

Background: C/EBPβ is a bZip transcription factor that triggers phosphorylation of p300.
Results: Protein kinase Hipk2 interacts with and phosphorylates the longest isoform of C/EBPβ, thereby facilitating recruitment and subsequent phosphorylation of p300.
Conclusion: C/EBPβ is a direct interaction partner and physiological substrate of Hipk2.
Significance: Hipk2 cooperates with C/EBPβ in an isoform-specific manner.
Keywords: p300, Phosphorylation, Protein Kinases, Protein-Protein Interactions, Transcription, C/EBPβ, Hipk2
Abstract
CCAAT box/enhancer-binding protein β (C/EBPβ) is a bZip transcription factor that plays crucial roles in important cellular processes such as differentiation and proliferation of specific cell types. Previously, we showed that C/EBPβ cooperates with the coactivator p300 through a novel mechanism that involves the C/EBPβ-induced phosphorylation of multiple sites in the carboxyl-terminal domain of p300 by protein kinase Hipk2. We have now examined the interaction and cooperation of C/EBPβ, p300, and Hipk2 in more detail. We show that Hipk2 and C/EBPβ are direct physical binding partners whose interaction is mediated by sequences located in the amino-terminal and central domains of Hipk2 and the amino-terminal part of C/EBPβ. In addition to phosphorylating p300 recruited to C/EBPβ, Hipk2 also phosphorylates C/EBPβ at sites that have previously been shown to plays key roles in the regulation of C/EBPβ activity. Silencing of Hipk2 expression disrupts adipocyte differentiation of 3T3-L1 cells, a physiological C/EBPβ-dependent differentiation process indicating that the cooperation of C/EBPβ and Hipk2 is functionally relevant. Finally, we demonstrate that C/EBPα, a related C/EBP family member whose amino-terminal sequences differ significantly from that of C/EBPβ, is unable to interact and cooperate with Hipk2. Instead, our data suggest that C/EBPα cooperates with the protein kinase Jnk to induce phosphorylation of p300. Overall, our data identify Hipk2 as a novel regulator of C/EBPβ and implicate different protein kinases in the cooperation of p300 with C/EBPβ and C/EBPα.
Introduction
C/EBPα and C/EBPβ are members of the CCAAT box/enhancer-binding protein (C/EBP)3 family of basic-region-leucine-zipper transcription factors, which play central roles in fundamental cellular processes including differentiation, proliferation, and growth arrest of specific cell types, such as adipocytes, keratinocytes, mammary epithelial cells, and myelomonocytic cells (1–9). The activities of C/EBPα and C/EBPβ are controlled in a complex manner on multiple levels, suggesting that they are key factors in the transcription network whose precise control is crucial for the cell. C/EBPα and C/EBPβ are encoded by intron-less genes that are transcribed into a single mRNA in each case, which is translated into different protein isoforms due to alternative translation initiation (10, 11). In case of C/EBPβ, two longer isoforms (LAP* and LAP) and a short isoform (LIP) are generated. LAP* and LAP act both as transcriptional activators, whereas LIP lacks a transactivation domain and functions as a transcriptional inhibitor (10–12). The relative amounts of the different isoforms vary between different cell types and are influenced via a short upstream open reading frame by the status of the cell (11, 13–15). In addition to differential isoform expression, C/EBPβ, is regulated by post-translational modifications, such as phosphorylation (16–18), acetylation (19–21), and sumoylation (22) as well as by the binding of specific cofactors via protein-protein interactions (23–27). Besides its function in normal cells, deregulation of C/EBPβ activity or isoform expression contributes to the development of cancer (28–31).
One of the binding partners known to interact with and stimulate the transactivation potential of C/EBPβ is the coactivator p300. C/EBPβ directly binds to the so-called E1A binding region in the carboxyl-terminal part of p300, leading to the acetylation of C/EBPβ and the stimulation of its transcriptional activity (19, 23). Binding of p300 to C/EBPβ is accompanied by a large electrophoretic mobility shift of p300, which is caused by the phosphorylation of multiple residues in the carboxyl-terminal part of p300 (32). The homeodomain interacting kinase 2 (Hipk2) has been identified as a key player in the C/EBPβ-induced phosphorylation of p300; Hipk2 interacts with C/EBPβ and stimulates the phosphorylation of C/EBPβ-bound p300 (33). The C/EBPβ-induced phosphorylation of p300 itself is subject to regulation by other factors; the transcription factor Myc inhibits the phosphorylation of C/EBPβ-bound p300 by binding itself to C/EBPβ and displacing Hipk2, thereby inhibiting the acetylation and the activity of C/EBPβ (33).
The observation that C/EBPβ recruits p300 and Hipk2 raised the question of whether C/EBPβ only acts as an adaptor between p300 and Hipk2 to facilitate the phosphorylation of p300 or whether C/EBPβ is itself also phosphorylated by Hipk2. Here, we have studied the cooperation of C/EBPβ and Hipk2 in more detail. Our work demonstrates that C/EBPβ is a direct binding partner and substrate for Hipk2. We show that Hipk2 expression is required for adipocyte conversion of 3T3-L1 cells, a physiological C/EBPβ-dependent differentiation process. Finally, we show that two related C/EBP family members, C/EBPβ and C/EBPα, differ in their ability to cooperate with Hipk2.
EXPERIMENTAL PROCEDURES
Cells
QT6 is a line of quail fibroblasts. Subclones of the mouse 3T3-L1 stably transfected with an expression vector for Hipk2-specific shRNA or with a control vector have been described (33). Adipocyte differentiation of 3T3-L1 cells was induced by published procedures (34).
RNA Interference
siRNA duplexes were obtained from Eurogentec. The Hipk2 siRNA target sequence was 5′-CAGCCCUGAUGAAACUCAA-3′. siRNA directed against Renilla luciferase (target sequence, 5′-AAACAUGCAGAAAAUGCUG-3′) was used as the negative control. Approximately 2.5 × 105 cells were plated the day before transfection in 6-cm plates and received 1.5 ml of fresh growth medium before transfection. siRNA was transfected using MetafecteneTM Pro (Biontex) or HiPerFect (Qiagen), according to manufacturers' protocols. Cells were harvested 48 h later and processed further for Western blotting.
Expression Vectors
Expression vectors for full-length and truncated C/EBPβ, pCDNA3-CCR, and pCDNA3-CCR-ΔN21 and for a mutant in which amino acids Phe-27, Tyr-28, and Tyr-29 of full-length C/EBPβ are replaced by alanine, pCDNA3-CCR-mut1, have been described (33, 35). A pCDNA3-based expression vector for full-length human C/EBPβ was obtained from G. Steger (Institute of Virology, University of Cologne). Phosphorylation site mutants of C/EBPβ S65A and T220A were generated by PCR using appropriate oligonucleotides as primers. An expression vector for full-length chicken C/EBPα was obtained by amplifying the C/EBPα coding region by PCR and inserting it into pCDNA3. The expression vector for a truncated form of p300 (p300/1751–2370) has been described (23). Expression vectors for HA-tagged Hipk2 (pLNCX-HA-Hipk2-wt) and a HA-tagged kinase-dead mutant of Hipk2, carrying a K221A mutation (pLNCX-HA-Hipk2-KD), were obtained from T. Hofmann (36). Carboxyl-terminal Hipk2 deletion constructs HA-Hipk2-(1–764) and HA-Hipk2-(1–541) were generated by truncating the Hipk2 coding region at AgeI or BglII restriction sites, respectively, and subcloning into pCDNA3. Myc-tagged Hipk2 deletion constructs Myc-Hipk2-(8–220), Myc-Hipk2-(8–170), Myc-Hipk2-(70–220), Myc-Hipk2Δ1–170, Myc-Hipk2-(170–764), and Myc-Hipk2-(170–548) were generated by amplifying the corresponding parts of the Hipk2 coding region by PCR and cloning them downstream of six copies of a Myc tag in pCDNA3.
A bacterial expression vector for a GST-Hipk2-(70–220) fusion protein was obtained by inserting the corresponding part of the Hipk2 coding region in-frame into plasmid pGex-5X3. The bacterial expression vector for a GST-p300-(1710–1891) fusion protein has been described (32). The coding region of wild-type C/EBPβ and C/EBPβ mutants S65A and T220A was inserted in-frame into pGEX-6P2.
Northern Blotting
MRP126, goose-type lysozyme gene #325, Hipk2, C/EBPα, GAPDH, and ribosomal protein S 17 mRNAs were detected using probes derived from the corresponding cDNA clones, as described previously (23).
