

Background: The fatty acid palmitate induces sphingosine kinase 1 (Sphk1) activity in skeletal muscle by an unknown mechanism.
Results: SphK1 is transcriptionally up-regulated by peroxisome proliferator-activated receptor α in lipid overload and promotes interleukin (IL)-6 expression and signaling.
Conclusion: Palmitate-induced IL-6 is SphK1-dependent and mediates paracrine/autocrine IL-6 signaling in muscle.
Significance: This study identifies a novel role for SphK1 in the context of obesity.
Keywords: Lipotoxicity, Obesity, Skeletal Muscle, Sphingolipid, Sphingosine-1-phosphate, IL-6, PPARα, Sphingosine Kinase 1
Abstract
We previously demonstrated that sphingosine kinase 1 (Sphk1) expression and activity are up-regulated by exogenous palmitate (PAL) in a skeletal muscle model system and in diet-induced obesity in mice; however, potential functions and in vivo relevance of this have not been addressed. Here, we aimed to determine the mechanism by which PAL regulates SphK1 in muscle, and to determine potential roles for its product, sphingosine-1-phosphate (S1P), in muscle biology in the context of obesity. Cloning and analysis of the mouse Sphk1 promoter revealed a peroxisome proliferator-activated receptor (PPAR) α cis-element that mediated activation of a reporter under control of the Sphk1 promoter; direct interaction of PPARα was demonstrated by chromatin immunoprecipitation. PAL treatment induced the proinflammatory cytokine interleukin (IL)-6 in a manner dependent on SphK1, and this was attenuated by inhibition of the sphingosine-1-phosphate receptor 3 (S1PR3). Diet-induced obesity in mice demonstrated that IL-6 expression in muscle, but not adipose tissue, increased in obesity, but this was attenuated in Sphk1−/− mice. Moreover, plasma IL-6 levels were significantly decreased in obese Sphk1−/− mice relative to obese wild type mice, and muscle, but not adipose tissue IL-6 signaling was activated. These data indicate that PPARα regulates Sphk1 expression in the context of fatty acid oversupply and links PAL to muscle IL-6 production. Moreover, this function of SphK1 in diet-induced obesity suggests a potential role for SphK1 in obesity-associated pathological outcomes.
Introduction
The past decades have witnessed an increase in the incidence of obesity and a subsequent increase in associated consequences (1–6). Although the mechanisms linking obesity to downstream pathology remain unclear, elevation in plasma lipids induced by obesity likely plays a role by overloading tissues with precursors for bioactive lipid synthesis including diacylglycerols and ceramides, thus perturbing cell homeostasis (7–9). This general process has been termed “lipotoxicity” (7). In obese individuals, plasma free fatty acids (FFA)2 can reach levels double those of lean individuals (10). Moreover, elevation of plasma triglycerides also leads to fatty acid oversupply in tissues that express endothelial lipoprotein lipase, including skeletal muscle (11). Skeletal muscle is particularly relevant in this context, as it accounts for ∼40% of total body mass and contributes significantly to whole body fatty acid uptake and oxidation (7, 12, 13).
PAL, the most abundant FFA in circulation, has been linked to lipotoxicity in skeletal muscle (4, 14, 15). PAL serves as a substrate for de novo sphingolipid synthesis and treatment with exogenous PAL induced de novo synthesis in skeletal muscle (6, 16–18). In addition to providing excess substrate for de novo synthesis, PAL has been demonstrated to drive sphingolipid synthesis through other mechanisms. These mechanisms include the activation of Toll-like receptor 4 (TLR4) (19) and regulation of the enzymes of sphingolipid metabolism. Specifically, we previously demonstrated aberrant regulation of dihydroceramide desaturase and SphK1 in fatty acid-treated cells and/or a mouse model of obesity (6, 20). We hypothesized that these events may play roles in pathophysiological outcomes of obesity. Supporting this notion, sphingolipids are implicated in obesity-induced insulin resistance, disruption of mitochondrial function, and other lipotoxic programs (7–9).
Two isoforms of sphingosine kinase, SphK1 and -2, catalyze phosphorylation of sphingosine to generate S1P. In addition to recently described intracellular targets of S1P (21), S1P signals through G protein-coupled receptors that mediate processes such as chemotaxis, cellular differentiation, proliferation, survival, and inflammation (6, 22–24). SphK1 activity is regulated by several factors including vascular endothelial growth factor (25), epidermal growth factor (26), cytokines, such as tumor necrosis factor α (27), and its metabolite S1P (27, 28); however, little is known regarding its transcriptional regulation. A previous study demonstrated its regulation in hypoxia in U87MG cells by HIF2α (29). However, mechanisms of Sphk1 transcriptional control in response to other stimuli remain undetermined.
Peroxisome proliferator-activated receptor (PPAR) α is a key transcription factor involved in regulating lipid catabolism including cellular fatty acid uptake, intracellular fatty acid transport, mitochondrial and peroxisomal fatty acid oxidation, and gluconeogenesis (30–32). PPARs are ligand-activated; whereas some controversy exists as to the specific endogenous ligands, studies have demonstrated activation of PPARα-dependent transcription by fatty acids (32–34). Interleukin-6 (IL-6), a cytokine that acts in both pro- and anti-inflammatory pathways (35, 36), is produced by cells of the immune system; however, recent studies have demonstrated that Il-6 expression and production increase in contracting skeletal muscle (37–39). IL-6 produced by skeletal muscle is the prototype for an emerging class of cytokines known as myokines, which may mediate cross-talk between muscle and other tissues (40, 41). IL-6 is also increased in plasma of obese individuals (42), and correlates with increased levels of plasma FFA (42). These findings suggest IL-6 may play a role in obesity-induced inflammation (41). IL-6 is known to be regulated by fatty acids, and has also been shown to be increased by S1P-induced signaling pathways, although not in the context of metabolic disease (43–45).
Importantly, whereas S1P was demonstrated to be elevated in mouse models of obesity (46, 47), a mechanistic role for SphK1/S1P in obesity remains unknown. Because PAL induced SphK1 in a skeletal muscle model, we hypothesized that SphK1 may regulate IL-6 in obesity. Here we demonstrate that transcriptional regulation of Sphk1 by PAL occurs through PPARα, placing SphK1 in the context of global lipid metabolism. Moreover, we demonstrate that in diet-induced obesity in mice, SphK1 is required for Il-6 muscle message, increased plasma IL-6, and muscle-specific activation of IL-6 signaling. These findings suggest a novel mechanistic role for SphK1 in the pathophysiological consequences of obesity.
EXPERIMENTAL PROCEDURES
Materials
Fetal bovine serum, horse serum, 0.5% trypsin EDTA, and Lipofectamine 2000 were from Invitrogen. DMEM with 4 mm l-glutamine was from ATCC (Manassas, VA); GW6471 and WY14,643 were from Sigma; palmitic acid salt (C16:0) was from Matreya, LLC (Pleasant Gap, PA); anti-PPARα antibody was from Abcam (Cambridge, MA); S1P was from Enzo Life Sciences (Plymouth Meeting, PA); JTE013 was from Cayman Chemical (Ann Arbor, MI); and VPC23019 was from Avanti Polar Lipids (Alabaster, AL).
Cell Culture
Mouse C2C12 myoblasts (ATCC) were maintained at 37 °C and 5% CO2 in DMEM containing 10% FBS. Primary myoblasts were isolated from mice as previously described in Ref. 48. In brief, muscles from the hind limbs of 8-week old C57Bl/6J (WT; The Jackson Laboratory, Bar Harbor, ME) and Sphk1−/− mice initially generated by Dr. Richard Proia (49) were maintained in house at the Veterans Affairs animal care facility on a 12-h light cycle. Tissues were dissociated in collagenase/dispase/CaCl2 solution (1.5 units/ml of collagenase D from Fisher Scientific, 2.4 units/ml of dispase II from Invitrogen, and 2.5 mm CaCl2) and incubated at 37 °C for 1 h. Cells were filtered and resuspended in F-10-based primary myoblast growth medium (F-10 nutrient mixture (Invitrogen)) supplemented with 20% FBS, 25 μg/ml of human basic fibroblast growth factor (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). Filtered cells were plated on calf collagen (Fisher Scientific)-coated plates and maintained in F-10-based primary myoblast growth medium for 7 days. Cells were then maintained in F-10/DMEM-based primary myoblast growth medium (40% F-10-based primary myoblast growth medium and 40% DMEM) for an additional 4 days. Confluent C2C12 and primary cells were both differentiated in DMEM supplemented with 10% horse serum for 7 days forming myotubes for utilization in experiments.
FFA Treatment
FFAs were prepared as previously described in Ref. 50. Briefly, palmitate salt (Matreya, LLC) dissolved in 100% ethanol to a concentration of 125 mm was added to serum-free DMEM containing 2% fatty acid-free BSA and 1% FBS to a concentration of 0.75 mm, briefly sonicated, incubated at 55 °C, and cooled to 37 °C. Myoblasts were treated for 8 h and primary myotubes were treated for 16 h.
Cloning of the Sphk1 Promoter
The mouse Sphk1 promoter was cloned based on sequence homology with the rat Sphk1 promoter (51, 52). Primers from Integrated DNA Technologies (Coralville, IA) beginning at the −2239 position upstream (5′-GAGGAGTCTCGAGGTTCTGTGTAACCGGA-3′) and the +1592 position downstream (5′-ATCGCTACCATGGTTCAGCTTATCGGT-3′) of the transcription initiation site were used to clone the promoter into the PGL3-basic vector (Promega) utilizing the XhoI and NcoI restriction sites.
