

Background: The role of cytoplasmic loops (CL) in ABCC4 expression at the plasma membrane is unknown.
Results: A conserved amino acid substitution in the CL3 of ABCC4 reduced expression and impaired membrane localization.
Conclusion: An α-helical domain in CL3 appears crucial for proper ABCC4 plasma membrane localization.
Significance: These findings reveal ABCC4 membrane localization requires a conserved domain in CL3.
Keywords: ABC Transporter, Homology Modeling, Molecular Modeling, Multidrug Transporters, Zebrafish, Mrp4, Cytoplasmic Loop 3
Abstract
The ABC transporter ABCC4 is recognized as an ATP-dependent exporter of endogenous substances as well as an increasing variety of anionic chemotherapeutics. A loss-of-function variant of zebrafish Abcc4 was identified with a single amino acid substitution in the cytoplasmic loop T804M. Because this substituted amino acid is highly conserved among ABCC4 orthologs and is located in cytoplasmic loop 3 (CL3), we investigated the impact of this mutation on human and zebrafish Abcc4 expression. We demonstrate that zebrafish Abcc4 T804M or human ABCC4 T796M exhibit substantially reduced expression, coupled with impaired plasma membrane localization. To understand the molecular basis for the localization defect, we developed a homology model of zebrafish Abcc4. The homology model suggested that the bulky methionine substitution disrupted side-chain contacts. Molecular dynamic simulations of a fragment of human or zebrafish CL3 containing a methionine substitution indicated altered helicity coupled with reduced thermal stability. Trifluoroethanol challenge coupled with circular dichroism revealed that the methionine substitution disrupted the ability of this fragment of CL3 to readily form an α-helix. Furthermore, expression and plasma membrane localization of these mutant ABCC4/Abcc4 proteins are mostly rescued by growing cells at subphysiological temperatures. Because the cystic fibrosis transmembrane conductance regulator (ABCC7) is closely related to ABCC4, we extended this by engineering certain pathogenic CFTR-CL3 mutations, and we showed they destabilized human and zebrafish ABCC4. Altogether, our studies provide the first evidence for a conserved domain in CL3 of ABCC4 that is crucial in ensuring its proper plasma membrane localization.
Introduction
The ATP-binding cassette transporter ABCC42 (also known as MRP4) was considered an orphan drug transporter (1) prior to 1999, when it was identified as the first transporter that exported nucleoside monophosphate derivatives (2). Because these acyclic monophosphate derivatives resemble cyclic nucleotides, this finding provided an opportunity to test the hypothesis that both natural (cyclic nucleotides) and drug-derived monophosphate derivatives were effluxed by a specific energy-dependent transporter (3, 4). Subsequent ABCC4 overexpression studies revealed that, in a variety of cell types, in both cellular systems (5) and in membrane vesicles, ABCC4 was capable of exporting nucleotide monophosphate derivatives (6, 7). Notably, in humans, impaired ABCC4 function is closely associated with increased hematopoietic toxicity secondary to thiopurine therapy, a finding attributed to elevated concentrations of thiopurine monophosphates in hematopoietic cells (8, 9). Although other ABCC4 variants have been described (10, 11), none have identified a particular domain that affects trafficking and processing of the protein.
ABCC4 has a domain organization typical of eukaryotic ABC transporters. This includes a core structure of two membrane-spanning domains (MSD1 and MSD2), each consisting of six transmembrane helices and two cytosolic nucleotide binding domains (NBDs) that bind and hydrolyze ATP to power substrate transport (12–14). The MSD are composed of six long transmembrane helices that penetrate the cytoplasm and are connected by cytoplasmic loops (CL). For ABC transporters, the location of the CL provides an opportunity for interaction with the NBDs, and current views suggest that CLs have an important role in both trafficking to the plasma membrane and in protein stability (15–17). It is unknown if the CL has a role in the trafficking and processing of ABCC4.
Recently, an ethylnitrosourea mutagenesis screen identified a zebrafish Abcc4 mutant (T804M) that produced developmental defects.3 In this study, we show that this mutation was in a highly conserved amino acid in CL3 (Fig. 1A). Our studies demonstrate that the analogous mutation in human ABCC4 (T796M) also reduces protein expression. To understand the molecular basis for the unstable protein, we assessed whether mutations in CL3 affect the conserved helices. By molecular dynamic simulations, we determined that this mutation in human and zebrafish (ZF) CL3 compromises the normal helicity of this conserved region. Moreover, by using circular dichroism, we demonstrated that the methionine substitution in a fragment of CL3 attenuated its capacity to adopt an α-helical conformation. Because CFTR (ABCC7) is a close relative of ABCC4 and this region is conserved between the two proteins, we introduced known pathogenic CL3 mutations from CFTR into ABCC4. Overall our studies show that a conserved α-helical domain within CL3 is important in ensuring precise ABCC4 plasma membrane localization.
FIGURE 1.
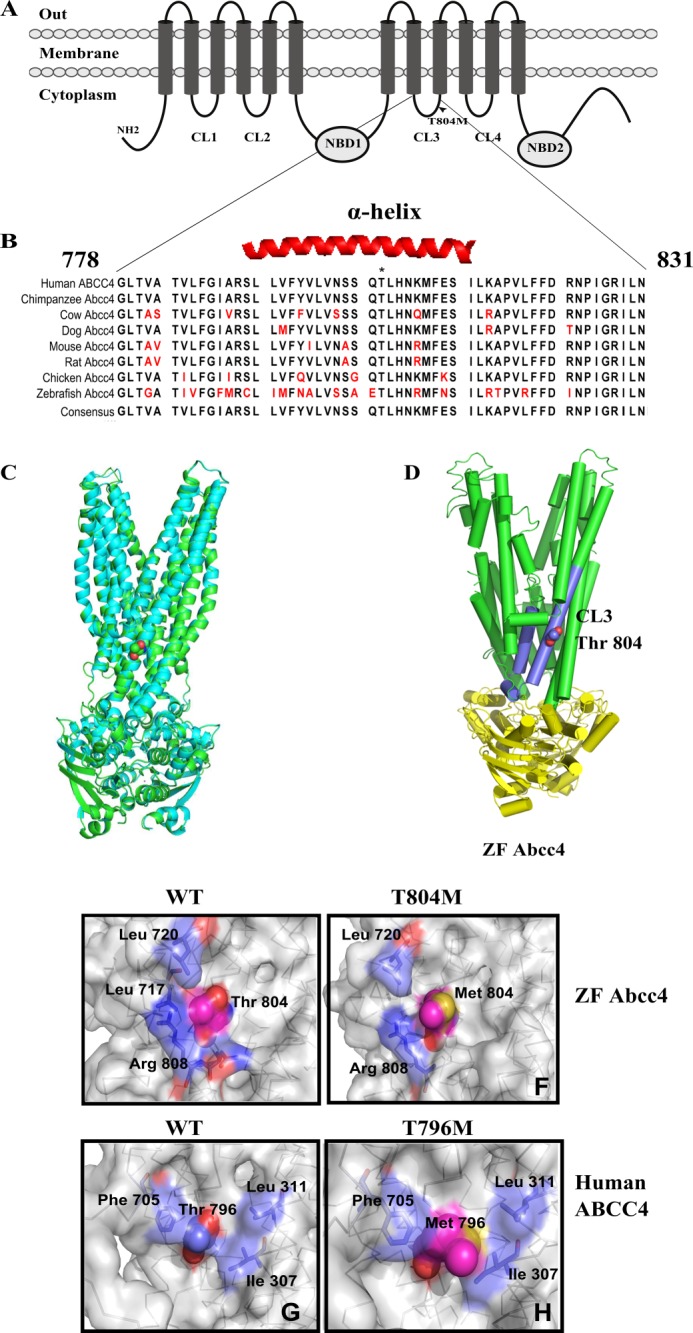
Abcc4 T804M mutation is localized to the cytoplasmic loop 3. A, schematic diagram of the protein secondary structure of Abcc4, including membrane spanning domains and NBD domains. Arrowhead, Abcc4 T804M mutation. B, ClustalW alignment of Abcc4 protein sequences from various species showing highly conserved CL3 region. C, superposition of homology model of zebrafish Abcc4 (green) with SAV1866 ABC transporter (cyan). Thr-804 is shown in sphere representation. D, structural homology model of zebrafish Abcc4 based on Sav1866 bacterial ABC transporter. E, intermolecular interactions of wild type zebrafish Abcc4 Thr-804 residue. F, loss of intermolecular interactions of T804M zebrafish Abcc4. G, intermolecular interactions of wild type human ABCC4 Thr-796 residue. H, loss of intermolecular interactions of T796M human ABCC4.
