

Background: The catalytically active species in dehaloperoxidase (DHP) contains both a ferryl heme and a tyrosyl radical.
Results: Radicals were shown to form on three tyrosines (Tyr-28, Tyr-34 and Tyr-38) in DHP.
Conclusion: Mutants that lacked tyrosines showed increases in the rates of both substrate oxidation and heme bleaching.
Significance: Tyrosyl radical formation is an evolutionary adaptation to protect the enzyme from irreversibly oxidizing itself.
Keywords: Hemoglobin, Iron, Peroxidase, Radicals, Tyrosine, Compound ES, Compound I, Compound II, Dehaloperoxidase, Ferryl
Abstract
Dehaloperoxidase (DHP) from Amphitrite ornata, having been shown to catalyze the hydrogen peroxide-dependent oxidation of trihalophenols to dihaloquinones, is the first oxygen binding globin that possesses a biologically relevant peroxidase activity. The catalytically competent species in DHP appears to be Compound ES, a reactive intermediate that contains both a ferryl heme and a tyrosyl radical. By simulating the EPR spectra of DHP activated by H2O2, Thompson et al. (Thompson, M. K., Franzen, S., Ghiladi, R. A., Reeder, B. J., and Svistunenko, D. A. (2010) J. Am. Chem. Soc. 132, 17501–17510) proposed that two different radicals, depending on the pH, are formed, one located on either Tyr-34 or Tyr-28 and the other on Tyr-38. To provide additional support for these simulation-based assignments and to deduce the role(s) that tyrosyl radicals play in DHP, stopped-flow UV-visible and rapid-freeze-quench EPR spectroscopic methods were employed to study radical formation in DHP when three tyrosine residues, Tyr-28, Tyr-34, and Tyr-38, were replaced either individually or in combination with phenylalanines. The results indicate that radicals form on all three tyrosines in DHP. Evidence for the formation of DHP Compound I in several tyrosine mutants was obtained. Variants that formed Compound I showed an increase in the catalytic rate for substrate oxidation but also an increase in heme bleaching, suggesting that the tyrosines are necessary for protecting the enzyme from oxidizing itself. This protective role of tyrosines is likely an evolutionary adaptation allowing DHP to avoid self-inflicted damage in the oxidative environment.
Introduction
The diversity of environmental haloaromatic toxins secreted as repellents by marine organisms, such as Notomastus lobatus (Polychaeta) (1–3) and Saccoglossus kowalevskii (Hemichordata) (4, 5), represents a significant challenge to other infaunal organisms that co-inhabit benthic ecosystems. Examples include mono-, di-, and tribromophenols, mono- and dibromovinylphenols, and bromopyrroles (6, 7). To overcome high levels of these volatile brominated secondary metabolites, a number of organisms employ detoxification enzymes. As an example, the terebellid polychaete Amphitrite ornata possesses a coelomic hemoglobin (8–10) called dehaloperoxidase (DHP)3 that has been suggested to function in this capacity (11, 12).
As a globin, DHP has a characteristic fold composed of the canonical 3/3 α-helical structure (11, 13–15) and binds oxygen reversibly when the heme iron is in the ferrous (Fe2+) oxidation state (8–10). Yet DHP is itself distinct from other globins; it has little sequence homology with other hemoglobins, possesses a crystallographically characterized internal substrate binding site for 2,4,6-trihalophenols (16, 17), and has been shown to utilize hydrogen peroxide to oxidize a range of substrates such as mono-, di-, and trisubstituted halophenols with bromine, chlorine, or fluorine as substituents (18, 19). Moreover, the DHP peroxidase cycle can be initiated from both the ferric and ferrous (or oxyferrous) oxidation states (20, 21). Thus, as a bifunctional globin-peroxidase capable of functioning from the reduced form, DHP may represent a new emerging class of peroxidases that shares neither sequence nor structural homology with any of the known peroxidases.
The mechanism for the oxidative dehalogenation of 2,4,6-trihalophenols into 2,6-dihaloquinones for both known isoenzymes of DHP (A and B, Fig. 1) has been investigated (13, 19, 22–37). Peroxidases generally function via the Poulos-Kraut mechanism (38) in which H2O2 reacts with a ferric heme to form Compound I, the iron(IV)-oxo porphyrin π-cation radical species that is formally two electrons oxidized relative to the ferric resting state. DHP also forms Compound I (Fig. 1) (36), but it rapidly converts to an iron(IV)-oxo heme center with an amino acid radical that has been termed Compound ES by analogy with cytochrome c peroxidase (39). The catalytic competency of Compound ES for oxidizing substrates has been demonstrated (22, 32, 36, 37). In the absence of a reducing 2,4,6-trihalophenol co-substrate, the formation of a species termed Compound RH, which has not been found in any other globin, was observed (21, 22, 37).
FIGURE 1.

Top, A. ornata DHP A (Protein Data Bank code 2QFK (13)) and DHP B (Protein Data Bank code 3IXF (14)). The heme group, the proximal and distal histidines (His-89 and His-55), and all tyrosines of the enzymes are shown. The distal His-55 is shown in the closed conformation. Note that Tyr-34 (DHP A) is not present in DHP B. Bottom, schematic representation of the dehaloperoxidase reaction (note: ferryl oxygen atoms are omitted for clarity). DXQ, dihaloquinone; TXP, trihalophenol.
With the help of the Tyrosyl Radical Spectra Simulation Algorithm (TRSSA) (40, 41), the free radical EPR signal reported for DHP A Compound ES was shown to be a pH-dependent superposition of two different free radical EPR signals assigned to different tyrosine residues, Tyr-34 and Tyr-38. We hypothesized at that time the following (40); (i) the dominant radical associated with the peroxidase function of Compound ES is located on Tyr-34, (ii) in the absence of substrate and when His-55 is in the open conformation (i.e. rotated away from the heme center), the Tyr-34 radical combines with a porphyrin radical formed upon ferryl heme autoreduction, which leads to formation of Compound RH, (iii) the Tyr-38 radical was interpreted as a result of the ferryl heme reduction when His-55 is in the closed conformation (i.e. rotated toward the heme), and (iv) Compound RH is a state that protects DHP against undesired peroxidase activity in the absence of substrate. Thus we proposed that the conformation of His-55, either open or closed, had an immediate effect on the evolution of Compound ES either to Compound RH or to a biradical state (40).
As the above hypotheses for the free radical chemistry of Compound ES were drawn from the TRSSA based radical site assignments, the focus of the present report was to obtain data on the DHP radicals using a more traditional mutagenesis approach. Of the five tyrosine residues in DHP A (Tyr16, Tyr-28, Tyr-34, Tyr-38, Tyr-107; Fig. 1), only three (Tyr-28, Tyr-34, and Tyr-38) are reasonably close (<10 Å) to the heme to reduce Compound I (the remaining two, Tyr-16 and Tyr-107, are at distances >15 Å away from the heme). As the DHP B isoenzyme bears an Asn at position 34 (42), only Tyr-28 and Tyr-38 were hypothesized as the likely site(s) for free radical formation (37). Thus, the experimental strategy employed herein focuses on altering radical formation in DHP via site-directed mutagenesis of three tyrosine residues, Tyr-28, Tyr-34, and Tyr-38, singly or in combinations, in both isoforms. We examined the reaction of the ferric state of the mutants with hydrogen peroxide using stopped-flow UV-visible and rapid-freeze-quench EPR spectroscopic methods. The data suggest a unique interplay between catalytic activity, protection from self-oxidation, and formation of Compound I, Compound ES, Compound RH, and tyrosyl radicals. Overall, the results reported herein help to understand what has been termed the dehaloperoxidase paradox (43), namely the phenomenon of a single heme protein being able to function both as an oxygen binding globin and as a peroxidase.
EXPERIMENTAL PROCEDURES
Materials and Methods
Buffer salts and acetonitrile (HPLC grade) were purchased from Fisher. All other reagents, unless otherwise specified, were from Sigma. EPR tubes were purchased from Norell (Landisville, NJ). Solutions of 2,4,6-trichlorophenol (TCP) were freshly prepared in 100 mm potassium phosphate (KPi) buffers and kept at 4 °C while protected from light. UV-visible spectra were recorded periodically to ensure that the substrate had not degraded. Hydrogen peroxide solutions were also freshly made before each experiment; initially, a 10 mm stock solution of H2O2 was prepared and maintained at 4 °C (typically for less than 15 min), during which time the protein/substrate solutions were loaded into the stopped-flow apparatus. The stock H2O2 solution did not exhibit any measurable degradation over this time as judged by its absorbance at 240 nm (ϵ240 = 43.6 m−1cm−1) (44). The stock H2O2 solution was then diluted to the appropriate premixing concentration and immediately loaded into the stopped-flow apparatus.
Plasmid Preparation, Protein Expression, and Purification
The following wild type (WT) and mutated proteins were expressed and purified as described previously (22, 24, 37) with minor modifications: WT DHP A, DHP A (Y34F), DHP A (Y38F), DHP A (Y34F/Y38F), WT DHP B, DHP B (Y28F), DHP B (Y38F), DHP B (Y28F/Y38F). The mutations were generated with the QuikChange® II site-directed mutagenesis kit (Stratagene, La Jolla, CA). The plasmids encoding WT DHP A or B with an N-terminal poly-His tag (pDHPA or pDHPB) were subjected to PCR amplification using mutagenic primers. Eighteen cycles of melting (95 °C, 50 s), annealing (60 °C, 50 s), and extension (68 °C, 6 min) were performed. The plasmids encoding WT DHP A and WT DHP B (both His6-tagged) were used as templates to generate the plasmids with mutations when using the following mutagenic primers synthesized by IDT DNA Technologies, Inc. (the mutagenic codons are underlined): pDHPA (Y38F), 5′-TTC AAA AAC TTT GTC GGC AAA TCT GAC CAA GAG CTC AAA TCG ATG GCC AAG-3′ (sense) and 5′-CTT GGC CAT CGA TTT GAG CTC TTG GTC AGA TTT GCC GAC AAA GTT TTT GAA G-3′ (antisense); pDHPA (Y34F), 5′-G CGC CGC TTC TTC AAA AAC TAT GTC-3′ (sense) and 5′-GAC ATA GTT TTT GAA GAA GCG GCG C-3′ (antisense); pDHPA (Y34F/Y38F), 5′-CCG GAC GAG CGC CGC TTC TTC AAA AAC TTT GTC GGC AAA TCT GAC-3′ (sense) and 5′-GTC AGA TTT GCC GAC AAA GTT TTT GAA GAA GCG GCG CTC GTC CGG-3′ (antisense); pDHPB (Y28F), 5′-C GCA TTT TTG AAT AAG TTT CCG GAC GAG AAA CGC A-3′ (sense) and 5′-T GCG TTT CTC GTC CGG AAA CTT ATT CAA AAA TGC G-3′ (antisense); pDHPB (Y38F), 5′-CGC AAC TTC AAA AAC TTC GTC GGC AAA TCT GAC-3′ (sense) and 5′-GTC AGA TTT GCC GAC GAA GTT TTT GAA GTT GCG-3′ (antisense).