Antibodies, Western Blotting and Immunoprecipitation
Rabbit antiserum against chicken C/EBPβ has been described (23). p300-specific antibodies (RW128) were obtained from Upstate Biotechnology, Inc. Antibodies against the HA and FLAG tags were from Hiss-Diagnostics and Sigma, respectively. Rabbit antiserum against phosphorylated Ser-2280 of p300 was raised against the peptide VQPNPMSPQQHMC where the underlined serine was phosphorylated (33). Antiserum against human C/EBPβ (H-7) was obtained from St. Cruz Biotechnology. Antiserum against phospho-C/EBPβ(T235) was obtained from Cell Signaling Technology. Antiserum against Hipk2 was kindly provided by Lienhard Schmitz (36). Total cell extracts were prepared by lysing the cells in ELB buffer (50 mm Tris-HCl, pH 7.5, 120 mm NaCl, 20 mm NaF, 1 mm EDTA, 6 mm EGTA, 15 mm sodium pyrophosphate, 1 mm PMSF, 0.1% Nonidet P-40) followed by centrifugation for 30 min at 14,000 × g. For endogenous immunoprecipitation, a high salt lysis buffer as described by Hofmann et al. (36) was used. Cleared lysates were analyzed directly by SDS-PAGE and Western blotting or were first subjected to immunoprecipitation. Immunoprecipitation was carried out in ELB buffer by supplementing aliquots of the total protein extract with the appropriate antibodies and incubating for 1 h at 4 °C. Protein A-Sepharose beads were then added and incubated further for 1 h at 4 °C under constant agitation. Immune complexes were then collected by centrifugation, washed 3 times with ELB buffer, and finally subjected to SDS-PAGE and Western blotting.
In Vitro Protein Kinase Assays
In vitro protein kinase assays using bacterially expressed GST proteins were performed as follows. HA-tagged wild-type or kinase-dead Hipk2 was isolated by immunoprecipitation with anti-HA antibodies from QT6 cells transfected with the appropriate expression vectors. As additional controls, untransfected cells were used. Cells were lysed in radioimmune precipitation assay buffer, and immunoprecipitates were washed extensively with the same buffer. GST proteins were purified from bacterial extracts by binding to GST-Sepharose as described (32). Glutathione-Sepharose beads loaded with the GST proteins were then mixed with protein G-Sepharose beads carrying the immunoprecipitated HA-Hipk2 and were incubated for 30 min at 37 °C in kinase buffer (25 mm Tris-HCl, pH 7.5, 5 mm β-glycerol phosphate, 2 mm DTT, 0.1 mm Na3VO4, 10 mm MgCl2, 20 μm ATP) containing 4 μCi of [γ-32P]ATP (Amersham Biosciences, specific activity 3000 Ci/mmol). The reactions were stopped by adding SDS sample buffer. The reaction mixture was then subjected to electrophoresis in a 10% SDS-polyacrylamide gel and analyzed with a phosphorimaging analyzer.
GST Pulldown
GST pulldown experiments using a GST-Hipk2 fusion protein and bacterially expressed C/EBPβ were performed as described previously (32). GST pulldown experiments with total eukaryotic cell extracts were performed as described (37).
RESULTS
Interaction of C/EBPβ with the Protein Kinase Hipk2
Our previous work has shown that the recruitment of the co-activator p300 by the transcription factor C/EBPβ is coupled to the phosphorylation of p300 at multiple sites in its carboxyl-terminal domain. These phosphorylations lead to a substantial electrophoretic mobility shift of p300 in an SDS-polyacrylamide gel and increase the activity of p300 as a coactivator (32). Subsequently, we have identified the protein kinase Hipk2 as the kinase that is responsible for these C/EBPβ-induced phosphorylation events. We have found that Hipk2 is itself recruited to the C/EBPβ to phosphorylate the C/EBPβ-bound p300 (33). To better understand the interplay between C/EBPβ, p300, and Hipk2, we were interested to know whether C/EBPβ serves mainly as a scaffold (or adapter) that allows Hipk2 and p300 to come into close proximity or whether C/EBPβ is itself also phosphorylated by Hipk2. As the first step to explore the cooperation of C/EBPβ and Hipk2, we investigated which domains of both proteins interact with each other and whether their binding is caused by direct protein-protein interactions. As illustrated in Fig. 1A, we generated a series Hipk2 deletion constructs and used them in combination with C/EBPβ expression vector to perform co-immunoprecipitations. Initial experiments using carboxyl-terminal Hipk2 deletions showed that truncation up to amino acid 764 increased the binding to C/EBPβ (Fig. 1B, lane 7). Further truncation up to amino acid 541 resulted in weaker, but still detectable binding (lane 5). This suggested that Hipk2 harbors two binding sites for C/EBPβ located in the amino-terminal part of the protein and between amino acids 541 and 764. The experiments shown in Fig. 1C confirmed that amino-terminal sequences are capable of binding to C/EBPβ and that a binding site for C/EBPβ most likely lies between amino acids 70 and 170 of Hipk2. That there must be a second binding site for C/EBPβ was demonstrated by the experiment in Fig. 1D, which shows that deletion of amino acids 1–170 of Hipk2 was not sufficient to abolish the binding to C/EBPβ. The combination of the amino-terminal deletion with carboxyl-terminal truncation finally confirmed the existence of a second binding site for C/EBPβ, located between amino acids 548 and 764 of Hipk2 (see Fig. 4E).
FIGURE 1.

Identification of the C/EBPβ binding region in Hipk2. A, shown is a schematic illustration of the Hipk2 constructs used for the binding experiments. KD refers to the kinase domain of Hipk2. B--E, QT6 cells were transfected with the indicated expression vectors for FLAG-C/EBPβ and various truncated forms of HA- or Myc-tagged Hipk2. 24 h after transfection the cells were harvested, and aliquots of the cell extracts were immunoprecipitated (IP) with antibodies against the FLAG tag followed by Western blotting (WB) analysis of the samples with antibodies against the HA tag (B) or Myc tag (C--E). The top and bottom parts of each panel show Western blots of the total cell extracts (TCE) stained with antibodies against the HA- or Myc-tag (top) or C/EBPβ (bottom). The middle parts show Western blots of anti-FLAG immunoprecipitates stained with the indicated antibodies. Molecular weight markers are indicated to the left of each panel. The asterisks mark background bands caused by immunoglobulin heavy chains.
FIGURE 4.

Identification of Hipk2 phosphorylation sites of C/EBPβ. A, QT6 cells were transfected with expression vectors for C/EBPβ and wild-type Hipk2, as indicated. 24 h later total cell extracts were analyzed by Western blotting with antibodies against C/EBPβ (left). Aliquots of the same extracts were immunoprecipitated with C/EBPβ-specific antibodies. The immunoprecipitates were then subjected to an in vitro kinase assay and analyzed by SDS-PAGE and autoradiography. B, extracts from QT6 cells transfected with an expression vector for HA-tagged Hipk2 (Hipk2-wt), the Ha-tagged kinase-dead mutant of Hipk2 (Hipk2-KD), or from mock-transfected QT6 cells (control) were immunoprecipitated with antibodies against the HA tag. The immunoprecipitates were then subjected to an in vitro kinase assay together with bacterially expressed GST or GST-C/EBPβ fusion protein. Radiolabeled proteins were analyzed by SDS-PAGE and autoradiography. The right part of the figure shows a Coomassie Blue-stained gel of the bacterially expressed proteins. C, shown is a similar experiment as in B except that bacterially expressed GST-C/EBPβ (S65A) and GST-C/EBPβ (T220A) fusion proteins were used in addition to the wild-type GST-C/EBPβ in the in vitro kinase assay. Only the part of the gel containing the GST fusion proteins is shown. The left panel shows a Coomassie Blue-stained gel to confirm that equal amounts of the GST-C/EBPβ fusion proteins were used in the kinase assay. D, QT6 cells were transfected with C/EBPβ and HA-Hipk2 expression vectors, as indicated. Total cell extracts were analyzed by Western blotting with antibodies against the HA tag, phospho-C/EBPβ (Thr-235), and C/EBPβ. E, extracts of QT6 fibroblasts transfected with expression vectors for wild-type or the T220A mutant of C/EBPβ were analyzed by Western blotting with antisera against C/EBPβ (left panel) or against phospho-C/EBPβ (Thr-235) (right panel). F, PC3 cells were transfected with Hipk2-specific or control siRNA. Cells were harvested after 48 h, and total cell extracts were analyzed by Western blotting using antibodies against Hipk2 (top), phospho-C/EBPβ (Thr-235) (middle), and C/EBPβ (bottom). The two bands seen in the bottom panel correspond to the LAP* and LAP isoforms of C/EBPβ.
