

Abstract
Dipyridamole (DPM) is widely used to prevent strokes and vascular thrombosis. Combination therapy of DPM and antimetabolites has shown synergistic anticancer activity. This study investigated the chemopreventive effects of DPM in the mouse mammary tumor virus promoter driven polyoma middle T oncoprotein (MMTV-PyMT) metastatic breast cancer model. We also investigated the effects of DPM on gene and miRNA expression. Chemopreventive activity was assessed by comparing the time to onset of palpable lesions, primary tumor growth kinetics and the number of lung metastases in transgenic mice treated with DPM or vehicle. Gene expression and microRNA (miRNA) expression profiles of mammary tumor tissues were then analyzed using the Affymetrix GeneChip® or miRNA 2.0 arrays. Real-time quantitative PCR (qPCR) was used to confirm changes in gene expression. Treatment with DPM beginning at the age of four weeks delayed the onset of palpable lesions, delayed tumor progression and suppressed lung metastasis. Microarray gene expression analysis identified 253 genes differentially expressed between DPM-treated and control mammary tumors. miRNA expression analysis revealed that 53 miRNAs were altered by DPM treatment. The results indicate that DPM has chemoprevention activity against breast cancer tumorigenesis and metastasis in mice. The array analyses provide insights into potential mechanisms of DPM’s chemopreventive effects, involving upregulation of several genes and miRNAs known to suppress cancer growth and/or metastasis and downregulation of genes known to promote cancer. Some of these genes have not been previously studied in breast cancer and may serve as novel molecular targets for breast cancer chemoprevention.
Keywords: Chemoprevention, Dipyridamole, breast cancer, MMTV-PyMT, microarray
Introduction
Cancer is the result of a multistep process of tumorigenesis in which there is a gradual transition from normal cells to increasing grades of dysplasia that culminate in an invasive and metastatic phenotype (1). Arresting one or several of these steps may impede or delay the development of carcinomas and provide opportunities for the development of clinical interventions aimed both at preventing cancer initiation and at treating premalignant lesions (1, 2). Cancer chemoprevention was first defined by Sporn in 1976 (3). It is an innovative area of cancer research that focuses on the development of pharmacological, biological, and nutritional interventions to prevent, reverse, or delay carcinogenesis, and it involves the primary prevention of initiation and the secondary prevention of delay, or reversal, of promotion and progression (3). There are at least two major mechanisms for cancer chemoprevention (4). One is antimutagenesis, which includes the inhibition of the uptake of carcinogens, the formation/activation of carcinogens, the deactivation/detoxification of carcinogens, and the other is antiproliferation/antiprogression.
Current chemoprevention drugs and potential chemopreventive agents under investigation are diverse in chemical structure and physiologic effects. The selective estrogen receptor modulator (SERM) drug tamoxifen, for example, reduces the risk of breast cancer in some women who are at risk of developing the disease or a recurrence. But adverse side effects, such as an increased risk of endometrial cancer and thromboembolic events, limit its use (5, 6). At present, there are very few options available for chemoprevention of cancer.
Breast cancer is the most common malignancy in women worldwide and is one of the most lethal carcinomas. It is the second leading cause of cancer related death in women in the United States (7). Advances in early detection and improved treatment for breast cancer have led to a steady decrease in the overall breast cancer mortality rate; however, breast cancer remains a significant cause of morbidity and mortality (8, 9). The SERMs tamoxifen and raloxifene reduce breast cancer incidence in high-risk women by approximately 50% (10, 11). However, they do not decrease the incidence of estrogen receptor (ER)-negative breast cancer, which accounts for 30–40% of all breast cancers. Furthermore as alluded to above, SERMs may cause severe side effects in patients. It is therefore imperative to develop safe and effective chemopreventive agents for both ER-positive and ER-negative breast cancers.
Dipyridamole (DPM) is a clinically tested nucleoside transport inhibitor and antithrombotic agent that is widely used to prevent strokes and vascular thrombosis due to its antiplatelet and vasodilatory activities as a nucleoside transporter and a non-selective phosphodiesterase inhibitor (12). Studies suggest that DPM possesses beneficial properties within the endothelium of the vasculature in addition to its anti-platelet effects, including direct and indirect effects such as inhibition of proliferation, antioxidant and anti-inflammatory properties, and downstream effects on cell signaling (13). DPM was previously reported to increase the cytotoxicity of antimetabolites like pemetrexed on human cancer cells (14, 15). Our in vitro study of nucleoside transport inhibitors as chemopreventive agents has found that DPM shows chemopreventive activity in the JB6 cell tumor promotion model (our unpublished results). Other studies have shown that DPM has preventive activity against prostate cancer (16) and pancreatic cancer (17) metastasis. Hence, we envisaged that DPM might be a novel primary and metastasis chemopreventive agent for breast cancer that may exhibit a better pharmacological profile for long-term prevention than the SERMs like tamoxifen. In support of this hypothesis, DPM has recently been shown to block growth of highly aggressive triple-negative breast cancer cell lines implanted in mice and to reduce cell proliferation and invasion in MCF10A cells that were chronically treated with three different chemical carcinogens (18, 19).
The mouse mammary tumor virus (MMTV) promoter-driven expression of the polyoma middle T oncoprotein (MMTV-PyMT) produces spontaneous, luminal-like breast cancer. It is widely utilized to model ER-negative late stage carcinomas, which are highly metastatic to the lung (20, 21). Therefore, we investigated the breast cancer chemopreventive potential of DPM in this highly aggressive model of breast cancer, which initiates from normal mammary epithelium.
