

Background: HP1a (Heterochromatin Protein 1a) discriminates among its partners, binding HP2 66-fold more tightly than PIWI.
Results: Leu-165 on the HP1a binding surface provides a contact site for HP2 that PIWI does not utilize.
Conclusion: The Leu-165 contact is critical for HP1a's partner discrimination.
Significance: Learning how HP1a selects its partners will help elucidate the function of HP1a in epigenetics.
Keywords: Amino Acid, Heterochromatin, Mutagenesis in vitro, Peptides, Protein-Protein Interactions, HP1, PIWI, Fluorescence Polarization
Abstract
Drosophila melanogaster Heterochromatin Protein 1a (HP1a) is an essential protein critical for heterochromatin assembly and regulation. Its chromo shadow domain (CSD) homodimerizes, a requirement for binding protein partners that contain a PXVXL motif. How does HP1a select among its many different PXVXL-containing partners? HP1a binds tightly to Heterochromatin Protein 2 (HP2), but weakly to PIWI. We investigated differences in homodimerization and the impact of the C-terminal extension (CTE) by contrasting HP1a to its paralogue, HP1b. HP1a and HP1b differ in the dimerization interface, with HP1a having an Arg at position 188 rather than Glu. We find that while this substitution reduces the dimerization constant, it does not impact the binding surface as demonstrated by unchanged partner binding affinities. However, the CTE (only 4 residues in HP1a as compared with 87 residues in HP1b) is critical; the charged residues in HP1a are necessary for tight peptide binding. Examining a panel of amino acid substitutions in the HP1a CSD, we find that Leu-165 in HP1a interacts with HP2 but not PIWI, supporting the conclusion that different sites in the binding surface provide discrimination for partner selection. Partner sequence is also critical for affinity, as the remaining difference in binding between HP2 and PIWI polypeptides is eliminated by swapping the PXVXL motifs between the two. Taken together, these studies indicate that the binding surface of the HP1a CSD plus its short CTE provide the needed discrimination among HP1a's partners, and that the CTE is important for differentiating the interactions of the Drosophila HP1 paralogs.
Introduction
Heterochromatin Protein 1 (HP1a)2 was first discovered in Drosophila melanogaster as a protein that localizes to the chromocenter, telomeres, and the fourth chromosome, all heterochromatic domains, as shown by immunofluorescent staining of polytene chromosomes (1, 2). Subsequent genetic analysis demonstrated that HP1a plays a critical role in heterochromatin formation, acting as a suppressor of variegation (showing loss of silencing of a heterochromatic reporter) when mutant (3). Four additional paralogs of HP1a are found in D. melanogaster: HP1b, HP1c, HP1d/Rhino, and HP1e (4). These proteins share an N-terminal chromodomain (CD) followed by a variable length hinge, a chromo shadow domain (CSD), and a variable length C-terminal extension (CTE). They do not, however, share target sites in the genome, as shown by their localization patterns (5–7). HP1a, HP1b, and HP1c all function in the somatic nucleus, while HP1d and HP1e are limited to the germline (4). Cytological analyses show that while HP1a is located primarily in heterochromatic domains, HP1c is primarily associated with euchromatic domains, and HP1b overlaps both (6). These distinctive distributions have been confirmed and mapped at higher resolution by ChIP-chip analysis (7). Thus while the recognizable domains, CD and CSD, are conserved, and the genes are all expressed in somatic cells, the HP1a, HP1b, and HP1c paralogs all have unique distributions and functions in the genome.
The chromo shadow domains of HP1a and HP1b share 58% sequence identity, whereas those of HP1a and HP1c share only 36% identity (8). All three proteins are predicted to dimerize (9). The chromo shadow domain, which can function independently of the chromo domain (10), plays a role in determining the genomic distribution patterns of the HP1 (6). To understand better what differences in the chromo shadow domain impart the differential roles of these proteins, we have compared the dimerization and partner binding properties of HP1a with those of HP1b.
CSD function is regulated not only by amino acid sequence, but also through its concentration. Concentration will determine the quantity of CSD dimers, and hence the quantity of partner protein binding sites, present in a certain location. HP1a is estimated to be present at micromolar concentrations in the nucleus (11). It dimerizes with a dissociation constant of 3.0 ± 0.2 μm (12). A phosphorylation mimic of the HP1a CSD, created by substituting glutamic acid for serine at residues 199 and 202, sites that appear constitutively phosphorylated (13), has an even weaker dimerization affinity (KD = 19 ± 1 μm) (12). We have proposed that a protein partner such as HP2, which binds with higher affinity than the dimerization constant, could “drive dimerization” by shifting the equilibrium from monomers of HP1a (84.5% in the absence of partner) to dimers. The HP1a orthologue in human and mouse, HP1β, was estimated to dimerize at a dissociation constant of 150 nm (14). If it is present in the mammalian nucleus at a concentration similar to that of HP1a in the fly nucleus, HP1β likely exists as a constitutive dimer in the cell. Swi6, the yeast Schizosaccharomyces pombe orthologue of HP1a, has been found to have a dimerization constant of about 17 nm (15), consistent with a constitutive dimer in the yeast nucleus. Partner binding under these circumstances would not alter the dimerization of HP1β or Swi6 (unless phosphorylation weakened the dimerization significantly). Thus it is of interest to understand why HP1a has a weaker dimerization interface, how the other somatic HP1a paralogs compare in dimerization properties, and what the implications might be for partner protein interaction.