To map the part of C/EBPβ that is responsible for the interaction with Hipk2, we performed further co-immunoprecipitation experiments with mutant C/EBPβ constructs. We found that full-length C/EBPβ interacts with Hipk2 (Fig. 2A), whereas the deletion mutant ΔN21, which lacks the first 21 amino-terminal amino acid residues of C/EBPβ, did not bind to Hipk2. C/EBPβ-ΔN21 corresponds to the natural LAP isoform of C/EBPβ, indicating that only the longest of the natural C/EBPβ isoforms, LAP*, is able to interact with Hipk2. Human C/EBPβ was also able to bind to Hipk2, indicating that the interaction of both proteins is conserved (Fig. 2C).
FIGURE 2.

Mapping of the Hipk2 binding region in C/EBPβ. Wild-type and two mutant versions of C/EBPβ are illustrated schematically at the top. A, QT6 cells were transfected with the indicated expression vectors for chicken C/EBPβ and HA-tagged kinase-dead Hipk2. After 24 h the cells were harvested, and aliquots of the cell extracts were immunoprecipitated (IP) with antibodies against C/EBPβ followed by Western blotting (WB) of the immunoprecipitates with antibodies against the HA tag (middle panel). The top and bottom panels show Western blots of the total cell extracts (TCE) stained with antibodies against the HA-tag (top) or C/EBPβ (bottom). B, QT6 fibroblasts were transfected with the indicated expression vectors. After 24 h total cell extracts were analyzed by Western blotting with the indicated antibodies. The arrowhead marks unphosphorylated p300/1751–2370, which is only weakly visible in lane 1 of this exposure. C, QT6 fibroblasts transfected with expression vectors for Hipk2 and human C/EBPβ (hC/EBPβ) were analyzed as described in panel A. All lanes are derived from the same exposure of the same blot.
As explained in more detail below, the interaction of C/EBPβ and Hipk2 triggers the phosphorylation of p300 when bound to C/EBPβ, thereby leading to a mobility shift of p300 (33). Because LAP failed to interact with C/EBPβ, we wished to confirm that Hipk2 was also no longer able to induce phosphorylation of p300 bound to C/EBPβ-ΔN21. The Hipk2-induced phosphorylation takes place at sites clustered in the carboxyl-terminal part of p300 and can be detected by an electrophoretic mobility shift of the carboxyl-terminal domain of p300 when expressed together with C/EBPβ and Hipk2. As shown in Fig. 2B, Hipk2 was indeed unable to affect the mobility of p300 bound to C/EBPβ-ΔN21, consistent with the lack of binding to the amino-terminal-truncated C/EBPβ. Interestingly, however, C/EBPβ-ΔN21 was still able to induce a basal mobility shift of p300, suggesting that an additional, as yet unknown protein kinase is able to induce phosphorylation of C/EBPβ-bound p300. Fig. 2A, lane 3, also shows that mutation of the FYY motif located in a conserved patch of amino acids residues close to the amino terminus of C/EBPβ also weakens the binding of Hipk2, demonstrating that the integrity of the amino-terminal domain of C/EBPβ is essential for the interaction with Hipk2. The sequence around the FYY motif resembles the well known ΦXXΦΦ protein-protein interaction motif, supporting the idea that it plays a role in the C/EBPβ-Hipk2 interaction.
To investigate whether C/EBPβ and Hipk2 are direct physical binding partners, we expressed both proteins in bacteria to perform a pulldown experiment in the absence of other eukaryotic proteins. We immobilized a purified bacterial GST-Hipk2-(70–220) fusion protein on GST-Sepharose beads and incubated them with bacterially expressed and purified C/EBPβ. Fig. 3A shows that purified GST-Hipk2 fusion protein is capable of binding to C/EBPβ expressed in bacteria. A control binding reaction showed that GST on its own did not bind to C/EBPβ. We, therefore, concluded that C/EBPβ and Hipk2 interact directly in the absence of other eukaryotic proteins.
FIGURE 3.
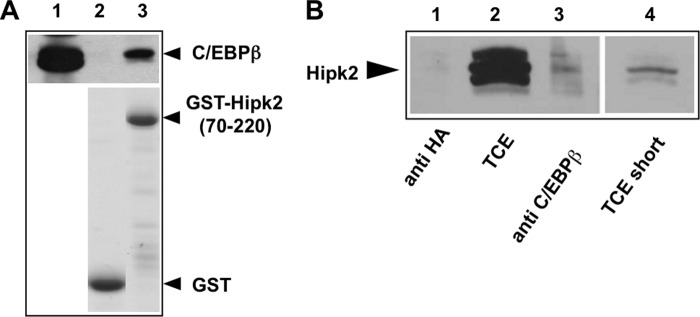
C/EBPβ and Hipk2 interact in untransfected cells and in the absence of other eukaryotic proteins. A, shown is an in vitro pulldown experiment. Similar amounts of bacterially expressed GST and GST-Hipk2-(70–220) fusion protein were bound to glutathione-Sepharose, incubated with bacterially expressed C/EBPβ, and subjected to a GST pulldown assay. Input (12% of the total amount, lane 1) and bound proteins (lanes 2 and 3) were analyzed by SDS-PAGE and Western blotting using C/EBPβ-specific antibodies. The bottom panel shows a Coomassie Blue-stained gel of the bacterial proteins to confirm equal loading of the Sepharose beads. B, total cell extract of untransfected PC3 cells was immunoprecipitated with a monoclonal antibody against human C/EBPβ (lane 3) or with the monoclonal anti-HA-antibody (lane 1). Lane 2 shows an aliquot (5%) of the total cell extract (TCE). Lane 4 shows a shorter exposure of lane 2. All samples were analyzed by Western blotting with polyclonal Hipk2-specific antiserum.
Finally, to confirm that the interaction of C/EBPβ and Hipk2 also occurs in untransfected cells that express both proteins at their endogenous levels, we performed co-immunoprecipitations with extracts of the human prostate cancer cell line PC3 (Fig. 3B). PC3 cells express detectable levels of Hipk2 and C/EBPβ. As shown in lane 3 of Fig. 3B, a small amount of endogenous Hipk2 was precipitated by antibodies against C/EBPβ but not by the control antibodies. Thus, this experiment showed that the interaction of C/EBPβ and Hipk2 also occurs in untransfected cells. Hipk2 is known to be highly modified by phosphorylation and acetylation; this might explain why the Hipk2-specific antiserum used in Fig. 3B detects several minor bands in addition to a major protein band.
Identification of Hipk2 Phosphorylation Sites of C/EBPβ
As a first step to investigate whether C/EBPβ is phosphorylated by Hipk2, we co-transfected fibroblasts with expression vectors for C/EBPβ and Hipk2. We then immunoprecipitated C/EBPβ from extracts of the transfected cells and subjected the immunoprecipitate to an in vitro kinase assay using γ-32P-labeled ATP. We reasoned that if Hipk2 remains bound to C/EBPβ during immunoprecipitation, it might phosphorylate C/EBPβ in the in vitro kinase reaction. Fig. 4A shows that the incorporation of radioactive phosphate into C/EBPβ was strongly increased when C/EBPβ was co-expressed with Hipk2, suggesting that C/EBPβ is a kinase substrate for Hipk2. C/EBPβ was also phosphorylated at a lower level when expressed in the absence of Hipk2; this phosphorylation might be due to the presence of endogenous Hipk2 or of other protein kinases known to phosphorylate C/EBPβ.
To substantiate that Hipk2 is able to phosphorylate C/EBPβ, we performed additional experiments in which we first immunoprecipitated Hipk2 from an extract of cells transfected with a HA-tagged version of Hipk2. We then incubated bacterially expressed GST-C/EBPβ with immunopurified Hipk2 in the presence of [γ-32P]ATP. As the control, we performed reactions with an equivalent amount of purified GST instead of GST-C/EBPβ. Fig. 4B shows that Hipk2 was able to phosphorylate GST-C/EBPβ but not GST. As additional controls, we immunopurified a kinase-dead mutant of Hipk2 from cells transfected with the corresponding expression vector or used extracts from untransfected cells for mock immunopurification of the kinase. In both cases C/EBPβ was phosphorylated at very low levels. Together, these data clearly demonstrate that Hipk2 is able to phosphorylate C/EBPβ in vitro.