Materials and Methods
Animals
MMTV-PyMT+ male mice (FVB/Nj strain) were derived from colonies managed by T.N.S. Transgenic females were obtained by breeding FVB/Nj females with PyMT+ transgenic males. All mice were housed in the Center for Adult Cancer Research animal facility, and were provided free access to food and water. The experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC). All ten mammary glands of transgenic females typically develop mammary tumors by the age of 14–16 weeks (21), therefore, whole body conditioning (BC) scoring was utilized in lieu of tumor burden to establish the criteria for experimental endpoints (22). At Week 11 of treatment, vehicle-treated mice developed a BC score of 2.0 or lower, therefore, the last drug treatment occurred at Week 11, and all mice were euthanized and tissues harvested 2–3 days after the final injection.
Formulation of DPM for the chemoprevention study
Dipyridamole (DPM) was purchased from Sigma Aldrich Chemicals (Supplementary Figure 1A) and reconstituted in the formulation below. All solvents were obtained from BASF. Each mouse received DPM intraperitoneally (IP) at a dose of 10 mg/kg body weight in a volume of 200 μl.
| Ingredient | Total % (w/w) |
|---|---|
| Ethanol | 10% |
| 2-Pyrolidone | 5% |
| Propylene glycol | 12–15% |
| Cremophor ELP | 10% |
| Saline | q.s. to 100% |
Preclinical chemoprevention experimental design
PyMT+ female mice were randomly divided into a DPM treatment group (n=10 mice, total of 100 glands) and a vehicle (control) group (n=7 mice, total of 70 glands). When mice were four weeks old, when hyperplasia lesions begin to develop in the FVB strain (21), DPM (10 mg/kg) or vehicle was injected IP into the peritoneum of each mouse on a 5 days on/2 days off dosing schedule for a total of 11 weeks (week one=injection one), until mice were 15 weeks of age. Each of the 10 mammary tumors per mouse was monitored until the study end point. Additional details are provided in the Supplementary materials.
RNA extraction for microarray or real time PCR studies
Total RNA was isolated from solid (non-necrotic) portions of breast tumors using the miRNeasy mini kit (Qiagen) and treated with Turbo DNase I (Ambion). RNA integrity was assayed by the Agilent Bioanalyzer 2100 (Agilent Technologies) at the UTHSC Molecular Resource Center (MRC). Samples with a RNA integrity number of >8.0 or >6.0 were compared by microarray analyses, or real-time quantitative PCR (qPCR), respectively.
Analysis of Gene Expression by Microarrays
Microarray analysis was performed by hybridization to Affymetrix GeneChip® Mouse Gene 1.0 ST array per manufacturer’s instructions (Affymetrix) at the MRC (100 ng of total RNA input). Expression across three biological replicate RNA preparations from tumors of the DPM or vehicle treatment groups were compared. CEL files generated by the Command Console (Affymetrix GeneChip® Operating Software) were uploaded into the Expression Console for normalization and probe set summarization using the RMA (Robust Multichip Analysis) algorithm (23). The resulting data of probe set intensities were exported to a .txt Excel file to compare mean expression profiles of DPM-treated samples to the vehicle control. Genes that changed by at least 1.5-fold between treatment groups (and with a p-value of ≤0.05 among biological replicates) were selected for qPCR validation, as described in the Supplementary materials.
miRNA Array Analysis
The miRNA 2.0 Array was hybridized using 500 ng of total RNA per to standard Affymetrix protocols. The same RNA preparations used in the mRNA microarray analysis were used in miRNA array analysis. Data extraction was performed using Affymetrix Command Console software. Raw data were analyzed by the following workflow: background detection, RMA global background correlation, quantile normalization, median polish and log2-transformed with miRNA QC tool software. qPCR validation was performed as described in the Supplementary materials.
Western Blot Analysis
Whole cell extract (WCE) was prepared of frozen tumor ground to powder in liquid nitrogen and lysed with RIPA buffer containing protease inhibitors. WCE (30 μg/lane) was resolved using SDS-PAGE electrophoresis and transferred onto nylon membrane (Bio-Rad). Blots were incubated for 1h at RT with previously validated antibodies to KLK10 (Bioss, bs-2531R, 1:50) or β-actin (Santa Cruz; 1:1,000), and complexes detected with HRP-conjugated goat anti-rabbit IgG secondary, or with an anti-LECT1 (Bioss Antibodies, bs-7561) HRP-conjugated antibody (1:500) and developed by enhanced chemiluminescence (Amersham).
miRNA Target and Ingenuity Pathway Analyses
Gene targets were predicted for qPCR-validated, upregulated miRNAs (mmu-miR-140, mmu-miR-140-3p, mmu-miR-455, mmu-miR-351, mmu-miR-214, and mmu-miR-1949) and the downregulated miRNA (mmu-miR-148a). The miRNA target prediction databases TargetScan and miRDB were queried. Potential target gene interactions were analyzed via networks generated in the Ingenuity Pathway Analysis® (IPA) tool v9.0 (Ingenuity Systems). The qPCR-validated, biologically active miRNAs and their predicted target genes were uploaded to the IPA software. Canonical pathways were identified from the standard IPA library. The probability that an association between genes in the dataset of interest and the canonical pathways in IPA was determined using built-in algorithms.
Statistical analysis
Prism 5.0 (GraphPad) was used for all statistical analyses and graphing. Differences in body weight over time were analyzed by two-way ANOVA. Palpation data were analyzed using Kaplan-Meier analysis, with glands that did not progress to the stage of interest censored (0 in Prism) and compared for significance using the log-rank test. Unless otherwise noted, all other data were analyzed using a two-way, unpaired student’s t test. All results were expressed as the mean ± S.E.M and a p ≤ 0.05 was considered statistically significant.