Generation of Sphk1 Promoter Deletions
The Sphk1 promoter was used as a PCR template to generate systematic deletion constructs. The forward primers −1869 (5′-CTCGAGTGTTTATCTCCACCGAAGCGCATAC-3′), −1691 (5′-CTCGAGCGATCATCCGCGGCAGGCAGCATCT-3′), −1333 (5′-CTCGAGTTCGAGGTTCAGTAAGCGCAGACCC-3′), −920 (5′-CTCGAGCCTTGGAGTCGGTGTCAGCCCAGG-3′), −519 (5′-CTCGAGTACGCGCCTCTCAATGCCAGTTCTG-3′), and −299 (5′-CTCGAGCAGGGGCTCTGGTTGGGCACTTTGT-3′) were used with the reverse primer, 5′-ATCGCTACCATGGTTCAGCTTCTTATCCGT-3′, from Integrated DNA Technologies (Coralville, IA). PCR products were cloned into pCR-2.1 TOPO vectors (Invitrogen) and subcloned into PGL3-basic vectors (Promega).
SphK1 Promoter Transfection and Luciferase Assay
The full-length Sphk1 promoter or the deletion constructs were transfected into C2C12 myoblasts using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. Cells were co-transfected with a LacZ-encoding plasmid as a control for transfection efficiency. Eighteen hours post-transfection, cells were treated with FFA for 8 h and luciferase activity was quantified utilizing the Luciferase Assay Kit (Agilent, Santa Clara, CA), following the manufacturer's instructions. Luciferase data were normalized to β-galactosidase activity as determined by the High Sensitivity β-Galactosidase Kit (Agilent).
Chromatin Immunoprecipitation Assay
C2C12 myoblasts were treated with FFA for 16 h and cross-linking was performed by treatment with 1% formaldehyde solution for 15 min at room temperature. Cross-linking was terminated by the addition of glycine (125 mm). Cells were lysed and chromatin was sheared in a sonicating water bath at ambient temperature, using a 6-s on and 10-s off protocol for 10 cycles. Chromatin was precleared with protein A-agarose/salmon sperm DNA beads (Millipore, Billerica, MA) at 4 °C for 2 h. Precleared chromatin samples were immunoprecipitated with mouse monoclonal anti-PPARα antibody (Abcam, Cambridge, MA) overnight at 4 °C. Immunoprecipitated chromatin was eluted and reverse cross-linked, purified, and subjected to real-time PCR. DNA removed from samples prior to preclearing served as input controls. Real-time primers were designed to amplify the region containing the PPARα responsive element located at the 5′ terminus of the Sphk1 promoter: sense, 5′-AGCTTCCTTGGGAGTTTGGTGTCT-3′ and antisense, 5′-TTTCATGGCAAGTGACCTGAGGGT-3′. Fatty acid-binding protein (FABP) was used as a comparison for PPARα as it is a known PPARα target using primers from SA Biosciences (Valencia, CA). Conventional PCR to amplify the region containing the PPARα responsive element was performed according to standard protocols using the same primers as those used for real-time PCR.
siRNA Transfection
Sixty percent confluent C2C12 myoblasts were transfected with either non-targeting control siRNA or specific siRNAs for PpaRα, S1pr1, or S1pr3 siRNAs using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Seventy-two hours post-transfection, cells were treated with BSA or 0.75 mm PAL for 16 h. Total RNA was isolated using the TRIzol reagent (Invitrogen) and utilized for quantitative PCR (qPCR).
Quantitative Real-time PCR
Total RNA from C2C12 myoblasts or primary myotubes was isolated using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. First-strand cDNA was synthesized from 4 μg of total RNA using the SuperscriptTM First-strand Synthesis System (Invitrogen). Quantitative real-time PCR was performed on an iCycler System as described in Ref. 6. Mouse Sphk1, Il-6, S1pr1, S1pr3, and suppressor of cytokine signaling 3 (Socs3) primers were from Integrated DNA Technologies (Coralville, IA). Gapdh was used as a reference gene.
Lipidomic Measurement by LC/MS
Lipidomic profiling was performed as previously described (53).
Diet-induced Obesity in Mice
Eight-week old male wild type (Jackson Laboratory, Bar Harbor, ME) and Sphk1−/− mice were maintained on control (10% calories from fat) and high-fat (HFD; 60% calories from fat) diets 12 weeks. At the culmination of 12 weeks, mice were sacrificed and skeletal muscle, adipose tissue, and plasma were isolated from the animals for use in future analyses.
Plasma IL-6 Measurements
Changes in plasma IL-6 levels were measured using the Bio-Plex 2200 system (Bio-Rad). Assays were carried out according to the manufacturer's instructions. Briefly, plasma samples from mice on control and a high-fat diet for 12 weeks were diluted 1:4 and incubated in a 96-well plate with magnetic antibody-coupled beads for 30 min at room temperature while shaking. The 96-well plates were washed and incubated with detection antibodies for 30 min, followed by the addition of streptavidin-PE for 10 min. IL-6 levels were measured using the Bio-Plex 2200 system. Each reaction was carried out in duplicate.
Statistics
Statistical significance was determined by Student's t test, with a value of p ≤ 0.05 considered significant.
RESULTS
PAL Induction of S1P Production Occurs through Transcriptional Regulation of SphK1
Our laboratory previously published that PAL increased SphK1 activity and S1P in C2C12 myotubes. In that study, the message for both Sphk1 and Sphk2 increased (6). Thus, to clarify which isoform mediated PAL-induced S1P production, we employed primary myoblasts isolated from male wild type (WT) or Sphk1−/− mice. Cells were isolated and differentiated to myotubes as described under “Experimental Procedures.” Myotubes were treated with 0.75 mm PAL for 16 h. S1P content was near the lower limit of detection, and thus, samples were pooled to determine S1P content by LC/MS. As demonstrated in Fig. 1A, S1P production was attenuated in pooled Sphk1−/− myotubes basally and in response to PAL treatment (p = 0.09), which suggested that S1P production by skeletal muscle derives largely from SphK1. Moreover, consistent with data in the C2C12 cell line, PAL treatment of primary myotubes increased Sphk1 expression ∼2-fold (Fig. 1B). Together, these data support that PAL induction of Sphk1 constitutes the primary route of S1P production in response to PAL treatment, and that this occurs in both immortalized and primary cells.
FIGURE 1.
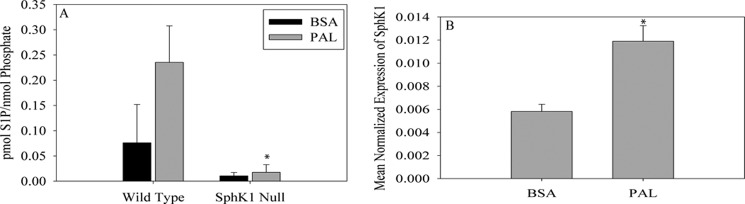
PAL-induced S1P production is SphK1 dependent. Myoblasts were isolated from C57Bl/6J and Sphk1−/− mice and differentiated to myotubes. Following a 16-h treatment with BSA or 0.75 mm PAL, cells were harvested and analyzed. A, three sets of isolated myotubes were pooled for the measurement of total S1P. S1P content was determined by LC/MS. Data are represented as mean ± S.E. (n = 2). *, p = 0.09 versus PAL WT. B, Sphk1 mRNA expression was determined in wild type myotubes by qPCR. Data are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus BSA-treated cells.
Studies have indicated that PAL perturbs cell sphingolipid profiles through multiple mechanisms including direct substrate supply (6, 20). A previous report implicated TLR4 in the regulation of the synthesis of ceramide in vivo (19); however, the regulation of S1P generation was not addressed. Because ceramides serve as metabolic precursors for S1P synthesis, we hypothesized that PAL may increase S1P through TLR4-mediated ceramide increase. To test this, we transfected C2C12 cells with siRNA targeted to Tlr4 or negative control siRNA. Knockdown of Tlr4 did not attenuate S1P production in response to PAL treatment (data not shown). Moreover, ceramides, previously suggested as downstream of TLR4 signaling in an in vivo model (19), were not attenuated in Tlr4 knockdown cells (data not shown), suggesting that at least some PAL-mediated regulation of sphingolipid metabolism in skeletal muscle may occur independently of TLR4 receptor signaling.
PAL Induces SphK1 Promoter Activity
Our previous studies indicated that the Sphk1 message increased upon treatment with PAL, which could be due to transcriptional regulation and/or mRNA stability. To distinguish between these possibilities, C2C12 myoblasts were treated with 0.75 mm PAL in the presence and absence of 1 μm actinomycin D for 16 h. Data demonstrated that the PAL-induced Sphk1 message increase was significantly attenuated in the presence of actinomycin D (Fig. 2A), indicating a requirement for transcription. Thus, we hypothesized that PAL caused Sphk1 promoter activation. To test this, the mouse Sphk1 promoter was cloned based on sequence homology with the published rat promoter sequence as described under “Experimental Procedures” (51, 52). The two sequences were compared using the BLAST program, using the 5-kb regions flanking the 5′ end of the SphK1 coding sequences. Although the overall sequences shared low homology, a number of key regulatory regions in the rat promoter were conserved in the mouse promoter sequence (Fig. 2B). For example, a 55-bp region of the rat promoter, which was shown to be necessary and sufficient for NGF-induced Sphk1 exon D expression, also appeared at −387 to −256 in the mouse sequence. Moreover, region III, an upstream region from the −3283 to −2991 position in the rat sequence, and which was demonstrated to be particularly sensitive to methylation, was also identified at positions −1543 to the transcription initiation site in the murine promoter sequence. Thus, these regions were used for orientation in cloning the mouse promoter through PCR amplification of wild type genomic DNA using the primers indicated under “Experimental Procedures.” Amplification resulted in a 2064-bp fragment that was cloned into TOPO-TA 2.1 vector and then subcloned into the PGL3-Basic luciferase reporter vector.