A number of phosphorylation sites for Hipk2 have been identified in different target proteins, including p53, Pax6, Groucho, HMGA1a, and AML1 (36, 38–42). All of these sites contain serine or threonine residues followed by a proline. To identify the sites of C/EBPβ phosphorylated by Hipk2, we therefore concentrated on Ser-65 and Thr-220, two previously identified phosphorylation sites of C/EBPβ for proline-directed protein kinases. We mutated both sites and performed similar in vitro kinase experiments as illustrated in Fig. 4B. A representative result of these experiments is shown in Fig. 4C. In contrast to wild-type C/EBPβ, the mutations strongly reduced (in case of S65A) or virtually completely eliminated (in case of T220A) the phosphorylation by Hipk2. We, therefore, concluded that Ser-65 and Thr-220 are potential phosphorylation sites for Hipk2. To further demonstrate that Thr-220 is a target site for Hipk2, we made use of an antibody that is specific for phosphorylated Thr-235 of human C/EBPβ, which (due to differences in the number of amino acids between chicken and human C/EBPβ) corresponds to Thr-220 of the chicken protein. Fig. 4D clearly shows that Hipk2 increased the phosphorylation of Thr-220. The specificity of the antibody was confirmed using the T220A mutant of C/EBPβ (Fig. 4E). Unfortunately, phosphorylation-specific antibodies are not available for Ser-65. The evidence for Ser-65 being a Hipk2-dependent phosphorylation site is, therefore, less strong than in the case of Thr-220.
Finally, to confirm the phosphorylation of C/EBPβ by Hipk2 when both proteins are expressed at their endogenous levels, we performed a Hipk2 knockdown experiment. We again made use of the phospho-Thr-235-specific antibody. The top panel of Fig. 4F shows that the expression level of Hipk2 was decreased in PC3 cells treated with Hipk2-specific siRNA relative to the control siRNA-treated cells. Western blotting with the phospho-Thr-235-specific antiserum showed that the phosphorylation of this residue decreases when the expression of Hipk2 is reduced. By contrast, the expression of C/EBPβ itself was not affected by the Hipk2 knockdown. As pointed out above, C/EBPβ is expressed in several isoforms, giving rise to truncated forms of the protein. The two bands seen in the bottom panels of Fig. 4F correspond to the full-length (or LAP*) and the LAP isoforms, respectively. Interestingly, only the LAP* isoform is recognized by the phosphorylation-specific antiserum, indicating that the smaller form is not phosphorylated at Thr-235. The smaller isoform lacks the first 21 amino acids of full-length C/EBPβ and corresponds to the C/EBPβ-ΔN21 construct used in Fig. 2, which failed to bind to Hipk2. The lack of phosphorylation of the LAP-isoform in PC3 cells is, therefore, consistent with the inability of this isoform to interact with Hipk2.
Hipk2 Is Required for Adipocyte Differentiation of 3T3-L1 Cells
The differentiation of 3T3-L1 pre-adipocytes into adipocytes is an established model of a C/EBPβ-dependent differentiation process. In these cells, the initial activation of C/EBPβ induces a cascade of gene activations, including the transcription of the C/EBPα gene, ultimately leading to a fat cell phenotype that is characterized by the appearance of lipid droplets in the cytoplasm of the cells (8, 34, 43). To investigate if Hipk2 is involved in the differentiation of these cells, we made use of a stable 3T3-L1 Hipk2 knockdown clone generated previously (33). Fig. 5A shows that Hipk2 expression is decreased in the Hipk2 shRNA-expressing cells compared with the control cells. As shown in the upper two panels of Fig. 5A, the phosphorylation of Ser-2280 of p300, one of the sites of Hipk2-dependent phosphorylation, is diminished upon Hipk2 knockdown, thus confirming previous results (33). The phosphorylation of C/EBPβ was not significantly altered in the Hipk2 knockdown cells. This might be due to the activation of other protein kinases, such as MAPK and cyclin A/Cdk2, which are known to induce phosphorylation of the relevant threonine residue at an early stage during adipocyte differentiation (44, 45), overriding the effect of the Hipk2 knockdown. To address whether silencing of Hipk2 also disrupts the adipocyte differentiation program, we subjected the 3T3-L1 cells expressing Hipk2-specific shRNA as well as the control cells to the adipocyte differentiation protocol. Fig. 5B shows that the Hipk2 knockdown cells failed to differentiate, whereas the control cells readily converted into adipocytes, as demonstrated by the appearance of lipid droplets in the cytoplasm and staining with Oil Red O. This demonstrated that Hipk2 is required for adipocyte differentiation. Northern blot analysis (Fig. 5C) confirmed that the induction of C/EBPα mRNA as a marker of fat cell differentiation was strongly diminished in the Hipk2 knockdown cells but not in the control cells. Northern blot analysis also showed that Hipk2 mRNA increases during differentiation. Furthermore, it is evident that the overall level of Hipk2 mRNA is reduced in the knockdown cells compared with the control cells, confirming the efficiency of the knockdown of Hipk2 expression. Taken together, these experiments indicate that Hipk2 is required for a physiological C/EBPβ-dependent process.
FIGURE 5.

Hipk2 is required for adipocyte differentiation of 3T3-L1 cells. A, total cell extracts of the 3T3-L1 cells stably transfected with expression vector for HIPK2-specific shRNA (pSuper-HIPK2) or with the empty vector (pSuper) were analyzed by Western blotting before (− lanes) or after 2 days of differentiation (diff., + lanes) using the indicated antibodies. B, Hipk2 knockdown or control 3T3-L1 cells were induced to differentiate for 5 days. The top panels show photomicrographs, and the bottom panels show Oil Red O staining of the cells. C, polyadenylated RNA isolated from 3T3-L1 HIPK2 knock-out and control cells before differentiation (− lanes) or after 2 days of differentiation (+ lanes) were analyzed by Northern blotting using probes specific for Hipk2, C/EBPα, and GAPDH.
Mutation of Hipk2 Phosphorylation Sites of C/EBPβ Impairs the C/EBPβ-induced Phosphorylation of p300
Our previous work has shown that the coactivator p300 is phosphorylated at several sites in its carboxyl-terminal domain upon being recruited to C/EBPβ (32). Furthermore, we showed that Hipk2 is responsible for these phosphorylation events (33). Together with the new data presented here, these findings suggest a model in which Hipk2 docks to the amino terminus of C/EBPβ and phosphorylates C/EBPβ as well as p300. Work by Aikawa et al. (40) on the regulation of transcription factor AML1 by Hipk2 has revealed a similar scenario; Hipk2 was shown to bind to the AML1-p300 complex and to phosphorylate p300 as well as AML1. By mutating the Hipk2-specific phosphorylation sites of AML1, Aikawa et al. (40) showed that phosphorylation of AML1 was required for the phosphorylation of p300. We were, therefore, interested to know whether the phosphorylation of C/EBPβ is a prerequisite for the subsequent phosphorylation of p300, as in the case of AML1, or whether the phosphorylations of C/EBPβ and p300 by Hipk2 are independent events. As shown in Fig. 6A, the C/EBPβ-induced phosphorylation of p300 can easily be visualized by the appearance of a substantial mobility shift in SDS-polyacrylamide gels of the carboxyl-terminal domain of p300 (amino acids 1751–2370) when it is co-expressed with C/EBPβ. We showed before that this mobility shift is entirely due to phosphorylation, as it is reverted by phosphatase treatment (32). Phosphorylation of p300 can also be monitored with an antiserum specific for phosphorylated Ser-2280, one of the amino acids of p300 whose phosphorylation is increased in the presence of C/EBPβ (Fig. 6A, middle panel). The top panel of Fig. 6A shows that the presence of C/EBPβ also leads to a substantial increase in the amount of the phosphorylated p300. This increase is due to a prolonged half-life of the phosphorylated p300 (data not shown).
FIGURE 6.

Mutation of Hipk2 phosphorylation sites affects the C/EBPβ-dependent phosphorylation of p300. A--C, QT6 cells were transfected with the expression vectors indicated below the lanes. Total cell extracts were prepared 24 h after transfection and were analyzed by SDS-PAGE and Western blotting with antibodies against p300, phospho-p300-(Ser-2280), C/EBPβ, and the HA tag, as indicated. The unphosphorylated and phosphorylated forms of p300 are marked by white and black arrows, respectively. D, total cell extracts from HepG2 cells were incubated with Sepharose beads carrying GST or a GST-300-(1710–1891) fusion protein. Bound proteins were analyzed by SDS-PAGE and Western blotting with antibodies against human C/EBPβ (left panel) or phospho-C/EBPβ (Thr-235) (middle panel). Control lanes show aliquots of the total cell extract (Input lanes). The LAP* and LAP isoforms of C/EBPβ are marked. The right panel shows a Coomassie Blue-stained gel to confirm equal loading of the Sepharose beads with GST and the GST-p300 fusion protein. E, total cell extract from human HepG2 cells and extracts from cells transfected with an expression vector for human LAP* were analyzed by SDS-Page and Western blotting with antiserum against C/EBPβ. F, cell extracts from human HepG2 cells were incubated with (+) or without (−) Fast AP phosphatase (Fermentas) for 2 h at 37 °C. Extracts were then analyzed by Western blotting (WB) using antiserum against C/EBPβ (lower panel). As control for the efficiency of the phosphatase treatment, we analyzed the phosphorylation of B-Myb using a phospho-specific antiserum against B-Myb phosphorylated at Thr-487 (top).