MATERIALS AND METHODS
Sequence Analysis and Alignment
Analysis of gene sequence conservation at the CL3 region was performed using CINEMA Version 1.4.5 software (The Advanced Interfaces Group) and CLC Workbench (Cambridge, MA).
Cloning and Site-directed Mutagenesis
ZF Abcc4 or human ABCC4 with N-terminal AcGFP fusion was generated by cloning the genes into the pAcGFP vector (Clontech) at XhoI and BamHI sites. Point mutations were introduced into pAcGFP ZF Abcc4 or human ABCC4 by using a QuikChange XL II site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions (primer sequences: T796M_S tacgtccttgttaactcttcacaaatgttgcacaacaaaatgtttgagtca- and T796M_AS tgactcaaacattttgttgtgcaacatttgtgaagagttaacaaggacgtag-; S794L_S gtattctacgtccttgttaactct ttacaaactttgcacaacaaaat- and S794L_AS 5′-attttgttgtgcaaagtttgtaaagagttaacaaggacgtagaatac-′; H798Y_S ′-gtccttgttaactcttcacaaactttgtataacaaaatgtttgagtcaattctgaaa- and H798Y_AS tttcagaattgactcaaacattttgttatacaaagtttgtgaagagtaacaaggac-; R815A_S ctgaaagctccggtattattctttgatgcaaatccaataggaagaattttaaatcg- and R815A_AS cgatttaaaattcttcctattggatttgcatcaaagaataataccgagctttcag-′; T804M_S ggtgagctcagcagagatgcttcacaaccg- and T804M_AS cggttgtgaagcatctctgctgagctcacc; A802L_S ttcaatgctctggtgagctcattagagacgcttcacaacc- and A802L_AS ggttgtgaagcgtctctaatgagctcaccagagcattgaa; H806Y_S ggtgagctcagcagagacgctttataaccgcatgttc- and H806Y_AS gaacatgcggttataaagcgtctctgctgagctcacc-3′). After the mutagenesis, gene sequences were confirmed by sequence analysis.
In Vitro Translation
In vitro translation reactions were performed with the following expression plasmids: pcDNA3.1 ZF Abcc4, Abcc4 T804M, ABCC4, ABCC4 T796M, and pLUC (luciferase vector) using the transcription- and translation-coupled (TnT) kit (Promega Inc., Madison, WI) in the presence of [35S]Met. The reaction was terminated by the addition of SDS-PAGE sample buffer. Subsequently, samples were resolved on a 10% SDS-polyacrylamide gel. The reaction products were visualized by exposure to a phospho screen using a phosphorimager from Molecular Devices.
Cell Culture
NIH3T3, HEK293, or the cells transiently expressing one of the following proteins, ZF Abcc4, ZF Abcc4 T804M, ABCC4, and ABCC4 T796M, were maintained in Dulbecco's DMEM containing 4500 mg/liter glucose, 10% FBS (HyClone, Logan, UT), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified 5% CO2 incubator at 37 °C. Where specified, NIH3T3 cells transiently expressing either pACGFP ABCC4 or T796M were incubated with 50 μg/ml cycloheximide for different time points.
Transient Transfection
NIH3T3 or HEK293 cells were transiently transfected with expression plasmids by using Lipofectamine LTX Plus reagent (Invitrogen) according to the manufacturer's protocols. Briefly, 24 h before the transfection, 200,000 cells were seeded per well in 6-well plates, and each well was transfected with 2.5 μg of expression plasmid.
Immunoblot Analysis
Twenty four hours after the transfection, cells were washed with PBS, scraped into 1 ml of cold PBS containing 1× protease inhibitor mixture (Roche Applied Science), and pelleted by centrifugation at 1000 × g for 6 min at 4 °C; the cell pellet was solubilized in M-PER reagent (Pierce) containing 1× protease inhibitor mixture. Following that, cell lysates were centrifuged at 20,000 × g for 30 min at 4 °C to remove cell debris. The total protein content of the protein lysates was quantified using a Bradford assay (18). For immunoblotting, the samples were fractionated by SDS-PAGE on a 10% gel. Proteins were transferred to a nitrocellulose membrane (GE Healthcare) and immunoblotted. The levels of AcGFP-tagged (5′) ABCC4 wild type and mutant proteins were determined using the AcGFP-specific rabbit polyclonal antibody pAvGFP (1:1000) that detects AcGFP and its tagged proteins as disclosed by Clontech. For secondary antibody, HRP-conjugated goat anti-rabbit (1:1000) (GE Healthcare) was used. Finally, chemiluminescence reagent from GE Healthcare (ECL) was used to detect the proteins. Nonspecific protein bands on immunoblot were used to confirm equal loading of protein.
Glycosidase Digestion
Glycosidase digestion of the protein lysates was performed as described earlier (19). Briefly, protein lysates from NIH3T3 cells transiently transfected with pAcGFP ZF Abcc4 and pAcGFP human ABCC4 were denatured in 1× denaturing buffer (0.5% SDS, 1% β-mercaptoethanol) and incubated at 37 °C for 1 h in reaction buffer (50 mm Na2PO4 (pH 7.5) and 1% Nonidet P-40) with or without PNGase F (New England Biolabs, Beverly, MA). Subsequently, samples were resolved on a 10% SDS-polyacrylamide gel and immunoblotted with pAvGFP antibody.
Confocal Microscopy
NIH3T3 or HEK293 cells were seeded on coverslips and chamber slides 24 h before transfection with the plasmid DNAs (250 ng) encoding pAcGFP ZF Abcc4, ZF Abcc4 T804M, ABCC4, ABCC4 T796M, and pCAL plasmid encoding RFP-tagged endoplasmic reticulum resident protein calreticulin encoding plasmid (pCAL (Origene) by using Lipofectamine LTX Plus reagent (Invitrogen))according to the manufacturer's protocols. Furthermore, cells were stained with Alexa 646-conjugated wheat germ agglutinin as a plasma membrane marker before the confocal microscopy. Leica spin disc confocal microscope with ×63 objective was used for the microscopy (Leica Microsystems, Heidelberg, Germany).