The DHP B (Y38F) plasmid was further used as a template for generating the double mutant DHP B (Y28F/Y38F) with the mutagenic primers for pDHPB (Y28F). The plasmids were extracted using the QIAprep® spin miniprep kit (Qiagen Sciences, Valencia, CA). DNA sequencing of the resulting mutated genes in their entirety confirmed the success of the site-directed mutagenesis and the absence of secondary mutations. Recombinant DHP proteins were obtained by expression in Escherichia coli as described elsewhere (22, 24, 37). The protein yield was greater than ∼9 mg/liter culture in all cases. A two-part purification strategy (immobilized metal affinity chromatography followed by ion-exchange chromatography) resulted in a purification level of >95% homogeneity, with the DHP mutants being indistinguishable by SDS-PAGE gel from their WT counterparts.
Molecular Weight Determination
The molecular weights of the proteins employed in this study were experimentally determined via electrospray ionization mass spectrometry in the positive-ion mode (Agilent Technologies 6210 LC-TOF, Santa Clara, CA). The protein samples were prepared in 50 mm ammonium acetate buffer, pH 7.0. The mobile phase consisted of a linear gradient using the following HPLC grade solvents: water + 0.1% formic acid (v/v) and water:acetonitrile (5:95) + 0.1% formic acid (v/v). The injection volume was 5 μl, and the flow rate was 300 μl/min. The experimentally determined and calculated monomeric molecular weights of the dehaloperoxidase mutants as determined by electrospray ionization mass spectrometry were as follows: DHP A(Y34F), 16,392.46 (calculated 16,392.50); DHP A(Y38F), 16,392.40 (calculated 16,392.50); DHP A(Y34F/Y38F), 16,376.39 (calculated 16,376.50); DHP B(Y28F), 16258.14 (calculated 16,258.37); DHP B(Y38F), 16258.39 (calculated 16,258.37); DHP B(Y28F/Y38F), 16242.47 (calculated 16,242.37).
Preparation of Ferric DHP
DHP as purified was treated with an excess of potassium ferricyanide to obtain a homogeneous solution of the enzyme in the ferric state. The ferricyanide was then removed using a PD-10 desalting column pre-packed with Sephadex G-25 medium. The protein was concentrated using an Amicon Ultra centrifugal filter equipped with a 10,000 kDa cutoff molecular mass membrane, and the purity of DHP was determined as previously published (22, 37). The concentrations of the mutant enzymes were determined spectrophotometrically based on the extinction coefficients for WT DHP A (ϵ406 = 116.4 mm−1 cm−1) (19) and WT DHP B (ϵ407 = 117.6 mm−1 cm−1) (37).
UV-Visible Spectroscopy and DHP Activity Assays
The enzymatic activity of DHP was assayed optically from the rate of TCP conversion to dichloroquinone (2,6-dichloro-1,4-benzoquinone) at variable H2O2 concentrations (10–200 μm, pH 7, 100 mm KPi). Although the enzyme and the starting TCP concentrations were kept constant in all assays (0.5 and 150 μm, respectively), the dependence of the rate on the H2O2 concentration was fit with a classical Michaelis-Menten model using the Grafit 4.0 software package. The kinetics parameters Km and kcat resulted from the optimization of the fitting procedure. Optical spectra were recorded at 25 °C using a Cary 50 UV-visible spectrophotometer equipped with thermostatted cell holders. The TCP concentration was measured by using the molar absorption coefficient ϵ312 = 3752 m−1 cm−1 (22).
Stopped-flow Kinetics Studies
Rapid changes in the optical spectra of the DHP variants upon reaction with H2O2 were studied by a Bio-Logic SFM-400 Triple Mixing Stopped-Flow instrument equipped with a diode array UV-visible spectrophotometer. The reactions were carried out at pH 7 in 100 mm KPi buffer at 20 °C. The temperature was maintained using a circulating water bath. Experiments were performed in single-mixing mode wherein enzyme at a final concentration of 10 μm was reacted with 2.5–25 eq of H2O2. Data were collected at variable intervals over three different time ranges using the BioKinet32 software package (Bio-Logic). All data were evaluated using the Specfit Global Analysis System software package (Spectrum Software Associates). Singular value decomposition (SVD) analysis was applied to the raw spectra sets when different mechanistic hypotheses were explored and fit to exponential functions as either one-step, two species or two-step, three species irreversible mechanisms, where applicable. Kinetics data were base-line-corrected using the Specfit autozero function.
Rapid Freeze Quenching of the Reaction Intermediates
A BioLogic SFM 400 Freeze-Quench apparatus was used to prepare samples for EPR spectroscopic analysis of the short-lived reaction intermediates. A 50 μm enzyme solution (final concentration) was reacted with a 10-fold excess of H2O2 in 100 mm KPi (pH 7) at 25 °C. Variable reaction times were achieved by means of different aging lines. To make a sample, a standard 4-mm outer diameter quartz EPR tube connected to a Teflon funnel was submersed in, and therefore filled with, isopentane cooled in a liquid nitrogen bath to −110 °C. The reaction mixture was sprayed into the funnel, and the resulting frozen material was then packed at the bottom of the quartz tube using a packing rod fitted with a Teflon tip. The sample was then transferred to a liquid nitrogen storage Dewar for subsequent spectroscopic analysis.
EPR Spectroscopy
EPR spectra were recorded on an X-band (9 GHz) E-9 EPR spectrometer (Varian, El Palo, CA). A quartz finger Dewar insert was used to measure the EPR spectra at 77 K. This required periodic refilling of the Dewar with liquid nitrogen due to its evaporation during longer acquisition runs. The typical spectrometer settings were as follows: field sweep 200 G, scan rate 3.33 Gauss/s, modulation frequency 100 KHz, modulation amplitude 4.0 G, and microwave power 2 milliwatts. The microwave frequency for each EPR experiment, typically close to 9.2977 GHz, was measured by an EIP-578 in-line microwave frequency counter (PhaseMatrix, San Jose, CA).
EPR Spectra Simulation and Assignment to Specific Site
The free radical EPR spectrum simulation was performed by using SIMPOW6 (45). TRSSA (41) was used to generate the EPR spectrum simulation parameters from an input of two variables, the phenoxyl ring rotation angle, and the spin density on atom C1 of the radical. The Phenol Ring Rotation Angle Database (46) was used to identify the tyrosine residues in the DHP structures (as Tyr-28) with the closest rotational conformation of the phenol group (the ring rotation angle) to the conformation determined from the simulation of the tyrosyl radical EPR spectrum.
RESULTS
UV-Visible Spectra of the Mutants in the Resting (Fe3+ Heme) State
The optical spectra of the two WT DHP isoforms in comparison with their corresponding tyrosine mutants are shown in Fig. 2. The spectra of these heme proteins in the oxidized state are mainly typical of the high spin (with reference to the electron spin heme iron) (S = 5/2) ferric heme forms; their slightly different optical characteristics are detailed in Table 1 as is their optical purity ratio (ASoret/A280, also termed the Reinheitzahl value, Rz) and the ratio of absorbances ASoret/A380 for all proteins studied. The latter ratio has been employed before in estimating the relative proportion of the five- and six-coordinated high spin ferric heme populations; the greater the ASoret/A380 ratio, the greater the six-coordinate to five-coordinate population ratio (47–49). Note that this ratio is smaller (indicating a greater relative proportion of the five-coordinate form) in all proteins that retain Tyr-38. Fig. 2 shows that whenever Tyr-38 is missing either in DHP A or DHP B, there is a characteristic deviation in the absorption spectrum, and the ASoret/A380 ratio is greater (Table 1), indicating a greater proportion of the six-coordinate ferric heme form as compared with the five-coordinate. Thus, it appears that the proteins that retain Tyr-38 have a greater proportion of the five-coordinate ferric heme state, which is likely to be associated with the His-55 “open” conformation. We propose that this indicates that Tyr-38 serves as the primary source of the proton for His-55 in regulating the balance of the open/closed conformations and, therefore, the route of free radical evolution (note the proximity of these two residues in Fig. 1).
FIGURE 2.
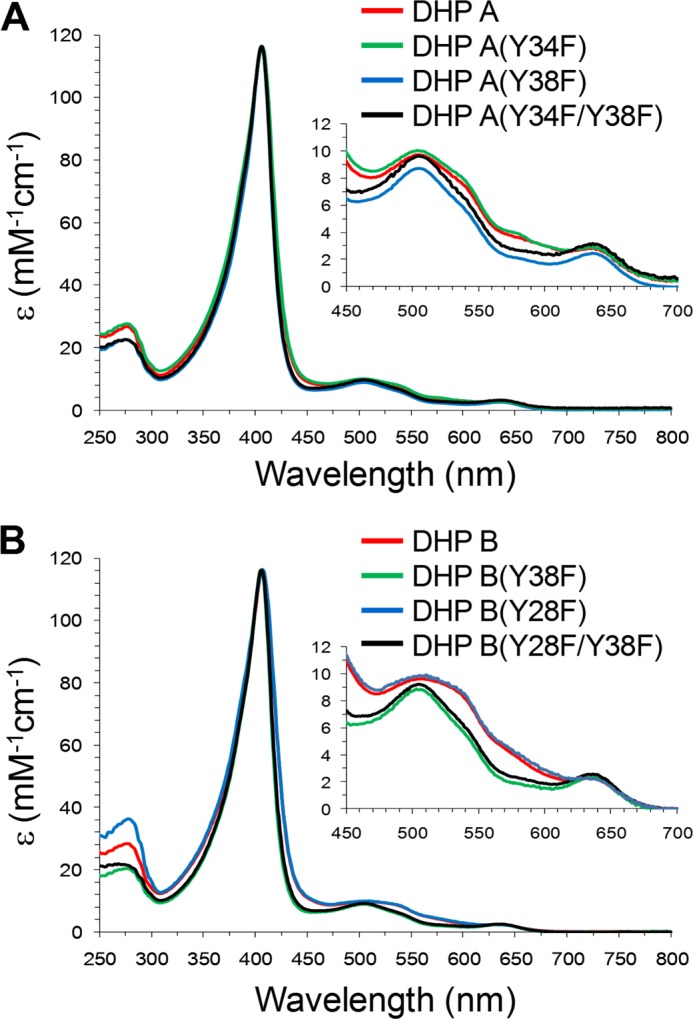
The UV-visible spectra of WT and mutants of DHP A (A) and DHP B (B) at pH 7. 0.