FIGURE 2.

PAL induces SphK1 through activation of the promoter. A, to evaluate the role of transcription in PAL-induced Sphk1 expression, C2C12 myoblasts were treated with 0.75 mm PAL in the presence or absence of 1 μm actinomycin D for 16 h. Following treatment, cells were harvested and the expression of Sphk1 was analyzed by qPCR. Data are presented as mean normalized expression ± S.E. (n = 3). *, p < 0.01 versus non-actinomycin D-treated cells. B, the murine Sphk1 promoter was cloned based on sequence homology with the rat Sphk1 promoter. The cloned promoter was cloned into a PGL3 basic vector for further use. The region of the promoter necessary for PAL-induced Sphk1 promoter activation was analyzed for transcription factor responsive elements using the transcription factor prediction model ALGGEN-PROMO (54, 56). C, C2C12 myoblasts were transfected with empty PGL3 or the PGL3-Sphk1 vector. Cells were treated with BSA or 0.75 mm PAL 18 h post-transfection for 8 h. Cells were assayed for relative luciferase activity and normalized to β-galactosidase activity. Data are presented as fold-change in relative normalized luciferase activity ± S.E. (n = 3). *, p < 0.05 versus PGL3-Sphk1 BSA cells. D, systematic deletions were generated of the Sphk1 promoter and cloned into the PGL3 vector. E, previously generated deletion constructs were transfected into C2C12 myoblasts. Eighteen hours post-transfection, cells were treated with BSA or 0.75 mm PAL for 8 h and assayed for relative luciferase activity. Measured luciferase activity was normalized to β-galactosidase activity. Data are presented as fold-change in normalized relative luciferase activity ± S.E., (n = 6). *, p < 0.05 versus PGL3-Sphk1.
C2C12 myoblasts were transfected with the PGL3-Sphk1 luciferase vector, treated for 8 h with 0.75 mm PAL, and luciferase activity was measured. As demonstrated in Fig. 2C, Sphk1 promoter activation occurred in these cells under basal conditions, reflected by a 4-fold increase in luciferase activity, which suggests that these cells basally transcribe Sphk1. Moreover, PAL treatment increased Sphk1 promoter activity 8.5-fold relative to BSA-treated PGL3-Sphk1-transfected cells, which further supported the hypothesis that PAL increases Sphk1 message and activity through increasing its transcription.
To identify potential regions of the Sphk1 promoter that mediated PAL-induced transcription, systematic promoter deletion constructs were generated in the PGL3 basic luciferase reporter vector; these constructs were used to transfect C2C12 skeletal muscle myoblasts (Fig. 2D). Transfected cells were treated with PAL or BSA as above and luciferase activity was quantified. Data indicated that deletion of −2068 to −1869 of the promoter prevented PAL-induced promoter activity (Fig. 2E), thus suggesting that this region may contain a PAL-responsive regulatory element. To identify potential cis-acting regulatory elements, the region was analyzed using the ALGGEN-PROMO transcription factor regulatory element prediction software (54), which yielded 3 potential transcription factor-binding domains in the region between the −2068 and −1869 positions (Fig. 2B). The potential cis-acting regulatory elements identified were c-Jun and c-Fos, which make up the AP-1 transcription factor, and a PPARα responsive element. To evaluate the contribution of these transcription factors to Sphk1 gene transcription, siRNA was used to knockdown c-Fos, c-Jun, and Pparα. Interestingly, knockdown of the Ap-1 complex diminished basal Sphk1 expression, perhaps suggesting a role for Ap-1 in basal transcription of Sphk1, but had no effect on PAL-induced Sphk1 expression (data not shown). In contrast, transfection with Pparα siRNA resulted in a 75% reduction in Pparα message (Fig. 3A), which was sufficient to result in a significant decrease in PAL-induced Sphk1 expression (Fig. 3B).
FIGURE 3.

PAL-induced Sphk1 expression is PPARα dependent. Analysis of the region indicated by the deletion model to be important in PAL-induced Sphk1 promoter activity indicated the presence of a Pparα responsive element. To assess the importance of this site in PAL-induced Sphk1 promoter activity. A, C2C12 myoblasts were transfected with 100 pmol of control or Pparα siRNA. Seventy-two hours post-transfection, cells were harvested and assessed for knockdown efficiency by qPCR. Data are presented as mean normalized expression ± S.E. (n = 3). *, p < 0.01 versus control. B, siRNA-transfected cells were treated with BSA or 0.75 mm PAL for 16 h. Cells were harvested and analyzed for Sphk1 mRNA expression. Data are presented as mean normalized expression ± S.E. (n = 3). *, p < 0.01 versus PAL control; **, p < 0.01 versus BSA control; #, p < 0.001 versus BSA Pparα. C, C2C12 myoblasts were transfected with the Sphk1 promoter vector and treated 18 h post-transfection with BSA or 0.75 mm PAL for 8 h in the presence of GW6471, a potent PPARα antagonist. Data are expressed as fold-change in normalized relative luciferase activity ± S.E. (n = 3); *, p < 0.05 versus non-GW6471 treated cells. D, myoblasts were isolated from WT mice and differentiated into myotubes. Myotubes were treated with 0.75 mm PAL in the presence of GW6471 for 8 h. Sphk1 expression was determined by qPCR. Data are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus non-GW6471 treated cells. E, to establish that PPARα associates with the Sphk1 promoter, C2C12 myoblasts were treated for 16 h with BSA or 0.75 mm PAL. The cells were utilized for ChIP assays as described under “Experimental Procedures.” Data are presented as the ratio of PPARα to background (IgG) ± S.E. (n = 5). *, p < 0.01 versus BSA. The presence of FABP was evaluated for comparison to PPARα pulldown. Data are presented as ratio of FABP to background (IgG) ± S.E. (n = 5). #, p < 0.05 versus BSA. F, DNA obtained from ChIP assays were utilized for conventional PCR. Following the PCR, the resulting products were visualized using a 10% native polyacrylamide gel. The gel pictured is a representative of 5 independent experiments.
To further test whether PPARα mediated the induction of Sphk1 by PAL, we treated myoblasts transfected with the Sphk1 promoter construct with PAL in the presence of GW6471, a potent PPARα antagonist. Data showed that GW6471 attenuated PAL-induced Sphk1 promoter activity at very low doses (Fig. 3C), further supporting that PPARα mediates PAL stimulation of Sphk1 transcription. To test whether this mechanism may also apply to activation of the endogenous Sphk1 promoter, we tested inhibition of the PAL-induced increase in a message encoding Sphk1 in isolated primary myotubes (Fig. 3D). These data showed a similar pattern to the luciferase data, suggesting that PPARα-dependent mechanisms were similar whether assessed by reporter gene strategies or direct measurement of endogenous Sphk1 message.
Although these data support that PAL stimulated PPARα-dependent activation of Sphk1 transcription, it was still unclear whether this occurred as a result of direct interaction of PPARα with the Sphk1 promoter, or through downstream PPARα-dependent events. Thus, we tested interaction of PPARα with the Sphk1 promoter in situ using a chromatin immunoprecipitation (ChIP) strategy. In brief, C2C12 myoblasts were treated with 0.75 mm PAL or BSA vehicle for 8 h. As described in detail under “Experimental Procedures,” cells were cross-linked, DNA was sheared, a 100-μl aliquot was retained for normalization, whereas the remainder of the chromatin was cleared and utilized for immunoprecipitation. DNA was precipitated from the immunoprecipitate and input samples and utilized for real-time PCR. Indeed, PAL induced a significant increase in the association of PPARα with the Sphk1 promoter (Fig. 3E; representative gel of conventional PCR products shown in Fig. 3F) relative to BSA alone treated cells. PPARα has been shown to regulate several genes, including those that participate in fatty acid metabolism. One such gene, Fabp, was utilized in the ChIP analysis to serve as positive control and as a comparison for PPARα pull down (Fig. 3E). Analysis of ChIP DNA demonstrated that PAL also promoted Fabp association with PPARα. Together, these data indicate activation of PPARα by PAL and establish the Sphk1 gene as a transcriptional target of PPARα in the context of PAL treatment.
PAL Induces Interleukin-6 Expression
Although these data establish the involvement of fatty acids in the induction of Sphk1 in skeletal muscle, potential roles for this event remain unknown. Given the emerging roles of IL-6 in pathophysiological processes associated with obesity (42, 55) as well as the key roles of IL-6 in muscle (37, 38, 57), we sought to test whether PAL treatment would induce muscle Il-6 through Sphk1. Consistent with previous findings (36, 58), treatment with PAL significantly increased Il-6 mRNA expression in both wild type primary and C2C12 myotubes (Fig. 4, , A and B). To test whether this occurred through a SphK1-dependent mechanism, we utilized primary myoblasts isolated from Sphk1−/− and WT mice as described under “Experimental Procedures.” Myotubes were treated with 0.75 mm PAL and Il-6 mRNA was measured using quantitative real-time PCR. As shown in Fig. 4C, PAL induced a significant increase in Il-6 expression in WT cells. In contrast, although Sphk1−/− cells basally expressed Il-6, its expression in response to PAL was significantly attenuated, indicating a requirement for Sphk1 for PAL-induced Il-6 expression under these conditions. Taken in total, these data suggest a novel role for SphK1/S1P in mediating the induction of Il-6 by PAL in skeletal muscle.