Cell Surface Biotinylation
Cell surface proteins were biotinylated with cell-impermeable biotin sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate (sulfo-NHS-SS-biotin) as described earlier (8). Briefly, NIH3T3 cells transiently expressing either pAcGFP human ABCC4 or T796M were rinsed with ice-cold PBS and incubated with 10 mm EZ-Link sulfo-NHS-SS-biotin solution in PBS (pH 8.0) (Pierce) for 30 min at room temperature. Following that, unreacted biotin molecules were quenched with ice-cold PBS containing 100 mm glycine for 10 min before cell lysis and streptavidin-agarose capture.
Homology Model of ZF Abcc4
The zebrafish Abcc4 structural model was built from the homology model using Sav1866 (Protein Data Bank code 2HYD) (20) as the template. Amino acid sequence alignment (supplemental Fig. 1) and loop optimization with the Prime Program from the Schrodinger suite were used (21, 22). The modeled structure was further minimized, and the mutations were made and minimized by Sybyl 8.0.
Circular Dichroism
Circular dichroism spectra were obtained with an Aviv 62DS CD spectrophotometer (Aviv) and processed by using Igor Pro software (Wavemetrics Inc). All experiments were performed at 37 °C by using a quartz cuvette with a 1-cm path length. We used 1 nm step resolution and 10 s average signaling time at 1 nm bandwidth. Concentration of sample was kept at 30 μm in PBS (pH 7.0). The CD spectra were expressed as millidegrees and percent of helicity as predicted by SOMA CD website (25).
RESULTS
ABCC4 and CL3 Mutation
Zhong and co-workers3 recently identified a single point mutation (T804M) in ZF Abcc4 (Fig. 1A) that produces developmental defects.3 Sequence alignment of Abcc4 from multiple species revealed that cytoplasmic loop 3 (CL3) of ZF Abcc4 is conserved among ABCC4 subfamily members that encompasses the T804M substitution (Fig. 1B). The boundaries of the ABCC4/Abcc4 CL3 vary depending upon the secondary structure prediction algorithm used. However, for purposes of this study, we define CL3 as extending from 778 to 831 in ZF Abcc4 and from 770 to 823 in human ABCC4 (Fig. 1B). Overall, the amino acid sequence in CL3 is over 70% identical to the orthologs of Abcc4 from eight different species. Prediction of the secondary structure of Abcc4 CL3 revealed that the region encompassing the methionine substitution is α-helical. This region in CL3 may be especially important because in the ABCC4 paralog, ABCC7 (CFTR), the few mutations reported in CL3 loop produce defects in protein maturation (26, 27).
The Sav1866 structure has been extensively used as a template to develop homology models for several ABC transporter family members (28–35). The Sav1866 homodimer structure is composed of two identical transmembrane domains and NBD (20). Analysis of the sequence alignment of Sav1866 and ZF Abcc4 protein demonstrates that Sav1866 is 23% identical to the N-terminal ABCC4 TMD1-NBD1 (amino acids 1–631) and 25% identical to the C-terminal ABCC4 TMD2-NBD2 (amino acids 632–1327) (supplemental Fig. 1). Notably, as shown for other ABC homology models, transmembrane domain topology is more important than sequence identity for modeling membrane proteins (37–39).
Our ZF Abcc4 model was superimposed upon the Sav1866 template (Fig. 1C). A few regions of the modeled Abcc4 conformation deviate from the template structure (including regions 142–146, 366–370, 846–848, 864–868), which appear to be due to sequence insertions. Nonetheless, the overall root mean square deviation is 0.244 angstrom, a finding demonstrating that the zebrafish Abcc4 homology model is highly consistent with the Sav1866 conformation. Furthermore, the superposition of Abcc4 and Sav1866 illustrates that the conformation of Abcc4 transmembrane helix containing 804 is highly similar between these proteins (Fig. 1C). Furthermore, to gain insight into the molecular changes produced by the T804M substitution, we used the homology model of ZF Abcc4 to predict the Thr-804 contacts as Leu-717, Leu-720, and Arg-808 (Fig. 1D).
PolyPhen and SIFT algorithms predicted that the methionine substitution will be benign and tolerated, respectively. This seemed unlikely because our model predicted that the side chain of Thr-804 forms a hydrogen bond with the carbonyl on Arg-808 (Fig. 1, E and F). Likewise, using a human ABCC4 homology model (32), we show that the orthologous threonine in human ABCC4 CL3 displays similar contacts with Phe-705, Leu-311, and Ile-307; however, replacing it with a bulky Met-796 residue appears to constrain the side-chain interactions as seen in the space filling model (Fig. 1, G and H).
Amino Acid Substation at Thr-804 Affects ZF Abcc4 Expression
The protein expression and subcellular localization of WT ZF Abcc4 and T804M mutant was determined using cDNA expression vectors encoding either full-length WT ZF Abcc4 or its T804M mutant, each fused with an AcGFP at the N terminus. The addition of the N-terminal GFP tag did not prevent ZF Abcc4 from exporting a well known substrate 9-(2-phosphonyl-methoxyethyl) adenine (data not shown). After transient transfection of NIH3T3 cells, the expression of WT ZF Abcc4 and T804M was assessed by probing immunoblots with an anti-AcGFP antibody, because currently available human or mouse Abcc4 antibodies do not cross-react with ZF Abcc4 (data not shown). These experiments showed that the expression of ZF Abcc4 T804M mutant was substantially lower (64%) than WT ZF Abcc4 (Fig. 2A). Although ZF Abcc4 appears to run as a diffuse single band, treatment with PNGaseF reveals it is glycosylated (Fig. 2B). We tested the possibility that ZF Abcc4 T804M had a defect in translation because mRNA folding analysis by the mfold webserver (40) suggested ZF Abcc4 T804M with a C nucleotide substitution was more stable with higher mRNA folding free energy of 196.8 kcal/mol compared with ZF Abcc4 WT of 193.8 kcal/mol. These results suggest the increased secondary structure of ZF Abcc4 T804M mRNA might disrupt translation as reported for CFTR (41). However, in vitro translation of ZF Abcc4 WT and T804M transcripts produced almost identical amounts of protein (Fig. 2C) indicating the nucleotide change producing the T804M substitution had no measurable effect on translation.
FIGURE 2.