TABLE 1.
UV-visible spectroscopic features of the DHP A and DHP B variants in the ferric heme state at pH 7
Rz, Reinheitzahl value. sh indicates “shoulder.”
| Protein | λmax | ASoret/A380 | Rz |
|---|---|---|---|
| nm | |||
| DHP A (WT) | 407, 504, 538 (sh), 635 | 1.89 | 4.35 |
| DHP A (Y34F) | 407, 507, 535 (sh), 639 | 1.92 | 4.20 |
| DHP A (Y38F) | 406, 505, 541 (sh), 638 | 2.17 | 5.16 |
| DHP A (Y34F/Y38F) | 406, 505, 540 (sh), 635 | 2.00 | 5.17 |
| DHP B (WT) | 407, 508, 540 (sh), 633 | 1.81 | 4.11 |
| DHP B (Y28F) | 407, 510, 538 (sh), 640 | 1.84 | 3.23 |
| DHP B (Y38F) | 406, 506, 540 (sh), 636 | 2.04 | 5.13 |
| DHP B (Y28F/Y38F) | 406, 504, 538 (sh), 636 | 2.04 | 5.62 |
We previously showed that the pH-dependent pattern of H2O2-induced free radical formation in DHP A (40) is linked to the balance of open/closed conformations of the distal histidine (His-55) (23, 28, 50), and the protonation of His-55 promotes its open conformation (associated with the five-coordinate ferric heme form). Thus, at lower pH values, H2O2 has easier access to the heme but a lower chance to be kept there long enough to react with the heme.
Kinetics of TCP Oxidation by Different DHP Variants
The kinetic parameters (kcat and Km) for the reaction of TCP oxidation to dichloroquinone as catalyzed by different DHP variants in the presence of H2O2 were determined for the following conditions: [enzyme] = 0.5 μm, [TCP]0 = 150 μm, and variable H2O2 concentration (10–200 μm); Table 2. Each mutant exhibited a H2O2 concentration dependence on the rate of TCP enzymatic oxidation as observed previously for WT DHP A (22). In the absence of DHP (non-enzymatic control), no product was observed under the conditions examined, in agreement with previous reports (19, 22). The catalytic efficiency, defined as the ratio kcat/Km, is also provided in Table 2 for the different DHP variants. The data presented in Table 2 allow for the following observations. (a) Removal of Tyr-34 or Tyr-38 in DHP A resulted in an increase of the maximal turnover number kcat (notably more so when both tyrosines were removed). A similar result was obtained for the DHP B mutants for Tyr-28 and Tyr-38. (b) The Km values for H2O2 for the mutants were not >3-fold greater than those of the WT enzymes with the exception of DHP B (Y28F). The latter 34-fold increase demonstrates that H2O2 binding can be affected significantly by replacing a residue that is 10 Å away from the heme, possibly due to the fact that this mutation occurs at the junction of the B and C helices. (c) The generally higher turnover numbers (kcat) and higher Km values in the mutants resulted in the catalytic efficiencies (kcat/Km) being only marginally different in the DHP A and B variants when compared with the WT isoenzymes.
TABLE 2.
Kinetics data for the oxidation of TCP as catalyzed by DHP in the ferric state in the presence of H2O2 at pH 7
| Protein | KmH2O2 | kcat | kcat/KmH2O2 |
|---|---|---|---|
| μm | s−1 | μm−1s−1 | |
| DHP A (WT) | 23 ± 1 | 0.61 ± 0.01 | 0.027 |
| DHP A (Y34F) | 27 ± 5 | 0.77 ± 0.02 | 0.029 |
| DHP A (Y38F) | 68 ± 5 | 1.09 ± 0.05 | 0.016 |
| DHP A (Y34F/Y38F) | 56 ± 7 | 4.9 ± 0.1 | 0.09 |
| DHP B (WT) | 22 ± 2 | 1.53 ± 0.03 | 0.070 |
| DHP B (Y28F) | 752 ± 48 | 5.13 ± 0.14 | 0.007 |
| DHP B (Y38F) | 60 ± 10 | 8.53 ± 0.33 | 0.143 |
| DHP B (Y28F/Y38F) | 47 ± 5 | 5.37 ± 0.07 | 0.114 |
Spectroscopic Characterization of the Mutants Reacting with H2O2
The intermediates of the reactions of the variants of DHP A and DHP B with hydrogen peroxide were characterized by UV-visible and EPR spectroscopies. Optical spectra were collected for the reaction of enzyme (10 μm) with H2O2 (2.5–25 eq), and the fast time-scale changes were subjected to SVD analyses. The rapid freeze-quench method was used to prepare samples for the EPR analysis of the free radicals rapidly formed and decayed upon reaction of enzyme (50 μm) with a 10-fold excess of H2O2. Typical for the EPR method, a higher concentration of H2O2, as compared with optical spectroscopy or activity assays, was employed to facilitate spectral acquisition. As the ferryl intermediates were identified in the stopped-flow studies at a significantly lower peroxide concentration (25 μm) and the biochemical assays demonstrated that DHP at a physiological concentration exhibits its enzymatic activity for a range of H2O2 concentrations (10–200 μm), we conclude that the chemistry observed at higher peroxide concentrations reflects the chemistry that occurs at more physiologically relevant concentrations. Table 3 summarizes these optical and EPR data related to the intermediates formed under H2O2 treatment for the different DHP A and DHP B variants.
TABLE 3.
Optically active and paramagnetic intermediates of DHP A and DHP B (and their mutants) formed upon activation by H2O2 at pH 7
| Protein | Compound I λmaxa | Compound II λmaxa | Protein bound radical concluded to be on residueb | Final observed species λmaxc |
|---|---|---|---|---|
| nm | nm | nm | ||
| DHP A (WT) | Not detected | 420, 545, 585 | Tyr-34 (mj); Tyr-38 (mn) | 411, 530, 564 (DHP A Compound RH) |
| DHP A (Y34F) | Not detected | 419, 545, 584 | Tyr-38 | 416, 560, 598 (Compound RH-like) |
| DHP A (Y38F) | Not detected | 419, 545, 585 | Tyr-34 | 418, 543, 578 (Compound III)d |
| DHP A (Y34F/Y38F) | 405, 505 (br sh), 643 | 414, 505, (sh), 540, 589 (sh) (Compound I/II mixture) | Tyr-28 | Bleached |
| DHP B (WT) | Not detected | 419, 545, 585 | Tyr-38 (mj); Tyr-28 (mn) | 411, 554, 599 (DHP B Compound RH) |
| DHP B (Y28F) | Not detected | 419, 545, 584 | Tyr-38 | 415, 552, 598 (Compound RH-like) |
| DHP B (Y38F) | 405, 520, 644 | 411, 518, 540 (sh), 587 (sh) (Compound I/II mixture) | Tyr-28 | Bleached |
| DHP B (Y28F/Y38F) | 406, 528, 645 | Not detected | Not detected | 404, 518, 542 (sh)d |
a sh indicates “shoulder,” and br sh indicates “broad shoulder.”
b mj indicates “major,” and mn indicates “minor” (free radical species).
c After 85 s.
d Slight bleaching was also observed.
A Note on the “Compound” Terminology
There is an ambiguity in the terminology adopted in the literature for describing the oxidation states of heme proteins and enzymes. We will use the Compound terminology when referring to the oxidation states of the heme as opposed to the case when it is referred to the whole protein molecule. In this paper the term “Compound I” describes a state when the heme iron is in a ferryl state (Fe4+=O2−) and the porphyrin macrocycle is one electron-oxidized, i.e. a cation radical state. The term “Compound II” refers to a ferryl heme (Fe4+=O2−) irrespective of whether a radical is present or not on the protein moiety of the enzyme. From this point of view, “Compound ES” is a particular case of Compound II, which becomes confusing, so we will try, whenever possible, to explicitly indicate the ferryl heme oxidation state (Fe4+=O2−) rather than use the term Compound II. We also feel that the term Compound ES should be only applicable to the WT enzyme to avoid ambiguity in describing Compound ES in different mutants where the protein radical might be located on different amino acids.
DHP A (Y34F); a Ferryl Heme + a Protein Radical
An SVD analysis of the rapid changes in the UV-visible spectra of this mutant resulted in a simple two component mechanism, a → b, wherein the starting ferric state was converted into an intermediate assignable to a ferryl (Fe4+=O2−) heme state, i.e. Compound II (Fig. 3A, Table 3). The rate constant of this pseudo first order conversion was found to be kobs = (3.56 ± 0.02) × 104 m−1s−1. In the WT enzymes of both isoforms, this state is associated with a Compound ES (22, 37), when the heme is in the ferryl oxidation state and a radical is formed on the protein moiety of the enzyme. The EPR spectra of DHP A (Y34F) reacting with H2O2, measured at different times after the initiation of the reaction, showed a time-dependent multicomponent radical signal (Fig. 4A). There was no evidence of formation of Compound RH within the first 3 s of the reaction; however, at a longer time (∼1 min) a species with optical characteristics (416, 560, 598 nm) reminiscent of Compound RH was observed (Table 3).
FIGURE 3.