Peptides from two HP1a-interacting nuclear proteins, Heterochromatin Protein 2 (HP2) and PIWI, have been used here because of their different binding characteristics; in some cases WOC (without children) was used as well, as it contains a peptide with a canonical PXVXL motif. HP2 colocalizes extensively with HP1a in heterochromatin; mutations resulting in loss of HP2 are suppressors of position effect variegation, resulting in loss of silencing of heterochromatin reporters (16). The stoichiometric response observed with different doses of the gene, and the lack of any enzymatic motifs, suggest that HP2 is a structural component of heterochromatin (Shaffer, Cenci et al. 2006). PIWI and HP1a are suggested to function together in germline heterochromatin formation (17). While both HP2 and PIWI contain the PXVXL pentapeptide HP1a binding motif (18), HP1a:HP2 binding is 66-fold stronger than HP1a:PIWI binding, the latter being typical of proteins with the PXVXL motif (12). The weaker association of HP1b with heterochromatin suggests that it may not bind HP2 as effectively. To begin to understand some of the features of HP1a that give it its unique distribution and partner interactions, we have 1) analyzed its dimerization properties compared with those of HP1b, and 2) used mutagenesis to determine the amino acids responsible for tight HP2 binding, in contrast to weak PIWI binding, to the chromo shadow domain. Our results demonstrate that the amino acid sequence of the binding partner together with the HP1a binding surface determines the strength of interaction.
EXPERIMENTAL PROCEDURES
Cloning/Site-directed Mutagenesis
D. melanogaster HP1b-CSD (residues 83–160) and HP1b-CSD-CTE (residues 83–240) were cloned into pET11a with restriction sites NdeI and BamHI. HP1a constructs are derived from D. melanogaster HP1a-CSD-CTE (residues 141–206) in pET 11a. In both HP1a and HP1b constructs, two Lys residues precede a 6-histidine tag at the N terminus, and the gene sequence is appended by two additional Lys residues at the C terminus. The Lys residues have been introduced to enhance solubility, while the 6-His tag is used for purification.
QuikChange PCR mutagenesis was used with the enzyme PfuUltra II Fusion HS DNA Polymerase. Mutagenesis was carried out using a two-step procedure. First a linear amplification was performed using only one of the two primers for 8 cycles. Then a logarithmic amplification was performed after mixing half of each reaction mix from the linear amplification together for 18 cycles (19). The PCR cycling conditions are based on the manufacturer's recommendations (Agilent Technologies).
Protein Expression
HP1a and HP1b constructs in pET11a were transformed into Escherichia coli BL21 cells. Cells were grown in lysogeny broth from an overnight starter culture 1:500 dilution to an optical density of 0.6 at 37 °C. The culture was then induced to express the transformed gene with 1 μm IPTG, typically for 22 h at room temp (20 °C). Cells were then harvested by centrifugation (4424 × g for 10 min) and frozen (−20 °C).
Protein Purification
Frozen cells from 2 liters of culture were resuspended in lysis buffer (50 mm phosphate pH 8.0, 0.5 m NaCl, 20 mm imidazole, 0.5 m benzamidine, 0.5 m PMSF). The suspension was mixed in a glass beaker with stirring and cooled using ice while being sonicated for two 15-min intervals of 15 s pulses and 15 s rests, with a 5 min rest between intervals. The lysate was spun down in an Optima l-80 XP Ultracentrifuge in a 45Ti rotor at 45,000 rpm for 45 min (Beckman Coulter). The supernatant was subsequently loaded on to a 2–4 ml of Ni-NTA column (Qiagen), and washed with 1L of wash buffer (50 mm phosphate, pH 8.0, 500 mm NaCl, 25 mm imidazole). The protein was eluted with elution buffer (50 mm phosphate, pH 8.0, 150 mm NaCl, 250 mm imidazole).
Fluorescence Polarization Binding Assays
Fluorescence polarization binding assays were carried out in a binding buffer (50 mm phosphate, pH 8.0 and 150 mm NaCl). The total concentration of protein was typically 270 μm as determined by UV absorbance at 280 nm using the HP1a-CSD-CTE extinction coefficient of 8480 m−1 cm−1, HP1b-CSD extinction coefficient of 12490 m−1 cm−1, and HP1b-CSD-CTE extinction coefficient of 12490 m−1 cm−1. Beginning with 300 μl of the concentrated protein sample, a serial dilution of 12 total samples was prepared, 150 μl each. To these samples 1 μl of 22 μm fluoresceinated peptide was added to obtain a final peptide concentration of 150 nm (12, 20). Samples were incubated at 15 °C, and their polarization measured in a Beacon 2000 Variable Temperature Fluorescence Polarization System (Invitrogen).
Calculation of Binding Affinities
The concentration of HP1a dimers was calculated based on the KD determined by AUC. That percentage was multiplied by the total protein concentration and then divided by 2 to obtain the concentration of dimer sites available for binding. That concentration was plotted as a function of the fraction of peptide bound (20). A one-to-one binding model was used to fit the data (20) and determine the dissociation constant using Kaleidagraph (Synergy Software). Error reflects 95% confidence intervals of the curve fit.