Mutation in the cytoplasmic loop3 region results in an unstable human and zebrafish ABCC4/Abcc4. A, immunoblots of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with ZF Abcc4 WT and Abcc4 T804M mutant. Blots are probed with anti-GFP antibody. B, immunoblot of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with ZF Abcc4 WT treated with PNGase F and probed with anti-AcGFP antibody. C, in vitro translation products (35S-labeled) from TnT (coupled transcription and translation) (20 μl/well) were run on 10% SDS-polyacrylamide gel and detected using phosphorimager. D, confocal microscopy; E, quantification of NIH3T3-transfected cells with GFP-tagged Abcc4, Abcc4 T804M mutant, and RFP-tagged ER marker (calreticulin). Cells were analyzed by Leica spin disc confocal microscopy (×63 magnification). Plasma membrane was stained with Alexa 646 wheat germ agglutinin (blue). Signals from the three channels were acquired independently, and the merged images are presented. Co-localization of Abcc4 WT and PM is indicated by a reddish blue and co-localization of GFP Abcc4 T804M and RFP-ER are indicated by a reddish yellow color. F, immunoblots of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with human ABCC4 WT and ABCC4 T796M mutant. G, immunoblots of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with human ABCC4 WT treated with PNGase F. H, in vitro translation products from TnT (coupled transcription and translation) (20 μl/well) were run on 10% SDS-polyacrylamide gel and detected using phosphorimager. I, confocal microscopy. J, quantification of HEK293 transfected cells with GFP-tagged human ABCC4 WT, ABCC T796M mutant, and RFP-tagged ER marker (calreticulin). Plasma membrane was stained with Alexa 646 wheat germ agglutinin (blue). Signals from the three channels were acquired independently, and the merged images are presented. Co-localization of ABCC4 WT and PM is indicated by a reddish blue, and co-localization of GFP ABCC4 T796M and RFP-ER is indicated by a yellow color. K, NIH3T3 cells were transfected with ABCC4 WT, T796M, and cycloheximide (final concentration, 50 μg/ml) was added 24 h post-transfection. Cells were harvested at the indicated times and analyzed for ABCC4 proteins using anti-GFP antibody. Intensity of the bands (c or b) was analyzed using densitometry and expressed as percentage of ABCC4 protein at 0 h for each construct. Values shown are the mean from two independent experiments with the range indicated by the error bars.
To determine whether subcellular localization is altered by T804M, we performed confocal microscopy on live cells. NIH3T3 cells were transiently co-transfected with either WT ZF Abcc4 or T804M and an expression plasmid for calreticulin (endoplasmic reticulum (ER) resident protein) tagged with a far-red fluorescent protein, and the plasma membrane was identified by wheat germ agglutinin conjugated with Alexa 646. WT ZF Abcc4 primarily localized to the plasma membrane (70%), as shown by co-localization with the plasma membrane marker. In contrast, T804M was diffusely distributed within the cell with a majority of the protein associated with the cytoplasm and ER/Golgi (80%) (Fig. 2, D and E).
Because the Thr at position 804 in ZF Abcc4 is conserved among Abcc4 subfamily members, we developed expression vectors encoding full-length human WT AcGFP-ABCC4 and produced an analogous mutation to T804M, T796M in PAcGFP ABCC4. We compared expression of these proteins in lysates from NIH3T3 cells transfected with these vectors. ABCC4 T796M exhibited a strong reduction in the core-glycosylated mature band c (Fig. 2F), which was confirmed by treating WT ABCC4-expressing lysates with the N-glycanase, PNGase F (Fig. 2G). Notably, WT human ABCC4 and T796M produced almost identical amounts of protein (Fig. 2H) when in vitro translated, indicating that, like ZF T804M, the T796M substitution in human ABCC4 has no effect on mRNA translation.
To determine T796M ABCC4 subcellular localization, live cell confocal microscopy was performed. Cells were transiently transfected with either ABCC4 WT or T796M expression plasmids along with an expression plasmid for calreticulin, an ER resident protein. The plasma membrane was detected by incubating the cells with Alexa 646-conjugated wheat germ agglutinin-lectin. Notably, 80% of cells showed ABCC4 localized to the plasma membrane with wheat germ agglutinin-lectin. In contrast, a majority (>95%) of ABCC4 T796M localized with the Golgi/ER marker, calreticulin, or more diffusely in the cytoplasm (Fig. 2, I and J).
We next determined if the T796M substitution in CL3 altered the stability of human ABCC4. NIH3T3 cells were transiently transfected with either WT ABCC4 or ABCC4 harboring T796M substitution. At 24 h post-transfection, cycloheximide was added to inhibit protein synthesis, and cells were harvested at the indicated times, and protein expression was evaluated by immunoblotting (Fig. 2K). The mature form of ABCC4 (band c) exhibits a bi-phasic curve. In contrast, ABCC4 T796M does not have a readily detectable mature ABCC4 (band c); however, the turnover of the immature ABCC4 (band b) for WT and T796M are almost identical, suggesting the immature form of ABCC4 and T796M ABCC4 has the same degradation rate.
Cytoplasmic Loop 3 Helicity Is Altered and Associated with Temperature-sensitive Rescue
We hypothesized that the T796M mutation in the human ABCC4 affects the helical conformation of the CL3 region. To test this idea, we used a 26-amino acid segment (residues 783–808) of CL3 and performed replica exchange molecular dynamics (MD) analysis (see under “Materials and Methods”). The helix containing T796M substitution was iteratively interrogated 16 times, and the structures were determined at temperatures ranging from 270 to 602 K. After MD, the conformation for each temperature was extracted, and the distribution of the backbone dihedral angle pair of residue 796 was calculated and plotted on the Ramachandran map. We observed that although in both simulations the proportions of β-sheet and polyproline II conformation arise when temperature increases, the substantial proportions of β-sheet and polyproline II conformation of residue 796 come out at the lowest temperature in the ABCC4 T796M mutant, indicating that the stability of Met-796 containing helix is worse than the wild type. The temperature-dependent α-helix also reveals that, at lower temperatures, the α-helical content of the peptide containing Met-796 strongly decreases between 300 and 350 K and is consistently lower than the wild type when the temperature is higher than 325 K (Fig. 3A). Based on the MD analysis, we hypothesized that expression of the ABCC4 T796M might be restored by reducing temperature to 28 °C.
FIGURE 3.

Human ABCC4 T796M peptide forms an unstable helix. A, percentage of α-helix is plotted versus temperature (K) from the data derived from in silico molecular dynamics analysis of WT human ABCC4 and T796M mutant peptide. B and C, immunoblot of surface-biotinylated streptavidin-bound lysates (100 μg of protein/lane) prepared from NIH3T3 cells exposed to 37 or 28 °C for 24 h after 24 h of transfection with WT (human ABCC4) and T796M and its quantification. D and E, circular dichroism spectra were obtained with an Aviv 62DS CD spectrophotometer (Aviv) and processed by using Igor Pro software (Wavemetrics Inc). All experiments were performed at 37 °C by using quartz cuvette with a 1-cm path length with 1-nm step resolution and 10 s of average signaling time at 1-nm bandwidth. The concentration of the peptide was 30 μm in 10 mm phosphate buffer (pH 7.0). The CD spectra were expressed as millidegrees and percent of helicity as predicted by the SOMA CD website (25).
We next investigated if subphysiological temperatures rescued expression and plasma membrane localization of ABCC4 T796M. NIH3T3 cells were transiently transfected with either ABCC4 or T796M expression vectors; 24 h post-transfection, one set of cells was transferred to 28 °C incubation, and the other remained at 37 °C. Following an additional 24 h of incubation at the indicated temperatures (Fig. 3, B and C), these cells were biotinylated with a non-cell-permeable NHS-SS-biotin as described previously (8). The surface biotinylated proteins were captured on streptavidin-agarose beads, and immunoblot analysis was performed on total lysate as well as streptavidin-captured surface proteins. For the total protein, T796M is reduced by 50% compared with ABCC4 at 37 °C, with the unexpected increase in the amount of the mature form ABCC4 T796M likely due to the additional 24-h incubation. Interestingly, the immature band b in T796M was dramatically reduced by this additional incubation period. However, at 28 °C, T796M expression and surface expression are strongly increased (Fig. 3, B and C). This result indicates that subphysiological temperatures can rescue T796M expression and restore plasma membrane localization.