Results
Effects of DPM treatment on body weight gain
Daily administration of DPM IP at a 10mg/kg dose on a five-day on and two-day off schedule was well-tolerated by all mice. No clinical signs of toxicity were present in either the group as no mice required removal from the study before week 11 of treatment. DPM administration did not result in a significant difference over time in mean body weight between vehicle and DPM-treated mice (two-way ANOVA, Figure 1A). Differences in body weight were individually significant by an unpaired t-test at Weeks 8, 9, 10 and 11. The increased body weight observed in the vehicle cohort is presumed to be due to the presence of more mammary tumors per mouse and the overall larger tumor sizes observed in the vehicle-treated group (Figures 1E, 2A).
Figure 1. DPM suppresses mammary tumor progression in PyMT mice without toxicity.

A, Mean body weight of MMTV-PyMT+ treated with vehicle or DPM on a 5 day on/2 day off schedule for 11 weeks. The effects of DPM treatment on the transition of normal epithelium to grainy (B), granule (C), pea-sized (D) or measurable tumors (E) as analyzed by Kaplan Meier curves plotting the percent of glands with the indicated phenotype as a function of time. Differences in curves between groups were analyzed by the log-rank test, and the median time to the palpable phenotype of interest, the hazard ratio, and the number of glands included in each curve are also indicated.
Figure 2. Mean tumor volume changes in transgenic mice treated with DPM or vehicle.

A, mean tumor volume observed from week 7 to 11 of treatment. B, Scatter plot of individual tumors and the mean tumor volume (dashed line) plus the S.E.M. derived from tumors measured in intact mice at week 11 of DPM treatment. C, Fraction of glands that developed measurable tumors over time; all comparisons at each week were significant by Fisher’s exact test.
Effects of DPM treatment on stage-specific progression
At four weeks of age, when hyperplasias are expected to be present in the FVB strain (21), the first sign of tumor initiation was the detection of grainy areas in glands during manual palpation. This was followed by progression of fine grains into larger granules that were not yet measurable with calipers, and then into pea-sized tumors (small early carcinomas <5 mm diameter), followed by transition into larger, measurable tumors (late stage carcinomas) with metastatic potential. These descriptors (grainy, granules, pea or tumor) were utilized to score the palpation results for all glands per mouse each week. Kaplan Meier curves were generated and differences between the curves analyzed by the log-rank test, which revealed that all comparisons for each stage of progression were significant (Figures 1B–E).
Palpable grainy lesions developed at a median of 3 weeks of treatment in both groups and 100% of glands per group were classified as grainy in the vehicle and DPM groups by week 6 and week 7, respectively (Figure 1B). The median time to 50% of glands classified as granule lesions was 5 weeks for the vehicle group and 7 weeks for the DPM group (Figure 1C), and 100% of glands transitioned to granules by Week 8 (vehicle) and Week 11 (DPM). The frequency of pea-sized tumors in the DPM-treated group was also significantly different compared to the vehicle group (Figure 1D). By Week 11, all 70 glands of the vehicle group were classified as pea-sized tumors, whereas 15 glands remained at the granule stage in the DPM group. Finally, treatment with DPM significantly delayed the onset of measurable tumors by approximately 3 weeks as compared to vehicle (Figure 1E). It should also be noted that 62 of 70 glands (89%) had formed measurable tumors by Week 11 in the vehicle group, whereas about one-half of the glands in the DPM group were still classified as non-measurable pea-sized lesions (52%).
Effects of DPM treatment on tumor growth and lung metastasis
DPM suppressed mammary tumor growth compared to the vehicle controls (Figures 2A–2C). The mean tumor volumes at Weeks 7, 8, 9, 10 and 11 of treatment are presented in Figure 2A. No tumors were observed in DPM-treated mice by Week 7, and only one tumor was observed by Week 8. Although there was a trend for DPM-treated tumors to be smaller at Weeks 9 and 10, differences did not approach statistical significance by an unpaired t-test. Tumor volume was significantly reduced in the DPM group at Week 11 of treatment; the average tumor volume was 406.3 ± 39.60 mm3 in the vehicle group (n=62 tumors) versus 286.8 ± 35.03 mm3 in the DPM group (n=52 tumors) (Figures 2A–2B). A comparison of the fraction of glands with measurable tumors over time revealed that the difference in measurable tumors was significantly different each week by Fisher’s exact test (Figure 2C).
All glands/tumors were harvested from anesthetized mice 2–3 days following the last injection at Week 11. More necrotic tissue and intratumoral fluid were observed during tumor resection in the vehicle group than in the DPM treatment group. Example images of all tumors harvested from either a vehicle- or DPM-treated mouse are included in Figure 3A. Since measurement of individual glands/tumors by calipers is somewhat hampered in intact transgenic mice as tumors enlarge, and since tumors were resected from mice 2–3 days following the last palpation of intact mice, ex vivo caliper measurements of individual tumors was also conducted at end point. There was a dramatic increase in the mean tumor volume as calculated based on ex vivo measurements for the vehicle group when compared to the mean volume calculated 2–3 days earlier at Week 11 (Figure 3B; increase from ~400mm3 to ~952 mm3). The mean tumor volume increased in the DPM group as well, from 268 mm3 to 476.5 mm3. These increases reflect both the additional growth of the tumor over a few days, as well as our ability to measure dissected tumors more carefully. DPM also significantly reduced mean tumor burden (tumor weight as a percentage of total body weight) from 42.4 % in the vehicle group to 33.2 % in the DPM group (Figure 3C), and decreased the mean tumor wet weight by almost half (16.21 g vs. 9.52 g, Figure 3D).
Figure 3. Effects of DPM treatment on tumor volume, tumor mass and pulmonary metastasis at experimental end point.