Kinetic data obtained by optical spectroscopy for the DHP A mutants. A, DHP A (Y34F): top panel, stopped-flow UV-visible spectra of DHP A (Y34F) (10 μm) reacting with a 10-fold excess of H2O2 at pH 7.0 (400 scans over 3 s); inset, the single wavelength (407 nm) dependence on time obtained from the raw spectra and its fit with a superposition of the calculated spectra components; middle panel, calculated spectra of the two reaction components derived from the SVD analysis: ferric heme state (black) and Compound II (red); bottom panel, time dependences of the relative concentrations for the three components shown in the middle panel as determined by fitting the spectra set in the top panel. B) DHP A (Y38F), top, middle, and bottom panels are similar to those in A. C, DHP A (Y34F/Y38F): top, middle, and bottom panels are similar to those in A except the calculated spectra derived from the SVD analysis were fit to three reaction components: ferric heme state (black), Compound I (blue), and the “Compound I/II mixture” species (red).
FIGURE 4.

Free radical EPR spectra collected at variable freeze-quench times (indicated) after mixing 50 μm DHP A mutants with 500 μm H2O2 (both concentrations are final): Y34F (A), Y38F (B), Y34F/Y38F (C). D, typical EPR spectra in different mutants are compared. In small boxes are the radicals thought to be responsible for the observed line shapes (see “Discussion”). The reaction was conducted in a 100 mm potassium phosphate buffer (pH 7) at 25 °C.
DHP A (Y38F); a Ferryl Heme + a Protein Radical
Similar to the Y34F variant above, the rapid changes in the optical spectra after mixing DHP A (Y38F) with peroxide were fitted with a two component model a → b, wherein the starting ferric state (Table 1, DHP A (Y38F)) converted (kobs = (2.78 ± 0.01) × 104 m−1s−1) into a ferryl heme state (Fig. 3B, Table 3). Unlike either WT DHP A or the Y34F variant, formation of Compound RH was not seen for DHP A (Y38F). Rather, the ferryl intermediate converted to a new, stable species whose spectral features strongly resembled those for the oxyferrous form of DHP, Compound III (Table 3). A free radical was also formed in the DHP A (Y38F) mutant upon H2O2 addition (Fig. 4B); however, the EPR spectrum of the radical is notably different from that in DHP A (Y34F) (see above).
DHP A (Y34F/Y38F); Formation of Compound I followed by a Species Reminiscent of a Ferryl Heme + a Protein Radical
The chemistry observed with the Y34F/Y38F double mutant of DHP A is markedly different from that observed for the Tyr-34 or Tyr-38 single mutation variants. An SVD analysis of the optical spectra (Fig. 3C) generated upon rapid mixing of DHP A (Y34F/Y38F) with an excess of peroxide yielded a three-component mechanism, a → b → c wherein the initial ferric heme enzyme, a, is converted (kobs = (1.13 ± 0.02) × 105 m−1s−1) to an intermediate, b, whose spectral characteristics are consistent with a Compound I state (51–57) (see the justification of this assignment under “Discussion”). This new intermediate further converted (kobs 5.4 ± 0.1 s−1) to a state, c, that exhibits spectral features (particularly the red shift of the Soret band) that are suggestive of a mixture of Compound I and II (Table 3) (48, 49, 53, 54, 58, 59). The EPR spectroscopic data also revealed that yet another type of free radical was formed in the Y34F/Y38F double mutant of DHP A upon reaction with H2O2 (Fig. 4C).
Mini Summary for the DHP A Mutants
The two single mutants of DHP A, Y34F and Y38F, when reacting in the ferric heme state with H2O2, showed a rapid transformation to the ferryl heme species (Compound II). No fast conversion of the latter to Compound RH was detected; rather, a species with spectral characteristics similar to those of Compound III was formed later in the reaction. In contrast to the single mutants, the first observed intermediate upon reaction of H2O2 with the double mutant DHP A (Y34F/Y38F) was Compound I, which rapidly converted to a mixture of Compounds I and II. In all three mutants, free radicals were detected under peroxide treatment (Fig. 5). Remarkably, the radicals were different in the three DHP A mutants. Their EPR spectra are shown for easy comparison on a common magnetic field axis in Fig. 4D.
FIGURE 5.
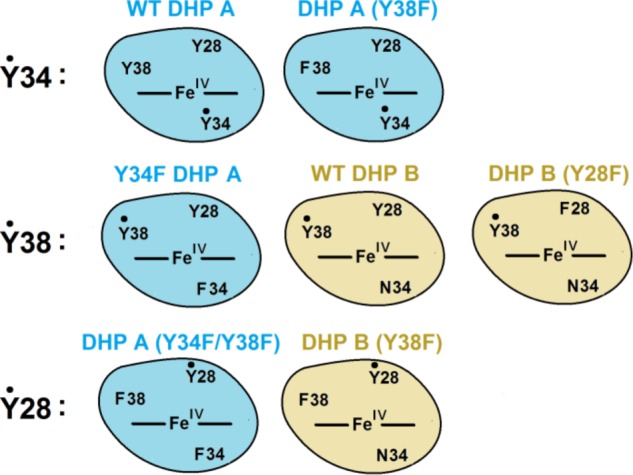
Summary of radical site assignments in WT and mutant DHP enzymes.
DHP B (Y28F); a Ferryl Heme + a Protein Radical followed by a Species Reminiscent of Compound RH
UV-visible characteristics of ferric DHP B (Y28F) are given in Table 1. Upon the addition of H2O2, spectral features consistent with a ferryl state (Compound II) were observed (kobs = (1.58 ± 0.08) × 105 m−1s−1; Fig. 6A) followed by decay (kobs = 6.2 ± 0.1 s−1) to a species reminiscent of Compound RH (Table 3). The EPR signal of the radical thus formed in the Y28F mutant of DHP B is similar to the “pH 5 radical” in DHP A (22, 40), previously assigned to a Tyr-38 radical (Fig. 7A and Table 4).
FIGURE 6.

Kinetic data obtained by optical spectroscopy for the DHP B mutants. A, DHP B (Y28F): top panel, stopped-flow UV-visible spectra of 10 μm enzyme reacting with a 10-fold excess of H2O2 at pH 7.0 (400 scans over 3 s); inset, the single wavelength (407 nm) dependence on time obtained from the raw spectra and its fit with a superposition of the calculated spectra components; middle panel, calculated spectra of the three reaction components derived from the SVD analysis: ferric heme state (black), ferryl heme or Compound II (red), and a species reminiscent of Compound RH (green); bottom panel, time dependences of the relative concentrations for the three components shown in the middle panel as determined by fitting the spectra set in the top panel. B, DHP B (Y38F): top, middle, and bottom panels are similar to those in A, whereas the three reaction components in the SVD analysis were ferric heme state (black), Compound I (blue), and a species reminiscent of Compound II (Compound I/II mixture, red). C, DHP B (Y28F/Y38F): top, middle, and bottom panels are similar to those in A. The three reaction components in the SVD analysis were ferric heme state (black), Compound I (blue), and a “Ferric-like” heme species (red).
FIGURE 7.

Comparison of the free radical EPR signals in different variants of DHP A and DHP B. The tyrosyl radicals mostly responsible for the line shapes presented in different panels are indicated in small boxes (see “Discussion” for these assignments). A, the DHP A signal previously obtained as the difference indicated and attributed to the Tyr-38 radical (pH 5 radical) (40) and the EPR spectrum detected in the Y28F mutant of DHP B. The two EPR spectra were obtained on different EPR spectrometers operating at slightly different microwave frequencies causing the same EPR signal to appear at slightly different magnetic field values. Therefore, the absolute values of the magnetic field are not indicated, and the signals were superimposed on the basis of the g-factors. The positions of the eight components of the EPR signal, common for both spectra, are indicated. B, the EPR signals of the free radicals formed in DHP A (Y34F/Y38F) and DHP B (Y38F). (note: DHP B does not have Tyr-34). The third signal of a similar line shape is from WT DHP B but freeze-quenched at a later time after the addition of peroxide (37). The bottom spectrum is a simulation of the tyrosyl radical EPR spectrum for the TRSSA (41) input parameters ρC1 = 0.38 and θ = 46.0°; the simulation parameters yielded by this two-value input are given in Table 4. C, the EPR spectra of the Tyr-34 radical in WT DHP A (as reported in Ref. 40) and in the DHP A Y38F mutant (as in Fig. 4). Similar to panel A, the two spectra were measured on different EPR spectrometers and the magnetic field values are not shown. D, EPR spectra show the presence of the Tyr-38 radical in WT DHP B (as a major species) (37), in WT DHP A (as a minor species but seen well at pH 5) (40) and in the Y34F mutant of DHP A at pH 7 (as in Fig. 4). Note that the free radical spectrum of WT DHP B at a later time point (see panel B) is consistent with the radical being localized on a different residue, Tyr-28. As in A and C, the magnetic field values are not shown.
TABLE 4.
The parameters generated by TRSSA for ρC1 = 0.38 and 0 = 46.0° and used to simulate the spectrum in Fig. 7B
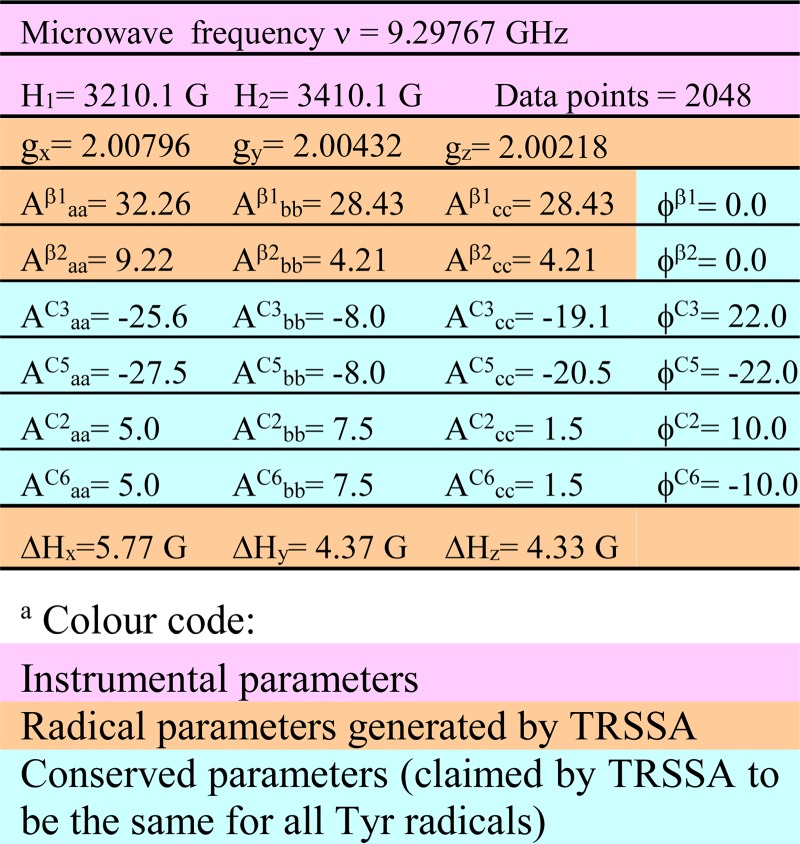
a The hyperfine interaction constants A are given in MHz, Euler angles φ are given in degrees.