Next, we directly determined the effect of T796M mutation on the helix forming capabilities of a fragment of CL3(783–808), using circular dichroism spectrophotometry on CL3 peptides from human WT ABCC4 or T796M CL3 using approaches we previously reported (42). The circular dichroism spectra of these peptides revealed mostly random coils in aqueous solution. However, titrating each peptide solution with increasing amounts of trifluoroethanol (TFE) readily demonstrated increased helicity of the ABCC4 peptide in direct proportion to the concentration of TFE (Fig. 3, D and E). In contrast, the ABCC4 peptide harboring the ABCC4 T796M mutation required substantially higher concentrations of TFE to elicit comparable changes in helicity. These results are compatible with our molecular dynamics simulations (Fig. 3A) and provide additional confirmation that the T796M mutation alters the helicity of this region of CL3 in ABCC4.
Structure-based Rescue of ZF Abcc4
We next investigated, by molecular dynamic simulations, the α-helical content of the same segments of CL3 in ZF Abcc4 and its mutant T804M. We observed that the helical content of T804M was consistently lower than the ZF Abcc4 at temperatures greater than 301 K (Fig. 4A), results that are similar to T796M (Fig. 3A). Based on these findings, we hypothesized that ZF Abcc4 T804M stability might be restored at reduced temperature. We tested this by transiently transfecting NIH3T3 cells with either expression vectors encoding WT ZF Abcc4 or T804M following incubations at either 37 or at 28 °C for 24 h. The Abcc4 and Abcc4 T804M proteins each increased expression at 28 °C; however, for Abcc4 T804M the mature band c is difficult to resolve (Fig. 4B), and instead we observed a strong increase in protein, a result that is qualitatively similar to human T796M.
FIGURE 4.

T804M peptide forms unstable helix compared with the ZF Abcc4 WT peptide. A, percentage of α-helix is plotted versus temperature (K) from the data derived from in silico molecular dynamics analysis of WT ZF Abcc4 and T804M mutant peptide. B, immunoblot of whole cell lysates (100 μg of protein/lane) prepared from NIH3T3 cells incubated at 28 °C for 24 h, 24 h after transfection with WT ZFAbcc4 and Abcc4 T804M, and blots were probed with anti-GFP antibody. C, zebrafish Abcc4 structural model was derived by homology based on the nucleotide-bound bacterial transporter Sav1866. D, Thr-804 at wild type zebrafish ABCC4 displays good contacts with Leu-717, Leu-720, and Arg-808; moreover, the hydroxyl group at the side chain of Thr-804 can form a hydrogen bond with the carbonyl group at Ser-800, which provides extra interaction for stabilizing the helical structure. E, side chain of Met-804 in ZF Abcc4 T804M, especially the position of Sulfur atom, seems highly solvent-accessible. The surrounding residues (Leu-719 and Arg-808) cannot effectively protect the sulfur atom in Met-804. E–G, three mutations of T804S, T804V, and T804D were modeled and analyzed. E, side chain of serine is shorter than methionine so that three hydrophobic residues (Ile-306, Leu-719, and Leu-720) and Arg-808 may protect it from the interruptions of the solvent molecules in T804S mutant. F, because of its hydrophobicity and relatively smaller size, the side chain of Val-804 in the T804V mutant shows good hydrophobic contacts with Arg-808, Leu-719, and Leu-720, and this may increase the stability of the protein structure. G, in T804D mutant, the side chain of Asp-804 can form a salt bridge with Arg-808, and this salt bridge is further stabilized by the contacts of Leu-719. H, molecular dynamic analysis of WT zebrafish Abcc4 and T804M, T804S, T804V, and T804D mutant predicted that valine or aspartic acid can restore the stability of the helix region of CL3. I and J, immunoblots of whole cell lysates (100 μg of protein/lane) prepared from NIH3T3 cells incubated at 37 °C 24 h after the transfection with WT ZF Abcc4 or its variants Abcc4 T804M, T804S, T804V, and T804D and probed with anti-GFP; as predicted by molecular dynamic analysis, T804V or T804D restores the expression almost equal to the WT. K, immunoblots of whole cell lysates (100 μg of protein/lane) prepared from NIH3T3 cells incubated at 37 °C 24 h after the transfection with WT zebrafish Abcc4 or T804G. L, confocal microscopy of NIH3T3 transfected cells with GFP-tagged Abcc4 and Abcc4 mutants, and RFP-tagged ER marker (calreticulin). Cells were analyzed by confocal microscopy. Plasma membrane was stained with Alexa 646 wheat germ agglutinin (blue). Signals from the three channels were acquired independently, and the merged images are presented. Co-localization of Abcc4 WT or its variants with plasma membrane is indicated by a reddish blue, and co-localization of GFP Abcc4 T804M and RFP-ER is indicated by a reddish yellow color.
Because the reduced temperature rescue was less robust for ZF Abcc4 T804M, we hypothesized that side-chain interactions were slightly different between human and zebrafish (see Fig. 1C). To investigate the role of the side-chain interactions for stability at position 804 in ZF ABCC4, we interrogated our molecular model to predict amino acids that, if substituted, might mimic the threonine contacts (Fig. 4, C–G). We next performed MD on each peptide containing the indicated amino acid substitutions (Fig. 4H). These studies suggested that the T804V and T804D were as thermostable as WT, whereas T804S was almost as unstable as T804M. To determine whether these predictions were recapitulated in the whole protein, we used site-directed mutagenesis to substitute Ser, Val, or Asp for Thr-804 in ZF Abcc4. These mutations were transfected into NIH3T3 cells, and protein expression was assessed by immunoblot analysis. The substitutions of either Asp or Val mostly rescued protein expression and gave strong membrane localization (Fig. 4, I and J), supporting the idea that the side-chain interactions are important. To confirm that the amino acid side-chain interactions in this region of CL3 are important to Abcc4 expression, we substituted Gly for Thr-804 and showed T804G was undetectable (Fig. 4K). Interestingly, subcellular localization (by confocal microscopy) demonstrated that T804S, T804V, and T804D substitutions mostly localized to the plasma membrane along with the plasma membrane marker wheat germ agglutinin (Fig. 4L).
Expression of ABCC4 with ABCC7 CL3 Mutations
Phylogenetically, ABCC4 is the closest paralog of ABCC7 (CFTR) with both appearing to share a common ancestor (43). It is notable that many amino acid residues are highly conserved in both CFTR and ABCC4 (supplemental Fig. 2) (43). Interestingly, the CL3 of CFTR is highly conserved and very similar to ABCC4 (from 70 to 80% across species) (Fig. 5A). Notably, disease-related point mutations in CFTR CL3 at positions S945L and H949Y (Fig. 5A) (44) affected maturation of CFTR (26). Because these two amino acids are adjacent to Thr-796 in ABCC4 and mostly conserved with CFTR, we hypothesized that this region of CL3 might be generally important to ABCC protein stability. Using site-directed mutagenesis, we generated the analogous mutations in human ABCC4 (S794L and H798Y) and ZF Abcc4 (A802L and H806Y). After transient transfection of NIH3T3 cells, we assessed expression of WT and mutant ABCC4 by immunoblot analysis. ABCC4 S794L and H796Y as well as ZF Abcc4 A802L and H806Y substitutions affected both protein expression and highly reduced the levels of the mature band c (Fig. 5, C and E). Notably, the expression pattern of these mutants with reduced mature glycosylated band c is similar to ABCC4 T796M (Fig. 5E). These results further confirm that this region of CL3 plays an important role in ABCC4 maturation. Moreover, MD analysis on the helical regions of CL3 encompassing these substitutions show a similar loss in helicity with elevated temperatures (Fig. 5, B and D). Interestingly, the His to Tyr substitution in CL3 for both human and zebrafish has profound reduction in α-helical content.