A, Representative image of tumors dissected from vehicle or DPM-treated females. B, Scatter plot of mean tumor volume (dashed line) plus the S.E.M. (error bars) as calculated from the ex vivo caliper measurements, analyzed by student’s t-test. Mean tumor burden at experimental end point analyzed by student’s t-test. C, Mean tumor burden at experimental end point analyzed by student’s t-test. D, Mean tumor wet weight per animal at experimental end point analyzed by student’s t-test. E, Representative images of H&E-staining of lungs of mice bearing end-stage mammary tumors, 200x magnification. D, Scatter plot of the mean number of lung metastases per cohort (dashed line) plus the S.E.M., analyzed by student’s t-test.
Comparison of histopathology of end stage tumors by H&E staining of tumor sections did not reveal any gross changes in tumor grade, nuclear morphology or the ratio of tumor cells to stroma in response to DPM treatment (Supplementary Figure 1B–1C). To assay for changes in cell proliferation or apoptosis, tumor sections were immunostained for both Ki67 and activated caspase-3, respectively. The percentage of tumor cells positive for Ki67 or caspase 3 was not significantly different in end-stage DPM-treated tumors compared to controls (Supplementary Figure 2A–B). We cannot rule out the possibility that DPM-regulated changes in proliferation or apoptosis occurred during progression over the course of treatment. Tumor sections were also stained for ERα to determine if DPM prevented the loss of ERα in late stage carcinomas (21). Virtually no ERα staining was observed in either vehicle- or DPM-treated tumors, although ERα was expressed in all ductal epithelial cells of nulliparous 6-week old FVB/Nj mice (Supplementary Figure 3A–3B vs. 3C). Therefore, it is not likely that the mechanism of DPM chemoprevention in the PyMT model is hormone-dependent. Representative H&E stained images of one lobe of the lung harvested from mice at experiment end point indicating metastases is presented in Figure 3E. DPM treatment significantly reduced the mean number of pulmonary metastases by 59 % (Figure 3F).
Gene expression profiling
To gain insight into the potential mechanisms of DPM’s chemoprevention activity, we performed gene expression profiling by whole-genome microarray analysis. End-stage breast tumors of DPM-treated or vehicle-treated mice were used with the Affymetrix GeneChip® Mouse Gene 1.0 ST array representing approximately 28,310 mouse transcripts and 21,041 protein-coding genes. Genes that were at least 1.5-fold down- or up-regulated among all biological replicates were selected as significantly differentially expressed genes. Significant changes in 253 transcripts were observed between DPM-treated and vehicle-treated tumors, of which 153 genes were upregulated and 100 genes were downregulated. Genes that were differentially expressed with > 2.0-fold change between vehicle- and DPM-treated tumors are shown in Supplementary Tables 1A and 1B, respectively. The GEO accession number for microarray data deposition is GSE43833.
Functional annotation of differentially expressed genes was performed using the DAVID Bioinformatics Database. Gene sets were grouped based on their biological functions (Supplementary Tables 2A and 2B). Genes that control proliferation and migration, two key hallmarks of cancer, and that were altered by DPM treatment are shown in Supplementary Table 3.
Validation of differentially expressed genes
The expression of the top up-regulated or down-regulated genes identified by array (Supplementary Table 1) was confirmed by qPCR using individual gene-specific primers after normalization for input based on GAPDH levels; results are summarized in Supplementary Table 4. The upregulation of Emb, Six1, Hpse2, Vcan, Loxl4, Wfdc5, Cpm, Klk10, Mme, Amtn, Mup5 and Lect1, and the downregulation of Gjb2, Fbp1, Cox8b, Thrsp and Pdk4 were validated by qPCR (Supplementary Table 4). In contrast, the increased expression of Smr2 and the decreased expression of Slc47a1, BC018465, Mup1, Mup2 (transcript variant 2), U46068, Mup2 (transcript variant 1), Vmn2r96, Mup7 and EG665955 was not confirmed by qPCR, suggesting these genes were false discoveries (Supplementary Table 4). To rule out the possibility that DPM treatment reduced the expression of the PyMT transgene, the relative levels of the PyMT cDNA expressed by vehicle- and DPM-treated tumors were compared by qPCR. No significant changes in transgene expression were observed, as the mean crossing point (Cp) values varied less than 10 % for either primer/probe set (Supplementary Figure S4).
We next assessed the levels of Klk10 and Lect1 proteins in individual end-stage tumors (n=3 DPM and n=4 vehicle). Western blot analysis confirmed that both Klk10 and Lect1 are up-regulated by DPM treatment (Figure 4). In fact, the expression of Klk10 is dramatically up-regulated by DPM treatment since Klk10 protein was not detectable in any of the vehicle treated tumors. Lect1 expression was more abundant in each of the DPM-treated tumors as compared to tumors in the vehicle treatment group.
Figure 4. Klk10 and Lect1 are up-regulated by DPM treatment.
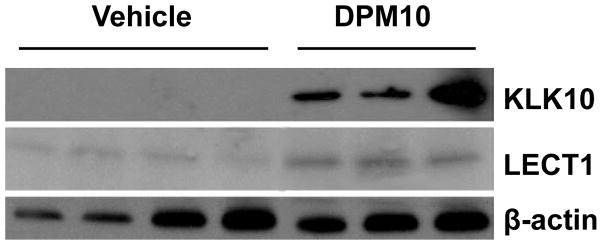
Western blotting for Klk10, Lect1 or β-actin (loading control) revealed a dramatic increase in Klk10 expression in DPM-treated tumors as compared to vehicle-treated tumors; a more modest increase is observed for Lect1 expression (n=3 tumors DPM, n=4 tumors vehicle).