Although the two spectra in Fig. 7, A, are not exactly superimposable (possibly because the coefficient of subtraction for the first spectrum, 0.0616, was estimated), it is important that all eight components in the line shapes of the two signals coincide with a very good accuracy, indicating that the same EPR signal is the main contributor to both spectra.
DHP B (Y38F); Formation of Compound I followed by a Species Reminiscent of a Ferryl Heme + a Protein Radical That Is the Same as in DHP A (Y34F/Y38F)
When the DHP B (Y38F) mutant in the ferric heme state (optical characteristics given in Table 1) reacted with hydrogen peroxide under the same concentration and pH conditions as employed above, a species was formed (kobs = (1.46 ± 0.08) × 105 m−1s−1; Fig. 6B) whose optical spectral features (Table 3) matched very well those reported for DHP A (Y34F/Y38F) Compound I. This compound in DHP B (Y38F) was rapidly converted (kobs = 6.5 ± 0.1 s−1) in the absence of an exogenously added substrate to a more stable state that had optical characteristics that resembled a mixture of Compounds I and II (note the red shift of the Soret band), again exhibiting similar behavior as DHP A (Y34F/Y38F).
Fig. 7B demonstrates that the double mutant of DHP A, in which Tyr-34 and Tyr-38 are replaced with phenylalanines, shows the same EPR spectrum as DHP B in which Tyr-38 is replaced and that lacks Tyr-34 naturally. In WT DHP B, the same EPR signal (37) is seen at a later time after the addition of a 10-fold excess of peroxide (Fig. 7B).
DHP B (Y28F/Y38F); Formation of Compound I followed by a Species Reminiscent of a Ferric Heme with No Detectable Protein Radical
Compound I was also formed (kobs = (1.64 ± 0.14) × 105 m−1s−1) in the double mutant Y28F/Y38F of DHP B when H2O2 was added (Fig. 6C; Table 3). Decay of this Compound I did not yield a detectable Compound II species (note the lack of a red shift in the Soret band) as was observed in the other DHP mutants, such as DHP A (Y34F/Y38F) or DHP B Y38F), but resulted in a final species that exhibited a ferric-like absorption spectrum (kobs = 6.4 ± 0.1 s−1; 404 (Soret), 518, 542 nm (sh)). At longer observation times, heme bleaching was noted (data not shown). No radical was detectable by the EPR spectroscopy in DHP B (Y28F/Y38F) at 77 K.
Mini Summary for the DHP B Mutants
DHP B naturally lacks Tyr-34, so it is not surprising that the single mutant DHP B (Y38F) demonstrates the same phenomenology upon reaction with H2O2 as does the double mutant DHP A (Y34F/Y38F); namely, formation of Compound I followed by its reduction to a state reminiscent of a ferryl heme with a protein radical. Importantly, the EPR spectra of the radicals in the two mutants are identical, indicating that the radical is formed on the same residue in the two proteins (Fig. 5). In the other single mutant of isoenzyme B, DHP B (Y28F), a more conventional ferryl heme state is formed with a protein radical that is likely to be the same as the “pH 5 radical” in WT DHP A, previously attributed to Tyr-38 (40). The ferryl heme + Tyr-38 radical state evolves (within 3 s) to a species that is reminiscent of Compound RH (37). The double mutant DHP B (Y28F/Y38F), which is three tyrosines down when compared with WT DHP A, shows Compound I as the first intermediate of its reaction with peroxide, but it does not show its transformation to a ferryl heme and/or a protein radical and evolves directly to a ferric-like heme form. The latter is likely to be a damaged enzyme as the bleaching of the protein follows.
DISCUSSION
Assignment of Radicals to Residues
The current study of the DHP mutants unequivocally supports our previous TRSSA-based radical assignment in DHP A (40); elimination of Tyr-38 results in the EPR spectrum previously attributed to the Tyr-34 radicals (Fig. 4), and elimination of Tyr-34, when Tyr-38 is kept intact, results in the spectrum of the pH 5 radical previously attributed to Tyr-38 (Fig. 7). As a further confirmation of such radical site assignment, the spectrum of the WT DHP B (37) (note that this isoenzyme has Asn-34 in place of Tyr-34 of DHP A) shows a spectrum of the Tyr-38 radical (Fig. 7D).
Interestingly, although WT DHP B and Y34F DHP A (having identical set of tyrosines) show the same Tyr-38 radical as the principal species, WT DHP A at pH 5, where the Tyr-34 radical is not seen, exhibits the same EPR line shape but at a lower resolution of the spectral components (Fig. 7D), which indicates that there might be another radical present in WT DHP A at pH 5. It is reasonable to suggest that this type of radical in DHP A is the one we see in the double mutant when both Tyr-34 and Tyr-38 are replaced (Figs. 4C and 7B). As expected, the Y38F DHP B mutant (in which Tyr-34 is missing naturally) shows the same EPR spectrum (Fig. 7B). This suggests that there is a third site of radical localization in addition to the already established Tyr-34 (in DHP A) and Tyr-38 (in both DHP A and B), which is common for both isoenzymes.
Which residue can this be? We suggest that the third site of radical stabilization is another tyrosine. It was possible to simulate the EPR line shape of the radical by using TRSSA (Fig. 7B), which means that a tyrosyl radical can indeed be responsible for the EPR signal. The simulation shown has been obtained for the TRSSA input parameter θ = 46.0° (the rotation angle of the tyrosyl ring). Now the question should be addressed, Which tyrosines in the DHP A and B have their ring rotation angles close to this value or to the complementing symmetrical angle of 74.0°? (note that complementing angles 46° and 74°, totaling 120°, will yield identical sets of the EPR spectrum simulation parameters.)
Table 5 shows the results of the analysis performed on the website of the Tyrosine Residues in Different Proteins database (46). Overall, the evidence unequivocally suggests that Tyr-28 is the most likely site of the radical, with the EPR spectral features presented in Fig. 7B in both DHP A (Y34F/Y38F) and DHP B (Y38F) (Table 3).
TABLE 5.
Rotation angle θ of the phenol ring in different tyrosine residues in Amphitrite ornata DHP A and DHP B, ranked by the closeness to the target values of 46 and 74°
| θtarget = 46° |
θtarget = 74° |
||||
|---|---|---|---|---|---|
| Tyrosinea | θ ° | |θ − θtarget|° | Tyrosine | θ ° | |θ − θtarget|° |
| DHP A, PDB ID 1EW6 (73) | |||||
| B Tyr-28 | 44.945 | 1.1 | B Tyr-107 | 65.66 | 8.3 |
| B Tyr-34 | 47.08 | 1.1 | A Tyr-107 | 62.15 | 11.9 |
| A Tyr-34 | 41.42 | 4.6 | B Tyr-34 | 47.08 | 26.9 |
| A Tyr-28 | 41.24 | 4.8 | B Tyr-28 | 44.945 | 29.1 |
| A Tyr-16 | 36.155 | 9.8 | A Tyr-34 | 41.42 | 32.6 |
| A Tyr-107 | 62.15 | 16.2 | A Tyr-28 | 41.24 | 32.8 |
| B Tyr-16 | 28.415 | 17.6 | A Tyr-16 | 36.155 | 37.8 |
| B Tyr-107 | 65.66 | 19.7 | B Tyr-16 | 28.415 | 45.6 |
| A Tyr-38 | −16.515 | 62.5 | A Tyr-38 | −16.515 | 90.5 |
| B Tyr-38 | −16.935 | 62.9 | B Tyr-38 | −16.935 | 90.9 |
| DHP B, PDB ID 3IXF (14) | |||||
| A Tyr-28 | 43.695 | 2.3 | A Tyr-107 | 55.945 | 18.1 |
| B Tyr-28 | 48.745 | 2.7 | B Tyr-107 | 55.275 | 18.7 |
| A Tyr-16 | 36.88 | 9.1 | B Tyr-28 | 48.745 | 25.3 |
| B Tyr-107 | 55.275 | 9.3 | A Tyr-28 | 43.695 | 30.3 |
| A Tyr-107 | 55.945 | 9.9 | A Tyr-16 | 36.88 | 37.1 |
| B Tyr-16 | 28.305 | 17.7 | B Tyr-16 | 28.305 | 45.7 |
| A Tyr-38 | −16.8 | 62.8 | A Tyr-38 | −16.8 | 90.8 |
| B Tyr-38 | −20.995 | 67.0 | B Tyr-38 | −20.995 | 95.0 |
a A and B indicate the chain ID of the two monomers of the protein in the asymmetric unit. The entries in bold are for the tyrosines that are most likely to host the radical (at the top of the table). Note that Tyr-34 cannot be the site responsible for the simulated spectrum as the same spectrum was observed in both DHP B, lacking Tyr-34 naturally, and in the DHP A mutant in which Tyr-34 is replaced (Fig. 7B).
Our hypothesis that the third radical site is Tyr-28 is further supported by the study of the DHP B (Y28F) mutant; the EPR spectrum of this protein when activated by H2O2 is consistent with the Tyr-38 radical (Fig. 7A), an obvious scenario for the protein that is naturally lacking Tyr-34. We also note that although the initial (0.3 s) radical site observed in WT DHP B is Tyr-38 (Fig. 7D), the radical character moves at a later time (2 s) to the nearby Tyr-28 (Fig. 7B), a significantly more solvent-exposed tyrosine residue.