FIGURE 5.

CL3 of ABCC4 and CFTR are highly conserved. A, sequence alignment of cytoplasmic loop 3 region of ABCC4 and CFTR across species shows highly conserved region that include two CFTR pathogenic mutations (S945L or H949Y) that were reported to cause the cystic fibrosis disease. B, percentage of α-helix is plotted versus temperature (K) from the data derived from in silico molecular dynamic analysis of WT ZF Abcc4 and Ala-802, Leu-802, His-806, and Tyr-806 mutant peptides. C, immunoblots of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with ZF Abcc4 WT and Abcc4 mutants (T804M, A802L, and H806Y); these blots were probed with anti-GFP antibody. D, percentage of α-helix is plotted versus temperature (K) from the data derived from in silico molecular dynamics analysis of WT human ABCC4 and Ser-794, Leu-794, His-798, and Tyr-798 mutant. E, immunoblot of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with human ABCC4 and ABCC4 mutants (T796M, S794L, and H798Y), and these blots were probed with anti-GFP antibody.
Mutation R815A in Human ABCC4 Cytoplasmic Region 3 (CL3) Destabilizes the Protein
Because CLs interact with NBDs in CFTR (26), we hypothesized that a mutation in an amino acid of CL3 in the “coupling helix” (a CL region near the NBD) might disrupt NBD and CL interdomain interactions producing an alteration in stability. Based upon the human ABCC4 homology model (32), we predicted that in CL3 of ABCC4 Arg-815 would interact with Asp-522 in the NBD1 domain by a salt bridge (Fig. 6A). To investigate this we mutated Arg-815 to Ala. The R815A mutation resulted in reduced ABCC4 expression and loss of mature glycosylated band c (Fig. 6, B and C). The reduction in mature ABCC4 is consistent with the prediction that CL3 Arg-815 interacts with the NBD to ensure proper folding of the protein
FIGURE 6.

A, representation of interdomain interaction between R815A of CL3 region and Asp-522 of NBD1 domain. B, immunoblot of whole cell lysates (100 μg of protein per lane) prepared from NIH3T3 cells transfected with human ABCC4 WT and ABCC4 mutant (R815A) and incubated at 37 or 28 °C for 24 h after 24 h of transfection. C, densitometric quantification of the blots.
To test if reduced temperature enhanced expression of R815A, NIH3T3 cells were transiently transfected with ABCC4, T796M-ABCC4, or R815A-ABCC4 and then incubated at 28 °C. The amount of R815A protein increased, which paralleled an increase in mature glycosylated protein band c (Fig. 6, B and C). Collectively, these studies support a role for CL3 interactions with NBD1 in promoting maturation of ABCC4.
DISCUSSION
We recently identified an ABCC4 orthologue in zebrafish (ZF) containing a point mutation that causes organ localization defects.3 This point mutation produced an amino acid substitution at T804M, a change that the computer algorithms PolyPhen and SIFT predicted as benign. However, in ZF and human ABCC4 (T796M, is the analogous position), protein expression was greatly reduced by this substitution. To explore the molecular basis for the instability of human and ZF ABCC4/Abcc4, we developed a homology model of ABCC4 based on the structure of the bacterial ABC transporter Sav1866 (20). The threonine in ZF Abcc4 is conserved from fish to humans and is located in a conserved α-helical domain in CL3. Our model indicated that the bulky methionine substitution disrupted amino acid contacts, suggesting such contacts are important for the helicity of CL3, an idea supported by molecular dynamic simulations under various temperatures. We propose that disrupting this region of CL3 in ABCC4 and its paralogs alters protein folding and assembly, which appears important for ABCC4 maturation. This proposition is supported by restored expression of ABCC4 at a temperature below physiological. Moreover, methionine-substituted human and zebrafish ABCC4/Abcc4 is retained intracellularly, a finding consistent with impaired maturation. Furthermore, we extended these studies to show that other mutations in CL3 (analogous to those in CFTR (S945L and H949Y)) that also disrupt its helical properties reduce protein expression. In toto these studies suggest this α-helical domain in CL3 is crucial for proper assembly and localization.
The reduced amounts of mutant ABCC4 protein (T804M in zebrafish and T796M in humans) are not related to an impaired translation of the ABCC4 mRNA, which is almost identical among WT and T796M mRNA despite an apparent alteration in the energetics of mRNA folding. It is likely that disrupted processing in the ER of ABCC4 is disrupted by the T796M substitution. Consistent with such a proposition are the findings showing disease-causing variations in the CL3 of the close relative of ABCC4, the cAMP-regulated CFTR/ABCC7, causes low protein expression secondary to impaired protein maturation (26, 27). Engineering these orthologous “CFTR CL3” mutations in both ZF and human ABCC4 strongly reduced protein expression for both species of ABCC4/Abcc4. Like the T796M for human and the orthologous T804M in zebrafish, these additional CL3 variants alter the helical properties of this region in CL3. Because these ABCC4 CL3 loop “CFTR-like” substitutions reduce stability, we propose that this region has a crucial role in regulating ABCC4 processing in the ER, and side-chain interactions are crucial to the interactions between CL3 and NBD1. Furthermore, it is likely that CL3, as a potential target of folding, might interact with NBD1 through an intermediary step requiring FKBP52/38 interactions (23).
Based upon studies with CFTR (26), we hypothesized that the interaction between the nucleotide-binding domain (NBD1) and CL3 is important for the proper folding of ABCC4 (15, 16, 24). These interdomain interactions between NBD and CL have been reported for CFTR, and it is likely the CL3/NBD1 interaction for ABCC4 is a prerequisite for proper folding of ABCC4. Based on our homology model of human ABCC4, arginine 815 in CL3 was predicted to contact aspartate 522 in NBD1. The reduced maturation and expression of alanine-substituted Arg-815 at physiological temperatures support the idea that Arg-815 in the coupling helix of CL3 is crucial for proper ABCC4 maturation. The increased formation of the mature c-form of ABCC4 R815A at the subphysiological temperature of 28 °C is consistent with the proposal that CL3/NBD1 interactions are necessary for proper folding of ABCC4. In further support of ABCC4 CL3 mutants being misfolded, preliminary studies show that that two small molecules that reduce the activity of the ER unfolded protein response (phenylbutyrate (29) and glycerol (36)) selectively rescue expression of the ABCC4 CL3 T796M (data not shown).