FIGURE 4.

PAL-induced Il-6 expression is Sphk1 dependent. The effect of PAL on IL-6, a downstream product of S1P-induced signaling pathways, was assessed in (A) primary myotubes and (B) C2C12 myotubes. Il-6 expression was determined by qPCR. Data are presented as mean ± S.E. (n = 3). *, p < 0.01 versus BSA; #, p < 0.05 versus BSA. C, to establish that PAL-induced Il-6 expression is Sphk1 dependent, myoblasts were isolated from C57Bl/6J and Sphk1−/− mice and differentiated into myotubes. Myotubes were treated with BSA or 0.75 mm PAL for 16 h. IL-6 expression was determined by qPCR. Data are presented as mean ± S.E. (n = 3). **, p < 0.05 versus WT BSA; *, p < 0.01 versus WT PAL; #, p < 0.05 versus Sphk1−/− BSA.
PAL-induced IL-6 Production Is Dependent on S1P Receptor 3
S1P mediates many processes including tissue inflammation (59). Most identified functions of S1P result from the activation of G protein-coupled S1P receptors of which there are 5 isoforms (S1PR1–S1PR5) (60). Previous studies indicate that skeletal muscle expresses S1PR1, S1PR2, and S1PR3 (61, 62). To determine which receptor may mediate SphK1-dependent IL-6 generation in response to PAL treatment, cells were pre-treated with 1 μm VPC23019, a S1PR1 and S1PR3 antagonist, or 1 μm JTE013, a S1PR2 antagonist, for 1 h. Cells were then treated with 0.5 μm S1P, harvested over time, and the Il-6 message was measured. As demonstrated in Fig. 5A, exogenously added S1P induced Il-6 expression, further supporting a role for S1P signaling through cell surface receptors in this process. Pretreatment with JTE013 had no effect on S1P-dependent Il-6 induction (data not shown); however, pre-treatment with VPC23019 blocked S1P induction of IL-6 (Fig. 5A). The expression of S1pr1 and S1pr3 in response to PAL treatment was evaluated and these data demonstrated that there was no significant difference in expression of either receptor in response to PAL treatment (data not shown).
FIGURE 5.

PAL-induced IL-6 expression is S1PR3 dependent. A, to evaluate if the activation of the S1P receptors expressed in skeletal muscle was required for PAL-induced Il-6 expression, C2C12 myoblasts were treated with S1P alone or pretreated for 1 h with 1 μm VPC23019, a S1P1,3 antagonist, followed by treatment with 0.5 μm S1P for 3 h. Data are presented as mean ± S.E. (n = 2). *, p < 0.05. B, to determine which of the two antagonized receptors is required for S1P-induced Il-6 expression, C2C12 myoblasts were transfected with 100 pmol of non-targeting control, S1pr1 or S1pr3 siRNA. Seventy-two hours post-transfection, cells were serum starved overnight and treated with 0.5 μm S1P for 3 h. The mRNA expression of both target genes was determined by qPCR. Data are expressed as mean normalized expression ± S.E. (n = 3). *, p < 0.01 versus control cells. C, upon confirmation of sufficient knockdown of both S1PRs, Il-6 expression in response to 0.5 μm S1P was evaluated was determined by qPCR. Data are expressed as fold-change in mean normalized expression as compared with the control ± S.E. (n = 3). *, p < 0.01 versus control.
These data suggested that PAL-induced Il-6 expression can be attributed to S1P activation of S1PR1 or S1PR3. To distinguish between these possibilities, we employed siRNA directed toward either receptor. Transfection with siRNAs resulted in approximately a 60 and 80% reduction in S1pr1 and S1pr3 message, respectively (Fig. 5B). Cells were treated with S1P and the Il-6 message was quantified by qPCR as described above. Data demonstrated that knockdown of the S1pr3 receptor, but not knockdown of the S1pr1 receptor, attenuated S1P-induced Il-6 (Fig. 5C), suggesting a role for this receptor in Il-6 induction by PAL.
Sphk1−/− Mice Are Protected from Obesity-induced Increase in Plasma IL-6
Fatty acid treatment is used to model lipid oversupply in obesity; however, whereas this may effectively facilitate mechanistic studies, it is limited in its relevance to obesity in vivo, which perturbs not only levels of fatty acids but also plasma triacylglycerols, an abundant source of fatty acids in organs such as heart and skeletal muscle, which contain abundant lipoprotein lipase in vascular endothelium (63). Moreover, plasma contains not only PAL, but complex mixtures of fatty acids; both amounts and profiles of these lipids are perturbed in obesity (10). Thus, to test the in vivo relevance of the findings in the cell studies above, we placed 8-week-old male WT and Sphk1−/− mice on HFD obesogenic or low-fat isocaloric control diets for 12 weeks. Sphk1−/− animals maintained on the obesogenic diets gained significantly more weight than their WT counterparts (Fig. 6A). To determine the underlying mechanisms for this, food intake was monitored and we found no differences in food intake between WT and Sphk1−/− animals. Thus, we analyzed locomotor activity in Sphk1−/−, which we found was only 25% of that observed in WT mice irrespective of diet (data not shown). Although the underlying mechanism for this remains unknown, it may explain the increased weight gain in the Sphk1−/− mice.
FIGURE 6.

Diet-induced obesity promotes Il-6 expression in skeletal muscle and is attenuated in Sphk1−/− mice. Eight-week-old male C57Bl/6J and Sphk1−/− mice were maintained on obesogenic or low-fat control diets for 12 weeks. A, mice maintained on the diets were weighed biweekly. Data are presented as average weight ± S.E., n ≥ 4 mice per diet group; #, p < 0.01 versus wild type control; *, p < 0.01 versus Sphk1−/− HFD; **, p < 0.05 versus Sphk1−/− control; &, p < 0.05 versus wild type HFD; $, p < 0.01 versus Sphk1−/− control. B, total skeletal muscle was removed from the hind limbs of diet-fed mice. Il-6 expression was evaluated using qPCR, *, p < 0.05 versus WT control; #, p < 0.01 versus WT HFD. C, adipose tissue was removed from the abdomen of mice maintained on the diet and Il-6 expression was determined by qPCR. All qPCR data in B and C are presented as mean normalized expression (n = 4) ± S.E. D, plasma was isolated post-mortem from diet-fed mice and analyzed for IL-6 content via Bioplex 2200 System assay. Data are presented as mean picograms of IL-6/ml of plasma ± S.E., n ≥ 4 mice per diet group; **, p < 0.05 versus WT HFD; #, p < 0.05 versus Sphk1−/− HFD; *, p < 0.05 versus WT control.
To determine whether a high-fat diet induced muscle Il-6 in a Sphk1-dependent manner in vivo, IL-6 mRNA was quantified in skeletal muscle from high-fat diet fed mice of each genotype. Data demonstrated that high-fat feeding induced IL-6 expression in skeletal muscle in WT, but not Sphk1−/− animals (Fig. 6B).
Adipose tissue and skeletal muscle constitute major tissue sources of plasma IL-6, and thus, to test whether obesity increased adipose IL-6 in a Sphk1-dependent manner, Il-6 mRNA was quantified in adipose tissue from high-fat diet fed WT and Sphk1−/− animals. As shown in Fig. 6C, high-fat feeding does not induce Il-6 expression in adipose tissue as there were no differences between the control and high-fat diet in the Sphk1−/− mice, suggesting a muscle-specific role for Sphk1 in Il-6 production in obesity. To test whether this change in Il-6 message in skeletal muscle had an impact on circulating IL-6, plasma was prepared and used for Bioplex analysis as described above to measure IL-6 content. These data demonstrated that plasma IL-6 was increased 3-fold in WT mice placed on the obesogenic diet relative to controls, consistent with the magnitude of Il-6 message increase in cultured skeletal muscle cells. Interestingly, although basal IL-6 was elevated 3.5-fold in Sphk1−/− mice, the obesogenic diet produced no further induction (Fig. 6D). These data together with the expression data suggest that IL-6 production in muscle in response to high-fat feeding is regulated by Sphk1, and the increase in plasma IL-6 associated with obesity may derive from skeletal muscle, but not adipose.
IL-6 plays roles in systemic inflammation, but a recently proposed hypothesis suggests that IL-6 from skeletal muscle mediates skeletal-adipose cross-talk (41). Specifically, in exercise, muscle releases IL-6, and the purpose of this activity has been speculated to be activation of signaling downstream for the IL-6 receptor in adipose, ultimately stimulating adipocyte lipolysis to temporarily increase plasma FFA, thus restoring energy to depleted tissues. Because obesity leads to chronic elevation of plasma FFA, we hypothesized that SphK1-dependent IL-6 may mediate a similar paradigm in cross-talk between muscle and adipose in this context. Thus, we tested whether adipose IL-6 signaling was activated in the diet-induced obese mice, and if so, whether this required SphK1. Classical signaling of IL-6 occurs through the IL-6 receptor and results in the transcription of Socs3 in both muscle and adipose (64). Thus, as a read-out of IL-6 signaling, the Socs3 message was evaluated in adipose tissue and skeletal muscle from mice maintained on the control and high-fat diets. In adipose tissue, obesity did not induce Socs3 expression (Fig. 7A), and Socs3 mRNA levels were similar in mice of both genotypes, suggesting that IL-6 signaling in response to high-fat feeding did not occur in this tissue. Analysis of IL-6 signaling targets in skeletal muscle, however, demonstrated that the obesogenic diet induced the expression of Socs3 in a SphK1-dependent manner (Fig. 7B), suggesting that IL-6 signaling was induced in muscle in high-fat feeding, and was attenuated by the absence of SphK1. These data suggest that under these conditions, muscle-derived IL-6 may act in a paracrine fashion by promoting IL-6 signaling in skeletal muscle. Together these data suggest that muscle constitutes a significant source of plasma IL-6 in response to high-fat feeding in mice, and that in this context, muscle IL-6 acts in a paracrine manner and is SphK1- and S1PR3-dependent, thus supporting relevance and tissue specificity of the PAL-SphK1-S1P-S1PR3-IL-6 pathway in obesity.
