

Background: Significance of Akt1 in myofibroblast differentiation is unknown.
Results: Akt1 mediates myofibroblast differentiation via serum response factor (SRF) and myocardin signaling leading to α-smooth muscle synthesis.
Conclusion: Akt1-myocardin-SRF signaling induces myofibroblast differentiation.
Significance: The role of Akt1 on myofibroblast differentiation highlights it as a potential target for the treatment of fibrotic diseases.
Keywords: Akt, Differentiation, Fibronectin, Fibrosis, Myofibroblast, Myocardin, Serum Response Factor
Abstract
Myofibroblast (MF) differentiation, marked by the de novo expression of smooth muscle α-actin (αSMA) stress fibers, plays a central role in wound healing and its persistence is a hallmark of fibrotic diseases. We have previously shown that Akt1 is necessary for wound healing through matrix regulation. However, the role of Akt1 in regulating MF differentiation with implications in fibrosis remains poorly defined. Here, we show that sustained activation of Akt1 was associated with a 6-fold increase in αSMA expression and assembly; an effect that is blunted in cells expressing inactive Akt1 despite TGFβ stimulation. Mechanistically, Akt1 mediated TGFβ-induced αSMA synthesis through the contractile gene transcription factors myocardin and serum response factor (SRF), independent of mammalian target of rapamycin in mouse embryonic fibroblasts and fibroblasts overexpressing active Akt1. Akt1 deficiency was associated with decreased myocardin, SRF, and αSMA expressions in vivo. Furthermore, sustained Akt1-induced αSMA synthesis markedly decreased upon RNA silencing of SRF and myocardin. In addition to its integral role in αSMA synthesis, we also show that Akt1 mediates fibronectin splice variant expression, which is required for MF differentiation, as well as total fibronectin, which generates the contractile force that promotes MF differentiation. In summary, our results constitute evidence that sustained Akt1 activation is crucial for TGFβ-induced MF formation and persistent differentiation. These findings highlight Akt1 as a novel potential therapeutic target for fibrotic diseases.
Introduction
Since its discovery in the early 1970s (1, 2), the myofibroblast (MF)2 has emerged as a central orchestrator of wound repair and pathologic hypertrophic scar formation characteristic of fibrotic diseases (3–6). Differentiated from fibroblasts, MFs exhibit a contractile phenotype marked by de novo expression of α-smooth muscle cell like actin (αSMA) stress fibers (7) and are primarily responsible for excessive extracellular matrix (ECM) production, cell to matrix adhesion, and resistance to apoptosis (3, 8–10). It is well established that accumulation of transforming growth factor β1 (TGFβ) is a major inducer of MF differentiation during wound healing and fibrosis (8, 11–13). Interestingly, basic fibroblast growth factor (bFGF) has been shown to control the extent of TGFβ-induced effects via stimulating de-differentiation of MFs (14, 15). This shows the intricate balance between these two cytokines in mediating physiological and pathophysiological fibrogenic responses.
Synthesis of αSMA, a principal component of MFs, is a highly regulated process that is controlled by TGFβ, splice variant ED-A fibronectin (ED-A FN), and mechanical tension (3, 5). Numerous lines of evidence indicate that the transcription of αSMA is reliant on RhoA-mediated activation and nuclear translocation of transcription factor serum response factor (SRF) (16–24). Furthermore, the interaction between the recently discovered myocardin, a transcription co-factor restricted to cardiac and smooth muscle cells, and SRF has been characterized in orchestrating contractile gene expression (18, 25–27). However, defining signaling pathways that regulate this transcription network in the hopes of devising targeted therapeutics remain incompletely understood.
Research from our laboratory has established the pivotal role of protein kinase Bα (Akt1) in wound healing, ECM remodeling, and vascular maturation (28). Furthermore, we have shown that Akt1 is instrumental for normal cellular processes including fibroblasts migration, proliferation, and cytoskeletal remodeling (29), and assembly of ECM proteins including fibronectin (30, 31). However, the precise role of Akt in MF differentiation remains unclear. Several studies have reported a possible positive correlation between Akt and αSMA (32–35), and have established its role in anoikis resistance (11, 36). Because Akt1 is the predominant isoform in fibroblasts, these studies suggest a role for Akt1 in MF differentiation. Collectively, this has prompted us to postulate that Akt1 modulates αSMA synthesis through myocardin and SRF leading to MF differentiation.
In the present study, our results demonstrate that Akt1 is critical for αSMA synthesis in MF differentiation. Our studies constitute evidence of a novel signaling cascade that links Akt1 to αSMA synthesis through enhanced expression of myocardin and SRF, a previously uncharacterized link in MFs. We also demonstrate the dual role of Akt1 in mediating αSMA synthesis and ED-A FN splice variant along with total fibronectin. In conclusion, our studies suggest that Akt1 is a potential therapeutic target in fibrotic diseases.
EXPERIMENTAL PROCEDURES
Cell Lines and Cell Culture
NIH 3T3 fibroblasts were obtained from ATCC (Manassas, VA) and stable transfected through retroviral infections using control pBabe plasmids and those expressing constitutively active CA-Akt1-GFP fusion (90 kDa) (also known as myristoylated-Akt1 (myr-Akt1), or DN-Akt1 (Akt1 K179M) vectors with puromycin resistance. Selection with antibiotic was carried out until 100% transfection efficiency was confirmed by GFP staining. To examine the role of Akt1: NIH 3T3, transfected with stable empty vector, myr-Akt1, or DN-Akt1 vector, were cultured on 6-well plates. After reaching 70% confluence, cells were subjected to serum starvation in the presence or absence of 100 pm TGFβ (a pre-determined dose (31)) for 72 h, a standardized time point that is associated with maximum MF differentiation. Cells were subjected to Western analyses and immunocytochemistry as described below. For the mechanistic pharmacologic inhibition studies: after reaching 70% confluence, NIH 3T3 and myr-Akt1 cells were treated with bFGF (20 ng/ml (29)) or TGFβ (100 pm) for 48 h. This was followed by co-treatment for 24 h (total 72 h) with inhibitors of PI3 kinase (25 μm LY294002), Akt (10 nm triciribine), mammalian target of rapamycin (mTOR) (25 nm rapamycin), or SRF/Rho (1 μm CCG1423). Cells were subjected to Western analyses as described below.
Antibodies
Anti-αSMA (catalog number SAB2500963) and anti-fibronectin (catalog number F6140) antibodies were purchased from Sigma. GAPDH (catalog number 2118), phospho-Akt (Ser-473) (catalog number 9271), and anti-SRF (catalog number 5147) antibodies were purchased from Cell Signaling (Boston, MA). Anti-myocardin (catalog number MAB 4028) antibodies were purchased from R&D Systems (Minneapolis, MN). Anti-ED-A-fibronectin (catalog number 6328) and anti-αSMA (catalog number 5694) antibodies were purchased from Abcam (Cambridge, MA).