To address if the phosphorylation of p300 by Hipk2 is influenced by the phosphorylation of C/EBPβ, we compared the ability of wild-type and a S65A/T220A mutant of C/EBPβ to trigger Hipk2-dependent phosphorylation of p300. Fig. 6B shows that Hipk2 strongly increased the mobility shift of p300 when co-expressed with wild-type C/EBPβ (compare lanes 2 and 3 of Fig. 6B), whereas the effect of Hipk2 was less strong in the presence of C/EBPβ mutated at Ser-65 and Thr-220 (compare lanes 3 and 5 of Fig. 6B). This was also evident when the phosphorylation of Ser-2280 was monitored with the phosphorylation-specific antiserum (middle panel of Fig. 6B). That mutation of the Hipk2 sites in C/EBPβ diminished the phosphorylation of p300 was even more obvious when only the endogenous kinase was present (Fig. 6C). Together, these data indicate that the phosphorylation of p300 in complex with C/EBPβ is stimulated by the phosphorylation of C/EBPβ itself.
To obtain insight into the mechanism by which phosphorylation of C/EBPβ stimulates the phosphorylation of p300, we investigated whether phosphorylation of C/EBPβ increases the recruitment of p300. We previously mapped the binding site for C/EBPβ to the E1A binding region of p300 (23, 32). To examine the effect of the phosphorylation of Thr-220 on the interaction between C/EBPβ and p300, we performed GST pulldown assays using a GST-p300 fusion protein containing amino acids 1710–1891 of p300. Total cell extract of HepG2 cells, which express C/EBPβ endogenously, was incubated with GST or GST-p300 immobilized on-Sepharose beads followed by analysis of the bound proteins by SDS-PAGE and Western blotting with antibodies against total C/EBPβ or against C/EBPβ phosphorylated at Thr-235 (corresponding to Thr-220 of chicken C/EBPβ). As shown in Fig. 6D, left panel, the C/EBPβ antibodies detected the LAP* and LAP isoforms of C/EBPβ. The binding of both of both isoforms was below the limit of detection under these exposure conditions. A longer exposure of the blot showed binding of a fraction of the proteins to GST-p300 (data not shown). Analysis of the same samples with antibodies against phosphorylated C/EBPβ, however, showed strong binding of the phosphorylated LAP* isoform to GST-p300 (Fig. 6D, middle panel). The absence of binding in the GST control lane showed that the strong binding of phosphorylated C/EBPβ to GST-p300 was specific. The LAP isoform was not detected with the phosphorylation-specific antiserum, indicating that it is not phosphorylated at detectable levels in HepG2 cells grown under normal conditions. Taken together, this experiment showed that the fraction of LAP* that is phosphorylated at Thr-235 (Thr-220 in the chicken protein) binds much better to p300 than the bulk of C/EBPβ, which appears to be not phosphorylated. To provide additional evidence that the slower-migrating isoform corresponds to LAP*, we compared its mobility to that of authentic human LAP* expressed from an expression vector. We observed that both proteins co-migrated on an SDS-polyacrylamide gel (Fig. 6E). Furthermore, we found that the mobility of the slower-migrating band was not increased to that of the LAP-isoform upon phosphatase treatment, indicating that the slower-migrating isoform is not simply a hyperphosphorylated form of LAP (Fig. 6F).
Hipk2 Cooperates with C/EBPβ but Not with the Related Transcription Factor C/EBPα
The results presented in Fig. 2 have shown that amino-terminal 21 amino acids of C/EBPβ that are specific for the LAP* isoform are essential for the interaction with Hipk2. As illustrated at the top of Fig. 7, the amino-terminal domain of the transcription factor C/EBPα differs strikingly from that of C/EBPβ; in particular, the first 21 LAP*-specific amino acids of C/EBPβ are missing completely in C/EBPα. Because of this difference we wondered whether or not C/EBPα is able to cooperate with Hipk2. To address this question we first investigated whether Hipk2 and C/EBPα are able to interact with each other. For convenience, we used the kinase-dead version of Hipk2 for this experiment. In contrast to wild-type Hipk2, which is autophosphorylated and tends to run as a somewhat fuzzy band on SDS-polyacrylamide gels, the kinase-dead version migrates as a sharp band and is, therefore, easier to detect in co-immunoprecipitation experiments. Fig. 7 shows that Hipk2 was not co-precipitated via C/EBPα, whereas binding of Hipk2 to C/EBPβ was clearly detectable under the same conditions. This suggested that C/EBPα might not be able to cooperate with Hipk2.
FIGURE 7.
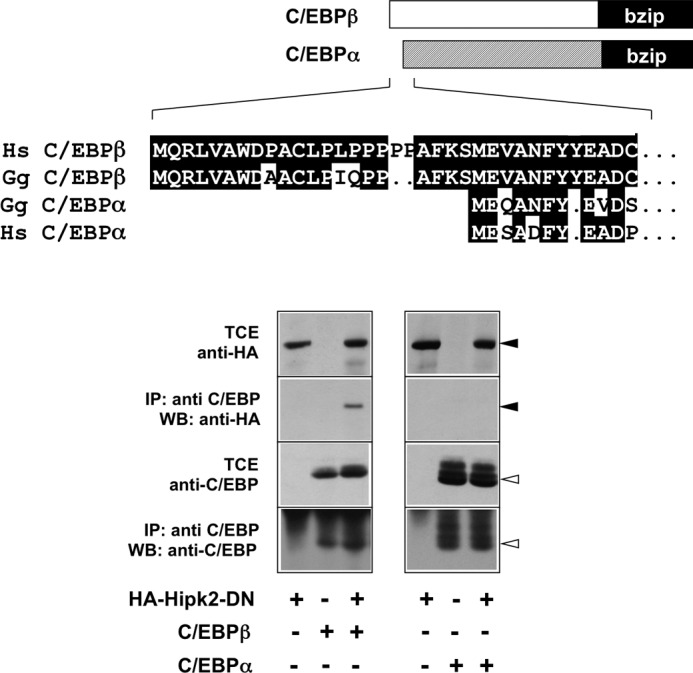
Co-immunoprecipitation of Hipk2 with C/EBPβ and C/EBPα. Upper panel, shown is a comparison of the amino-terminal amino acid sequences of human (Hs) and chicken (Gg) C/EBPβ and C/EBPα. Lower panel, shown is a co-immunoprecipitation experiment. QT6 cells were transfected with expression vectors for vectors for HA-tagged kinase-dead Hipk2, C/EBPα, and C/EBPβ, as indicated at the bottom. After 24 h the cells were harvested, and aliquots of the total cell extracts (TCE) were immunoprecipitated (IP) with antibodies against C/EBPβ or C/EBPα followed by Western blotting (WB) of the immunoprecipitate samples with antibodies against the HA tag or against the C/EBP proteins. The black and white arrowheads mark Hipk2 and the C/EBP proteins, respectively.
We previously showed that C/EBPα induces a similar phosphorylation-dependent mobility shift of p300 as C/EBPβ (32). If Hipk2 does not bind to C/EBPα, we would expect the C/EBPα-induced phosphorylation of p300 not to be influenced by Hipk2. We, therefore, tested whether expression of active or kinase-dead versions of Hipk2 affects the C/EBPα-induced phosphorylation of p300. Co-transfection of expression vectors for the carboxyl-terminal domain of p300 and C/EBPα confirmed that C/EBPα also triggers the phosphorylation of p300, as shown by the mobility shift of p300 in the presence of C/EBPα (lanes 1 and 2 of Fig. 8A). Co-expression of active (lane 3) or kinase-dead (lane 4) Hipk2 did not significantly influence the C/EBPα-induced mobility shift of p300. By contrast, the mobility shift induced by C/EBPβ was strongly augmented by active Hipk2 and diminished by kinase-dead Hipk2. These results are consistent with earlier work by Aikawa et al. (40) who have suggested that the phosphorylation of p300 induced by C/EBPα is independent of Hipk2. In an additional experiment we analyzed the effect of the kinase-dead version of Hipk2 on the ability of C/EBPα and C/EBPβ to stimulate the transcription of the Mrp126 and the goose-type lysozyme gene #325, two endogenous C/EBP target genes whose expression is often used to monitor the activity of C/EBP transcription factors. Fig. 8B shows that transcription of Mrp126 and #325 mRNAs was induced by C/EBPα as well as C/EBPβ (lanes 1 and 5). Kinase-dead Hipk2 clearly inhibited the C/EBPβ-dependent transcription of the Mrp126 and #325 genes, whereas the activity of C/EBPα was not affected. In addition, we assessed the effect of the kinase-dead Hipk2 on the activity of C/EBPα and C/EBPβ when both were expressed together with p300. These experiments showed that the activity of both C/EBPs was strongly augmented by p300, confirming that they both cooperate with p300 (Fig. 8C). However, expression of kinase-dead Hipk2 only diminished the activity of C/EBPβ-p300, whereas the activity of C/EBPα-p300 was not affected (Fig. 8, D and E). Taken together, the data shown in Figs. 7 and 8 strongly suggest that Hipk2 regulates the activity of C/EBPβ but not that of C/EBPα via p300.