FIGURE 7.
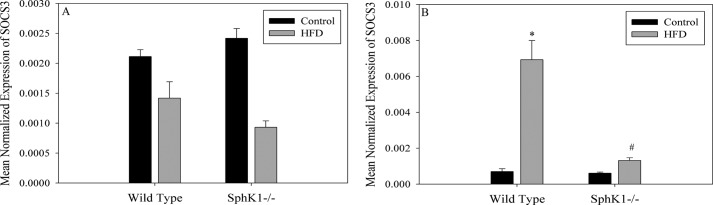
Diet-induced obesity induces the expression of Socs3 in skeletal muscle. A, adipose tissue was taken from the abdomen of wild type and Sphk1−/− mice maintained on the high fat and control diets. Total RNA was extracted and cDNA was synthesized. Socs3 expression was determined by qPCR. Data are presented as mean normalized expression ± S.E. (n = 4). B, Socs3 mRNA expression in skeletal muscle from mice maintained on the diets was determined by qPCR. Data are presented as mean normalized expression ± S.E. (n = 4); *, p < 0.05 versus WT control; #, p < 0.05 versus Sphk1−/− control.
DISCUSSION
Studies from our laboratory have demonstrated that treatment of mouse skeletal muscle cells with exogenous PAL increased Sphk1 message (6). PAL-induced SphK1 has also been recently demonstrated to occur similarly in INS-1 pancreatic β cells (65), and thus, this activity of palmitate may be relevant to numerous tissues and organs in vivo that succumb to pathology upon fatty acid oversupply.
Post-transcriptional and post-translational regulation of SphK1 has been well characterized (23); for example, SphK1 activation is mediated by ligands of G-protein-coupled receptors (23, 66), phosphorylation of tyrosine receptors (67), TNFα (27), vitamin D3 (68), FBS (66), and the product of SphK1 activity, S1P (23). Moreover, whereas a few studies have revealed transcriptional regulation of Sphk1 by cis-elements within the human and rat promoters, specifically, binding sites for HIF-1, AP-2, and Sp1 (52, 69), regulation of SphK1 in the context of obesity has not been addressed. The purpose of this study was to determine underlying mechanisms for fatty acid-mediated induction of SphK1 and potential functions in skeletal muscle in obesity.
The data presented in this article demonstrates that PAL-induced S1P production is Sphk1 dependent and that PAL promotes transcription of Sphk1 through the activation of its promoter. We have also demonstrated that PPARα is necessary for PAL-induced Sphk1 transcription. Additionally, the data demonstrated that PAL induces expression of Il-6 through S1PR3. Moreover, we demonstrated that a target of IL-6 signaling, Socs3, is increased in skeletal muscle, but not adipose tissue of diet-induced obese mice, supporting a muscle-specific role for this pathway in vivo.
Pparα expression has been found in tissues with high levels of fatty acid catabolism. These tissues include liver, heart, kidney, the small and large intestine, and skeletal muscle (70, 71). There are several known ligands of PPARα; in the context of metabolic syndrome and obesity, fatty acids have been shown to serve as biological, or endogenous, PPARα ligands (70, 72). Polyunsaturated fatty acids have been shown to be potent PPARα activators; however, it has come to light that saturated fatty acids can also induce PPARα activation (73, 74). However, data here do not preclude a metabolite of PAL in PPARα activation, for example, PAL-derived phosphatidylcholine species, which were recently reported as endogenous ligands of PPARα in liver (75), may also increase in muscle in this model. PPARα-dependent transcriptional regulation results in increased lipid oxidation and decreased triacylglycerol accumulation (76, 77). As PPARα is a central regulator of lipid metabolism, these data place SphK1 in the broad context of lipid metabolism.
IL-6 has been shown to act as a proinflammatory cytokine, playing a role in several diseases including inflammatory bowel disease (78, 79), colorectal cancer (79, 80), and type 2 diabetes (42, 55). In recent years, skeletal muscle has been identified as a site for cytokine, including IL-6, production (37), placing IL-6 in a class of cytokines secreted by skeletal muscle and thus termed “myokines” (40, 41). Previous studies have demonstrated that the expression and production of IL-6 is sensitive to elevations of FFA, more specifically PAL, in skeletal muscle (36, 58, 81). PAL has been shown to promote the activation of NFκB to induce IL-6 generation in rat skeletal muscle (83). In C2C12 myotubes, PAL treatment was shown to robustly induce IL-6 as early as 8 h with maximal IL-6 production achieved following 16 h of treatment (84). Consistent with our findings that high-fat feeding induces Il-6 in the skeletal muscle of WT mice, Reyna et al. (85) demonstrated that IL-6 expression was increased in the skeletal muscle of obese individuals. These studies support that data presented here may bear relevance to pathophysiological outcomes of obesity in humans.
Adipose tissue-derived IL-6 accounts for a large proportion of circulating IL-6 (86), with visceral or omental adipose tissue accounting for a larger proportion than that of subcutaneous fat depots (87). Consistent with our data, studies have demonstrated that 12 weeks of high-fat feeding did not induce Il-6 mRNA expression in adipose tissue (88, 89), but rather occurred at time points beyond 16 weeks (90). Whether this would occur in a manner dependent on SphK1 remains unknown. However, because our data demonstrated that circulating IL-6 was elevated in response to high-fat feeding (Fig. 6D), concomitant with Il-6 message induction of skeletal muscle, another major tissue source of IL-6, we conclude that muscle constitutes a major determinant of plasma IL-6 under our experimental conditions. We show here that a well established target of IL-6 signaling, Socs3, is elevated in muscle but not adipose tissue in high-fat feeding (Fig. 7A), and thus conclude that IL-6 may signal in an autocrine/paracrine manner in this context. This conclusion gains further support from studies that demonstrate that, whereas adipose tissue accounts for a large proportion of circulating IL-6, IL-6 signaling in adipose tissue is not well established (91).
In a similar vein, in vivo induction of skeletal muscle IL-6 by high-fat feeding (Fig. 6) was of a higher magnitude than in palmitate-treated isolated cells or cell lines (Fig. 4). We suspect that some of the additional increase observed in tissue homogenates may derive from other cell types in the homogenates including myoblasts and/or immune cells. However, because muscle and adipose tissue are major tissues determining plasma IL-6 levels, and that Il-6 message in adipose did not change in a manner dependent on SphK1, muscle IL-6 likely underlies the findings in plasma, which demonstrated an increase in high-fat feeding that was attenuated in Sphk1−/− animals. This is a consistent with the observation that changes in message level in muscles of 2–3-fold are similar to the magnitude of the increase we observed in plasma.
IL-6 signaling occurs in part through increased phosphorylation of STAT3, a transcription factor that regulates the expression of IL-6 target genes including Socs3, which prevents the phosphorylation of STAT3 by JAK, and thus constitutes a negative feedback loop for IL-6 signaling (92, 93). Assessment of STAT3 phosphorylation in mouse muscle was inconclusive under our conditions (not shown), which may occur because STAT3 serves as a target of many signaling pathways (94–96). In this animal model, which was derived from high-fat feeding over time, SOCS3-dependent inhibition of STAT3 phosphorylation precluded conclusions about IL-6 signaling based on the phosphorylation status of STAT3, i.e. loss of STAT3 phosphorylation induced by IL-6 receptor activation could indicate decreased IL-6 signaling or, in contrast, increased IL-6 signaling, which would up-regulate SOCS3 and thus inhibit STAT3 phosphorylation. Due to these complicating factors, we utilized Socs3 expression as a read-out of IL-6 signaling. We found that Socs3 was elevated in muscle in response to high-fat feeding and attenuated in the absence of SphK1.
Muscle-derived IL-6 was proposed to mediate cross-talk between adipose tissue and skeletal muscle during exercise (40, 41); however, the activation of IL-6 signaling, as evidenced by the increase in Socs3 message in skeletal muscle, but not adipose tissue, suggests that in obesity, this proposed cross-talk may not occur.
Previous studies have demonstrated that Sphk1 expression is required for the differentiation of myoblasts to myotubes; however, in the absence of SphK1, myogenesis can be induced through the addition of S1P (24). Here, our media for differentiation contained 10% horse serum, and thus, whereas the Sphk1−/− myotubes differentiated in the presence of serum, they differentiated at a slower rate than their WT counterparts. The differences in the degree of differentiation were similar to those shown by Meacci et al. (24).