Western Blot Analysis
Cell lysates were prepared using lysis buffer (20 mm Tris-HCl, pH 7.4, 1% Triton X-100, 3 mm EGTA, 5 mm EDTA, phosphatase inhibitors (10 mm sodium pyrophosphate, 5 mm sodium orthovanadate, 5 mm sodium fluoride, and 10 μm okadaic acid), protease inhibitor mixture (Roche Diagnostics) and 1 mm PMSF). SDS-PAGE and Western blotting were performed as described previously (31).
siRNA Transfection
For myocardin siRNA (100 nm) and SRF siRNA (150 nm), a pool of two target-specific siRNAs (Qiagen) were designed to knockdown mouse myocardin and SRF gene expression, respectively. A non-silencing oligonucleotide sequence (non-silencing siRNA) that does not recognize any known homology to mammalian genes was obtained as a negative control. NIH 3T3 and myr-Akt1 fibroblasts were transfected with siRNAs using a FuGENE transfection kit (Qiagen) for 24–48 h with TGFβ (100 pm) treatment for 72 h (supplemental Fig. S1). Cells were subjected to Western analyses and immunocytochemistry to detect αSMA expression and assembly, respectively.
Three-dimensional Collagen Gel Contraction Assay
Prior to preparing collagen gels as described below, fibroblasts were detached by 0.05% trypsin in 0.53 mm EDTA and suspended in 10 ml of serum-free DMEM containing soybean trypsin inhibitor. The cell number was then counted with Coulter Counter. Collagen gels were prepared according to the manufacturer's protocol (Cytoskelton, CO) by mixing rat tail tendon collagen, distilled water, 4× DMEM, and cells. The final concentration was 1× DMEM, 0.75 mg/ml of collagen, and fibroblasts were present at 3 × 105 cells/ml. Following this, 500 μl of the mixture was cast into each well of a 24-well culture plate. The solution was then allowed to polymerize for 1 h. After polymerization, the gels were allowed to remain attached to the plates for 48 h to form stress in the presence of DMEM without FBS, then gels were gently released from the plates to mimic free-floating. The optimum concentrations of bFGF, TGFβ, and triciribine were standardized in the laboratory. The area of each gel was measured daily and imaged. Data are expressed as the percentage of area compared with the initial gel area.
Immunocytochemistry
Immunofluorescence staining was performed as described previously (29). Briefly, NIH 3T3, transfected with stable empty vector, myr-Akt1, or DN-Akt1 vector, were plated on 8-well chamber slides. After reaching 70% confluence, cells were subjected to serum starvation in the presence or absence of TGFα for 72 h. Next, cells were fixed with 4% paraformaldehyde in 1× PBS followed by permeabilization with 0.1% Triton X-100 in 1× PBS. The nonspecific staining was blocked with 2% BSA for 1 h at room temperature. The fixed and permeabilized cells were incubated with primary anti-αSMA antibody (Abcam) (dilution 1:1000) overnight at 4 °C and washed. Secondary Alexa Fluor 488-labeled antibody was applied for 1 h followed by standardized dilution of Alexa Fluor 555-labeled phalloidin (Invitrogen) for 40 min. The slides were mounted with Vectashield (Vector Laboratories, PA), and imaged by a Zeiss fluorescent microscope.
Animals
All experiments were performed with approval by the Charlie Norwood Veterans Affairs Medical Center Institutional Animal Care and Use Committees. Akt1−/− mice were generated as previously described (28) and were maintained in the C57BL/6 background. Sex and age-matched wild-type and Akt1−/− were randomized to normobaric hypoxia (10% O2) (Biospherix, New York) or room air. To establish the hypoxic environment, the chamber was flushed with nitrogen. The chamber was opened once per week for no more than 1 h to clean the cages and replenish food and water supplies. After 14 days, mice were euthanized; lungs were isolated and subjected to Western analyses.
Statistical Analysis
All data are presented as mean ± S.D. To determine significant differences between treatment and control values, we used the Student's two-tailed t test. The significance was set at 0.05 levels (marked with symbols wherever data are statistically significant).
RESULTS
Akt1 Inactivation Abolishes TGFβ-induced Myofibroblast Differentiation
We first determined that 72 h is the optimal time for TGFβ-induced MF differentiation in NIH 3T3 fibroblasts, as measured by a 5-fold increase in αSMA expression). To examine whether Akt1 activation is required for αSMA expression, the marker for MF differentiation, fibroblasts transfected with constitutively active Akt1 (myr-Akt1) and inactive dominant-negative-Akt1 (DN-Akt1; Akt1 K179M), respectively, were serum starved and treated with control PBS or TGFβ (100 pm) for 72 h. The results showed that despite the absence of TGFβ stimulation, sustained hyperactivation of Akt1 significantly enhanced αSMA expression (∼6-fold) and assembly compared with control fibroblasts; this effect was further amplified with TGFβ stimulation (Fig. 1, A and B). In contrast, inactivation of Ak1 blunted the stimulatory effects of TGFβ on αSMA expression and assembly (Fig. 1, A and B). Thus, the absence of Akt1 impedes TGFβ-induced αSMA expression, implying that Akt1 is an important modulator of MF differentiation.
FIGURE 1.