FIGURE 8.

Influence of Hipk2 on the C/EBPα- and C/EBPβ-induced phosphorylation of p300 and the activation of an endogenous C/EBP target gene. A, QT6 cells were transfected with the expression vectors indicated at the bottom. Cells were harvested after 24 h, and total cells extracts were analyzed by Western blotting using antibodies against the HA tag, p300, phospho- p300-(Ser-2280), or the C/EBP proteins. Because of autophosphorylation, wild-type Hipk2 migrates slower than kinase-dead Hipk2. The unphosphorylated and highly phosphorylated forms of p300-(1751–2370) are marked by white and black arrowheads, respectively. B, QT6 cells were transfected with the expression vectors indicated at the bottom and harvested 24 h after transfection. Polyadenylated RNA isolated from the cells was analyzed by Northern blotting for the expression of MRP126, the goose-type lysozyme gene #325 and S 17 mRNAs (upper panels). Additionally, cells were subjected to Western blotting using antibodies specific for Hipk2 and the C/EBP proteins (lower panels). C--E, QT6 fibroblasts were transfected with expression vectors for C/EBPα, C/EBPβ, p300, and kinase-dead Hipk2, as indicated at the bottom. Polyadenylated RNA isolated from the cells was analyzed by Northern blotting for the expression of MRP126 and S 17 mRNAs.
Interestingly, although C/EBPα does not appear to bind or cooperate with Hipk2, it nevertheless is able to induce the phosphorylation of p300 (compare lanes 1 and 2 of Fig. 8A). This suggests that instead of Hipk2, a different protein kinase must be involved in the C/EBPα-induced phosphorylation of p300. Previously, it has been shown that C/EBPα interacts with Jnk1 (46); we, therefore, tested the effect of the Jnk inhibitor SP600125 on the C/EBPα- and C/EBPβ-induced phosphorylation of p300. Fig. 9A shows that SP600125 significantly inhibited the C/EBPα-induced mobility shift of p300, whereas the mobility shift induced by C/EBPβ was virtually not affected. The p38 inhibitor SB203580 used in parallel had no effect on the C/EBPα- or C/EBPβ-induced phosphorylation of p300. It has been reported previously that ERK1/2 binds to and phosphorylates C/EBPα through an FXFP docking motif in the amino-terminal region of C/EBPα (47). The ERK phosphorylation site is not conserved in chicken C/EBPα; when we checked the effect of the MEK1 inhibitor U0126, no inhibition of the C/EBPα-induced phosphorylation of p300 was observed (Fig. 9A), indicating that ERK1/2 are not involved. To substantiate the notion that Jnk is involved in the C/EBPα-induced phosphorylation of p300, we examined the influence of co-expression of a kinase-dead Jnk mutant on the phosphorylation of p300 (Fig. 9B). This showed that the C/EBPα-induced phosphorylation of p300 was diminished by the kinase-dead Jnk, whereas the phosphorylation induced by C/EBPβ remained unaffected. Taken together, these observations suggest that the C/EBPα-induced phosphorylation of p300 is mediated by Jnk kinase. Fig. 9, C and D, shows hypothetical models of how C/EBPβ and C/EBPα cooperate with different protein kinases to induce the phosphorylation of p300.
FIGURE 9.

Effect of kinase inhibitors on the C/EBPα- and C/EBPβ-induced phosphorylation of p300. A and B, QT6 cells were transfected as indicated at the bottom. Cells in panel A were additionally treated with the Jnk inhibitor SP600125 (40 μm), the p38 inhibitor SB203580 (15 μm), or the Mek1 inhibitor U0126 (10 μm) overnight before they were harvested. In panel B, Jnk-apf refers to a kinase-dead mutant of Jnk. Total cell extracts were analyzed after 24 h by Western blotting with antibodies against the p300 and the C/EBP proteins. The unphosphorylated and highly phosphorylated forms of p300-(1751–2370) are marked by white and black arrowheads, respectively. C, shown is a hypothetical model of the C/EBPβ-induced phosphorylation of p300. Hipk2 binds to the amino-terminal domain of C/EBPβ and phosphorylates C/EBPβ as well as p300 bound to C/EBPβ. Phosphorylation of C/EBPβ stimulates binding of p300; however, it is not known whether the phosphorylation affects the binding of p300 directly (as shown arbitrarily in this model) or indirectly, i.e. by inducing conformational changes in C/EBPβ. D, shown is a hypothetical model of the C/EBPα-induced phosphorylation of p300. C/EBPα triggers the phosphorylation of p300 by Jnk kinase. It is not known if Jnk binds directly to a specific domain of C/EBPα (as arbitrarily shown here) and whether Jnk also phosphorylates C/EBPα.
DISCUSSION
The work presented here identifies protein kinase Hipk2 as a novel regulator of transcription factor C/EBPβ. We have shown that Hipk2 interacts with C/EBPβ in cells transfected with expression vectors for C/EBPβ and Hipk2 as well as in untransfected cells expressing these proteins at their endogenous levels. We have also demonstrated the interaction of C/EBPβ and Hipk2 when both proteins are expressed in bacteria (i.e. in the absence of other eukaryotic proteins), indicating that they are direct physical interaction partners. Furthermore, we have shown that Hipk2 induces the phosphorylation of C/EBPβ at sites whose phosphorylation has previously been demonstrated to play important roles in the regulation of C/EBPβ activity. Together, these data provide strong evidence that Hipk2 is an important regulator of the activity of C/EBPβ. In agreement with this, knock-down experiments have shown that decreased expression of Hipk2 diminishes the adipocytic differentiation of 3T3-L1 cells, an established differentiation model that is crucially dependent on the activity of C/EBPβ (4, 8, 34, 43).
In vitro kinase assays with immunopurified Hipk2 indicate that Hipk2 phosphorylates Thr-220 and possibly also Ser-65 of C/EBPβ. Both sites are target sites for other protein kinases as well and have been strongly implicated in regulating the activity of C/EBPβ. Ser-65 was identified as a site for a cyclin-dependent kinase whose phosphorylation is required for the ability of C/EBPβ to promote transformation of 3T3 fibroblasts by oncogenic ras (48). Thr-235 in human C/EBPβ and Thr-188 in rat C/EBPβ (which correspond to Thr-220 of chicken C/EBPβ) are also phosphorylated by the Ras/MAP kinase signaling pathway (16, 24) as well as by cyclin-dependent protein kinase (45). Phosphorylation of Thr-220 appears to play a key role in controlling the activity of C/EBPβ. It was shown that phosphorylation of Thr-220 results in conformational changes of C/EBPβ that lead to the derepression of the transactivation potential of the protein (24). Phosphorylation of Thr-220 was already shown to be required for adipocyte differentiation (44, 45), a finding that is consistent with our observation that diminished Hipk2 expression hampers adipocyte differentiation. Recent work has shown that phosphorylation of Thr-220 triggers a cascade of events involving abrogation of the binding of the lysine methylase G9a and arginine methyl transferase PRMT4, thereby altering the pattern of covalent post-translational modifications, in particular the PRMT4-dependent methylation of arginine 3 of C/EBPβ. This, in turn, facilitates the recruitment of SWI/SNF chromatin remodeling and Mediator complexes by C/EBPβ to ultimately increase the transcription of C/EBPβ target genes (26, 27, 49). Our work suggests increased binding and phosphorylation of p300 as an alternative or additional mechanism by which the phosphorylation of C/EBPβ at Thr-220 stimulates the transactivation potential of C/EBPβ. Furthermore, our work suggests that Hipk2-induced phosphorylation provides an additional pathway, alternative to Ras/MAPK-signaling, for derepressing and activating C/EBPβ.