Fluoresceinated Peptides
HP2 and PIWI peptides were generated by the Yale University Keck Facility. Fluorescein was added to the N terminus of the following peptides: HP2 peptide Cys to Ser mutant used for all assays: NH2-QNISPRKLSVKINRRPYNKWLR-COOH, HP2 peptide with PIWI pentamer: NH2-QNISPRKPRVKVNRRPYNKWLR-COOH, PIWI peptide: PIWI NH2-TSRGSGDPRVKVFRGSSSGDY-COOH, PIWI peptide with HP2 pentamer: NH2-TSRGSGDLSVKIFRGSSSGDY-COOH.
Circular Dichroism
The protein secondary structure was assessed by circular dichroism with a Jasco J-815 spectrometer at a scan speed of 50 nm/min and temperature of 15 °C. The data pitch was 0.2 nm, the bandwidth 1 nm, and the response time 1 s, with a continuous scanning mode. The protein samples were 20 μm in binding buffer (50 mm phosphate, pH 8.0 and 150 mm NaCl).
Gel Filtration
Gel Filtration was performed on a Superdex 75 column (GE Healthcare Life Sciences). A 50 μl sample was loaded on the column in binding buffer (50 mm phosphate, 150 mm NaCl, pH 8.0). The sample concentrations were HP1a-CSD-L147K, 92 μm; -L153K 167 μm; -L165K 114 μm; -V171K 99 μm; -V177A 260 μm; -V181K 71 μm; -E205Q 198 μm; and -D206N 106 μm.
Analytical Ultracentrifugation
HP1a-CSD-CTE-R188Q was prepared in binding buffer (50 mm phosphate pH 8.0, 150 mm NaCl) at 6 different concentrations ranging from 5–91 μm. Samples were spun at 25, 35, and 45 krpm until equilibrium was reached as judged by two scans separated by 2 h overlaying each other. Equilibrated curves were analyzed using Ultrascan (21). The molecular mass of HP1a-CSD-CTE-R188Q is 9989 Da. When performing a global analysis of the equilibrium AUC data, the molecular mass was fixed to that value, and the equilibrium constant fit to the data.
The same procedure was followed with HP1b-CSD (5–60 μm) and HP1b-CSD-CTE (3.5–60 μm). The extinction coefficient for both samples is 12490 m−1 cm−1, and their respective molecular masses are 9711 Da and 17,281 Da. HP1b-CSD was spun at 25, 35, 45 krpm and HP1b-CSD-CTE at 15, 25, 35 krpm. 1 mm TCEP was included in the binding buffer for both HP1b samples. The error analysis was performed using a Monte Carlo simulation, estimating an error of 8% to ensure that we were sampling possible equivalent fits; 95% confidence intervals were obtained.
RESULTS
The D. melanogaster Dimerization Interface of HP1a Is Weaker Than That of Several Homologues
We previously determined the crystal structure of the HP1a chromo shadow domain, and found that the dimerization interface composed of nine residues (Fig. 1A) (12). Seeking to understand the relatively weak dimerization constant, we compared the critical residues with those of HP1β. Among them, Arg-188 and Ser-199 in Drosophila (numbering from Dm) are not identical to the mouse/human sequence, where the corresponding residues are Gln and Thr (Fig. 1A). The structure of HP1β (PDB 2FMM) (22) indicates that there is a hydrophobic interaction between Gln-188 and Asn-183, while the structure of HP1a (PDB 3P7J) (12) indicates that R188 forms an electrostatic interaction with the neighboring residue Glu-184 (Fig. 1B). Given this structural information, and noting that the difference between Arg and Gln is the more significant change (compared with a Ser to Thr change), we investigated the contribution of this residue to the stability of the dimer. A Gln at this position is more common among the HP1 homologues (Fig. 1A); the substitution of an Arg appears to be a change exclusive to the Drosophila family (melanogaster, schelia, and simulans).3
FIGURE 1.

The dimerization interface of HP1a-CSD-CTE has undergone a recent change that reduces its stability. A, multiple sequence alignment of the chromo shadow domains of Drosophila HP1a, HP1b, HP1c, Human/Mouse HP1β, and yeast Swi6. Residues with a blue triangle above them are found in the dimerization interface (9, 12). Residues with a rectangle above or around them were mutated in this study. Brown rectangles: residues important for differences in partner binding observed between HP1a and HP1b. Blue rectangles: mutation impacts chromoshadow domain folding. Red rectangle: mutation has a differential impact on HP2 and PIWI binding in HP1a. Boxed residue: mutation has an impact on CSD dimer stability. Residues that are underlined indicate the last residue of the peptide used for analytical ultracentrifugation (AUC) studies. HP1a sequence from the CSD to the end was used since the last three residues improve its affinity. This length is comparable to that used for HP1b, HP1β, and Swi6. B, top: the crystal structure of Dm HP1a (1s4z) with Arg-188 highlighted together with its pairing amino acid Glu-184. Bottom: the crystal structure of HP1β highlighting the position of Gln-188 (numbering of Dm) and its interacting amino acid Asn-183. C, data collected from equilibrium AUC of HP1a-CSD-CTE-R188Q showing that a monomer/dimer fit to the Lamm equation approximates that data well, as judged by the random distribution of the residuals about the mean.