Compound I Is Detected When Both Tyr-34 and Tyr-38 Are Missing
The optical spectral features of DHP A (Y34F/Y38F) (Fig. 3C), DHP B (Y38F) (Fig. 6B), and DHP B (Y28F/Y38F) (Fig. 6C) that are observed upon their reaction with peroxide, particularly the distinct hypochromicity of the Soret band as well as the appearance of a visible absorbance at ∼640–650 nm, are highly reminiscent of Compound I in other hemoproteins, including WT sperm whale myoglobin (51) and its His-64 mutants (52), horseradish peroxidase (HRP) (53), Arthromyces ramosus peroxidase (54), peanut peroxidase (55), soybean peroxidase (56), and Rhodnius prolixus nitrophorin 2 (57). Thus, we assign the first detectable peroxide-induced intermediates in DHP A (Y34F/Y38F), DHP B (Y38F), and DHP B (Y28F/Y38F) to Compound I (Table 3). In the absence of an exogenously added substrate, this species for the variants that retain Tyr-28, i.e. not in DHP B (Y28F/Y38F), rapidly decays to a more stable state with spectroscopic features that are similar to, but do not quite match those of, a typical Compound II/Compound ES. It is likely that this state, with a red shift of the Soret band, represents an equilibrium mixture of Compounds I and II that has been noted before for other peroxidases, including HRP (53, 58), A. ramosus peroxidase (54), and catalase-peroxidase (48, 49, 59). At longer reaction times (up to 85 s), this state undergoes bleaching.
Thus, Compound I was observable only when the two primary sites of radical localization, Tyr-34 and Tyr-38, were both absent, as in DHP A (Y34F/Y38F), DHP B (Y38F), and DHP B (Y28F/Y38F). For the first two of these three mutants, Compound I was found to partially convert to Compound II, and a tyrosyl radical was formed on the residue identified as Tyr-28. In DHP B (Y28F/Y38F), neither a free radical nor Compound II was observed. This might be explained by a very fast two-electron reduction of Compound I via concerted or two rapid sequential one-electron transfer events, which brings the heme to a ferric state but results in some oxidative damage to the protein.
Peroxidase Activity of the Mutants
Removal of both Tyr-34 and Tyr-38 not only makes Compound I detectable but also increases the turnover number in both isoforms (Table 2). Using the peroxidase kinetic model described by Ma et al. (60), we observe that, with the exception of DHP B(Y28F), the mutants studied follow general steady-state enzyme kinetics where high kcat values lead to an observed increase in Km. Removal of Tyr-28 in DHP B dramatically increases Km. As this mutation occurs at the junction of the B and C helices, we surmise that the resulting change in the vicinity of the active site leads to the altered Km value for H2O2. Strangely enough, removal of Tyr-28 together with Tyr-38 in the double mutant DHP B (Y28F/Y38F) does not have that effect. It is possible that the second mutation, which occurs between helices C and D also near the active site, “rescues” the negative effect of the first one, but this was not further studied.
Compound RH in DHP A Is Not the Same as in DHP B
In the absence of a reducing substrate, a species unique to DHP, termed Compound RH, was observed to form upon the decay of Compound ES for both WT DHP A and WT DHP B (22, 37). The four variants that appear to form a Compound RH-like species can be grouped by their optical spectra into two classes; the three that lack Tyr-34, i.e. DHP A (Y34F), WT DHP B, and DHP B (Y28F), all exhibit Q bands ∼552–560 and 598–599 nm, and the one that has Tyr-34 present (WT DHP A) exhibits Q bands at 530 and 564 nm (Table 3). The observation of these differences parallels our earlier conclusion about the formation of a covalent cross-link between the heme and Tyr-34 in WT DHP A Compound RH (40). Obviously, such a cross-link cannot be formed in the other three aforementioned variants that lack Tyr-34.
Although the exact nature of the Compound RH species will require further elaboration, the present study provides additional evidence that Compound RH is a protected state of DHP, the formation of which is mediated/regulated by tyrosyl radicals. As pointed out above, in an environmental situation when the aggressive oxidant H2O2 is present but the substrates meant to be oxidized are absent, most globins would start oxidizing themselves (and other cell components). Formation of Compound RH in DHP is a two-way solution to this problem. First, the enzyme, although remaining in the ferric heme state, is significantly deactivated in terms of reactivity toward H2O2, possibly via a mechanism in which the distal histidine is arrested in a conformation that prevents the heme from further interacting with H2O2 (40). Second, the oxygen binding function of DHP can be rescued from Compound RH as the oxyferrous form can be generated from Compound RH under mild reducing conditions (i.e. sodium dithionite in vitro) (22, 37).
Why Nature Employs Tyrosyl Radicals in DHP
The data presented in Tables 2 and 3 show that the peroxidase reaction is generally faster when catalyzed by the DHP mutants that lack the ability to form a stable and long-lived radical on Tyr-34 or Tyr-38. An alternative way to look at it is that those mutants that give rise to Compound I is more active toward substrate oxidation. The question thus arises as to why nature utilizes tyrosyl radical formation in DHP, as it appears that the presence of a tyrosyl radical diminishes the ability of the enzyme to catalyze substrate oxidation. We propose that this is likely a protective mechanism that is unique to DHP as a dual function globin-peroxidase. Specifically, in the absence of reducing substrate, Compound I rapidly (<1 s) undergoes heme bleaching (DHP A (Y34F/Y38F) or DHP B (Y38F)) or forms an inactive ferric-like state (DHP B (Y28F/Y38F)) that also exhibits some bleaching (Table 3). This represents the loss of both peroxidase and globin functions of DHP. By invoking a tyrosyl radical, the peroxidase activity of DHP is attenuated, but the oxidized (activated) form of the enzyme (as Compound ES) is nearly 2 orders of magnitude longer lived than Compound I, increasing the likelihood that the enzyme will react with substrate to regenerate the ferric resting form without heme bleaching.
Why is Compound I so much more prone to bleaching than Compound ES? As a globin, DHP possesses a relatively high redox potential (∼+200 mV (37 and 61) for the Fe3+/Fe2+ couple in comparison to those for peroxidases (HRP, −266 mV (62), cytochrome c peroxidase, −182 mV (63), myeloperoxidase, +5 mV (64)) and even other globins (sperm whale Mb, +43 mV (65), horse heart Mb, +46 mV (66), horse heart Hb, +152 mV (67), human Hb, +158 mV (68)). The high redox potential is to ensure that the oxygen binding function is maintained, as only a heme in the ferrous oxidation state can reversibly bind molecular oxygen. In Mb and Hb, which both possess similarly high redox potentials as DHP, Compound I is rapidly reduced to form ferryl species with protein-centered radicals, likely to avoid heme degradation. For DHP, it appears that a similar protective mechanism is in place; as the redox potential for the ferryl/Fe3+ couple correlates and trends with the Fe3+/Fe2+ couple (69), the unusually high redox potential for the Fe3+/Fe2+ couple in DHP leads to an estimated redox potential of ∼1.6 V for the ferryl/Fe3+ couple, which would rank among the highest for any peroxidase. It is likely that it is for this reason that the Compound I intermediate in the DHP tyrosine mutants rapidly undergoes heme bleaching. By introducing tyrosine-mediated electron transfer pathways (70) in DHP, nature has provided an endogenous reducing substrate that effectively quenches Compound I, leveling the redox potential of one of the two oxidizing equivalents to that of tyrosine (∼900 mV) (71, 72) and protecting the enzyme from irreversible heme bleaching.
Conclusions
The present study provides support for our explanation of the structure-function relationship that enables the dual activity in the dehaloperoxidase hemoglobins. To sustain its oxygen binding function, DHP possesses a high redox potential that is atypical of monofunctional peroxidases because it would imply a fast autoreduction of Compound I that could cause the enzyme's self-oxidation and heme degradation. To function as a peroxidase without self-damaging, DHP employs tyrosines as endogenous reducing co-factors that allow reduction of Compound I to Compound II, which also leads to formation of a radical on the tyrosine. The employment of tyrosines in the catalytic chemistry of DHP allows for Compound ES to live long enough to have a chance to encounter a substrate to oxidize. Such a purpose for an active site tyrosine does not need to be invoked in the classical monofunctional peroxidases, such as HRP, given that their ferryl/Fe3+ redox potentials are generally around 1 V (69), which is similar to that of tyrosine itself. As a hallmark of the globin-peroxidases, tyrosine radical formation in DHP is, therefore, nature's adaptation that enables peroxidase activity within a globin framework.
Acknowledgments
EPR instrumentation employed in this work was supported by National Institutes of Health Grant S10RR023614, National Science Foundation Grant CHE-0840501, and North Carolina Biotechnology Center Grant 2009-IDG-1015. Mass spectra were obtained at the Mass Spectrometry Facility for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation.
This work was supported, in whole or in part, by the North Carolina State University Molecular Biotechnology Training Program through a National Institutes of Health T32 Biotechnology Traineeship grant (to J. D.). This work was also supported by Army Research Office Grant 57861-LS (to R. G.), a National Science Foundation CAREER Award (CHE-1150709; to R. G.), and a National Science Foundation Research Experience for Undergraduates Summer Fellowship (to A. D.-L., CHE-0552604).
- DHP
- dehaloperoxidase
- Compound I
- the state of enzyme with a ferryl heme (Fe4+=O2−) and a porphyrin π-cation radical
- Compound II
- deprotonated or protonated ferryl heme state Fe4+=O2− or Fe4+-OH−
- Compound III
- oxyferrous [Fe2+-O2 or Fe3+-( O2˙̄)] state of the enzyme
- Compound ES
- the state of enzyme with a ferryl heme iron [Fe4+=O2−] and an amino acid radical (usually tryptophanyl or tyrosyl)
- Compound RH
- “Reversible Heme” state of dehaloperoxidase, formed from in the process of Compound ES decay in the absence of substrate
- SVD
- singular value decomposition
- TCP
- 2,4,6-trichlorophenol
- TRSSA
- tyrosyl radical spectra simulation algorithm.