miRNA expression profiling and qPCR validation of differentially-expressed miRNAs
Since several miRNAs are dysregulated during tumor progression and the metastatic switch, we next compared miRNA expression levels between DPM- and vehicle-treated tumors. The miRNA 2.0 Array represents 1,411 mouse microRNA probesets. Only the miRNAs that increased or decreased by more than 1.5-fold were considered as significantly up- or down-regulated. Supplementary Table 5 summarizes the miRNAs identified as enriched or suppressed in tumors following DPM treatment, which include 53 significantly differentially expressed (>1.5-fold) miRNAs. Of these, 23 miRNAs were upregulated by DPM, whereas 25 miRNAs were downregulated. In addition, two V11-mmu (microRNA sequence from Sanger miRNA database v11) and two hp-mmu (Pre-mir probe) were upregulated and one V11-mmu was downregulated. Differential expression of those miRNAs with high signals from the microarray was also validated by qPCR. mmu-miR-140-star, mmu-miR-1937b, mmu-miR-140, mmu-miR-1937a, mmu-miR-455, mmu-miR-214 and mmu-miR-351 were confirmed to be significantly upregulated by DPM treatment. v11_mmu-miR-685_st, mmu-miR-690_st, mmu-miR-762_st, mmu-miR-1949_st and the four down-regulated miRNAs identified by array analysis either did not produce significant differences or may have been false positives.
Canonical pathways affected by DPM-regulated miRNAs
To investigate the biological relevance of DPM-regulated miRNAs, we performed Ingenuity Pathway Analysis (IPA) of differentially expressed genes that are putative targets of these miRNAs. The putative targets of one downregulated miRNA (mmu-miR-148a) and five upregulated miRNAs (mmu-miR-140, mmu-miR-140-star, mmu-miR-455, mmu-miR-351, mmu-miR-214, and mmu-miR-1949) that were detected by both microarray and qPCR were identified by using miRNA target filter of Ingenuity Pathway Analysis program. The first query was to identify associations between those genes up-regulated in tumors that are also predicted targets of the downregulated mmu-miR-148a, which revealed 22 canonical pathways as significantly enriched (Figure 5). In contrast, the down-regulated genes that are also putative targets of miRNAs upregulated by DPM were significantly associated with 42 canonical pathways (Figure 6).
Figure 5. Ingenuity Canonical Pathways affected by DPM-downregulated miRNAs.

The putative targets of the DPM-downregulated mmu-miR-148a that were also upregulated by DPM according to the mRNA expression array were used for IPA analysis. The −log(p value) of the significantly enriched pathways (P,0.05) are presented in the bar graph.
Figure 6. Ingenuity Canonical Pathways affected by DPM-upregulated miRNAs.

The putative targets of the DPM-upregulated miRNAs that were also downregulated by DPM according to the mRNA expression array were used for IPA analysis. The −log(p value) of the significant enriched pathways (p<0.05) are presented in the bar graph.
Discussion
We have shown that DPM has significant in vivo chemopreventive effects in the MMTV-PyMT model of metastatic breast cancer. A distinct advantage of this model of spontaneous breast cancer is that it can be seamlessly used to study both primary tumor development and lung metastasis. Almost 100% of PyMT mice backcrossed to the FVB/Nj strain develop metastases in the lungs by four months of age. In addition, PyMT tumors are initiated as ER-positive lesions, but eventually progress to estrogen-independent adenocarcinomas (21), therefore the model can be used to study both ER-positive and ER-negative tumorigenesis. Our studies are unique in that they describe for the first time the primary chemopreventive potential of DPM on mammary carcinogenesis in vivo in animals with a normal immune system.
Lesions that developed in the DPM-treated cohort remained at the earlier stages of PyMT-induced mammary cancer initiation and progression longer than lesions in the control group. Importantly, DPM treatment led to a significant reduction in the total mammary tumor burden and lung metastases. Together, these results indicate that DPM treatment may be useful in the chemoprevention of either ER+ or ER-negative metastatic breast cancer. It is remarkable that the dose of DPM used in the present mouse study (10 mg/kg/day), a relatively low dose compared to the maximum tolerated dose in patients (23.1 mg/kg/72h by IV) (24), clearly exerted significant tumorigenesis and metastasis prevention activity in PyMT mice, one of the most aggressive pre-clinical models of breast cancer used in the research community. These results suggest that DPM is likely to have potent chemoprevention activity in additional models of breast cancer.
Microarray gene expression profiling, has improved our understanding of breast cancer biology and allowed the development of multigene ‘signatures’ to predict outcome and response to systemic therapies (25). Through comprehensive array profiling and analysis of gene and miRNA expression levels, we have identified putative candidate genes and pathways that mediate DPM’s chemoprevention activity. Genes differentially expressed in response to DPM treatment included those involved in cell growth, maintenance, proliferation and metastasis, as well as apoptosis and autophagy.
The most highly related functions of genes altered by DPM treatment included cell signaling and cell adhesion (Table 2A). The latter has a major influence in metastasis, stimulating local invasion and intravasation to facilitate cell dissemination to, and colonization of distant organs (26). Metalloendopeptidase (Mme) was upregulated by DPM treatment. High expression of Mme occurs in some cancers such as renal cell and hepatocellular carcinomas. In addition, membrane metalloendopeptidase inhibits prostate cancer cell migration (27), while transfection of the Mme gene into B16-BL6 melanoma cells suppresses tumor growth (28). In contrast, Claudin-2 (Cldn2), which promotes breast cancer cell adhesion to the extracellular matrix (ECM) and metastasis to the liver (29), was down-regulated by DPM. Our results also implicate regulation of the mitogen-activated protein kinases (MAPKs), which can either be protective or damaging to cells depending on the context and strength of activation (30). For example, up-regulated Map3k3 has been reported to cause a reversion of Ras-induced cellular transformation, suggesting possible tumor suppressor functions (31), whereas Map3k1 has been associated with breast cancer susceptibility (32).