To elucidate the contribution of Arg-188 toward the dimerization dissociation constant of HP1a, we mutated this residue to Gln. This recombinant HP1a chromo shadow domain expressed well and eluted as a single peak on the size exclusion column Superdex 75, similar to the WT construct. Interestingly, as measured by equilibrium analytical ultracentrifugation (AUC), there is a 2.4-fold improvement in the dimerization constant (Fig. 1C). HP1a R188Q has a dimerization constant in the high nanomolar range. Thus the difference in dimerization constants between HP1a and HP1β is largely explained by this difference in amino acid sequence.
To determine the impact that this mutation has on partner binding, we performed fluorescence polarization assays with three peptides derived from chromo shadow domain binding partners. WOC (without children), which co-precipitates with HP1c (23, 24), has a canonical PXVXL sequence (PHVLL, centered on Val-1538, which we refer to as position 0); HP2 has a non-canonical PXVXL sequence (LSVKI, centered on Val-2468); and PIWI has a near-canonical sequence, but lacks large hydrophobic residues in the plus or minus 2 position (PRVKV, centered on Val-30). We found no change in the dissociation constant on binding to WT or R188Q HP1a dimers (Fig. 2), indicating that this modest shift in dimerization does not affect Dm HP1a's ability to interact with its partners. (Note that the change is not near the binding surface.) This is in contrast to the effects observed when the dimer is abolished with the I191E substitution, or when the PXVXL binding surface is disrupted with the W200A substitution (12).
FIGURE 2.
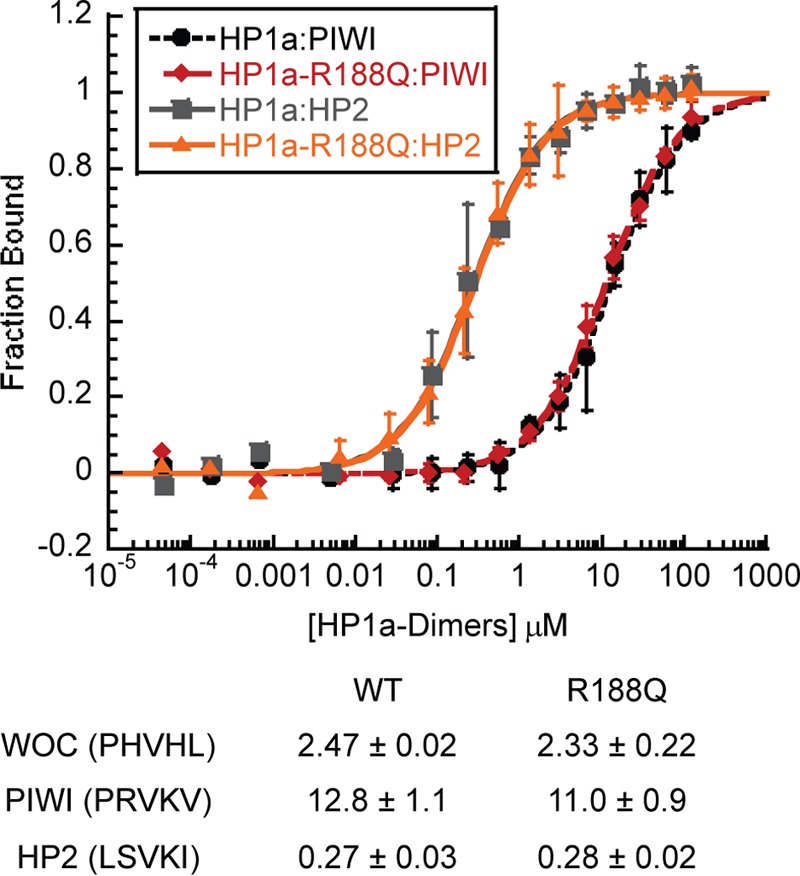
Peptide binding to HP1a-CSD-CTE-R188Q is not altered from WT. Fluorescence Polarization data of HP1a-CSD-CTE WT and the R188Q mutant binding to PIWI or HP2. The fraction of peptide bound is plotted as a function of HP1a-CSD-CTE dimer. Below is a table of the Kd values of the peptides binding to the WT or R188Q mutant.
HP1b CSD Exhibits Tighter Dimer Formation, Which Is Reduced by the 87 Residue CTE
HP1b shares all of its residues in the dimerization interface with mouse/human HP1β. Unlike HP1β, however, it has an 87 residue CTE. Our previous study of HP1a showed that the chromo shadow domain with the C-terminal extension has the same dimerization constant as was found for the full-length protein, indicating that the chromo shadow domain acts independently of the chromodomain and hinge regions that are N-terminal to it. Since our previous investigation of HP1a indicated a 0.7-fold tighter affinity for the chromo shadow domain per se, compared with when the CTE (4 residues) were included, we performed equilibrium AUC on HP1b chromo shadow domain with and without its CTE (Fig. 3A). We find that the chromo shadow domain alone (aa 87–160; see Fig. 3A) has a dimerization constant of less than 50 nm (Fig. 3B), which more closely resembles what is found for HP1β and Swi6 rather than HP1a. The HP1b chromo shadow domain with the full CTE exhibits a significantly reduced dimerization constant of 5.2 ± 0.2 μm (Fig. 3C). This weakened dimerization in the presence of the CTE suggests that in vivo, HP1b is similar to HP1a, in that peptide binding could influence dimerization.