REFERENCES
- 1. Chen Y. P., Lincoln D. E., Woodin S. A., Lovell C. R. (1991) Purification and properties of a unique flavin-containing chloroperoxidase from the capitellid polychaete Notomastus lobatus. J. Biol. Chem. 266, 23909–23915 [PubMed] [Google Scholar]
- 2. Roach M. P., Chen Y. P., Woodin S. A., Lincoln D. E., Lovell C. R., Dawson J. H. (1997) Notomastus lobatus chloroperoxidase and Amphitrite ornata dehaloperoxidase both contain histidine as their proximal heme iron ligand. Biochemistry 36, 2197–2202 [DOI] [PubMed] [Google Scholar]
- 3. Lincoln D. E., Fielman K. T., Marinelli R. L., Woodin S. A. (2005) Bromophenol accumulation and sediment contamination by the marine annelids Notomastus lobatus and Thelepus crispus. Biochem. Syst. Ecol. 33, 559–570 [Google Scholar]
- 4. King G. M. (1986) Inhibition of microbial activity in marine sediments by a bromophenol from a hemichordate. Nature 323, 257–259 [Google Scholar]
- 5. Fielman K. T., Targett N. M. (1995) Variation of 2,3,4-tribromopyrrole and its sodium sulfamate salt in the hemichordate Saccoglossus kowalevskii. Mar. Ecol.-Prog. Ser. 116, 125–136 [Google Scholar]
- 6. Woodin S. A., Walla M. D., Lincoln D. E. (1987) Occurrence of brominated compounds in soft-bottom benthic organisms. J. Exp. Mar. Biol. Ecol. 107, 209–217 [Google Scholar]
- 7. Woodin S. A. (1991) Recruitment of infauna. Positive or negative cues. Am. Zool. 31, 797–807 [Google Scholar]
- 8. Weber R. E., Mangum C., Steinman H., Bonaventura C., Sullivan B., Bonaventura J. (1977) Hemoglobins of two terebellid polychaetes. Enoplobranchus sanguineus and Amphitrite ornata. Comp. Biochem. Physiol. A Comp. Physiol. 56, 179–187 [DOI] [PubMed] [Google Scholar]
- 9. Chiancone E., Ferruzzi G., Bonaventura C., Bonaventura J. (1981) Amphitrite ornata erythrocruorin. 2. Molecular controls of function. Biochim. Biophys. Acta 670, 84–92 [DOI] [PubMed] [Google Scholar]
- 10. Chiancone E., Brenowitz M., Ascoli F., Bonaventura C., Bonaventura J. (1980) Amphitrite ornata erythrocruorin. 1. Structural properties and characterization of subunit interactions. Biochim. Biophys. Acta 623, 146–162 [DOI] [PubMed] [Google Scholar]
- 11. Lebioda L., LaCount M. W., Zhang E., Chen Y. P., Han K., Whitton M. M., Lincoln D. E., Woodin S. A. (1999) An enzymatic globin from a marine worm. Nature 401, 445. [DOI] [PubMed] [Google Scholar]
- 12. Zhang E., Chen Y. P., Roach M. P., Lincoln D. E., Lovell C. R., Woodin S. A., Dawson J. H., Lebioda L. (1996) Crystallization and initial spectroscopic characterization of the heme-containing dehaloperoxidase from the marine polychaete Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 52, 1191–1193 [DOI] [PubMed] [Google Scholar]
- 13. de Serrano V., Chen Z., Davis M. F., Franzen S. (2007) X-ray crystal structural analysis of the binding site in the ferric and oxyferrous forms of the recombinant heme dehaloperoxidase cloned from Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 63, 1094–1101 [DOI] [PubMed] [Google Scholar]
- 14. de Serrano V., D'Antonio J., Franzen S., Ghiladi R. A. (2010) Structure of dehaloperoxidase B at 1.58 A resolution and structural characterization of the AB dimer from Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 66, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murzin A. G., Brenner S. E., Hubbard T., Chothia C. (1995) SCOP. A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540 [DOI] [PubMed] [Google Scholar]
- 16. Zhao J., de Serrano V., Zhao J., Le P., Franzen S. (2013) Structural and kinetic study of an internal substrate binding site in dehaloperoxidase-hemoglobin A from Amphitrite ornata. Biochemistry 52, 2427–2439 [DOI] [PubMed] [Google Scholar]
- 17. Wang C., Lovelace L. L., Sun S., Dawson J. H., Lebioda L. (2013) Complexes of dual-function hemoglobin/dehaloperoxidase with substrate 2,4,6-trichlorophenol are inhibitory and indicate binding of halophenol to compound I. Biochemistry 52, 6203–6210 [DOI] [PubMed] [Google Scholar]
- 18. Chen Y. P., Woodin S. A., Lincoln D. E., Lovell C. R. (1996) An unusual dehalogenating peroxidase from the marine terebellid polychaete Amphitrite ornata. J. Biol. Chem. 271, 4609–4612 [PubMed] [Google Scholar]
- 19. Osborne R. L., Taylor L. O., Han K. P., Ely B., Dawson J. H. (2004) Amphitrite ornata dehaloperoxidase: enhanced activity for the catalytically active globin using MCPBA. Biochem. Biophys. Res. Commun. 324, 1194–1198 [DOI] [PubMed] [Google Scholar]
- 20. D'Antonio J., Ghiladi R. A. (2011) Reactivity of deoxy- and oxyferrous dehaloperoxidase B from Amphitrite ornata. Identification of compound II and its ferrous-hydroperoxide precursor. Biochemistry 50, 5999–6011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du J., Sono M., Dawson J. H. (2010) Functional switching of Amphitrite ornata dehaloperoxidase from O2-binding globin to peroxidase enzyme facilitated by halophenol substrate and H2O2. Biochemistry 49, 6064–6069 [DOI] [PubMed] [Google Scholar]
- 22. Feducia J., Dumarieh R., Gilvey L. B., Smirnova T., Franzen S., Ghiladi R. A. (2009) Characterization of dehaloperoxidase compound ES and its reactivity with trihalophenols. Biochemistry 48, 995–1005 [DOI] [PubMed] [Google Scholar]
- 23. Chen Z., de Serrano V., Betts L., Franzen S. (2009) Distal histidine conformational flexibility in dehaloperoxidase from Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 65, 34–40 [DOI] [PubMed] [Google Scholar]
- 24. Davis M. F., Gracz H., Vendeix F. A., de Serrano V., Somasundaram A., Decatur S. M., Franzen S. (2009) Different modes of binding of mono-, di-, and trihalogenated phenols to the hemoglobin dehaloperoxidase from Amphitrite ornata. Biochemistry 48, 2164–2172 [DOI] [PubMed] [Google Scholar]
- 25. Smirnova T. I., Weber R. T., Davis M. F., Franzen S. (2008) Substrate binding triggers a switch in the iron coordination in dehaloperoxidase from Amphitrite ornata. HYSCORE experiments. J. Am. Chem. Soc. 130, 2128–2129 [DOI] [PubMed] [Google Scholar]
- 26. Miksovská J., Horsa S., Davis M. F., Franzen S. (2008) Conformational dynamics associated with photodissociation of CO from dehaloperoxidase studied using photoacoustic calorimetry. Biochemistry 47, 11510–11517 [DOI] [PubMed] [Google Scholar]
- 27. Nienhaus K., Nickel E., Davis M. F., Franzen S., Nienhaus G. U. (2008) Determinants of substrate internalization in the distal pocket of dehaloperoxidase hemoglobin of Amphitrite ornata. Biochemistry 47, 12985–12994 [DOI] [PubMed] [Google Scholar]
- 28. Franzen S., Gilvey L. B., Belyea J. L. (2007) The pH dependence of the activity of dehaloperoxidase from Amphitrite ornata. Biochim. Biophys. Acta 1774, 121–130 [DOI] [PubMed] [Google Scholar]
- 29. Franzen S., Jasaitis A., Belyea J., Brewer S. H., Casey R., MacFarlane A. W., 4th, Stanley R. J., Vos M. H., Martin J. L. (2006) Hydrophobic distal pocket affects NO-heme geminate recombination dynamics in dehaloperoxidase and H64V myoglobin. J. Phys. Chem. B 110, 14483–14493 [DOI] [PubMed] [Google Scholar]
- 30. Franzen S., Belyea J., Gilvey L. B., Davis M. F., Chaudhary C. E., Sit T. L., Lommel S. A. (2006) Proximal cavity, distal histidine, and substrate hydrogen-bonding mutations modulate the activity of Amphitrite ornata dehaloperoxidase. Biochemistry 45, 9085–9094 [DOI] [PubMed] [Google Scholar]
- 31. Belyea J., Belyea C. M., Lappi S., Franzen S. (2006) Resonance Raman study of ferric heme adducts of dehaloperoxidase from Amphitrite ornata. Biochemistry 45, 14275–14284 [DOI] [PubMed] [Google Scholar]
- 32. Osborne R. L., Coggins M. K., Raner G. M., Walla M., Dawson J. H. (2009) The mechanism of oxidative halophenol dehalogenation by Amphitrite ornata dehaloperoxidase is initiated by H2O2 binding and involves two consecutive one-electron steps. Role of ferryl intermediates. Biochemistry 48, 4231–4238 [DOI] [PubMed] [Google Scholar]
- 33. Osborne R. L., Coggins M. K., Walla M., Dawson J. H. (2007) Horse heart myoglobin catalyzes the H2O2-dependent oxidative dehalogenation of chlorophenols to DNA-binding radicals and quinones. Biochemistry 46, 9823–9829 [DOI] [PubMed] [Google Scholar]
- 34. Osborne R. L., Raner G. M., Hager L. P., Dawson J. H. (2006) C. fumago chloroperoxidase is also a dehaloperoxidase: oxidative dehalogenation of halophenols. J. Am. Chem. Soc. 128, 1036–1037 [DOI] [PubMed] [Google Scholar]
- 35. Osborne R. L., Sumithran S., Coggins M. K., Chen Y. P., Lincoln D. E., Dawson J. H. (2006) Spectroscopic characterization of the ferric states of Amphitrite ornata dehaloperoxidase and Notomastus lobatus chloroperoxidase. His-ligated peroxidases with globin-like proximal and distal properties. J. Inorg. Biochem. 100, 1100–1108 [DOI] [PubMed] [Google Scholar]