Among the genes validated by qPCR (Supplementary Table 4), the gene with the highest induction by DPM was Kallikrein-10 (Klk10); upregulation of this gene was also confirmed by Western blotting. Klk10 is of particular interest because it is associated with suppression of tumorigenesis in breast and prostate cancers (33). Moreover, stable expression of Klk10 in the Klk10-negative MDA-MB-231 triple negative breast cancer cells suppresses oncogenicity, supporting the tumor suppressor function (34). Notably, White et al. found that down-regulation of miRNAs let-7f, miR-224 and miR-516a are associated with Klk10 protein up-regulation and cell proliferation in ovarian cancer (35). In agreement, our miRNA array results showed that let-7f was down-regulated by 20% by DPM treatment, whereas expression of miR-224 and miR-516a were not detectable. The upregulation of the Lect1 gene, which encodes chondromodulin-1, and was previously found to suppress tumor growth and angiogenesis in vivo (36, 37), was also confirmed by qPCR and western blotting.
Likewise, a subset of genes was identified as down-regulated by DPM treatment, which was validated by qPCR analysis. For example, Thrsp, which encodes for the thyroid hormone-inducible hepatic protein, was significantly down-regulated by DPM. Thrsp activates genes required for fatty acid synthesis, and is found expressed in lipogenic breast cancers. Notably, low levels of Thrsp are associated with prolonged disease-free survival in invasive breast cancers (38). Another example is the Fbp1 gene, which encodes the far upstream element-binding protein 1, a member of the FBP family that modulates c-myc expression. Inhibition or loss of FBP function arrests cellular proliferation and abrogates c-myc expression (39). MCF-7 breast cancer cells have elevated FBP1 and FBP2 compared to the normal breast cell line MCF10-A, which may increase glycolysis to drive cancer cell growth in hypoxia (40). Another interesting down regulated gene is Pdk4, which encodes for pyruvate dehydrogenase kinase, and is up-regulated in a variety of cancers, allowing tumor cells to thrive in a hypoxic environment (41). The down-regulatory effects of DPM on these genes may synergize to contribute to the breast cancer chemopreventive effects. Interestingly, some of these differentially expressed genes have hitherto not been reported to be involved in cancer growth or metastasis, or at least not in breast cancer, and therefore constitute novel candidate genes of high priority for further studies pertinent to breast cancer tumorigenesis and progression.
microRNAs (miRNAs) play central roles in physiological and pathological processes by either inducing mRNA degradation or by regulating the translational efficiency of mRNA (42). Our results demonstrate that miR-140-star, miR-1937b, miR-140, miR-1937a, miR-455, miR-214 and miR-351 were significantly upregulated by DPM treatment. In addition, our observations that DPM up-regulates miR-31 and that lung metastasis is significantly reduced in DPM treated mice are consistent with prior studies that have shown that miR-31 that represses multiple steps of metastasis via downregulation of a group of pro-metastatic genes in breast cancer (43), including colonization, the final and rate-limiting step of metastasis (44). miR-140-star, miR-455, miR-1274a, and miR-31, were highly enriched with DPM treatment. It has also been recently reported that levels of miR-140, a putative tumor suppressor, were decreased in all breast tumor samples studies (45). miR-1274a, was recently found by Zhou et al. to be up-regulated after sorafenib treatment of hepatocellular cancer cells, revealing a possible new miRNA-based mechanism of sorafenib antitumor activity (46).
Finally, we observed that miRNA-10b was down-regulated by DPM to levels ~40% of control. miRNA-10b is highly expressed in metastatic breast cancer cells and enhances migration and invasion; with its silencing markedly suppresses formation of lung metastases (43, 47). Thus, miR10b down-regulation may also contribute to the chemopreventive activity of DMP. We propose that these miRNAs are among the most interesting up-regulated candidates for subsequent investigation into the mechanism of action of DPM.
Since the same miRNA may target diverse genes in different pathways, and each gene may be targeted by several miRNAs (48), one miRNA change could have a big impact on the regulation of target genes that impact cancer phenotypes. Our miRNA target prediction analysis using IPA showed that some of the downregulated pathways included those involved in cell cycle, NGF signaling, tissue factor signaling, mTOR signaling, and p38 MAPK signaling. In the future, it will be of interest to determine if these same sets of miRNAs could be biomarkers that are associated with chemoprevention efficacy.
Since cancer is still a leading cause of death worldwide, second only to heart disease, the use of preventive medicine approaches is becoming increasingly important. Identification of novel targets and development of effective cancer preventive agents will be necessary to decrease cancer mortality; and our study is important in that respect, being the first to demonstrate in vivo primary chemopreventive effects of DPM. We have also shown that DPM treatment significantly alters the expression pattern of several genes and miRNAs that may be potential novel targets for breast cancer chemoprevention. Together, our results demonstrate that DPM is a candidate or a lead compound for future translational development of new chemoprevention therapies.
Supplementary Material
Acknowledgments
Grant support from the NCI, CA-125850 (J.K.B.) and CA-138488 (L.P.S. and T.N.S.) is gratefully acknowledged. PyMT+ mice were provided to T.N.S. by Dr. Kent Hunter (NCI). The authors appreciate the skillful assistance of Richard C. Cushing, Danielle L. Peacock, and Alexander Croft, and data recording assistance by Shan Sun and Hilaire Playa.
Footnotes
Disclosure of Potential Conflicts of Interest: no potential conflicts of interest were disclosed.