FIGURE 3.
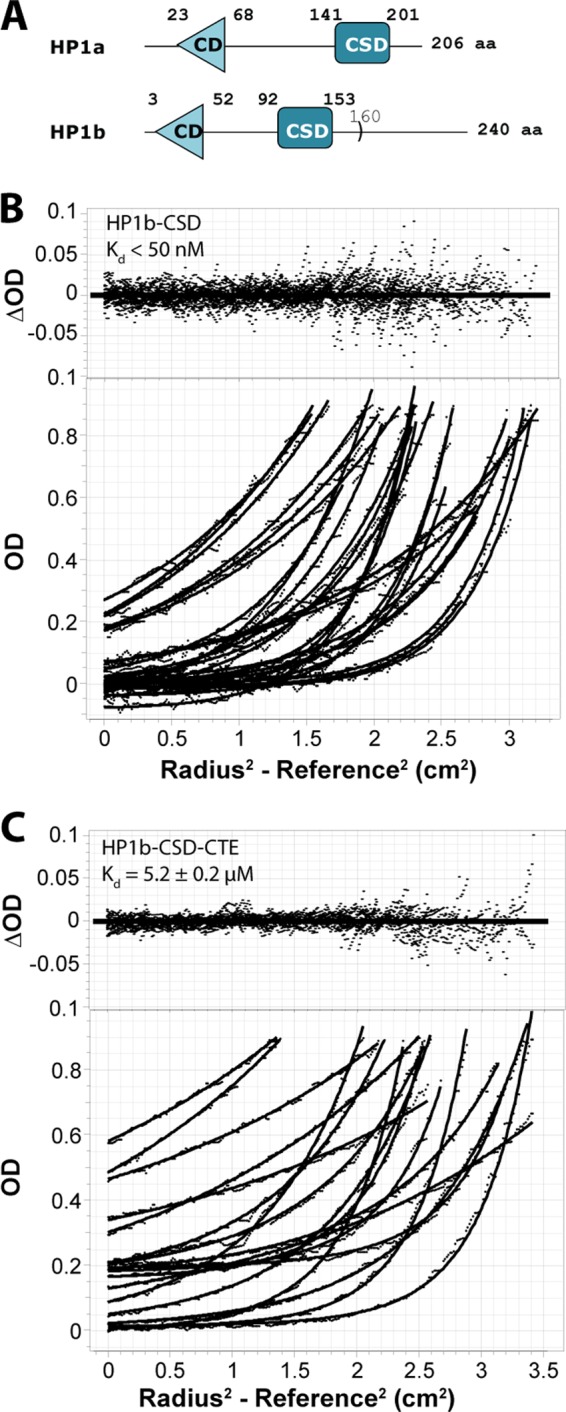
HP1b chromo shadow domain self-association is opposed by its 87 residue C-terminal extension. A, domain diagram of HP1a and HP1b. The chromoshadow domain construct for HP1b extends from residue 92 to 160. B and C, global analysis of HP1b-CSD (B) and HP1b-CSD-CTE (C) by equilibrium AUC. A monomer/dimer fit to the Lamm equation approximates that data well, as judged by the random distribution of the residuals about the mean (upper panels; raw data in the lower panels), and shows weaker dimerization in the presence of the C-terminal extension.
HP1a and HP1b are both predicted to bind PXVXL containing sequences (9), and it is of interest to compare their affinity for a variety of partners. We found that the WOC peptide, with an exact PXVXL sequence, bound with highest affinity to the HP1b CSD, with a KD of 1.98 ± 0.16 μm, while the HP2 peptide bound with a KD of 5.2 ± 0.5 μm. In the presence of the CTE the affinity of WOC decreased (3.2-fold) (Fig. 4) while there was no change in HP2 affinity. Thus the presence of the CTE reduces dimerization propensity, and enhances the ability of the HP1b chromo shadow domain to discriminate among potential partners. We find that the binding of HP2 to HP1b is about 10-fold weaker than its binding to HP1a.
FIGURE 4.