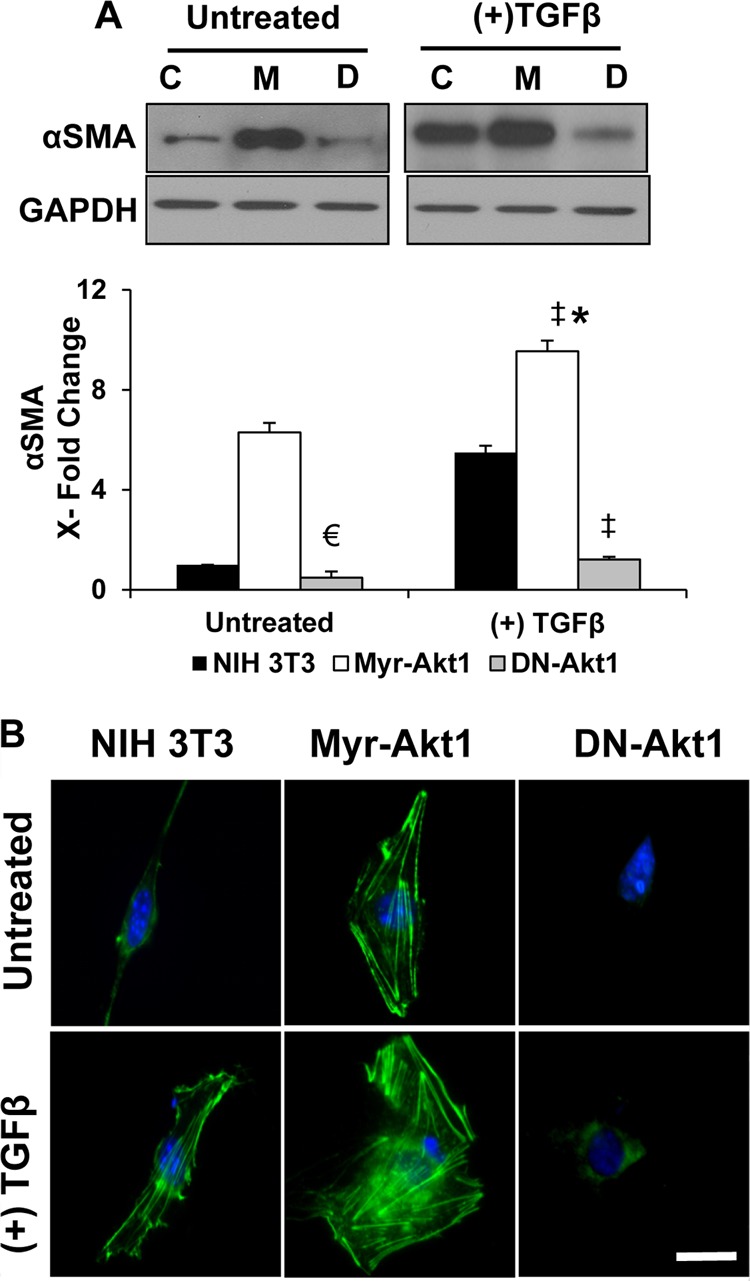
Akt1 is critical for αSMA expression in response to TGFβ. A, control vector (pBabe-Puro), myr-Akt1, and DN-Akt1 expressing NIH 3T3 fibroblasts grown to 80% confluence were subjected to serum-free medium in the presence and absence of 100 pm TGFβ for 72 h. Lysates were prepared and subjected to Western analyses with antibodies against αSMA (n = 3) (C, control NIH 3T3; M, myr-Akt1; D, DN-Akt1) (▴, p = 0.0001 and €, p = 0.02 compared with control untreated NIH 3T3; *, compared with myr-Akt1 control; ‡, compared with TGFβ-treated NIH 3T3 cells). B, cells, subjected to serum-free medium in the presence and absence of 100 pm TGFβ for 72 h, were fixed using 4% paraformaldehyde and stained using anti-αSMA antibodies. Fluorescent images of αSMA assembly were visualized using a fluorescence microscope and photographed. Scale bar, 50 μm.
Akt1-mediated Dynamic Switch between Fibroblast and MFs Is Stimuli Dependent
Because physiological modulators of fibroblasts such as bFGF also activate Akt1 (Fig. 2A), we examined the effect of bFGF on αSMA expression and MF differentiation. To do this, we subjected control and myr-Akt1 expressing fibroblasts to serum starvation and treated the fibroblasts with control PBS, TGFβ (100 pm), or bFGF (20 ng/ml). In contrast to the stimulatory effect of TGFβ, bFGF was associated with diminished αSMA expression in control and myr-Akt1 expressing NIH 3T3 fibroblasts (Fig. 2B). This suggested a molecular see-saw of bFGF and TGFβ, clearly demonstrating a reciprocal regulation of physiological and pathological fibrogenic events by these two cytokines via differential regulation of Akt1 activity (Fig. 2, A and B).
FIGURE 2.
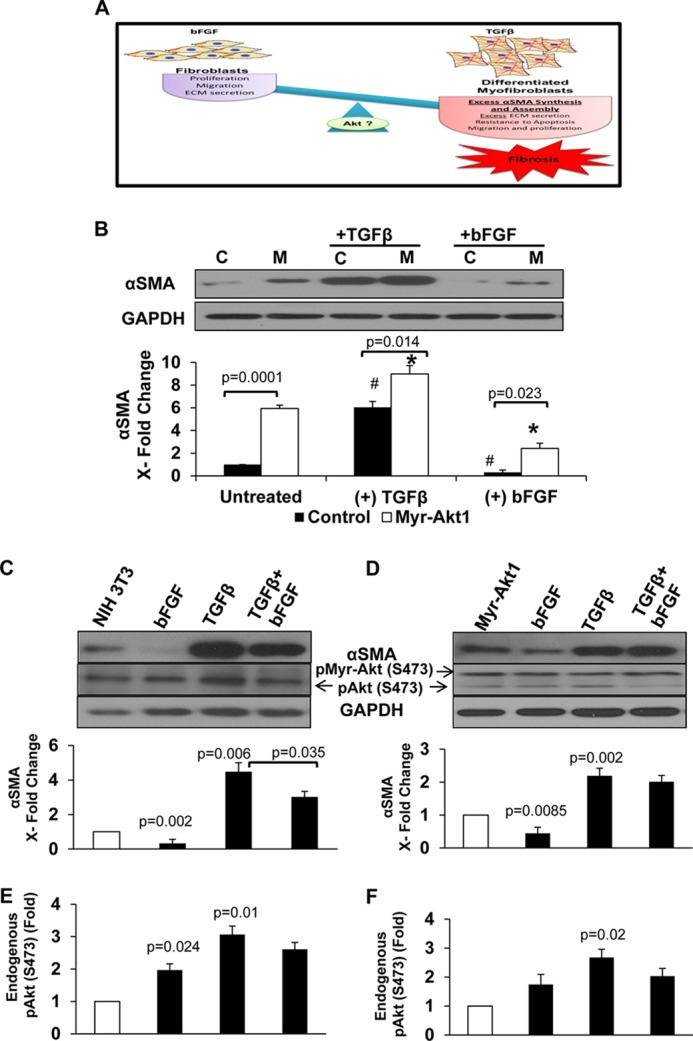
Akt1 is differentially utilized by TGFβ and bFGF in fibroblast to MF differentiation. A, a schematic representation of the hypothesis that the stimuli-dependent dynamic switch between fibroblasts (bFGF) and MFs (TGFβ) is mediated by Akt1. Control vector (B) and myr-Akt1 (C) (M) expressing fibroblasts were subjected to serum starvation alone or plus treatment with 20 ng/ml of bFGF or 100 pm TGFβ for 72 h. Lysates were prepared and subjected to Western analyses with antibodies against αSMA (n = 3) (C, control NIH 3T3; M, myr-Akt1; D, DN-Akt1) (*, compared with myr-Akt1 control; #, compared with control; ♦, bFGF compared with TGFβ). C–F, to examine the de-differentiation potential, control and Myr-Akt1 expressing NIH 3T3 cells were treated with TGFβ for 72 h plus bFGF (for an additional 24 h) for a total of 96 h. Lysates were prepared and subjected to Western analyses with antibodies against αSMA and phospho-Ser-473 Akt (n = 3).
