. E., Prestwich G. D., Plettner E. (2003) Structure-activity studies with pheromone-binding proteins of the gypsy moth, Lymantria dispar. Chem. Senses 28, 479–489 [DOI] [PubMed] [Google Scholar]
- 24. Ban L., Scaloni A., D'Ambrosio C., Zhang L., Yahn Y., Pelosi P. (2003) Biochemical characterization and bacterial expression of an odorant-binding protein from Locusta migratoria. Cell. Mol. Life Sci. 60, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sandler B. H., Nikonova L., Leal W. S., Clardy J. (2000) Sexual attraction in the silkworm moth: structure of the pheromone-binding-protein-bombykol complex. Chem. Biol. 7, 143–151 [DOI] [PubMed] [Google Scholar]
- 26. Kruse S. W., Zhao R., Smith D. P., Jones D. N. (2003) Structure of a specific alcohol-binding site defined by the odorant-binding protein LUSH from Drosophila melanogaster. Nat. Struct. Biol. 10, 694–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mao Y., Xu X., Xu W., Ishida Y., Leal W. S., Ames J. B., Clardy J. (2010) Crystal and solution structures of an odorant-binding protein from the southern house mosquito complexed with an oviposition pheromone. Proc. Natl. Acad. Sci. U.S.A. 107, 19102–19107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andronopoulou E., Labropoulou V., Douris V., Woods D. F., Biessmann H., Iatrou K. (2006) Specific interactions among odorant-binding proteins of the African malaria vector Anopheles gambiae. Insect Mol. Biol. 15, 797–811 [DOI] [PubMed] [Google Scholar]
- 29. Biessmann H., Walter M. F., Dimitratos S., Woods D. (2002) Isolation of cDNA clones encoding putative odourant binding proteins from the antennae of the malaria-transmitting mosquito, Anopheles gambiae. Insect Mol. Biol. 11, 123–132 [DOI] [PubMed] [Google Scholar]
- 30. Boix E. (2001) Eosinophil cationic protein. Methods Enzymol. 341, 287–305 [DOI] [PubMed] [Google Scholar]
- 31. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 33. French S., Wilson K. (1978) On the treatment of negative intensity observations. Acta Crystallogr. Sect. A 34, 517–525 [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strong M., Sawaya M. R., Wang S., Phillips M., Cascio D., Eisenberg D. (2006) Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 8060–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McDonald I. K., Thornton J. M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793 [DOI] [PubMed] [Google Scholar]
- 42. Hubbard S. J., Thornton J. M. (1993) NACCESS Computer Program, Version 2.1.1–Atomic Solvent Accessible Area Calculations, Department of Biochemistry and Molecular Biology, University College London [Google Scholar]
- 43. Medek P., Benes P., Sochor J. (2007) Computation of tunnels in protein molecules using Delaunay triangulation. J. Wscg, 15, 107- 114 [Google Scholar]
- 44. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 45. Lee R. A., Razaz M., Hayward S. (2003) The DynDom database of protein domain motions. Bioinformatics 19, 1290–1291 [DOI] [PubMed] [Google Scholar]
- 46. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.5.0.4, DeLano Scientific, Palo Alto, CA [Google Scholar]
- 47. Barone G., Delvecchio P., Fessas D., Giancola C., Graziano G. (1992) Theseus–a new software package for the handling and analysis of thermal denaturation data of biological macromolecules. J. Therm. Anal. 38, 2779–2790 [Google Scholar]
- 48. D'auria S., Barone R., Rossi M., Nucci R., Barone G., Fessas D., Bertoli E., Tanfani F. (1997) Effects of temperature and SDS on the structure of β-glycosidase from the thermophilic archaeon Sulfolobus solfataricus. Biochem. J. 323, 833–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fessas D., Iametti S., Schiraldi A., Bonomi F. (2001) Thermal unfolding of monomeric and dimeric β-lactoglobulins. Eur. J. Biochem. 268, 5439–5448 [DOI] [PubMed] [Google Scholar]
- 50. Fessas D., Staiano M., Barbiroli A., Marabotti A., Schiraldi A., Varriale A., Rossi M., D'Auria S. (2007) Molecular adaptation strategies to high temperature and thermal denaturation mechanism of the d-trehalose/d-maltose-binding protein from the hyperthermophilic archaeon Thermococcus litoralis. Proteins 67, 1002–1009 [DOI] [PubMed] [Google Scholar]
- 51. Ausili A., Pennacchio A., Staiano M., Dattelbaum J. D., Fessas D., Schiraldi A., D'Auria S. (2013) Amino acid transport in thermophiles: characterization of an arginine-binding protein from Thermotoga maritima. 3. Conformational dynamics and stability. J. Photochem. Photobiol. B 118, 66–73 [DOI] [PubMed] [Google Scholar]
- 52. Press W. H., Flannery B. P., Teukolsky S. A., Vetterling W. T. (1989) in Numerical Recipes: The Art of Scientific Computing (Press C. U., ed) pp. 521–538, Cambridge, UK [Google Scholar]
- 53. Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 54. Campanacci V., Krieger J., Bette S., Sturgis J. N., Lartigue A., Cambillau C., Breer H., Tegoni M. (2001) Revisiting the specificity of Mamestra brassicae and Antheraea polyphemus pheromone-binding proteins with a fluorescence binding assay. J. Biol. Chem. 276, 20078–20084 [DOI] [PubMed] [Google Scholar]
- 55. Leatherbarrow R. J. (2007) GraFit, Version 6.0, Erithakus Software, Staines, UK [Google Scholar]
- 56. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tegoni M., Ramoni R., Bignetti E., Spinelli S., Cambillau C. (1996) Domain swapping creates a third putative combining site in bovine odorant-binding protein dimer. Nat. Struct. Biol. 3, 863–867 [DOI] [PubMed] [Google Scholar]
- 58. Pesenti M. E., Spinelli S., Bezirard V., Briand L., Pernollet J. C., Campanacci V., Tegoni M., Cambillau C. (2009) Queen bee pheromone binding protein pH-induced domain swapping favors pheromone release. J. Mol. Biol. 390, 981–990 [DOI] [PubMed] [Google Scholar]
- 59. Bennett M. J., Schlunegger M. P., Eisenberg D. (1995) 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 4, 2455–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schultze A., Schymura D., Forstner M., Krieger J. (2012) Expression pattern of a 'Plus-C' class odorant-binding protein in the antenna of the malaria vector Anopheles gambiae. Insect Mol. Biol. 21, 187–195 [DOI] [PubMed] [Google Scholar]