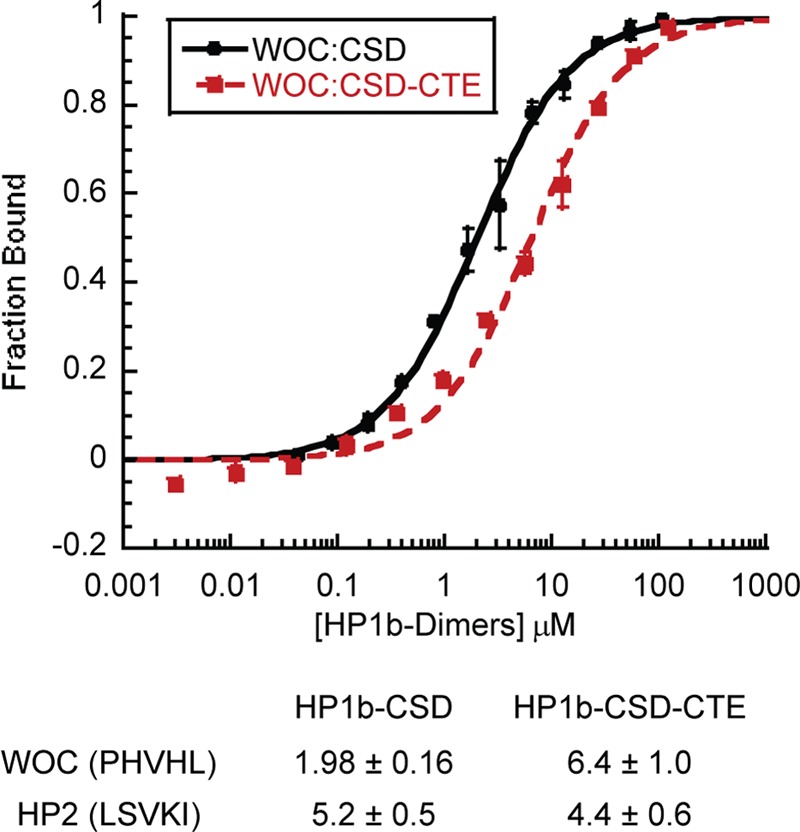
The HP1b carboxyl tail extension enhances chromo shadow domain discrimination in some cases. Fluorescence Polarization data of HP1b-CSD and HP1b-CSD-CTE binding to WOC. A table of the Kd values for WOC and HP2 peptides binding to HP1b-CSD or HP1b-CSD-CTE is shown below.
Weaker HP1b Peptide Binding May Reflect a Difference in CTE Interactions
58% of the residues in the chromo shadow domain of HP1a and HP1b are identical, but their CTEs are entirely different. In HP1a, CTE residues 203, 205, and 206 are negatively charged, whereas the corresponding residues in HP1b are neutral. Since deleting HP1a's last three residues (the CTE) had an impact on both HP2 and PIWI binding (12), we wondered whether the charge present in these residues contributed to the difference observed between HP1a and HP1b binding to HP2 and PIWI. To test the impact of charged CTE residues on partner binding, we made charge neutralization mutations in HP1a-CSD-CTE, changing E205Q and D206N. These mutations did not significantly alter the secondary structure of HP1a-CSD-CTE or its ability to dimerize (25), but they both significantly reduced binding to each peptide from 2.2–4.6-fold (Fig. 5). In contrast, HP1b-CSD-CTE showed weaker binding of the WOC peptide, with no appreciable change in HP2 peptide binding, in comparison to HP1b-CSD (Fig. 4).
FIGURE 5.
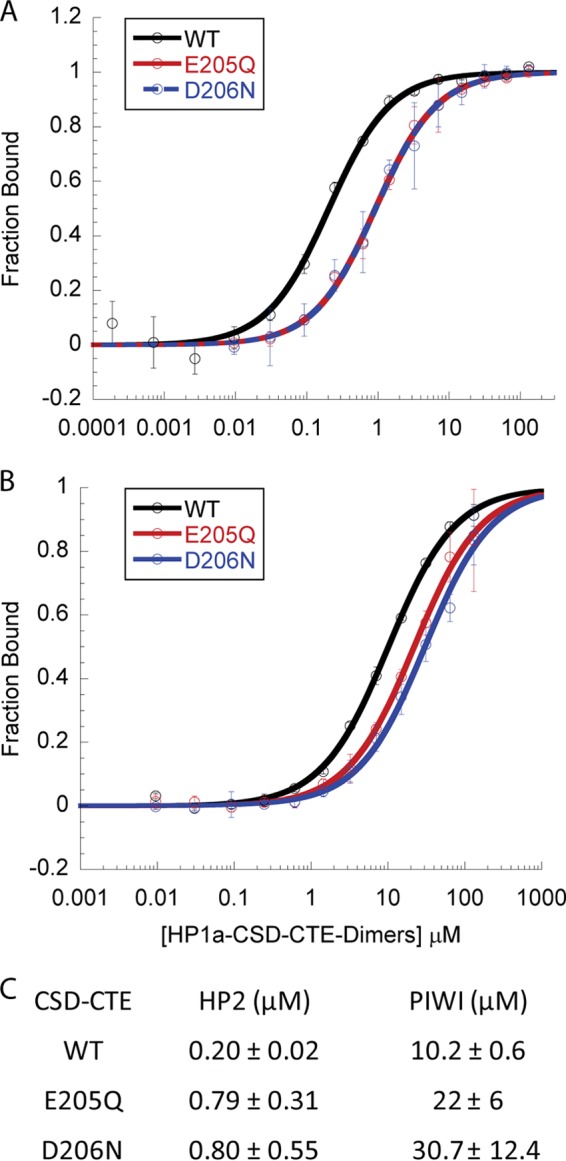
HP1a CTE charged residues enhance binding of both HP2 and PIWI. A and B, fluorescence Polarization data assessment of HP1a-CSD-CTE WT binding of HP2 (A) or PIWI (B) compared with that of CSD mutations E205Q or D206N. The fraction of peptide bound is plotted as a function of HP1a-CSD-CTE dimer. C, table of the KD values of the peptides binding to the WT, E205Q, or D206N forms of HP1a-CSD-CTE. Loss of either charged residue in the CSD results in weaker binding of both peptides.