This differential regulation of αSMA expression by bFGF and TGFβ prompted us to investigate whether Akt1 is involved in the de-differentiation effect of bFGF. Because maximum MF differentiation occurred at 72 h, to study the de-differentiation effect, we treated control and myr-Akt1 NIH 3T3 fibroblasts with bFGF (or control PBS) for 24 h after a 72-h TGFβ stimulation (total 96 h). Although treatment with bFGF partially, but significantly, inhibited TGFβ-induced αSMA expression in fibroblasts (Fig. 2C), it did not show any discernible de-differentiation in myr-Akt1 expressing cells (Fig. 2D). Interestingly, although bFGF treatment increased Akt phosphorylation in NIH 3T3 cells, in combination with TGFβ, it decreased the TGFβ-induced Akt phosphorylation (Fig. 2, C and E). Furthermore, although myr-Akt1 expressing cells were resistant to bFGF-induced de-differentiation, a decrease in endogenous Akt phosphorylation was observed in these cells (Fig. 2, D and F), thus indicating that the resistance to bFGF treatment in these cells was purely due to the expression of myr-Akt1. Together, these data indicate the need for sustained Akt1 activation in fibroblasts to drive αSMA synthesis and MF differentiation. This supports our previous findings that, whereas transient activation of Akt1 in response to bFGF mainly regulates fibroblast proliferation, matrix adhesion, and assembly (29), sustained Akt1 activation promotes TGFβ-induced MF differentiation.
Akt1 Modulates TGFβ-induced MF Differentiation through SRF
Once we identified Akt1 as a key modulator of αSMA expression, we sought to define and characterize the mechanisms by which Akt1 mediates TGFβ-induced αSMA expression and MF differentiation. Because studies have shown that the transcription factor SRF is integral for αSMA synthesis, we explored whether SRF is also a potential target of the PI3K/Akt/mTOR pathway during MF differentiation. To investigate this, we utilized inhibitors of PI3K (25 μm LY294002), Akt (10 nm triciribine), mTOR (25 nm rapamycin), and Rho signaling-responsive SRF (1 μm CCG1423) (Calbiochem) in control and myr-Akt1 expressing fibroblasts stimulated with TGFβ, and determined αSMA and SRF expressions, and Akt phosphorylation.
TGFβ-induced αSMA expression was correlated with elevated SRF expression and phosphorylation of Akt in NIH 3T3 cells (Fig. 3, A–D) and myr-Akt1 cells (Fig. 3, E–H). Although inhibiting PI3K resulted in a moderate decrease in αSMA, SRF, and Akt phosphorylation compared with TGFβ-treated NIH 3T3 cells, targeting Akt abolished TGFβ-induced αSMA expression and was associated with a marked decrease in SRF expression and Akt phosphorylation (Fig. 3, A–D). TGFβ-stimulated myr-Akt1 expressing fibroblasts while exhibiting resistance upon PI3K inhibition, inhibiting Akt resulted in a significant decrease in αSMA and SRF expression, as well as Akt phosphorylation compared with TGFβ-treated myr-Akt1 fibroblasts (Fig. 3, E–H). Inhibiting RhoA produced a modest but significant inhibition of αSMA that was associated with a significant reduction of SRF expression and Akt phosphorylation compared with TGFβ-treated fibroblasts in NIH 3T3 (Fig. 3, A–D) and myr-Akt cells (Fig. 3, E–H). However, a significant effect of mTOR inhibitor rapamycin on αSMA expression was not observed (Fig. 3, A, B, E, and F). Interestingly, in bFGF-stimulated cells, inhibiting mTOR promoted αSMA and SRF expressions, implying that mTOR is a key factor in maintaining fibroblast stability under bFGF and suppress its differentiation to MFs (Fig. 4, A–H). Taken together, these results show that TGFβ-induced αSMA expression is mediated by the PI3K/Akt/SRF pathway, independent of mTOR.
FIGURE 3.

TGFβ-induced αSMA expression is Akt1-dependent through SRF, independent of mTOR. NIH 3T3 fibroblasts (A–D) and myr-Akt1 expressing fibroblasts (E–H) grown to 70% confluence were treated with TGFβ for 72 h in the absence of FBS. To examine pathways governing MF differentiation, inhibitors were applied before significant differentiation occurred: cells were treated with TGFβ for 48 h and then co-treated with inhibitors for an additional 24 h targeting the PI3K/Akt/mTOR pathway and responsive SRF pathway (LY294002, triciribine (TCBN), rapamycin, and CCG1423, respectively). Lysates were prepared and subjected to Western analyses with antibodies against αSMA (B and F), SRF (C and G), and pAkt (Ser-473) (D and H) (n = 4). *, compared with TGFβ; ‡, compared with control untreated cells.
FIGURE 4.

bFGF-induced αSMA down-regulation is mTOR dependent. NIH 3T3 fibroblasts (A–D) and myr-Akt1 expressing fibroblasts (E–H) grown to 70% confluence were treated with bFGF for 72 h in the absence of FBS. To examine pathways governing bFGF-mediated effects on MF differentiation, cells were treated with bFGF for 48 h and then co-treated with inhibitors for an additional 24 h targeting the PI3K/Akt/mTOR pathway and responsive SRF pathway (LY294002, triciribine (TCBN), rapamycin, and CCG1423, respectively). Lysates were prepared and subjected to Western analyses with antibodies against αSMA (B and F), SRF (C and G), and pAkt (Ser-473) (D and H) (n = 4). *, compared with bFGF; ‡, compared with control untreated cells.
Akt1 Regulates αSMA Synthesis through SRF and Myocardin
Because SRF is ubiquitous, not contractile gene specific, and because myocardin is a critical transcriptional co-activator of smooth muscle cell genes (37), we considered whether the regulatory effect of Akt1 on αSMA expression is mediated through myocardin in conjunction with SRF. We first evaluated the expression of myocardin and SRF in relationship to TGFβ-induced αSMA expression during MF differentiation. The results show that at 72 h, αSMA expression correlates with a significant increase in myocardin and SRF levels (∼6- and ∼3-fold, respectively) in NIH 3T3 cells (Fig. 5, A–D). Interestingly, in the absence of TGFβ stimulation, myr-Akt1-induced αSMA expression correlated with a 3.5-fold increase in myocardin and a 1.8-fold increase in SRF expressions, compared with control fibroblasts (Fig. 5, E–H).
FIGURE 5.

Akt1 induces MF differentiation through enhanced expression of myocardin and SRF. NIH 3T3 fibroblasts treated with TGFβ for 72 h (A–D) and myr-Akt1 fibroblasts (E–H) were subjected to Western analysis for SMA, myocardin, and SRF expression (n = 4).
We argued that if Akt1 acts at least in part through myocardin and SRF, then silencing either protein should result in reduced αSMA. To test this, we performed gene knockdown experiments with myocardin siRNA and SRF siRNA. NIH 3T3 and myr-Akt1 fibroblasts were transfected with control vector, myocardin, or SRF siRNA (siMyoc and siSRF, respectively) for 48 h in the absence or presence of TGFβ; cells were pretreated with TGFβ to promote MF differentiation. Although sustained activation of Akt1 resulted in increased αSMA expression and assembly, knockdown of myocardin and SRF significantly reduced αSMA levels in myr-Akt1 expressing fibroblasts both in the absence (Fig. 6, B and E) and presence of TGFβ (Fig. 6, D and F). To validate our in vitro observations, we utilized a chronic hypoxia model as it has been shown to induce fibroblast to MF differentiation in animal models thus playing a major role in fibrotic response (38, 39). Hence, as a proof of concept and further confirm the role of Akt1 in αSMA synthesis in vivo, we subjected wild-type (WT) and Akt1 null (Akt1−/−) mice to continuous hypoxia exposure for 14 days and lung tissues were subjected for Western analysis. As expected, Akt1−/− mice had significantly lower αSMA, myocardin, and SRF expressions in the lung tissue compared with WT mice (Fig. 6, G and H). Taken together, our data demonstrate that Akt1 plays a crucial permissive role in αSMA synthesis mediated, in part, by regulating myocardin and SRF expressions.