References
- 1.Crowell JA. The chemopreventive agent development research program in the division of cancer prevention of the US National Cancer Institute: An overview. European Journal of Cancer. 2005;41:1889–910. doi: 10.1016/j.ejca.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 2.Tsao AS, Kim ES, Hong WK. Chemoprevention of Cancer. CA: A Cancer Journal for Clinicians. 2004;54:150–80. doi: 10.3322/canjclin.54.3.150. [DOI] [PubMed] [Google Scholar]
- 3.Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer research. 1976;36:2699–702. [PubMed] [Google Scholar]
- 4.Kelloff GJ. Perspectives on cancer chemoprevention research and drug development. Advances in Cancer Research, Vol 78. 2000;78:199–334. doi: 10.1016/s0065-230x(08)61026-x. [DOI] [PubMed] [Google Scholar]
- 5.Decensi A, Maisonneuve P, Rotmensz N, Bettega D, Costa A, Sacchini V, et al. Effect of tamoxifen on venous thromboembolic events in a breast cancer prevention trial. Circulation. 2005;111:650–6. doi: 10.1161/01.CIR.0000154545.84124.AC. [DOI] [PubMed] [Google Scholar]
- 6.Taylor AL, Adams-Campbell LL, Wright JT., Jr Risk/benefit assessment of tamoxifen to prevent breast cancer-still a work in progress? J Natl Cancer Inst. 1999;91:1792–3. doi: 10.1093/jnci/91.21.1792. [DOI] [PubMed] [Google Scholar]
- 7.Jemal A, Siegel R, Ward E, Murray T, Xu JQ, Thun MJ. Cancer statistics, 2007. Ca-a Cancer Journal for Clinicians. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 8.Espey DK, Wu XC, Swan J, Wiggins C, Jim MA, Ward E, et al. Annual report to the nation on the status of cancer, 1975–2004, featuring cancer in American Indians and Alaska natives. Cancer. 2007;110:2119–52. doi: 10.1002/cncr.23044. [DOI] [PubMed] [Google Scholar]
- 9.Thomsen A, Kolesar JM. Chemoprevention of breast cancer. American journal of health-system pharmacy: AJHP: official journal of the American Society of Health-System Pharmacists. 2008;65:2221–8. doi: 10.2146/ajhp070663. [DOI] [PubMed] [Google Scholar]
- 10.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 study. Journal of the National Cancer Institute. 1998;90:1371–88. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 11.Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA: the journal of the American Medical Association. 1999;281:2189–97. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- 12.Jones L, Griffin S, Palmer S, Main C, Orton V, Sculpher M, et al. Clinical effectiveness and cost-effectiveness of clopidogrel and modified-release dipyridamole in the secondary prevention of occlusive vascular events: a systematic review and economic evaluation. Health Technology Assessment. 2004;8:iii–iv. 1–196. doi: 10.3310/hta8380. [DOI] [PubMed] [Google Scholar]
- 13.Chakrabarti S, Freedman JE. Dipyridamole, cerebrovascular disease, and the vasculature. Vascular pharmacology. 2008;48:143–9. doi: 10.1016/j.vph.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Saravanan K, Barlow HC, Barton M, Calvert AH, Golding BT, Newell DR, et al. Nucleoside transport inhibitors: structure-activity relationships for pyrimido[5,4-d]pyrimidine derivatives that potentiate pemetrexed cytotoxicity in the presence of alpha1-acid glycoprotein. Journal of Medicinal Chemistry. 2011;54:1847–59. doi: 10.1021/jm101493z. [DOI] [PubMed] [Google Scholar]
- 15.Smith PG, Marshman E, Newell DR, Curtin NJ. Dipyridamole potentiates the in vitro activity of MTA (LY231514) by inhibition of thymidine transport. British Journal of Cancer. 2000;82:924–30. doi: 10.1054/bjoc.1999.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drago JR, Curley RM, Sipio JC. Nb rat prostate adenocarcinoma model: metastasis. Anticancer Res. 1985;5:193–6. [PubMed] [Google Scholar]
- 17.Tzanakakis GN, Agarwal KC, Vezeridis MP. Prevention of human pancreatic cancer cell-induced hepatic metastasis in nude mice by dipyridamole and its analog RA-233. Cancer. 1993;71:2466–71. doi: 10.1002/1097-0142(19930415)71:8<2466::aid-cncr2820710807>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 18.Choudhary S, Sood S, Wang HC. Dipyridamole intervention of breast cell carcinogenesis. Mol Carcinog. 2012 doi: 10.1002/mc.21970. [DOI] [PubMed] [Google Scholar]
- 19.Spano D, Marshall JC, Marino N, De Martino D, Romano A, Scoppettuolo MN, et al. Dipyridamole prevents triple-negative breast-cancer progression. Clin Exp Metastasis. 2013;30:47–68. doi: 10.1007/s10585-012-9506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin EY, Jones JG, Li P, Zhu UY, Whitney KD, Muller WJ, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. American Journal of Pathology. 2003;163:2113–26. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ullman-Cullere MH, Foltz CJ. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim Sci. 1999;49:319–23. [PubMed] [Google Scholar]
- 23.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 24.Willson JK, Fischer PH, Tutsch K, Alberti D, Simon K, Hamilton RD, et al. Phase I clinical trial of a combination of dipyridamole and acivicin based upon inhibition of nucleoside salvage. Cancer research. 1988;48:5585–90. [PubMed] [Google Scholar]
- 25.Colombo PE, Milanezi F, Weigelt B, Reis-Filho JS. Microarrays in the 2010s: the contribution of microarray-based gene expression profiling to breast cancer classification, prognostication and prediction. Breast Cancer Research. 2011;13:212. doi: 10.1186/bcr2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zetter BR. Adhesion Molecules in Tumor-Metastasis. Seminars in Cancer Biology. 1993;4:219–29. [PubMed] [Google Scholar]