A Hydrophobic Patch on the CSD Impacts HP1a Secondary Structure and Peptide Binding
To determine how the residues in the chromo shadow domain surface participate in peptide discrimination, contributing to the very effective binding of HP2 by HP1a, we created a panel of mutations in the HP1a binding surface. Because the binding affinity of HP2 or PIWI to HP1a-CSD-CTE changes very little under different salt concentrations (25, 150, 500 mm NaCl) (data not shown), we tested hydrophobic residues as a potential source of discrimination between HP2 and PIWI peptide binding (L147K, L153K, V177A, V181K) (Fig. 6, A–D). However, we found that these mutations all result in perturbation of the structure of the chromo shadow domain and induce aggregation (Fig. 6E). We anticipated that mutations in residues such as Leu-147 and Val-177, which were previously identified to be important for the fold (tertiary structure) of the chromo shadow domain (9), would potentially impact the secondary structure (Figs. 1 and 6A). However, we also observed that residues Leu-153 and Val-181, which were not previously identified as being important for tertiary structure, are important for proper folding (Fig. 6E). In all of these cases, although the data suggest a weakened affinity for the HP2 and PIWI peptides (Fig. 6, B and C), the actual binding affinity cannot be determined because there is a lack of homogeneous, well-folded HP1a protein as a result of these mutations.
FIGURE 6.
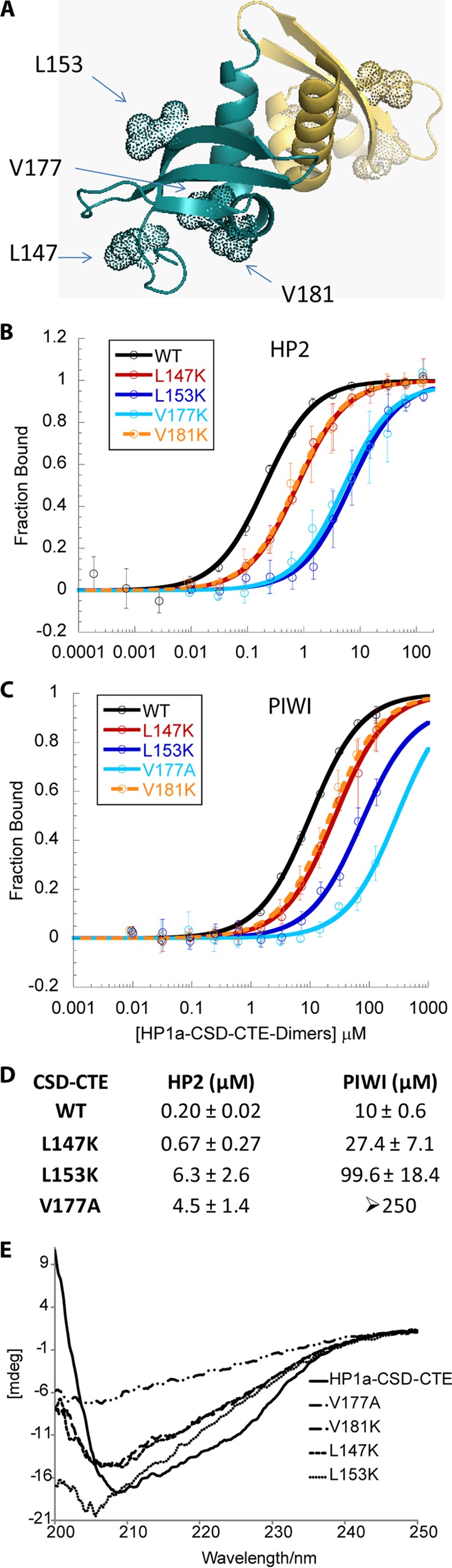
HP1a-CSD-CTE mutations L147K, L153K, V177A, and V181K all cause a decrease in binding to both HP2 and PIWI concomitant with a change in secondary structure. A, crystal structure of HP1a-CSD-CTE (PDB 3P7J). WT residues that were mutated are shown as dotted spheres. B and C, fluorescence polarization binding curves for HP2 (B) and PIWI (C) for the WT and mutant forms of the HP1a-CSD-CTE. The fraction of peptide bound is plotted as a function of HP1a-CSD-CTE dimer. D, table of the KD values of the peptides binding to HP1a-CSD-CTE WT or mutated forms. E, circular dichroism far-UV spectra of HP1a-CSD-CTE WT and mutants V147, V153, V177, and V181. These mutants exhibit a change in their CD spectrum that indicates a change in secondary structure relative to WT.