The roles of IL-6 and its paracrine signaling in muscle in obesity remain unaddressed in this study; however, data have shown that muscle-derived IL-6 was associated with the induction of satellite cells following muscle damage in humans (97, 98). Thus, we propose that in the context of obesity, myotubes may sense damage due to steatosis, increased oxidative stress, or other metabolic disruptions, which may increase IL-6 production to initiate repair mechanisms, and SphK1 may play a role in this process. This notion is consistent with other studies that suggest roles for SphK1 in muscle regeneration (99). Thus, it is still undetermined whether the function of S1P-induced IL-6 plays a pathological role in this context, or rather, a response to repair muscle damage that may occur in obesity.
S1PR3 is a G-protein-coupled receptor that is expressed in the brain, heart, spleen, liver, lung, kidney, and most important to our study, skeletal muscle (100, 101). Activation of the S1PR3 results in coupling with several G-proteins and the activation of downstream targets including Rho GTPase and MAP kinases (82, 102). Although the involvement of S1PR3 in the production of pro-inflammatory cytokines is not fully understood, it has been implicated in generation of IL-6 in fibroblast-like synoviocytes in rheumatoid arthritis models in response to S1P treatment (102). These studies support the data shown here that S1PR3 plays an important role in S1P-induced IL-6 production in skeletal muscle.
In conclusion, our data demonstrated that in the context of fatty acid oversupply, Sphk1 is transcriptionally regulated through PPARα, which constitutes a novel mechanism of SphK1 regulation. Moreover, as a PPARα target, SphK1/S1P might be considered a potential effector of signaling by PPARα, a key regulator of whole body lipid metabolism and homeostasis. Additionally, these data indicate that SphK1 plays a mechanistic role in induction of muscle IL-6 in obesity, suggesting that SphK1/S1P mediate some pathophysiological changes associated with obesity. We propose that these data may broaden the known disease contexts in which SphK1/S1P signaling plays roles.
Acknowledgment
We thank Dr. Richard Proia (National Institute of Health, NIDDK, Bethesda, MD) for supplying the Sphk1−/− mice.
This work was supported, in whole or in part, by National Institutes of Health COBRE award P20 RR017677 (to L. A. C.), a Veterans Affairs Merit award, and a GAANN fellowship in Lipidomics and Systems Biology (to J. S. R.). Lipidomic analysis was performed by the Medical University of South Carolina Lipidomic Core supported by the National Center for Research Resources and the Office of the Director of the National Institutes of Health through Grant C06 RR018823. Research was supported in part by the Lipidomics Shared Resource, Hollings Cancer Center, Medical University of South Carolina Grant P30 CA138313, and the Lipidomics Core in the South Carolina Lipidomics and Pathobiology COBRE Grant P20 RR017677.
- FFA
- plasma free fatty acid
- SphK1
- sphingosine kinase 1
- IL-6
- interleukin-6
- PAL
- palmitate
- S1P
- sphingosine-1-phosphate
- S1PR
- S1P receptor
- TLR4
- Toll-like receptor 4
- SOCS3
- suppressor of cytokine signaling 3
- HFD
- high-fat diet
- STAT3
- signal transducer and activator of transcription 3
- FABP
- fatty acid-binding protein
- qPCR
- quantitative PCR.
REFERENCES
- 1. Hansen B. C. (1999) The metabolic syndrome X. Ann. N.Y. Acad. Sci. 892, 1–24 [DOI] [PubMed] [Google Scholar]
- 2. Lempiäinen P., Mykkänen L., Pyörälä K., Laakso M., Kuusisto J. (1999) Insulin resistance syndrome predicts coronary heart disease events in elderly nondiabetic men. Circulation 100, 123–128 [DOI] [PubMed] [Google Scholar]
- 3. Modan M., Halkin H., Almog S., Lusky A., Eshkol A., Shefi M., Shitrit A., Fuchs Z. (1985) Hyperinsulnemia. A link between hypertension, obesity, and glucose intolerance. J. Clin. Invest. 75, 809–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Summers S. A. (2006) Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 45, 42–72 [DOI] [PubMed] [Google Scholar]
- 5. Straczkowski M., Kowalska I., Baranowski M., Nikolajuk A., Otziomek E., Zabielski P., Adamska A., Blachnio A., Gorski J., Gorska M. (2007) Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 50, 2366–2373 [DOI] [PubMed] [Google Scholar]
- 6. Hu W., Bielawski J., Samad F., Merrill A. H., Jr., Cowart L. A. (2009) Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via up-regulation of sphingosine kinase message and activity. J. Lipid Res. 50, 1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turpin S. M., Ryall J. G., Southgate R., Darby I., Hevener A. L., Febbraio M. A., Kemp B. E., Lynch G. S., Watt M. J. (2009) Examination of “lipotoxicity” in skeletal muscle of high-fat fed and ob/ob mice. J. Physiol. 587, 1593–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schrauwen P. (2007) High-fat diet, muscular lipotoxicity and insulin resistance. Proc. Nutr. Soc. 66, 33–41 [DOI] [PubMed] [Google Scholar]
- 9. Schrauwen-Hinderling V. B., Kooi M. E., Hesselink M. K., Moonen-Kornips E., Schaart G., Mustard K. J., Hardie D. G., Saris W. H., Nicolay K., Schrauwen P. (2005) Intramyocellular lipid content and molecular adaptations in response to a 1-week high-fat diet. Obes. Res. 13, 2088–2094 [DOI] [PubMed] [Google Scholar]
- 10. Belfort R., Mandarino L., Kashyap S., Wirfel K., Pratipanawatr T., Berria R., Defronzo R. A., Cusi K. (2005) Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 54, 1640–1648 [DOI] [PubMed] [Google Scholar]
- 11. Wang H., Eckel R. H. (2009) Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 297, E271–E288 [DOI] [PubMed] [Google Scholar]
- 12. Cowart L. A. (2009) Sphingolipids. Players in the pathology of metabolic disease. Trends Endocrinol. Metab. 20, 34–42 [DOI] [PubMed] [Google Scholar]
- 13. Vusse G. V. D., Reneman R. (1996) in Handbook of Physiology (Rowell L., Shepherd J., eds) pp. 952–994, Oxford University Press, New York [Google Scholar]
- 14. Sparagna G. C., Hickson-Bick D. L., Buja L. M., McMillin J. B. (2000) A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 279, H2124–H2132 [DOI] [PubMed] [Google Scholar]
- 15. Wei Y., Wang D., Topczewski F., Pagliassotti M. (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 291, E275–E281 [DOI] [PubMed] [Google Scholar]
- 16. Shimabukuro M., Higa M., Zhou Y. T., Wang M. Y., Newgard C. B., Unger R. H. (1998) Lipoapoptosis in beta cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J. Biol. Chem. 273, 32487–32490 [DOI] [PubMed] [Google Scholar]
- 17. Zheng W., Kollmeyer J., Symolon H., Momin A., Munter E., Wang E., Kelly S., Allegood J. C., Liu Y., Peng Q., Ramaraju H., Sullards M. C., Cabot M., Merrill A. H., Jr. (2006) Ceramides and other bioactive sphingolipid backbones in health and disease. Lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling, and autophagy. Biochim. Biophys. Acta. 1758, 1864–1884 [DOI] [PubMed] [Google Scholar]
- 18. Hannun Y. A., Obeid L. M. (2002) The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem. 277, 25847–25850 [DOI] [PubMed] [Google Scholar]
- 19. Holland W. L., Bikman B. T., Wang L. P., Yuguang G., Sargent K. M., Bulchand S., Knotts T. A., Shui G., Clegg D. J., Wenk M. R., Pagliassotti M. J., Scherer P. E., Summers S. A. (2011) Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest. 121, 1858–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu W., Ross J., Geng T., Brice S. E., Cowart L. A. (2011) Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids. Implications for insulin resistance. J. Biol. Chem. 286, 16596–16605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Strub G. M., Maceyka M., Hait N. C., Milstien S., Spiegel S. (2010) Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol. 688, 141–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pettus B. J., Chalfant C. E., Hannun Y. A. (2004) Sphingolipids in inflammation. Roles and implications. Curr. Mol. Med. 4, 405–418 [DOI] [PubMed] [Google Scholar]
- 23. Taha T. A., Hannun Y. A., Obeid L. M. (2006) Sphingosine kinase. Biochemical and cellular regulation and role in disease. J. Biochem. Mol. Biol. 39, 113–131 [DOI] [PubMed] [Google Scholar]
- 24. Meacci E., Nuti F., Donati C., Cencetti F., Farnararo M., Bruni P. (2008) Sphingosine kinase activity is required for myogenic differentiation of C2C12 myoblasts. J. Cell. Physiol. 214, 210–220 [DOI] [PubMed] [Google Scholar]
- 25. Shu X., Wu W., Mosteller R. D., Broek D. (2002) Sphingosine kinase mediates vascular endothelial growth factor-induced activation of ras and mitogen-activated protein kinases. Mol. Cell. Biol. 22, 7758–7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sarkar S., Maceyka M., Hait N. C., Paugh S. W., Sankala H., Milstien S., Spiegel S. (2005) Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 579, 5313–5317 [DOI] [PubMed] [Google Scholar]