N-Glycosylation is an important post-translational modification that occurs during protein synthesis in the ER with subsequent enzymatic modifications revealed by an electrophoretic mobility shift as the glycoprotein matures. In cells harboring ABCC4, a T796M substitution, reduced amounts of the mature glycosylated form are observed. These findings, and the internal mislocalization of T796M, suggest that either ER glycosylation of ABCC4 is impaired or the protein is mostly degraded before maturation is complete. We show that T796M has reduced mature ABCC4 (c-form) coupled with decreased abundance at the plasma membrane. We favor the idea that the T796M substitution does not generally produce defective glycosylation of ABCC4 because plasma membrane localization and ABCC4 glycosyl maturation are mostly restored at reduced temperatures. However, we cannot rule out the possibility that the increased expression of T796M at subphysiological temperatures might be due, in part, to reduced protease activity (it seems unlikely that reduced protease activity modifies glycosylation).
Our studies highlight an ABCC4 CL3 mutation that appears to disrupt CL3 helix stability. Using TFE-induced protein folding, we demonstrate that the peptide fragment of CL3 harboring a methionine substituted for the conserved threonine has an impaired ability to form an α-helix in the presence of TFE. These findings buttress our molecular dynamics simulations showing that the region containing T796M has less thermal stability, a finding consistent with this region and with CL3's reduced ability to form an α-helix.
Among the mammalian ABCC4 proteins, CL3 is conserved and characterized by several highly conserved amino acid residues. In CL3, a highly conserved block of amino acids (TLHN) contains the threonine that appears to have a vital role in the expression, stability, and localization of human and ZF ABCC4/Abcc4. Our results highlight that substitutions at and around this block of amino acids reduce the ABCC4 expression (see the CFTR-CL3 substitutions) and uncover an important but previously unknown role of CL3 in ensuring ABCC4 expression. Our studies suggest that amino acid substitutions perturbing the inherent helicity of this region of CL3 affect ABCC4 plasma membrane localization. Although it is unknown if helicity was changed, similar mutations in a paralog of ABCC4 CFTR also affected its maturation (26). We speculate, based on studies with CFTR (16), that CL3 mutations in this region ultimately affect contacts between CL3 and NBD1. In human CFTR, the threonine 966 in CL3 has been shown to contact Glu-543 in the NBD domain (15, 16). Likewise, we show that the predicted contact between CL3 Arg-815 and NBD1 Asp-522 when disrupted by an R815A substitution reduces the amount of mature ABCC4. This supports the idea that these CL3/NBD1 contacts are also important for the maturation of both ABCC4 and CFTR. It is intriguing to speculate that CL3 mutations are especially deleterious to both ABCC4 and CFTR and perhaps other full-length ABC proteins. An analysis of the frequency of CL mutations (26, 27) reveals a far greater number of mutations in CL4 region compared with the CL3 region. These observations suggest that, considering CL3 and CL4 are of similar length (57 and 67 amino acids, respectively), the lower frequency of CL3 mutations is because CL3 mutations are either less likely or more deleterious to ABCC4 expression. If it is the latter case, then we might expect a lower frequency because CL3 mutations might be selected against either ABCC4 or CFTR. Based on our studies, we propose key residues in the membrane-proximal region of the cytoplasmic loop ensure proper interdomain interaction. This new knowledge may ultimately help pave the way for development of agents that correct cytoplasmic loop helicity, perhaps by restoring interactions between the CL3 and NBD1.
Acknowledgment
We thank Jama Temirov for assistance with immunofluorescence microscopy from the microscopy core facility.
This work was supported, in whole or in part, by National Institutes of Health Grants 2R01GM60904, P30CA21745, and CA21865 (to J. D. S.) and DK080834 and DK093045 (to A. P. N.). This work was also supported by ALSAC.

This article contains supplemental Figs. 1 and 2.
D. Jin, T. T. Ni, J. Sun, G. Yu, H. Wan, J. D. Amack, S. Cheepala, D. Nachagari, J. Fleming, H. Ma, C. Chiang, G. Conseil, S. P. C. Cole, I. Drummond, J. D. Schuetz, and T. P. Zhong, manuscript in preparation.
- ABCC4
- ATP-binding cassette protein C4
- CL
- cytoplasmic loop
- ZF
- zebrafish
- CFTR
- cystic fibrosis transmembrane conductance regulator
- NBD
- nucleotide binding domain
- RFP
- red fluorescent protein
- ER
- endoplasmic reticulum
- PNGase F
- peptide:N-glycosidase F
- MD
- molecular dynamics
- TFE
- trifluoroethanol.
REFERENCES
- 1. Kool M., de Haas M., Scheffer G. L., Scheper R. J., van Eijk M. J., Juijn J. A., Baas F., Borst P. (1997) Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res. 57, 3537–3547 [PubMed] [Google Scholar]
- 2. Schuetz J. D., Connelly M. C., Sun D., Paibir S. G., Flynn P. M., Srinivas R. V., Kumar A., Fridland A. (1999) MRP4: A previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat. Med. 5, 1048–1051 [DOI] [PubMed] [Google Scholar]
- 3. Barber R., Ray K. P., Butcher R. W. (1980) Turnover of adenosine 3′,5′-monophosphate in WI-38 cultured fibroblasts. Biochemistry 19, 2560–2567 [DOI] [PubMed] [Google Scholar]
- 4. Brunton L. L., Buss J. E. (1980) Export of cyclic AMP by mammalian reticulocytes. J. Cyclic Nucleotide Res. 6, 369–377 [PubMed] [Google Scholar]
- 5. Cheepala S., Hulot J. S., Morgan J. A., Sassi Y., Zhang W., Naren A. P., Schuetz J. D. (2013) Cyclic nucleotide compartmentalization: contributions of phosphodiesterases and ATP-binding cassette transporters. Annu. Rev. Pharmacol. Toxicol. 53, 231–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kruh G. D., Belinsky M. G., Gallo J. M., Lee K. (2007) Physiological and pharmacological functions of Mrp2, Mrp3, and Mrp4 as determined from recent studies on gene-disrupted mice. Cancer Metastasis Rev. 26, 5–14 [DOI] [PubMed] [Google Scholar]
- 7. Wielinga P. R., Reid G., Challa E. E., van der Heijden I., van Deemter L., de Haas M., Mol C., Kuil A. J., Groeneveld E., Schuetz J. D., Brouwer C., De Abreu R. A., Wijnholds J., Beijnen J. H., Borst P. (2002) Thiopurine metabolism and identification of the thiopurine metabolites transported by MRP4 and MRP5 overexpressed in human embryonic kidney cells. Mol. Pharmacol. 62, 1321–1331 [DOI] [PubMed] [Google Scholar]
- 8. Krishnamurthy P., Schwab M., Takenaka K., Nachagari D., Morgan J., Leslie M., Du W., Boyd K., Cheok M., Nakauchi H., Marzolini C., Kim R. B., Poonkuzhali B., Schuetz E., Evans W., Relling M., Schuetz J. D. (2008) Transporter-mediated protection against thiopurine-induced hematopoietic toxicity. Cancer Res. 68, 4983–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ban H., Andoh A., Imaeda H., Kobori A., Bamba S., Tsujikawa T., Sasaki M., Saito Y., Fujiyama Y. (2010) The multidrug-resistance protein 4 polymorphism is a new factor accounting for thiopurine sensitivity in Japanese patients with inflammatory bowel disease. J. Gastroenterol. 45, 1014–1021 [DOI] [PubMed] [Google Scholar]