The LAP* and LAP isoforms of C/EBPβ differ only by the first 21 amino-terminal amino acids, which are essential for the binding of Hipk2. Our finding that Hipk2 does not bind to the LAP isoform suggests that Hipk2 is involved in isoform-specific regulation of C/EBPβ activity. Previous work has shown that the LAP isoform can be activated by various signals, including growth factors and expression of oncogenic Ras. Because we have analyzed the interaction with Hipk2 only in untreated cells, we cannot exclude the possibility that Hipk2 and LAP can be induced to bind under specific circumstances. The significance of the functional differences between the LAP* and LAP isoforms of C/EBPβ is not fully understood at present. Both forms show different expression profiles under certain circumstances, for example in normal versus tumorigenic mammary epithelial cells, and have been implicated in mammary tumorigenesis (22, 29). Functional differences between the LAP* and LAP isoforms have been implicated in senescence (50) and the differentiation of specific cell types (51). On the molecular level, LAP* but not LAP interacts with the SWI/SNF chromatin remodeling proteins and the mediator complex (49, 52), explaining differential effects of the isoforms on the expression of specific target genes, for example in myeloid cells. Another interesting difference between the isoforms is the ability of LAP* but not of LAP to be sumoylated (22). The identification of Hipk2 as a LAP*-specific interaction partner adds a novel aspect to the functional differences of the C/EBPβ isoforms.
In addition to phosphorylating C/EBPβ, Hipk2 also plays a role in the cooperation of C/EBPβ and p300. As we showed before, the recruitment of p300 by C/EBPβ triggers the phosphorylation of p300 and Hipk2 has been identified as a kinase that mediates these phosphorylations (32, 33). Previous work showed that the phosphorylation of p300 increases its activity as a coactivator (32), presumably by stimulating the acetyl transferase activity of p300, as shown by Aikawa et al. (40). Our data show that the role of C/EBPβ is more complex than that of a simple scaffold that brings Hipk2 and p300 into close proximity and thereby enables Hipk2 to efficiently phosphorylate p300 recruited to C/EBPβ. We showed that the mutation of the Hipk2 phosphorylation sites of C/EBPβ hamper the phosphorylation of p300, suggesting that the phosphorylation of p300 is facilitated by the phosphorylation of C/EBPβ. As a possible explanation for this observation, the pulldown experiment in Fig. 6 showed that phosphorylation of C/EBPβ stimulates the binding of p300. Hence, phosphorylation of C/EBPβ might lead to increased recruitment of p300 and its subsequent phosphorylation by Hipk2. It is interesting to note that phosphorylation events similar to those described here for C/EBPβ have been shown to occur upon interaction of p300 with AML1 (40). AML1 recruits Hipk2, which then phosphorylates AML1 itself as well as the associated p300. As in the case of C/EBPβ, mutation of the Hipk2 phosphorylation sites of AML1 decreases the phosphorylation of p300.
As a final aspect our work has provided new insight into the cooperation of C/EBPα and 300. The activity of C/EBPβ and C/EBPα is stimulated by p300 (23, 53), and both C/EBPs induce the phosphorylation of the carboxyl-terminal domain of p300 (23, 32). Despite these similarities, our data show that C/EBPβ and C/EBPα differ strikingly in how they trigger the phosphorylation of p300. We found that C/EBPα does not interact with Hipk2, an observation that clearly underlines the role of the amino-terminal LAP*-specific amino acid sequences for the interaction with Hipk2. Our data show that the phosphorylation of p300, which is induced by C/EBPα, is not mediated by Hipk2. Instead, C/EBPα appears to employ Jnk kinase to induce the phosphorylation of p300. Thus, our data reveal an additional level of complexity in the cooperation of two related C/EBP family members with p300.
Acknowledgments
We thank B. Berkenfeld for excellent technical assistance, A. Plachetka for initial cloning of phosphorylation-site mutants, M. Denningmann for performing initial experiments on C/EBPα, and T. Hofmann, I. Kitabayashi, L. Schmitz, and G. Steger for plasmids and antibodies.
This work was supported by Deutsche Forschungsgemeinschaft Grant KL461/12-1.
- C/EBP
- CCAAT box/enhancer-binding protein
- Hipk2
- homeodomain-interacting kinase 2.
REFERENCES
- 1. Cao Z., Umek R. M., McKnight S. L. (1991) Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 5, 1538–1552 [DOI] [PubMed] [Google Scholar]
- 2. Lin F.-T., Lane M. D. (1992) Antisense CCAAT/enhancer-binding protein RNA suppresses coordinate gene expression and triglyceride accumulation during differentiation of 3T3-L1 preadipocytes. Genes Dev. 6, 533–544 [DOI] [PubMed] [Google Scholar]
- 3. Müller C., Kowenz-Leutz E., Grieser-Ade S., Graf T., Leutz A. (1995) NF-M (chicken C/EBPβ) induces eosinophilic differentiation and apoptosis in a hematopoietic progenitor cell line. EMBO J. 14, 6127–6235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yeh W.-C., Cao Z., Classon M., McKnight S. L. (1995) Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 9, 168–181 [DOI] [PubMed] [Google Scholar]
- 5. Zhang D. E., Zhang P., Wang N. D., Hetherington C. J., Darlington G. J., Tenen D. G. (1997) Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer-binding protein α-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 94, 569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pedersen T. A., Kowenz-Leutz E., Leutz A., Nerlov C. (2001) Cooperation between C/EBPα, TBP/TFIIB, and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev. 15, 3208–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramji D. P., Foka P. (2002) CCAAT/enhancer-binding proteins. Structure, function, and regulation. Biochem. J. 365, 561–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang Q. Q., Otto T. C., Lane M. D. (2003) CCAAT/enhancer-binding protein β is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 850–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nerlov C. (2007) The C/EBP family of transcription factors. A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 17, 318–324 [DOI] [PubMed] [Google Scholar]
- 10. Descombes P., Schibler U. (1991) A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67, 569–579 [DOI] [PubMed] [Google Scholar]
- 11. Calkhoven C. F., Müller C., Leutz A. (2000) Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev. 14, 1920–1932 [PMC free article] [PubMed] [Google Scholar]
- 12. Ossipow V., Descombes P., Schibler U. (1993) CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc. Natl. Acad. Sci. U.S.A. 90, 8219–8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raught B., Gingras A. C., James A., Medina D., Sonenberg N., Rosen J. M. (1996) Expression of a translationally regulated, dominant-negative CCAAT/enhancer-binding protein β isoform and up-regulation of the eukaryotic translation initiation factor 2α are correlated with neoplastic transformation of mammary epithelial cells. Cancer Res. 56, 4382–4386 [PubMed] [Google Scholar]
- 14. Lincoln A. J., Monczak Y., Williams S. C., Johnson P. F. (1998) Inhibition of CCAAT/enhancer-binding protein α and β translation by upstream open reading frames. J. Biol. Chem. 273, 9552–9560 [DOI] [PubMed] [Google Scholar]
- 15. Wethmar K., Bégay V., Smink J. J., Zaragoza K., Wiesenthal V., Dörken B., Calkhoven C. F., Leutz A. (2010) C/EBPβδ uORF Mice. A genetic model for uORF-mediated translational control in mammals. Genes Dev. 24, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakajima T., Kinoshita S., Sasagawa T., Sasaki K., Naruto M., Kishimoto T., Akira S. (1993) Phosphorylation at threonine 235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc. Natl. Acad. Sci. U.S.A. 90, 2207–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trautwein C., Caelles C., van der Geer P., Hunter T., Karin M., Chojkier M. (1993) Transactivation by NF-IL6/LAP is enhanced by phosphorylation of its activation domain. Nature 364, 544–547 [DOI] [PubMed] [Google Scholar]