FIGURE 6.

siRNA-mediated knockdown of myocardin and SRF ablates Akt1-induced SMA expression and MF differentiation. Control NIH 3T3 fibroblasts (A and C) and myr-Akt1 fibroblasts (B and D) were transfected with control, myocardin, or SRF siRNA (siMyoc, 100 nm and siSRF, 150 nm), and 24 to 48 h later, cells were treated with 100 pm TGFβ for 72 h. Lysates were prepared and subjected to Western analyses, quantification was normalized to control (*, p < 0.001, p < 0.01) (n = 3). E and F, cells were fixed using 4% paraformaldehyde and stained using anti-αSMA antibodies. Fluorescent images of αSMA assembly were visualized using a fluorescence microscope and photographed. Scale bar, 50 μm. G and H, as a proof of concept, wild type (WT) Akt null (Akt1−/−) mice were subjected to chronic hypoxia (a well established model of pulmonary hypertension that induces marked MF differentiation). After 14 days of continuous exposure to 10% oxygen, mice were euthanized and whole lungs collected, homogenized, and analyzed using Western blotting for αSMA, myocardin, SRF, and Akt1 expressions. Data were normalized to GAPDH (n = 4–5).
Akt1 Plays a Dual Role as a Modulator of TGFβ-induced αSMA and Fibronectin
Because both MF differentiation and excess ECM secretion constitute key hallmark events of deregulated tissue remodeling that occurs in fibrotic conditions such as idiopathic pulmonary fibrosis and cirrhosis (3, 9, 34, 40, 41) and having seen that Akt1 is crucial for αSMA synthesis, we asked whether it could be involved in pathologic ECM regulation as well. We have previously shown that Akt1 regulates the synthesis of ECM proteins such as fibronectin via activation of mTOR pathway (31). Fibronectin splice variant ED-A FN, which is not typically expressed in undifferentiated fibroblasts, is mandatory for TGFβ-induced αSMA synthesis and MF differentiation (3). Furthermore, total fibronectin, which is secreted excessively by differentiated MFs, also exerts mechanical tension on MFs as part of a positive feedback loop (3). Therefore, to determine whether Akt1 is involved in expression of the ED-A FN splice variant in the regulation of MF differentiation, we subjected control and myr-Akt1 expressing fibroblasts to TGFβ for 48 h and co-treated with inhibitors of Akt1 and mTOR for an additional 24 h. Expression of αSMA, ED-A FN, total fibronectin and phosphorylated Akt was determined. Inhibiting Akt significantly reduced the stimulatory effect of TGFβ on αSMA, ED-A FN, and fibronectin expressions. However, treatment with the mTOR inhibitor rapamycin only inhibited total fibronectin expression with no discernible decrease in αSMA expression in both control NIH 3T3 (Fig. 7, A–D) and myr-Akt1 expressing fibroblasts (Fig. 7, A and E-G). It is noteworthy that rapamycin reduced ED-A FN expression only in myr-Akt1 cells implying that under physiological conditions mTOR does not regulate its expression. Herein, we demonstrate that whereas both Akt1 and mTOR are needed for total fibronectin synthesis (31), Akt1 also modulates the expression of specialized ED-A FN, which sheds light on the mechanisms underlying the dynamic reciprocal mechanical interactions between cells and the ECM during MF differentiation.
FIGURE 7.

Akt1-mediated expression αSMA and ED-A FN in response to TGFβ is independent of mTOR activity. Control NIH 3T3 (A–D) and myr-Akt1 expressing fibroblasts (E–H) grown to 70% confluence were subjected to serum-free medium and treated with TGFβ for 72 or 48 h and then co-treated with inhibitors for an additional 24 h targeting the Akt/mTOR pathway (triciribine (TCBN) and rapamycin, respectively). Lysates were prepared and subjected to Western analyses with antibodies against αSMA, ED-A FN, total fibronectin, and pAkt(Ser-473) (n = 3). *, compared with TGFβ; ‡, compared with control untreated cells.
Targeted Akt Inhibition Reverses TGFβ-mediated Contraction of Fibroblast-populated Collagen Lattices
To validate the observation with regards to the functional characteristics of myofibroblasts, we performed a collagen gel contraction assay and examined the degree of contraction of a three-dimensional collagen lattice, an accepted model system of MF contractility (42). We compared the de-differentiation effect of inhibiting Akt using triciribine to bFGF in TGFβ-stimulated gels. Control and bFGF-treated lattice exhibited a slight contraction as a result of physiological proliferation of the fibroblasts, but the effects were not significant. At 96 h, the marked TGFβ-induced contraction, whereas moderately reduced by bFGF treatment, was markedly ameliorated upon Akt inhibition (Fig. 8, A and B). It is noteworthy that compared with the contractility baseline at 72 h, bFGF had no discernible effect on gel contraction (Fig. 8C) suggesting that TGFβ-mediated Akt activation is necessary for the contractile force generated by MFs. Collectively, these morphological, biochemical, and functional data confirm a role of Akt1 in mediating, at least in part, TGFβ effects on MF differentiation.
FIGURE 8.