- 36. Davydov R., Osborne R. L., Shanmugam M., Du J., Dawson J. H., Hoffman B. M. (2010) Probing the oxyferrous and catalytically active ferryl states of Amphitrite ornata dehaloperoxidase by cryoreduction and EPR/ENDOR spectroscopy. Detection of compound I. J. Am. Chem. Soc. 132, 14995–15004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. D'Antonio J., D'Antonio E. L., Thompson M. K., Bowden E. F., Franzen S., Smirnova T., Ghiladi R. A. (2010) Spectroscopic and mechanistic investigations of dehaloperoxidase B from Amphitrite ornata. Biochemistry 49, 6600–6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Poulos T. L., Kraut J. (1980) The stereochemistry of peroxidase catalysis. J. Biol. Chem. 255, 8199–8205 [PubMed] [Google Scholar]
- 39. Sivaraja M., Goodin D. B., Smith M., Hoffman B. M. (1989) Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science 245, 738–740 [DOI] [PubMed] [Google Scholar]
- 40. Thompson M. K., Franzen S., Ghiladi R. A., Reeder B. J., Svistunenko D. A. (2010) Compound ES of dehaloperoxidase decays via two alternative pathways depending on the conformation of the distal histidine. J. Am. Chem. Soc. 132, 17501–17510 [DOI] [PubMed] [Google Scholar]
- 41. Svistunenko D. A., Cooper C. E. (2004) A new method of identifying the site of tyrosyl radicals in proteins. Biophys. J. 87, 582–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han K., Woodin S. A., Lincoln D. E., Fielman K. T., Ely B. (2001) Amphitrite ornata, a marine worm, contains two dehaloperoxidase genes. Mar. Biotechnol. 3, 287–292 [DOI] [PubMed] [Google Scholar]
- 43. Franzen S., Thompson M. K., Ghiladi R. A. (2012) The dehaloperoxidase paradox. Biochim. Biophys. Acta 1824, 578–588 [DOI] [PubMed] [Google Scholar]
- 44. Beers R. F., Jr., Sizer I. W. (1952) A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 195, 133–140 [PubMed] [Google Scholar]
- 45. Nilges M. J., Mattson K., Bedford R. L. (2007) in ESR Spectroscopy in Membrane Biophysics (Hemminga M. A., Berliner L. eds) p. 379, Springer Springer-Verlag New York Inc., New York [Google Scholar]
- 46. Svistunenko D. A. (2004) Tyrosine residues in different proteins. Phenol ring rotation angle database. http://privatewww.essex.ac.uk/∼svist/lev1/tyrdb/home.shtml
- 47. Chouchane S., Girotto S., Kapetanaki S., Schelvis J. P., Yu S., Magliozzo R. S. (2003) Analysis of heme structural heterogeneity in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 278, 8154–8162 [DOI] [PubMed] [Google Scholar]
- 48. Ghiladi R. A., Medzihradszky K. F., Rusnak F. M., Ortiz de Montellano P. R. (2005) Correlation between isoniazid resistance and superoxide reactivity in Mycobacterium tuberculosis KatG. J. Am. Chem. Soc. 127, 13428–13442 [DOI] [PubMed] [Google Scholar]
- 49. Ghiladi R. A., Medzihradszky K. F., Ortiz de Montellano P. R. (2005) The role of the Met-Tyr-Trp crosslink in Mycobacterium tuberculosis catalase-peroxidase (KatG) as revealed by KatG(M255I). Biochemistry 44, 15093–15105 [DOI] [PubMed] [Google Scholar]
- 50. Nicoletti F. P., Thompson M. K., Howes B. D., Franzen S., Smulevich G. (2010) New insights into the role of distal histidine flexibility in ligand stabilization of dehaloperoxidase-hemoglobin from Amphitrite ornata. Biochemistry 49, 1903–1912 [DOI] [PubMed] [Google Scholar]
- 51. Egawa T., Shimada H., Ishimura Y. (2000) Formation of compound I in the reaction of native myoglobins with hydrogen peroxide. J. Biol. Chem. 275, 34858–34866 [DOI] [PubMed] [Google Scholar]
- 52. Matsui T., Ozaki S. i., Watanabe Y. (1997) On the formation and reactivity of compound I of the His-64 myoglobin mutants. J. Biol. Chem. 272, 32735–32738 [DOI] [PubMed] [Google Scholar]
- 53. Hewson W. D., Hager L. P. (1979) Oxidation of horseradish peroxidase compound II to compound I. J. Biol. Chem. 254, 3182–3186 [PubMed] [Google Scholar]
- 54. Farhangrazi Z. S., Copeland B. R., Nakayama T., Amachi T., Yamazaki I., Powers L. S. (1994) Oxidation-reduction properties of compounds I and II of Arthromyces ramosus peroxidase. Biochemistry 33, 5647–5652 [DOI] [PubMed] [Google Scholar]
- 55. Rodríguez Marañón M. J., Mercier D., van Huystee R. B., Stillman M. J. (1994) Analysis of the optical absorption and magnetic-circular-dichroism spectra of peanut peroxidase. Electronic structure of a peroxidase with biochemical properties similar to those of horseradish peroxidase. Biochem. J. 301, 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nissum M., Schiødt C. B., Welinder K. G. (2001) Reactions of soybean peroxidase and hydrogen peroxide pH 2.4–12.0 and veratryl alcohol at pH 2.4. Biochim. Biophys. Acta 1545, 339–348 [DOI] [PubMed] [Google Scholar]
- 57. Singh R., Berry R. E., Yang F., Zhang H., Walker F. A., Ivancich A. (2010) Unprecedented peroxidase-like activity of Rhodnius prolixus nitrophorin 2. Identification of the [FeIV=O Por*]+ and [FeIV=O Por](Tyr-38*) intermediates and their role(s) in substrate oxidation. Biochemistry 49, 8857–8872 [DOI] [PubMed] [Google Scholar]
- 58. Adediran S. A. (1984) Effect of pH on the formation of compounds II and III of horseradish peroxidase. J. Protein Chem. 3, 437–444 [Google Scholar]
- 59. Ghiladi R. A., Knudsen G. M., Medzihradszky K. F., Ortiz de Montellano P. R. (2005) The Met-Tyr-Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG). Autocatalytic formation and effect on enzyme catalysis and spectroscopic properties. J. Biol. Chem. 280, 22651–22663 [DOI] [PubMed] [Google Scholar]
- 60. Ma H., Thompson M. K., Gaff J., Franzen S. (2010) Kinetic analysis of a naturally occurring bioremediation enzyme. Dehaloperoxidase-hemoglobin from Amphitrite ornata. J. Phys. Chem. B 114, 13823–13829 [DOI] [PubMed] [Google Scholar]
- 61. D'Antonio E. L., Bowden E. F., Franzen S. (2012) Thin-layer spectroelectrochemistry of the Fe(III)/Fe(II) redox reaction of dehaloperoxidase-hemoglobin. J. Electroanal. Chem. 668, 37–43 [Google Scholar]
- 62. Tanaka M., Nagano S., Ishimori K., Morishima I. (1997) Hydrogen bond network in the distal site of peroxidases: spectroscopic properties of Asn-70 → Asp horseradish peroxidase mutant. Biochemistry 36, 9791–9798 [DOI] [PubMed] [Google Scholar]
- 63. Goodin D. B., McRee D. E. (1993) The Asp-His-Fe triad of cytochrome c peroxidase controls the reduction potential, electronic structure, and coupling of the tryptophan free radical to the heme. Biochemistry 32, 3313–3324 [PubMed] [Google Scholar]
- 64. Battistuzzi G., Bellei M., Zederbauer M., Furtmüller P. G., Sola M., Obinger C. (2006) Redox thermodynamics of the Fe(III)/Fe(II) couple of human myeloperoxidase in its high-spin and low-spin forms. Biochemistry 45, 12750–12755 [DOI] [PubMed] [Google Scholar]
- 65. Battistuzzi G., Bellei M., Casella L., Bortolotti C. A., Roncone R., Monzani E., Sola M. (2007) Redox reactivity of the heme Fe3+/Fe2+ couple in native myoglobins and mutants with peroxidase-like activity. J. Biol. Inorg. Chem. 12, 951–958 [DOI] [PubMed] [Google Scholar]
- 66. Heineman W. R., Meckstroth M. L., Norris B. J., Su C.-H. (1979) Optically transparent thin layer electrode techniques for the study of biological redox systems. J. Electroanal. Chem. 104, 577–585 [Google Scholar]
- 67. Conant J. B., Pappenheimer A. M., Jr. (1932) A redetermination of the oxidation potential of the hemoglobin-methemoglobin system. J. Biol. Chem. 98, 57–62 [Google Scholar]
- 68. McKinley A. B., Kenny C. F., Martin M. S., Ramos E. A., Gannon A. T., Johnson T. V., Dorman S. C. (2004) Applications of absorption spectroelectrochemistry in artificial blood research. Spectrosc. Lett. 37, 275–287 [Google Scholar]
- 69. Ayala M., Roman R., Vazquez-Duhalt R. (2007) A catalytic approach to estimate the redox potential of heme-peroxidases. Biochem. Biophys. Res. Commun. 357, 804–808 [DOI] [PubMed] [Google Scholar]
- 70. Reeder B. J., Svistunenko D. A., Cooper C. E., Wilson M. T. (2012) Engineering tyrosine-based electron flow pathways in proteins. The case of aplysia myoglobin. J. Am. Chem. Soc. 134, 7741–7749 [DOI] [PubMed] [Google Scholar]
- 71. DeFelippis M. R., Murthy C. P., Broitman F., Weinraub D., Faraggi M., Klapper M. H. (1991) Electrochemical properties of tyrosine phenoxy and tryptophan indolyl radicals in peptides and amino acid analogues. J. Phys. Chem. 95, 3416–3419 [Google Scholar]
- 72. DeFelippis M. R., Murthy C. P., Faraggi M., Klapper M. H. (1989) Pulse radiolytic measurement of redox potentials: the tyrosine and tryptophan radicals. Biochemistry 28, 4847–4853 [DOI] [PubMed] [Google Scholar]
- 73. LaCount M. W., Zhang E., Chen Y. P., Han K., Whitton M. M., Lincoln D. E., Woodin S. A., Lebioda L. (2000) The crystal structure and amino acid sequence of dehaloperoxidase from Amphitrite ornata indicate common ancestry with globins. J. Biol. Chem. 275, 18712–18716 [DOI] [PubMed] [Google Scholar]