- 18. Buck M., Poli V., van der Geer P., Chojkier M., Hunter T. (1999) Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBP β is required for hepatocyte proliferation induced by TGFα. Mol. Cell 4, 1087–1092 [DOI] [PubMed] [Google Scholar]
- 19. Ceseña T. I., Cardinaux J. R., Kwok R., Schwartz J. (2007) CCAAT/enhancer-binding protein (C/EBP) β is acetylated at multiple lysines. Acetylation of C/EBPβ at lysine 39 modulates its ability to activate transcription. J. Biol. Chem. 282, 956–967 [DOI] [PubMed] [Google Scholar]
- 20. Joo M., Park G. Y., Wright J. G., Blackwell T. S., Atchison M. L., Christman J. W. (2004) Transcriptional regulation of the cyclooxygenase-2 gene in macrophages by PU. 1. J. Biol. Chem. 279, 6658–6665 [DOI] [PubMed] [Google Scholar]
- 21. Xu M., Nie L., Kim S. H., Sun X. H. (2003) STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPβ. EMBO J. 22, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eaton E. M., Sealy L. (2003) Modification of CCAAT/enhancer-binding protein-β by the small ubiquitin-like modifier (SUMO) family members, SUMO-2 and SUMO-3. J. Biol. Chem. 278, 33416–33421 [DOI] [PubMed] [Google Scholar]
- 23. Mink S., Haenig B., Klempnauer K.-H. (1997) Interaction and functional collaboration of p300 and C/EBPβ. Mol. Cell. Biol. 17, 6609–6617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kowenz-Leutz E., Twamley G., Ansieau S., Leutz A. (1994) Novel mechanism of C/EBPβ (NF-M) transcriptional control. Activation through derepression. Genes Dev. 8, 2781–2791 [DOI] [PubMed] [Google Scholar]
- 25. Johnson P. F. (2005) Molecular stop signs. Regulation of cell-cycle arrest by C/EBP transcription factors. J. Cell Sci. 118, 2545–2555 [DOI] [PubMed] [Google Scholar]
- 26. Pless O., Kowenz-Leutz E., Knoblich M., Lausen J., Beyermann M., Walsh M. J., Leutz A. (2008) G9a-mediated lysine methylation alters the function of CCAAT/enhancer-binding protein-β. J. Biol. Chem. 283, 26357–26363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kowenz-Leutz E., Pless O., Dittmar G., Knoblich M., Leutz A. (2010) Crosstalk between C/EBPβ phosphorylation, arginine methylation, and SWI/SNF/Mediator implies an indexing transcription factor code. EMBO J. 29, 1105–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zahnow C. A., Cardiff R. D., Laucirica R., Medina D., Rosen J. M. (2001) A role for CCAAT/enhancer-binding protein β-liver-enriched inhibitory protein in mammary epithelial cell proliferation. Cancer Res. 61, 261–269 [PubMed] [Google Scholar]
- 29. Bundy L. M., Sealy L. (2003) CCAAT/enhancer-binding protein β (C/EBPβ)-2 transforms normal mammary epithelial cells and induces epithelial to mesenchymal transition in culture. Oncogene 22, 869–883 [DOI] [PubMed] [Google Scholar]
- 30. Nerlov C. (2004) C/EBPα mutations in acute myeloid leukemias. Nat. Rev. Cancer 4, 394–400 [DOI] [PubMed] [Google Scholar]
- 31. Gomis R. R., Alarcón C., Nadal C., Van Poznak C., Massagué J. (2006) C/EBPβ at the core of the TGFβ cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 10, 203–214 [DOI] [PubMed] [Google Scholar]
- 32. Schwartz C., Beck K., Mink S., Schmolke M., Budde B., Wenning D., Klempnauer K.-H. (2003) Recruitment of p300 by C/EBPβ triggers phosphorylation of p300 and modulates coactivator activity. EMBO J. 22, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steinmann S., Schulte K., Beck K., Chachra S., Bujnicki T., Klempnauer K.-H. (2009) v-Myc inhibits C/EBPβ activity by preventing C/EBPβ-induced phosphorylation of the co-activator p300. Oncogene 28, 2446–2455 [DOI] [PubMed] [Google Scholar]
- 34. Lane M. D., Tang Q. Q., Jiang M. S. (1999) Role of the CCAAT enhancer-binding proteins (C/EBPs) in adipocyte differentiation. Biochem. Biophys. Res. Commun. 266, 677–683 [DOI] [PubMed] [Google Scholar]
- 35. Plachetka A., Chayka O., Wilczek C., Melnik S., Bonifer C., Klempnauer K.-H. (2008) C/EBPβ induces chromatin opening at a cell-type-specific enhancer. Mol. Cell. Biol. 28, 2102–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hofmann T. G., Möller A., Sirma H., Zentgraf H., Taya Y., Dröge W., Will H., Schmitz M. L. (2002) Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4, 1–10 [DOI] [PubMed] [Google Scholar]
- 37. Schubert S., Horstmann S., Bartusel T., Klempnauer K.-H. (2004) The cooperation of B-Myb with the coactivator p300 is orchestrated by cyclins A and D1. Oncogene 23, 1392–1404 [DOI] [PubMed] [Google Scholar]
- 38. D'Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G., Piaggio G., Fanciulli M., Appella E., Soddu S. (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser-46 and mediates apoptosis. Nat. Cell Biol. 4, 11–19 [DOI] [PubMed] [Google Scholar]
- 39. Choi C. Y., Kim Y. H., Kim Y. O., Park S. J., Kim E. A., Riemenschneider W., Gajewski K., Schulz R. A., Kim Y. (2005) Phosphorylation by the DHIPK2 protein kinase modulates the corepressor activity of Groucho. J. Biol. Chem. 280, 21427–22136 [DOI] [PubMed] [Google Scholar]
- 40. Aikawa Y., Nguyen L. A., Isono K., Takakura N., Tagata Y., Schmitz M. L., Koseki H., Kitabayashi I. (2006) Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 25, 3955–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim E. A., Noh Y. T., Ryu M. J., Kim H. T., Lee S. E., Kim C. H., Lee C., Kim Y. H., Choi C. Y. (2006) Phosphorylation and transactivation of Pax6 by homeodomain-interacting protein kinase 2. J. Biol. Chem. 281, 7489–7497 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Q., Wang Y. (2007) Homeodomain-interacting protein kinase-2 (HIPK2) phosphorylates HMGA1a at Ser-35, Thr-52, and Thr-77 and modulates its DNA binding affinity. J. Proteome Res. 6, 4711–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otto T. C., Lane M. D. (2005) Adipose development. From stem cell to adipocyte. Crit. Rev. Biochem. Mol. Biol. 40, 229–242 [DOI] [PubMed] [Google Scholar]
- 44. Tang Q. Q., Grønborg M., Huang H., Kim J.-W., Otto T. C., Pandey A., Lane M. D. (2005) Sequential phosphorylation of CCAAT enhancer-binding protein β by MAPK and glycogen synthase kinase 3b is required for adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 9766–9771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li X., Kim J. W., Grønborg M., Urlaub H., Lane M. D., Tang Q. Q. (2007) Role of cdk2 in the sequential phosphorylation/activation of C/EBPβ during adipocyte differentiation. Proc. Natl. Acad. Sci. U.S.A. 104, 11597–11602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Trivedi A. K., Bararia D., Christopeit M., Peerzada A. A., Singh S. M., Kieser A., Hiddemann W., Behre H. M., Behre G. (2007) Proteomic identification of C/EBP-DBD multiprotein complex. JNK1 activates stem cell regulator C/EBPα by inhibiting its ubiquitination. Oncogene 26, 1789–1801 [DOI] [PubMed] [Google Scholar]
- 47. Ross S. E., Radomska H. S., Wu B., Zhang P., Winnay J. N., Bajnok L., Wright W. S., Schaufele F., Tenen D. G., MacDougald O. A. (2004) Phosphorylation of C/EBPα inhibits granulopoiesis. Mol. Cell. Biol. 24, 675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shuman J. D., Sebastian T., Kaldis P., Copeland T. D., Zhu S., Smart R. C., Johnson P. F. (2004) Cell cycle-dependent phosphorylation of C/EBPβ mediates oncogenic cooperativity between C/EBPβ and H-RasV12. Mol. Cell. Biol. 24, 7380–7391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mo X., Kowenz-Leutz E., Xu H., Leutz A. (2004) Ras induces mediator complex exchange on C/EBP β. Mol. Cell 13, 241–250 [DOI] [PubMed] [Google Scholar]
- 50. Atwood A. A., Sealy L. (2010) Regulation of C/EBPβ1 by Ras in mammary epithelial cells and the role of C/EBPβ1 in oncogene-induced senescence. Oncogene 29, 6004–6015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smink J. J., Leutz A. (2012) Instruction of mesenchymal cell fate by the transcription factor C/EBPβ. Gene 497, 10–17 [DOI] [PubMed] [Google Scholar]
- 52. Kowenz-Leutz E., Leutz A. (1999) A C/EBP β isoform recruits the SWI/SNF complex to activate myeloid genes. Mol. Cell 4, 735–743 [DOI] [PubMed] [Google Scholar]
- 53. Erickson R. L., Hemati N., Ross S. E., MacDougald O. A. (2001) p300 coactivates the adipogenic transcription factor CCAAT/enhancer-binding protein α. J. Biol. Chem. 276, 16348–16355 [DOI] [PubMed] [Google Scholar]