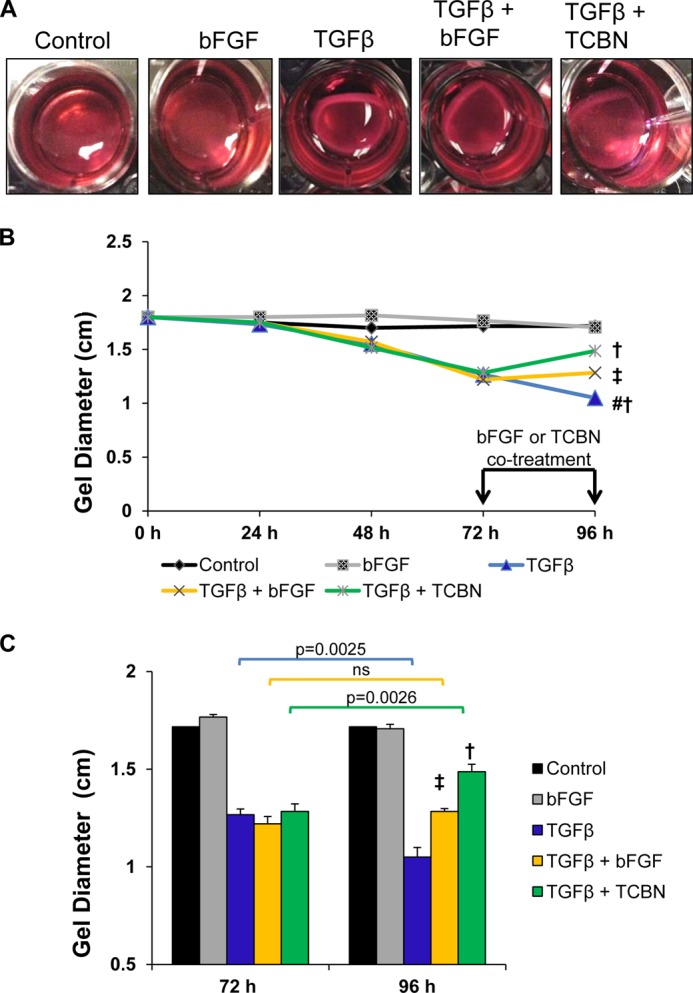
Akt inhibition reverses TGFβ-mediated contraction of fibroblast-populated collagen lattices. A, after seeding NIH 3T3 fibroblasts in neutralized collagen solution, cells were serum starved and incubated with bFGF or TGFβ for 96 h or TGFβ for 72 h, and co-treated with bFGF or triciribine (TCBN) for an additional 24 h (total 96 h) to examine the de-differentiation effect. Degree of gel contraction was assessed daily. B, degree of contraction was compared at 96 h between groups. C, degree of de-differentiation/relaxation compared with 72 h (n = 3). #, compared with control; †, p = 0.0001 compared with TGFβ; ‡, p = 0.001 compared with TGFβ.
DISCUSSION
In the present study, we provide the first conclusive evidence on the significance of sustained Akt1 activation in persistent MF differentiation with potential pathophysiological implications in tissue fibrosis. First, whereas sustained activation of Akt1 mimicked TGFβ stimulation and induced an ∼6-fold increase in αSMA, inactivation of Akt1 blunted TGFβ-induced αSMA expression and assembly. Second, Akt1 mediates αSMA synthesis, in part, through modulation of myocardin and SRF expression, independent of mTOR, a novel and previously uncharacterized link in MFs. Third, in addition to its control on total fibronectin, Akt1 induces ED-A FN expression, which is mandatory for MF differentiation. Fourth, TGFβ-mediated Akt1 activation is necessary for the functional contractile force generated by MFs. Collectively, our study identifies the critical role of Akt1 and defines novel mechanisms by which it mediates MF differentiation, and sheds light on the dynamic mechanical interactions between the MF and ECM. Together, this creates a deregulated continuous positive feedback loop resulting in persistent TGFβ-induced MF differentiation with implication in progression of fibrotic diseases (supplemental Fig. S2).
Several lines of evidence have identified and characterized multiple inter-cellular networks dictating MF differentiation. However, defining key signaling pathways leading to MF differentiation with the hope of devising targeted therapeutics remains elusive. Thus far, extensive research has been geared toward RhoA-mediated αSMA synthesis in smooth muscle cells and MFs. Literature also suggests a correlation between Akt activation and αSMA expression during MF differentiation (32–35). Additionally, it has been shown that deficiency in phosphatase and tensin homologue deleted on chromosome 10 that normally inactivate Akt resulted in a markedly high proliferation rate and increase in αSMA and collagen expressions (43). This led us to investigate the role of Akt, Akt1 in particular, as a mediator of persistent MF differentiation. We show that inactivation of Akt1 abolished TGFβ-induced αSMA expression. Noteworthy, by conducting our studies in NIH 3T3 fibroblasts treated with TGFβ, fibroblasts expressing myr-Akt1 with sustained (constitutively active) Akt1 activity, fibroblasts expressing DN-Akt1 (inactive), and Akt1 knock-out mice, we confirm our findings and demonstrate the clinical relevance of the association between Akt1 and MF differentiation with implications in fibrotic diseases.
RhoA-dependent signaling has been shown to involve SRF, which plays a major role in αSMA synthesis (4, 16–24, 26, 44, 45). Notably, SRF, a ubiquitous transcription factor, is not smooth muscle-specific. Furthermore, Olson and colleagues (25) discovered that myocardin, the transcription co-activator of SRF, is a critical master activator of αSMA gene expression. Although fibroblasts are not reported to constitutively express myocardin, which is assumed to be specific to cardiac and smooth muscle cells, forced expression of myocardin has been demonstrated to drive the expression of αSMA in fibroblasts (46). Our studies showed that the expression of both SRF and myocardin were up-regulated during TGFβ-induced MF differentiation in NIH 3T3 fibroblasts. Interestingly, we observed a positive correlation between Akt1 activity and expression levels of SRF and myocardin. To further delineate this potential molecular cross-talk between Akt1 and SRF-myocardin complex in αSMA synthesis and MF differentiation, we utilized an siRNA-mediated gene knockdown approach to target myocardin and SRF. Our studies revealed that in un-stimulated myr-Akt1 fibroblasts, myocardin and SRF knockdown resulted in ∼50% attenuation of αSMA synthesis and MF differentiation. Additionally, genetic ablation of Akt1 correlated with decreased SRF and myocardin in vivo, which further confirms this association. Earlier, our experiments utilizing the RhoA/SRF inhibitor CCG-1423 exhibited ∼50% decrease in MF differentiation, which led us to postulate that Akt1 and RhoA, two parallel signaling pathways might reconcile their effects in modulating TGFβ-induced αSMA synthesis. The results from the myocardin and SRF siRNA studies lend support to our hypothesis that both Akt1 and RhoA cooperate in the regulation of αSMA synthesis, the former regulating the expression of SRF and myocardin, and the latter leading to phosphorylation and nuclear translocation of the SRF-myocardin complex. Further investigation into this signaling network will likely shed light on the intricate dynamic network governing this phenotypic behavior.
Another intriguing finding from our studies is the paradoxical utilization of Akt1 in modulating αSMA synthesis. We have previously demonstrated that Akt1 promotes bFGF-induced migration and proliferation (29). Indeed, Greenberg et al. (47) have demonstrated that bFGF negatively regulates TGFβ-induced αSMA synthesis thus maintaining fibroblast phenotype and promoting MF de-differentiation. The interesting finding that sustained Akt1 activation is sufficient to drive a marked increase in αSMA expression and resistance to bFGF-induced de-differentiation reflects the preferential effect of Akt1 in mediating cell differentiation as opposed to proliferation. The differential utilization of Akt1, in addition to being dictated by growth factors, appears to also be dependent on the strength and duration of the stimuli and, importantly, the cooperation of Akt1 with RhoA in response to TGFβ, but not with bFGF, thereby highlighting the importance of Akt1 in physiologic regulation and, importantly, pathologic differentiation deregulation.