- 27.Sumitomo M, Shen RQ, Walburg M, Dai J, Geng YP, Navarro D, et al. Neutral endopeptidase inhibits prostate cancer cell migration by blocking focal adhesion kinase signaling. Journal of Clinical Investigation. 2000;106:1399–407. doi: 10.1172/JCI10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorrin-Rivas MJ, Arii S, Furutani M, Mizumoto M, Mori A, Hanaki K, et al. Mouse macrophage metalloelastase gene transfer into a murine melanoma suppresses primary tumor growth by halting angiogenesis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:1647–54. [PubMed] [Google Scholar]
- 29.Tabaries S, Dong Z, Annis MG, Omeroglu A, Pepin F, Ouellet V, et al. Claudin-2 is selectively enriched in and promotes the formation of breast cancer liver metastases through engagement of integrin complexes. Oncogene. 2011;30:1318–28. doi: 10.1038/onc.2010.518. [DOI] [PubMed] [Google Scholar]
- 30.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 31.Ellinger-Ziegelbauer H, Kelly K, Siebenlist U. Cell cycle arrest and reversion of Ras-induced transformation by a conditionally activated form of mitogen-activated protein kinase kinase kinase 3. Molecular and Cellular Biology. 1999;19:3857–68. doi: 10.1128/mcb.19.5.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebbeck TR, DeMichele A, Tran TV, Panossian S, Bunin GR, Troxel AB, et al. Hormone-dependent effects of FGFR2 and MAP3K1 in breast cancer susceptibility in a population-based sample of post-menopausal African-American and European-American women. Carcinogenesis. 2009;30:269–74. doi: 10.1093/carcin/bgn247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu XL, Wazer DE, Watanabe K, Band V. Identification of a novel serine protease-like gene, the expression of which is down-regulated during breast cancer progression. Cancer research. 1996;56:3371–9. [PubMed] [Google Scholar]
- 34.Goyal J, Smith KM, Cowan JM, Wazer DE, Lee SW, Band V. The role for NES1 serine protease as a novel tumor suppressor. Cancer research. 1998;58:4782–6. [PubMed] [Google Scholar]
- 35.White NMA, Chow TFF, Mejia-Guerrero S, Diamandis M, Rofael Y, Faragalla H, et al. Three dysregulated miRNAs control kallikrein 10 expression and cell proliferation in ovarian cancer. British Journal of Cancer. 2010;102:1244–53. doi: 10.1038/sj.bjc.6605634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayami T, Shukunami C, Mitsui K, Endo N, Tokunaga K, Kondo J, et al. Specific loss of chondromodulin-I gene expression in chondrosarcoma and the suppression of tumor angiogenesis and growth by its recombinant protein in vivo. Febs Letters. 1999;458:436–40. doi: 10.1016/s0014-5793(99)01201-6. [DOI] [PubMed] [Google Scholar]
- 37.Shukunami C, Hiraki Y. Expression of cartilage-specific functional matrix chondromodulin-1 mRNA in rabbit growth plate chondrocytes and its responsiveness to growth stimuli in vitro. Biochemical and Biophysical Research Communications. 1998;249:885–90. doi: 10.1006/bbrc.1998.9233. [DOI] [PubMed] [Google Scholar]
- 38.Wells WA, Schwartz GN, Morganelli PM, Cole BF, Gibson JJ, Kinlaw WB. Expression of “Spot 14” (THRSP) predicts disease free survival in invasive breast cancer: immunohistochemical analysis of a new molecular marker. Breast Cancer Research and Treatment. 2006;98:231–40. doi: 10.1007/s10549-005-9154-z. [DOI] [PubMed] [Google Scholar]
- 39.Chung HJ, Levens D. c-myc Expression: Keep the noise down! Molecules and Cells. 2005;20:157–66. [PubMed] [Google Scholar]
- 40.Drabovich AP, Pavlou MP, Dimitromanolakis A, Diamandis EP. Quantitative analysis of energy metabolic pathways in MCF-7 breast cancer cells by selected reaction monitoring assay. Molecular & cellular proteomics: MCP. 2012;11:422–34. doi: 10.1074/mcp.M111.015214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sameen S, Khalid Z, Malik SI. Role of pyruvate dehydrogenase kinases (PDK’s) and their respective microRNA’s in human ovarian cancer. J Med Genet Genomics. 2011;3:115–21. [Google Scholar]
- 42.Yamamoto Y, Yoshioka Y, Minoura K, Takahashi RU, Takeshita F, Taya T, et al. An integrative genomic analysis revealed the relevance of microRNA and gene expression for drug-resistance in human breast cancer cells. Molecular Cancer. 2011;10:135. doi: 10.1186/1476-4598-10-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nature Biotechnology. 2010;28:341–7. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZGC, et al. A Pleiotropically Acting MicroRNA, miR-31, Inhibits Breast Cancer Metastasis. Cell. 2009;137:1032–46. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Persson H, Kvist A, Rego N, Staaf J, Vallon-Christersson J, Luts L, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer research. 2011;71:78–86. doi: 10.1158/0008-5472.CAN-10-1869. [DOI] [PubMed] [Google Scholar]
- 46.Zhou CC, Liu JB, Li Y, Liu L, Zhang XJ, Ma CY, et al. microRNA-1274a, a modulator of sorafenib induced a disintegrin and metalloproteinase 9 (ADAM9) down-regulation in hepatocellular carcinoma. Febs Letters. 2011;585:1828–34. doi: 10.1016/j.febslet.2011.04.040. [DOI] [PubMed] [Google Scholar]
- 47.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer (vol 449, pg 682, 2007) Nature. 2008;455:256. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 48.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.