- 27. Xia P., Gamble J. R., Rye K. A., Wang L., Hii C. S., Cockerill P., Khew-Goodall Y., Bert A. G., Barter P. J., Vadas M. A. (1998) Tumor necrosis factor-α induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 14196–14201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olivera A., Kohama T., Edsall L., Nava V., Cuvillier O., Poulton S., Spiegel S. (1999) Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Cell Biol. 147, 545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anelli V., Gault C. R., Cheng A. B., Obeid L. M. (2008) Sphingosine kinase 1 is up-regulated during hypoxia in U87MG cells. Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 283, 3365–3375 [DOI] [PubMed] [Google Scholar]
- 30. Ringseis R., Eder K. (2011) Regulation of genes involved in lipid metabolism by dietary oxidized fat. Mol. Nutr. Food Res. 55, 109–121 [DOI] [PubMed] [Google Scholar]
- 31. Mandard S., Müller M., Kersten S. (2004) Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci. 61, 393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kersten S., Seydoux J., Peters J. M., Gonzalez F. J., Desvergne B., Wahli W. (1999) Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ferré P. (2004) The biology of peoxisome proliferator-activated receptors. Relationship with lipid metabolism and insulin sensitivity. Diabetes 53, S43–S50 [DOI] [PubMed] [Google Scholar]
- 34. Ren B., Thelen A., Jump D. B. (1996) Peroxisome proliferator-activated receptor alpha inhibits hepatic S14 gene transcription. Evidence against the peroxisome proliferator-activated receptor α as the mediator of polyunsaturated fatty acid regulation of S14 gene transcription. J. Biol. Chem. 271, 17167–17173 [DOI] [PubMed] [Google Scholar]
- 35. Akira S., Taga T., Kishimoto T. (1993) Interleukin-6 in biology and medicine. Adv. Immunol. 54, 1–78 [DOI] [PubMed] [Google Scholar]
- 36. Weigert C., Brodbeck K., Staiger H., Kausch C., Machicao F., Häring H. U., Schleicher E. D. (2004) Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor kappa B. J. Biol. Chem. 279, 23942–23952 [DOI] [PubMed] [Google Scholar]
- 37. Steensberg A., van Hall G., Osada T., Sacchetti M., Saltin B., Klarlund Pedersen B. (2000) Production of interleukin-6 in contracting human skeletal can account for the exercise-induced increase in plasma interleukin-6. J. Physiol. 529, 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keller C., Steensberg A., Pilegaard H., Osada T., Saltin B., Pedersen B. K., Neufer P. D. (2001) Transcriptional activation of the IL-6 gene in human contracting skeletal muscle. Influence of muscle glycogen content. FASEB J. 15, 2748–2750 [DOI] [PubMed] [Google Scholar]
- 39. Steensberg A., Keller C., Starkie R. L., Osada T., Febbraio M. A., Pedersen B. K. (2002) IL-6 and TNF-α expression in, and release from, contracting human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 283, E1272–E1278 [DOI] [PubMed] [Google Scholar]
- 40. Pedersen B. K. (2011) Muscles and their myokines. J. Exp. Biol. 214, 337–346 [DOI] [PubMed] [Google Scholar]
- 41. Pedersen B. K., Febbraio M. A. (2008) Muscle as an endocrine organ. Focus on muscle-derived interleukin-6. Physiol. Rev. 88, 1379–1406 [DOI] [PubMed] [Google Scholar]
- 42. Kern P. A., Ranganathan S., Li C., Wood L., Ranganathan G. (2001) Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 280, E745–E751 [DOI] [PubMed] [Google Scholar]
- 43. Benamer N., Fares N., Bois P., Faivre J. (2011) Electrophysiological and functional effects of sphingosine-1-phosphate in mouse ventricular fibroblasts. Biochem. Biophys. Res. Commun. 408, 6–11 [DOI] [PubMed] [Google Scholar]
- 44. Gurgui M., Broere R., Kalff J. C., van Echten-Deckert G. (2010) Dual action of sphingosine-1-phosphate in eliciting proinflammatory responsones in primary cultured rat intestinal smooth muscle cells. Cell. Signal. 22, 1727–1733 [DOI] [PubMed] [Google Scholar]
- 45. Ammit A. J., Hastie A. T., Edsall L. C., Hoffman R. K., Amrani Y., Krymskaya V. P., Kane S. A., Peters S. P., Penn R. B., Spiegel S., Panettieri R. A., Jr. (2001) Sphingosine-1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 15, 1212–1214 [DOI] [PubMed] [Google Scholar]
- 46. Samad F., Badeanlou L., Shah C., Yang G. (2011) Adipose tissue and ceramide biosynthesis in the pathogenesis of obesity. Adv. Exp. Med. Biol. 721, 67–86 [DOI] [PubMed] [Google Scholar]
- 47. Yang G., Badeanlou L., Bielawski J., Roberts A. J., Hannun Y. A., Samad F. (2009) Central role of ceramide biosynthesis in body weight regulation, energy, metabolism, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 297, E211–E224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Springer M., Rando T., Blau H., (1997) in Current Protocols in Human Genetics (Boyle A., ed) pp. 13.4.1–13.4.19, John Wiley & Sons, New York: [DOI] [PubMed] [Google Scholar]
- 49. Allende M. L., Sasaki T., Kawai H., Olivera A., Mi Y., van Echten-Deckert G., Hajdu R., Rosenbach M., Keohane C. A., Mandala S., Spiegel S., Proia R. L. (2004) Mice deficient in sphingosine kinase 1 are rendered lyphopenic by FTY720. J. Biol. Chem. 279, 52487–52492 [DOI] [PubMed] [Google Scholar]
- 50. Chavez J. A., Holland W. L., Bär J., Sandhoff K., Summers S. A. (2005) Acid Ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J. Biol. Chem. 280, 20148–20153 [DOI] [PubMed] [Google Scholar]
- 51. Imamura T., Ohgane J., Ito S., Ogawa T., Hattori N., Tanaka S., Shiota K. (2001) CpG island of rat sphingosine kinase-1 gene. Tissue dependent DNA methylation status and multiple alternative first exons. Genomics 76, 117–125 [DOI] [PubMed] [Google Scholar]
- 52. Sobue S., Hagiwara K., Banno Y., Tamiya-Koizumi K., Suzuki M., Takagi A., Kojima T., Asano H., Nozawa Y., Murate T. (2005) Transcription factor specificity protein 1 (Sp1) is the main regulator of nerve growth factor-induced sphingosine kinase 1 gene expression of the rat pheochromocytoma cell line. PC12. J. Neurochem. 95, 940–949 [DOI] [PubMed] [Google Scholar]
- 53. Bielawski J., Szulc Z. M., Hannun Y. A., Bielawska A. (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high performance liquid chormatography-tandem mass spectrometry. Methods 39, 82–91 [DOI] [PubMed] [Google Scholar]
- 54. Messeguer X., Escudero R., Farré D., Núñez O., Martínez J., Albà M. M. (2002) PROMO. Detection of known transcription factor regulatory elements using sepcies-tailored searches. Bioinformatics 18, 333–334 [DOI] [PubMed] [Google Scholar]
- 55. Mitrou P., Lambadiari V., Maratou E., Boutati E., Komesidou V., Papakonstantinou A., Raptis S. A., Dimitriadis G. (2011) Skeletal muscle insulin resistance in morbid obesity. The role of interleukin-6 and leptin. Exp. Clin. Endocrinol. Diabetes 119, 484–489 [DOI] [PubMed] [Google Scholar]
- 56. Farré D., Roset R., Huerta M., Adsuara J. E., Roselló L., Albà M. M., Messeguer X. (2003) Idenfication of patterns in biological sequences at the ALGEEN server. PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Steensberg A., Keller C., Starkie R. L., Osada T., Febbraio M. A., Pedersen B. K. (2002) IL-6 and TNFα expression in, and release from, contracting human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 283, E1272–E1278 [DOI] [PubMed] [Google Scholar]
- 58. Jové M., Planavila A., Laguna J. C., Vázquez-Carrera M. (2005) Palmitate-induced interleukin 6 production is mediated by protein kinase C and nuclear-factor κB activation and leads to glucose transporter 4 down-regulation in skeletal muscle cells. Endocrinology 146, 3087–3095 [DOI] [PubMed] [Google Scholar]
- 59. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling. Lessons from sphingolipids. Nat. Rev. Mol. Cell. Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 60. Pyne S., Pyne N. J. (2000) Sphingosine 1-phosphate signalling in mammalian cells. Biochem. J. 349, 385–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meacci E., Cencetti F., Donati C., Nuti F., Farnararo M., Kohno T., Igarashi Y., Bruni P. (2003) Down-regulation of EDG5/S1P2 during myogenic differentiation results in the specific uncoupling of sphingosine 1-phosphate signalling to phospholipase D. Biochim. Biophys. Acta 1633, 133–142 [DOI] [PubMed] [Google Scholar]
- 62. Zanin M., Germinario E., Dalla Libera L., Sandonà D., Sabbadini R. A., Betto R., Danieli-Betto D. (2008) Trophic action of S1P in denervated rat soleus muscle. Am. J. Physiol. Cell Physiol. 294, C36–C46 [DOI] [PubMed] [Google Scholar]
- 63. Unger R. H., Clark G. O., Scherer P. E., Orci L. (2010) Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 1801, 209–214 [DOI] [PubMed] [Google Scholar]
- 64. Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Véret J., Coant N., Gorshkova I. A., Giussani P., Fradet M., Riccitelli E., Skobeleva A., Goya J., Kassis N., Natarajan V., Portha B., Berdyshev E. V., Le Stunff H. (2013) Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 β-cell survival. Biochim. Biophys. Acta 1831, 251–262 [DOI] [PubMed] [Google Scholar]