We have previously shown that the Akt1-mTOR pathway regulates FN translation and ECM secretion and assembly (29, 31). Recent studies have suggested that Akt can modulate alternative splicing of FN extra domain III (ED-A FN), which plays a major role in promoting MF differentiation (48–50). We found that unlike Akt1, targeting mTOR did not infer discernible effects on TGFβ-induced αSMA synthesis. This prompted us to investigate their effects on total FN and ED-A FN in MFs. Our data show that, in addition to its profound effects on αSMA expression and assembly, targeting Akt markedly decreased both ED-A FN and total FN, which further highlights the pro-differentiation potential of the Akt1 pathway. Interestingly, targeting mTOR did not influence ED-A FN (or did so marginally), which suggests that mTOR primarily regulates total FN as part of ECM regulation not MF differentiation per se. However, in the presence of robust sustained activation of Akt1 along with TGFβ stimulation, rapamycin treatment did infer some effect on ED-A FN in addition to total FN in myr-Akt1 fibroblasts. The results from NIH 3T3 studies are in line with recent reports that suggested that rapamycin, whereas primarily blocks FN translation via regulation of S6K, can enhance the activity of Akt1 via a positive feedback loop (50, 51). It has been suggested that elevated basal levels of Akt can directly mediate alternative splicing of ED-A FN (50, 51). However, our findings in myr-Akt1 fibroblasts that rapamycin modestly affects both ED-A FN and FN raise the possibility that constitutive activation of Akt1 may, partly, engage mTOR in the translational regulation of ED-A FN. Further studies are warranted to confirm this hypothesis.
In conclusion, we identify Akt1 as an integral modulator of αSMA synthesis, and found it to be mediated, in part, through interaction with myocardin and SRF, independent of mTOR activity. Moreover, we shed light on the dual and functional role of Akt1 in regulating ECM fibronectin as well as ED-A FN splice variant driving persistent MF differentiation and contractile force generation. We also characterize the paradoxical use of Akt1 in differentially mediating physiological and pathological effects in response to bFGF and TGFβ up-regulation, respectively. Collectively, these findings highlight the importance of Akt1 as a novel therapeutic target for fibrotic diseases.
This work was supported, in whole or in part, by National Institutes of Health Grant R01HL103952, American Heart Association Scientist Development Grant 0830326N (to P. R. S.), American Heart Association Predoctoral Fellowship 13PRE17100070 (to M. A.), and grants from the University of Georgia Research Foundation, University of Georgia College of Pharmacy Dean's Foundation (to P. R. S.).

This article contains supplemental Figs. S1 and S2.
- MF
- myofibroblast
- Akt
- protein kinase B
- αSMA
- α-smooth muscle like actin
- TGFβ
- transforming growth factor β
- bFGF
- basic fibroblast growth factor
- DN-Akt
- dominant-negative Akt
- Myoc
- myocardin
- myr-Akt
- myristoylated (active) Akt
- SRF
- serum response factor
- mTOR
- mammalian target of rapamycin
- FN
- fibronectin.
REFERENCES
- 1. Gabbiani G., Ryan G. B., Majne G. (1971) Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549–550 [DOI] [PubMed] [Google Scholar]
- 2. Majno G., Gabbiani G., Hirschel B. J., Ryan G. B., Statkov P. R. (1971) Contraction of granulation tissue in vitro. Similarity to smooth muscle. Science 173, 548–550 [DOI] [PubMed] [Google Scholar]
- 3. Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., Brown R. A. (2002) Nature Rev. 3, 349–363 [DOI] [PubMed] [Google Scholar]
- 4. Hinz B., Gabbiani G. (2003) Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 14, 538–546 [DOI] [PubMed] [Google Scholar]
- 5. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 [DOI] [PubMed] [Google Scholar]
- 6. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology. Paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hinz B., Celetta G., Tomasek J. J., Gabbiani G., Chaponnier C. (2001) α-Smooth muscle actin expression up-regulates fibroblast contractile activity. Mol. Biol. Cell 12, 2730–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang H. Y., Phan S. H. (1999) Inhibition of myofibroblast apoptosis by transforming growth factor β1. Am. J. Respir. Cell Mol. Biol. 21, 658–665 [DOI] [PubMed] [Google Scholar]
- 9. Darby I. A., Hewitson T. D. (2007) Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 257, 143–179 [DOI] [PubMed] [Google Scholar]
- 10. Phan S. H. (2002) The myofibroblast in pulmonary fibrosis. Chest 122, 286S-289S [DOI] [PubMed] [Google Scholar]
- 11. Thannickal V. J., Lee D. Y., White E. S., Cui Z., Larios J. M., Chacon R., Horowitz J. C., Day R. M., Thomas P. E. (2003) Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 278, 12384–12389 [DOI] [PubMed] [Google Scholar]
- 12. Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. (1993) Transforming growth factor-β 1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rønnov-Jessen L., Petersen O. W. (1993) Induction of α-smooth muscle actin by transforming growth factor-β1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest. 68, 696–707 [PubMed] [Google Scholar]
- 14. Mattey D. L., Dawes P. T., Nixon N. B., Slater H. (1997) Transforming growth factor β1 and interleukin 4 induced α-smooth muscle actin expression and myofibroblast-like differentiation in human synovial fibroblasts in vitro. Modulation by basic fibroblast growth factor. Ann. Rheum. Dis. 56, 426–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maltseva O., Folger P., Zekaria D., Petridou S., Masur S. K. (2001) Fibroblast growth factor reversal of the corneal myofibroblast phenotype. Invest. Ophthalmol. Vis. Sci. 42, 2490–2495 [PubMed] [Google Scholar]
- 16. Zhao X. H., Laschinger C., Arora P., Szászi K., Kapus A., McCulloch C. A. (2007) Force activates smooth muscle α-actin promoter activity through the Rho signaling pathway. J. Cell Sci. 120, 1801–1809 [DOI] [PubMed] [Google Scholar]
- 17. Akhmetshina A., Dees C., Pileckyte M., Szucs G., Spriewald B. M., Zwerina J., Distler O., Schett G., Distler J. H. (2008) Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum. 58, 2553–2564 [DOI] [PubMed] [Google Scholar]
- 18. Wang D. Z., Li S., Hockemeyer D., Sutherland L., Wang Z., Schratt G., Richardson J. A., Nordheim A., Olson E. N. (2002) Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. U. S. A. 99, 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fan L., Sebe A., Péterfi Z., Masszi A., Thirone A. C., Rotstein O. D., Nakano H., McCulloch C. A., Szászi K., Mucsi I., Kapus A. (2007) Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phosphomyosin pathway. Mol. Biol. Cell 18, 1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miralles F., Posern G., Zaromytidou A. I., Treisman R. (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342 [DOI] [PubMed] [Google Scholar]