- 10. Abla N., Chinn L. W., Nakamura T., Liu L., Huang C. C., Johns S. J., Kawamoto M., Stryke D., Taylor T. R., Ferrin T. E., Giacomini K. M., Kroetz D. L. (2008) The human multidrug resistance protein 4 (MRP4, ABCC4): functional analysis of a highly polymorphic gene. J. Pharmacol. Exp. Ther. 325, 859–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janke D., Mehralivand S., Strand D., Gödtel-Armbrust U., Habermeier A., Gradhand U., Fischer C., Toliat M. R., Fritz P., Zanger U. M., Schwab M., Fromm M. F., Nürnberg P., Wojnowski L., Closs E. I., Lang T. (2008) 6-Mercaptopurine and 9-(2-phosphonyl-methoxyethyl) adenine (PMEA) transport altered by two missense mutations in the drug transporter gene ABCC4. Hum. Mutat. 29, 659–669 [DOI] [PubMed] [Google Scholar]
- 12. Borst P., de Wolf C., van de Wetering K. (2007) Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 453, 661–673 [DOI] [PubMed] [Google Scholar]
- 13. Deeley R. G., Westlake C., Cole S. P. (2006) Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 86, 849–899 [DOI] [PubMed] [Google Scholar]
- 14. Sauna Z. E., Nandigama K., Ambudkar S. V. (2004) Multidrug resistance protein 4 (ABCC4)-mediated ATP hydrolysis: effect of transport substrates and characterization of the post-hydrolysis transition state. J. Biol. Chem. 279, 48855–48864 [DOI] [PubMed] [Google Scholar]
- 15. He L., Aleksandrov L. A., Cui L., Jensen T. J., Nesbitt K. L., Riordan J. R. (2010) Restoration of domain folding and interdomain assembly by second-site suppressors of the ΔF508 mutation in CFTR. FASEB J. 24, 3103–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He L., Aleksandrov A. A., Serohijos A. W., Hegedus T., Aleksandrov L. A., Cui L., Dokholyan N. V., Riordan J. R. (2008) Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J. Biol. Chem. 283, 26383–26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serohijos A. W., Hegedus T., Aleksandrov A. A., He L., Cui L., Dokholyan N. V., Riordan J. R. (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 19. Fukuda Y., Aguilar-Bryan L., Vaxillaire M., Dechaume A., Wang Y., Dean M., Moitra K., Bryan J., Schuetz J. D. (2011) Conserved intramolecular disulfide bond is critical to trafficking and fate of ATP-binding cassette (ABC) transporters ABCB6 and sulfonylurea receptor 1 (SUR1)/ABCC8. J. Biol. Chem. 286, 8481–8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 21. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., Friesner R. A. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 [DOI] [PubMed] [Google Scholar]
- 22. Jacobson M. P., Friesner R. A., Xiang Z., Honig B. (2002) On the role of crystal packing forces in determining protein sidechain conformations. J. Mol. Biol. 320, 597–608 [DOI] [PubMed] [Google Scholar]
- 23. Banasavadi-Siddegowda Y. K., Mai J., Fan Y., Bhattacharya S., Giovannucci D. R., Sanchez E. R., Fischer G., Wang X. (2011) FKBP38 peptidylprolyl isomerase promotes the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum. J. Biol. Chem. 286, 43071–43080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El Hiani Y., Linsdell P. (2012) Role of the juxtamembrane region of cytoplasmic loop 3 in the gating and conductance of the cystic fibrosis transmembrane conductance regulator chloride channel. Biochemistry 51, 3971–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Unneberg P., Merelo J. J., Chacón P., Morán F. (2001) SOMCD: method for evaluating protein secondary structure from UV circular dichroism spectra. Proteins 42, 460–470 [DOI] [PubMed] [Google Scholar]
- 26. Seibert F. S., Linsdell P., Loo T. W., Hanrahan J. W., Riordan J. R., Clarke D. M. (1996) Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. J. Biol. Chem. 271, 27493–27499 [DOI] [PubMed] [Google Scholar]
- 27. Seibert F. S., Linsdell P., Loo T. W., Hanrahan J. W., Clarke D. M., Riordan J. R. (1996) Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J. Biol. Chem. 271, 15139–15145 [DOI] [PubMed] [Google Scholar]
- 28. Rosenberg M. F., O'Ryan L. P., Hughes G., Zhao Z., Aleksandrov L. A., Riordan J. R., Ford R. C. (2011) The cystic fibrosis transmembrane conductance regulator (CFTR): three-dimensional structure and localization of a channel gate. J. Biol. Chem. 286, 42647–42654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayashi H., Sugiyama Y. (2007) 4-Phenylbutyrate enhances the cell surface expression and the transport capacity of wild type and mutated bile salt export pumps. Hepatology 45, 1506–1516 [DOI] [PubMed] [Google Scholar]
- 30. Loo T. W., Bartlett M. C., Detty M. R., Clarke D. M. (2012) The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem. 287, 26806–26816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seeger M. A., van Veen H. W. (2009) Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta 1794, 725–737 [DOI] [PubMed] [Google Scholar]
- 32. Ravna A. W., Sager G. (2008) Molecular model of the outward facing state of the human multidrug resistance protein 4 (MRP4/ABCC4). Bioorg. Med. Chem. Lett. 18, 3481–3483 [DOI] [PubMed] [Google Scholar]
- 33. Ravna A. W., Sager G. (2009) Molecular modeling studies of ABC transporters involved in multidrug resistance. Mini. Rev. Med. Chem. 9, 186–193 [DOI] [PubMed] [Google Scholar]
- 34. Ravna A. W., Sylte I., Sager G. (2008) A molecular model of a putative substrate releasing conformation of multidrug resistance protein 5 (MRP5). Eur. J. Med. Chem. 43, 2557–2567 [DOI] [PubMed] [Google Scholar]
- 35. Hazai E., Bikádi Z. (2008) Homology modeling of breast cancer resistance protein (ABCG2). J. Struct. Biol. 162, 63–74 [DOI] [PubMed] [Google Scholar]
- 36. Kanki K., Kawamura T., Watanabe Y. (2009) Control of ER stress by a chemical chaperone counteracts apoptotic signals in IFN-γ-treated murine hepatocytes. Apoptosis 14, 309–319 [DOI] [PubMed] [Google Scholar]
- 37. Chang C., Swaan P. W. (2006) Computational approaches to modeling drug transporters. Eur. J. Pharm. Sci. 27, 411–424 [DOI] [PubMed] [Google Scholar]
- 38. Vardy E., Arkin I. T., Gottschalk K. E., Kaback H. R., Schuldiner S. (2004) Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci. 13, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walters R. F., DeGrado W. F. (2006) Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 13658–13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartoszewski R. A., Jablonsky M., Bartoszewska S., Stevenson L., Dai Q., Kappes J., Collawn J. F., Bebok Z. (2010) A synonymous single nucleotide polymorphism in ΔF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J. Biol. Chem. 285, 28741–28748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bertolucci C. M., Guibao C. D., Zheng J. (2005) Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly. Protein Sci. 14, 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jordan I. K., Kota K. C., Cui G., Thompson C. H., McCarty N. A. (2008) Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 18865–18870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Claustres M., Laussel M., Desgeorges M., Giansily M., Culard J. F., Razakatsara G., Demaille J. (1993) Analysis of the 27 exons and flanking regions of the cystic fibrosis gene: 40 different mutations account for 91.2% of the mutant alleles in southern France. Hum. Mol. Genet. 2, 1209–1213 [DOI] [PubMed] [Google Scholar]