- 66. Maceyka M., Payne S. G., Milstien S., Spiegel S. (2002) Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 1585, 193–201 [DOI] [PubMed] [Google Scholar]
- 67. Olivera A., Edsall L., Poulton S., Kazlauskas A., Spiegel S. (1999) Platelet-derived growth factor-induced activation of sphingosine kinase requires phosphorylation of PDGF receptor tyrosine residue responsible for binding PLCγ. FASEB J. 13, 1593–1600 [DOI] [PubMed] [Google Scholar]
- 68. Kleuser B., Cuvillier O., Spiegel S. (1998) α1,25-Dihydroxyvitamin D3 inhibits programmed cell death in HL-60 cells by activation of sphingosine kinase. Cancer Res. 58, 1817–1824 [PubMed] [Google Scholar]
- 69. Nakade Y., Banno Y., T-Koizumi K., Hagiwara K., Sobue S., Koda M., Suzuki M., Kojima T., Takagi A., Asano H., Nozawa Y., Murate T. (2003) Regulation of sphingosine kinase 1 gene expression by protein kinase C in a human leukemia cell line, MEG-O1. Biochim. Biophys. Acta 1635, 104–116 [DOI] [PubMed] [Google Scholar]
- 70. Pyper S. R., Viswakarma N., Yu S., Reddy J. K. (2010) PPARα. Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 8, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ferré P. (2004) The biology of peroxisome proliferatory-activated receptors. Relationship with lipid metabolism and insulin sensitivity. Diabetes 53, S43–S50 [DOI] [PubMed] [Google Scholar]
- 72. Forman B. M., Chen J., Evans R. M. (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors a and d. Proc. Natl. Acad. Sci. U.S.A. 94, 4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Forman B. M., Chen J., Evans R. M. (1996) The peroxisome proliferator-activated receptors. Ligands and activators. Ann. N.Y. Acad. Sci. 804, 266–275 [DOI] [PubMed] [Google Scholar]
- 74. Göttlicher M., Widmark E., Li Q., Gustafsson J. (1992) Fatty acids activate a chimera of the clofibric acid-activated receptor and glucocorticoid receptor. Proc. Natl. Acad. Sci. U.S.A. 89, 4653–4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chakravarthy M. V., Lodhi I. J., Yin L., Malapaka R. R., Xu H. E., Turk J., Semenkovich C. F. (2009) Identification of a physiologically relevant endogenous ligand of PPARα in Liver. Cell. 138, 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Muoio D. M., Way J. M., Tanner C. J., Winegar D. A., Kliewer S. A., Houmard J. A., Kraus W. E., Dohm G. L. (2002) Peroxisome proliferator-activated receptor-α regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 51, 901–909 [DOI] [PubMed] [Google Scholar]
- 77. Djouadi F., Aubey F., Schlemmer D., Bastin J. (2005) Peroxisome proliferator activated δ (PPARδ) agonist but not PPARα corrects carnitine palmitoyl transferase 2 deficieny in human muscle cells. J. Clin. Endocrinol. Metab. 90, 1791–1797 [DOI] [PubMed] [Google Scholar]
- 78. Drastich P., Frolova-Brizova L., Zanvit P., Tlasakalova-Hogenova H. (2011) Spontaneous in vitro IL-6 production in various intestinal segments in patients with inflammatory bowel disease. Folia Microbiol. (Praha) 56, 185–190 [DOI] [PubMed] [Google Scholar]
- 79. Li Y., de Haar C., Chen M., Deuring J., Gerrits M. M., Smits R., Xia B., Kuipers E. J., van der Woude C. J. (2010) Disease-related expression of the IL6/STAT3/SOCS3 signaling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. GUT 59, 227–235 [DOI] [PubMed] [Google Scholar]
- 80. Cammarota R., Bertolini V., Pennesi G., Bucci E. O., Gottardi O., Garlanda C., Laghi L., Barberis M. C., Sessa F., Noonan D. M., Albini A. (2010) The tumor microenvironment of colorectal cancer. Stromal TLR-4 expression as a potential prognostic marker. J. Transl. Med. 8, 112–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Varma V., Yao-Borengasser A., Rasouli N., Nolen G. T., Phanavanh B., Starks T., Gurley C., Simpson P., McGehee R. E., Jr., Kern P. A., Peterson C. A. (2009) Muscle inflammatory response and insulin resistance. Synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am. J. Physiol. Endocrinol. Metab. 296, E1300–E1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Paik J. H., Chae Ss., Lee M. J., Thangada S., Hla T. (2001) Sphingosine-1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of αvβ3 and β1-containing intergrins. J. Biol. Chem. 276, 11830–11837 [DOI] [PubMed] [Google Scholar]
- 83. Green C. J., Macrae K., Fogarty S., Hardie D. G., Sakamoto K., Hundal H. S. (2011) Counter-modulation of fatty acid-induced pro-inflammatory nuclear factor κB signaling in rat skeletal muscle calls by AMP-activated protein kinase. Biochem. J. 435, 463–474 [DOI] [PubMed] [Google Scholar]
- 84. Coll T., Palomer X., Blanco-Vaca F., Escolà-Gil J. C., Sánchez R. M., Laguna J. C., Vázquez-Carrera M. (2010) Cyclooxygenase 2 inhibition excerbates palmitate-induced inflammation and insulin resistance in skeletal muscle cells. Endocrinology 151, 537–548 [DOI] [PubMed] [Google Scholar]
- 85. Reyna S. M., Ghosh S., Tantiwong P., Meka C. S., Eagan P., Jenkinson C. P., Cersosimo E., Defronzo R. A., Coletta D. K., Sriwijitkamol A., Musi N. (2008) Elevated Toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57, 2595–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fernández-Real J. M., Ricart W. (2003) Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr. Rev. 24, 278–301 [DOI] [PubMed] [Google Scholar]
- 87. Fried S. K., Bunkin D. A., Greenberg A. S. (1998) Omental and subcutaneous adipose tissues and obese subjects release interleukin-6. Depot differences and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 83, 847–850 [DOI] [PubMed] [Google Scholar]
- 88. Betanzos-Cabrera G., Estrada-Luna D., Belefant-Miller H., Cancino-Díaz J. (2012) Mice fed high fat diet show a decrease in the expression of “Toll-like receptor” (TLR) 2 and TLR6 mRNAs in adipose and hepatic tissues. Nutr. Hosp. 27, 1196–1203 [DOI] [PubMed] [Google Scholar]
- 89. DeOliveira C. C., Acedo S. C., Gotardo E. M., Carvalho Pde O., Rocha T., Pedrazzoli J., Jr., Gambero A. (2012) Effects of methotrexate of inflammatory alterations induced by obesity. An in vivo and in vitro study. Mol. Cell. Endocrinol. 361, 92–98 [DOI] [PubMed] [Google Scholar]
- 90. Matsubara T., Mita A., Minami K., Hosooka T., Kitazawa S., Takahashi K., Tamori Y., Yokoi N., Watanabe M., Matsuo E., Nishimura O., Seino S. (2012) PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15, 38–50 [DOI] [PubMed] [Google Scholar]
- 91. Mohamed-Ali V., Goodrick S., Rawesh A., Katz D. R., Miles J. M., Yudkin J. S., Klein S., Coppack S. W. (1997) Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-α, in vivo. J. Clin. Endocrinol. Metab. 82, 4196–4200 [DOI] [PubMed] [Google Scholar]
- 92. Hoene M., Runge H., Häring H. U., Schleicher E. D., Weigert C. (2013) Interleukin-6 promotes myogenic differentiation of mouse skeletal muscle cells. Role of the STAT3 pathway. Am. J. Physiol. Cell Physiol. 304, C128–C136 [DOI] [PubMed] [Google Scholar]
- 93. Takeda K., Akira S. (2000) STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 11, 199–207 [DOI] [PubMed] [Google Scholar]
- 94. Levy D. E., Lee C. K. (2002) What does Stat3 do? J. Clin. Invest. 109, 1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Vaisse C., Halaas J. L., Horvath C. M., Darnell J. E., Jr., Stoffel M., Friedman J. M. (1996) Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice by not db/db mice. Nat Genet. 14, 95–97 [DOI] [PubMed] [Google Scholar]
- 96. Grandis J. R., Drenning S. D., Chakraborty A., Zhou M. Y., Zeng Q., Pitt A. S., Tweardy D. J. (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J. Clin. Invest. 102, 1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Toth K. G., McKay B. R., De Lisio M., Little J. P., Tarnopolsky M. A., Parise G. (2011) IL-6 induced STAT3 signalling is associated with the proliferation of human muscle satellite cells following acute muscle damage. PLoS One 6, e17392–e17403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. McKay B. R, De Lisio M., Johnston A. P., O'Reilly C. E., Phillips S. M., Tarnopolsky M. A., Parise G. (2009) Association of interleukin-6 signalling with the stem cell response following muscle-lengthening contractions in humans. PLoS One 4, e6027–e6039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bruni P., Donati C. (2008) Pleiotropic effects of sphingolipids in skeletal muscle. Cell. Mol. Life Sci. 65, 3725–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rosen H., Gonzalez-Cabrera P. J., Sanna M. G., Brown S. (2009) Sphingosine 1-phosphate receptor signaling. Ann. Rev. Biochem. 78, 743–768 [DOI] [PubMed] [Google Scholar]
- 101. Kluk M. J., Hla T. (2002) Signaling of sphingosine-1-phosphate via S1P/EDG-family of G-protein-coupled receptors. Biochim. Biophys. Acta 1582, 72–80 [DOI] [PubMed] [Google Scholar]
- 102. Zhao C., Fernandes M. J., Turgeon M., Tancrède S., Di Battista J., Poubelle P. E., Bourgoin S. G. (2008) Specific and overlapping sphingosine-1-phosphate receptor functions in human synoviocytes. Impact of TNF-α. J. Lipid Res. 49, 2323–2337 [DOI] [PubMed] [Google Scholar]