- 21. Jeon E. S., Moon H. J., Lee M. J., Song H. Y., Kim Y. M., Bae Y. C., Jung J. S., Kim J. H. (2006) Sphingosylphosphorylcholine induces differentiation of human mesenchymal stem cells into smooth-muscle-like cells through a TGF-β-dependent mechanism. J. Cell Sci. 119, 4994–5005 [DOI] [PubMed] [Google Scholar]
- 22. Qiu P., Feng X. H., Li L. (2003) Interaction of Smad3 and SRF-associated complex mediates TGF-β1 signals to regulate SM22 transcription during myofibroblast differentiation. J. Mol. Cell. Cardiol. 35, 1407–1420 [DOI] [PubMed] [Google Scholar]
- 23. Sandbo N., Qin Y., Taurin S., Hogarth D. K., Kreutz B., Dulin N. O. (2005) Regulation of serum response factor-dependent gene expression by proteasome inhibitors. Mol. Pharmacol. 67, 789–797 [DOI] [PubMed] [Google Scholar]
- 24. Sandbo N., Lau A., Kach J., Ngam C., Yau D., Dulin N. O. (2011) Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-β. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L656–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A., Olson E. N. (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862 [DOI] [PubMed] [Google Scholar]
- 26. Small E. M., Thatcher J. E., Sutherland L. B., Kinoshita H., Gerard R. D., Richardson J. A., Dimaio J. M., Sadek H., Kuwahara K., Olson E. N. (2010) Myocardin-related transcription factor-α controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 107, 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Z. P., Wang Z., Yanagisawa H., Olson E. N. (2005) Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev. Cell 9, 261–270 [DOI] [PubMed] [Google Scholar]
- 28. Chen J., Somanath P. R., Razorenova O., Chen W. S., Hay N., Bornstein P., Byzova T. V. (2005) Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 11, 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Somanath P. R., Byzova T. V. (2009) 14–3-3β-Rac1-p21 activated kinase signaling regulates Akt1-mediated cytoskeletal organization, lamellipodia formation and fibronectin matrix assembly. J. Cell. Physiol. 218, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Somanath P. R., Kandel E. S., Hay N., Byzova T. V. (2007) Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J. Biol. Chem. 282, 22964–22976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goc A., Choudhary M., Byzova T. V., Somanath P. R. (2011) TGFβ- and bleomycin-induced extracellular matrix synthesis is mediated through Akt and mammalian target of rapamycin (mTOR). J. Cell. Physiol. 226, 3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi-Wen X., Chen Y., Denton C. P., Eastwood M., Renzoni E. A., Bou-Gharios G., Pearson J. D., Dashwood M., du Bois R. M., Black C. M., Leask A., Abraham D. J. (2004) Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol. Biol. Cell 15, 2707–2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jun J. B., Kuechle M., Min J., Shim S. C., Kim G., Montenegro V., Korn J. H., Elkon K. B. (2005) Scleroderma fibroblasts demonstrate enhanced activation of Akt (protein kinase B) in situ. J. Investig. Dermatol. 124, 298–303 [DOI] [PubMed] [Google Scholar]
- 34. Guo W., Shan B., Klingsberg R. C., Qin X., Lasky J. A. (2009) Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L864–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He Z., Zhu Y., Jiang H. (2009) Toll-like receptor 4 mediates lipopolysaccharide-induced collagen secretion by phosphoinositide 3-kinase-Akt pathway in fibroblasts during acute lung injury. J. Recept. Signal. Transduct. Res. 29, 119–125 [DOI] [PubMed] [Google Scholar]
- 36. Horowitz J. C., Rogers D. S., Sharma V., Vittal R., White E. S., Cui Z., Thannickal V. J. (2007) Combinatorial activation of FAK and AKT by transforming growth factor-β1 confers an anoikis-resistant phenotype to myofibroblasts. Cell. Signal. 19, 761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Z., Wang D. Z., Pipes G. C., Olson E. N. (2003) Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. U.S.A. 100, 7129–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stenmark K. R., Fagan K. A., Frid M. G. (2006) Hypoxia-induced pulmonary vascular remodeling. Cellular and molecular mechanisms. Circ. Res. 99, 675–691 [DOI] [PubMed] [Google Scholar]
- 39. Short M., Nemenoff R. A., Zawada W. M., Stenmark K. R., Das M. (2004) Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am. J. Physiol. Cell Physiol. 286, C416–425 [DOI] [PubMed] [Google Scholar]
- 40. Abdalla M., Goc A., Somanath P. R. (2012) NOVA Publisher Chapter 7 [Google Scholar]
- 41. Nicholson A. G., Fulford L. G., Colby T. V., du Bois R. M., Hansell D. M., Wells A. U. (2002) The relationship between individual histologic features and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 166, 173–177 [DOI] [PubMed] [Google Scholar]
- 42. Meyer-Ter-Vehn T., Gebhardt S., Sebald W., Buttmann M., Grehn F., Schlunck G., Knaus P. (2006) p38 inhibitors prevent TGF-β-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest. Ophthalmol. Vis. Sci. 47, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 43. White E. S., Atrasz R. G., Hu B., Phan S. H., Stambolic V., Mak T. W., Hogaboam C. M., Flaherty K. R., Martinez F. J., Kontos C. D., Toews G. B. (2006) Negative regulation of myofibroblast differentiation by PTEN (phosphatase and tensin homolog deleted on chromosome 10). Am. J. Respir. Crit. Care Med. 173, 112–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sandbo N., Kregel S., Taurin S., Bhorade S., Dulin N. O. (2009) Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-β. Am. J. Respir. Cell Mol. Biol. 41, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson S., DiCesare L., Tan I., Leung T., SundarRaj N. (2004) Rho-mediated assembly of stress fibers is differentially regulated in corneal fibroblasts and myofibroblasts. Exp. Cell Res. 298, 574–583 [DOI] [PubMed] [Google Scholar]
- 46. Pipes G. C., Creemers E. E., Olson E. N. (2006) The myocardin family of transcriptional coactivators. Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 20, 1545–1556 [DOI] [PubMed] [Google Scholar]
- 47. Greenberg R. S., Bernstein A. M., Benezra M., Gelman I. H., Taliana L., Masur S. K. (2006) FAK-dependent regulation of myofibroblast differentiation. FASEB J. 20, 1006–1008 [DOI] [PubMed] [Google Scholar]
- 48. Kohan M., Muro A. F., White E. S., Berkman N. (2010) EDA-containing cellular fibronectin induces fibroblast differentiation through binding to α4β7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 24, 4503–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blaustein M., Pelisch F., Tanos T., Muñoz M. J., Wengier D., Quadrana L., Sanford J. R., Muschietti J. P., Kornblihtt A. R., Cáceres J. F., Coso O. A., Srebrow A. (2005) Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 12, 1037–1044 [DOI] [PubMed] [Google Scholar]
- 50. White E. S., Sagana R. L., Booth A. J., Yan M., Cornett A. M., Bloomheart C. A., Tsui J. L., Wilke C. A., Moore B. B., Ritzenthaler J. D., Roman J., Muro A. F. (2010) Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp. Cell Res. 316, 2644–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karni R., Hippo Y., Lowe S. W., Krainer A. R. (2008) The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc. Natl. Acad. Sci. U.S.A. 105, 15323–15327 [DOI] [PMC free article] [PubMed] [Google Scholar]