HP2 Peptide but Not PIWI Interacts with Leu-165
Leu-165 along with Met-176 and Ser-156 have been identified as a site of interaction for residues −7/−6 and +5/+6 away from the central Val in the partner protein (9). Leu-165 is the only residue among these to be absolutely conserved among D. melanogaster HP1a, HP1b, and mouse/human HP1β. We also note that the HSQC map of HP1a in the presence of HP2 exhibits a split peak at Leu-165, indicating that each Leu-165 in the homodimer engages in a distinct interaction with the HP2 peptide. In contrast, binding by PIWI results in a single Leu-165 shifted peak, indicating a less specific response of this residue to the bound peptide (12). Following mutation of Leu-165 to Lys, fluorescence polarization binding assays showed that HP2 has a 22-fold reduction in affinity for HP1a, while PIWI shows a 1.6-fold change in affinity (Fig. 7). In contrast, mutation of Val-171, located in a loop that connects the β2 and β3 domains (Fig. 1A), has a 1.2–1.6-fold impact on either HP2 or PIWI binding to HP1a. Neither mutation significantly alters secondary structure (analyzed by circular dichroism, Fig. 7E) or induces aggregation (25). Thus Leu-165 gives HP1a the ability to form a more stable interaction with a specific subset of its partners.
FIGURE 7.
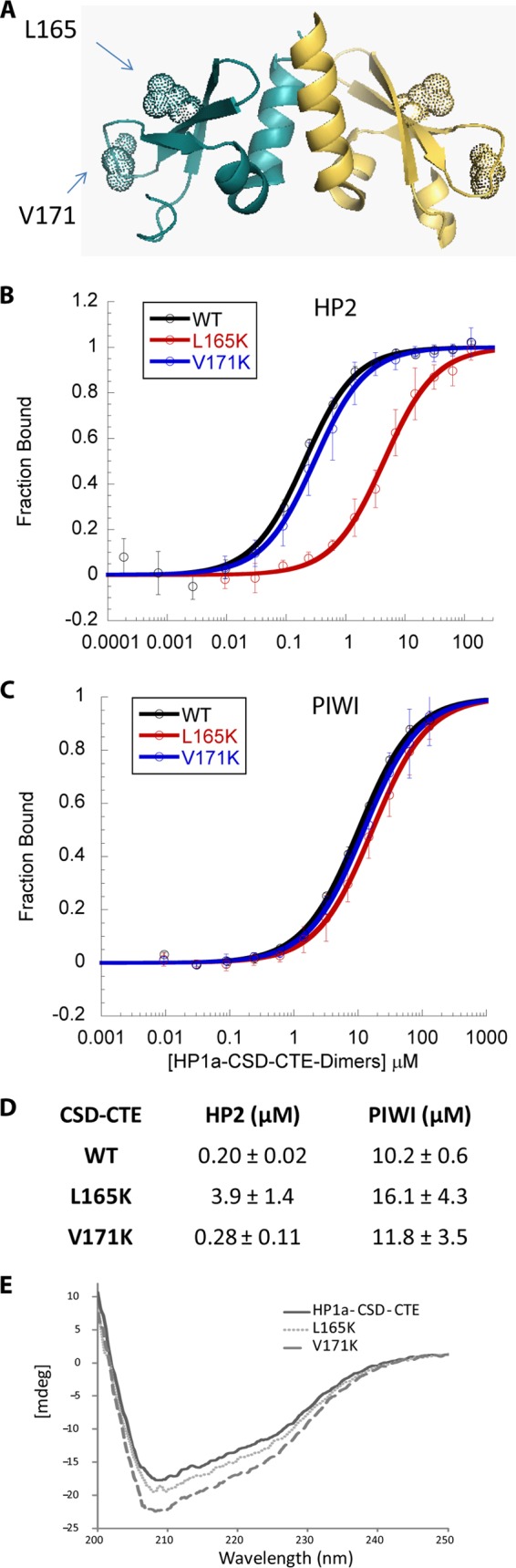
HP1a-CSD-CTE residue Leu-165 is critical for selective high-affinity binding of HP2. A, x-ray structure of HP1a-CSD-CTE (PDB 3P7J) with dotted spheres highlighting the positions of Leu-165 and Val-171. B and C, fluorescence polarization binding curves for HP2 (B) and PIWI (C) for the WT and mutant forms of the HP1a-CSD-CTE. The fraction of peptide bound is plotted as a function of HP1a-CSD-CTE dimer. D, table of the dissociation constant values demonstrating the selective impact that L165K has on binding. E, far UV circular dichroism spectra of HP1a-CSD-CTE WT, L165K, and V171K, show that these residues when mutated do not significantly alter secondary structure.
The PXVXL Pentamer of the Binding Partner Has an Important Role
Previously we found that the LXVXI motif in HP2 contributes to HP2 tight binding, but swapping this pentamer into the PIWI peptide does not change its affinity significantly (12). To test whether the remaining enhanced affinity of the HP2 polypeptide is due to its pentamer sequence (the PXVXL motif), we swapped those sequences between HP2 and PIWI and measured the binding to the HP1a-L165K mutant. Under these circumstances, the affinity of both the HP2 and PIWI peptides decreases to the same value within the error, 15 ± 2 μm versus 14.1 ± 1.4 μm (Fig. 8). Thus, the pentamer sequence in HP2, LSVKI, and the contact with the HP1a residue Leu-165, are sufficient to account for the difference in binding between HP2 and PIWI. This result argues that both binding of the central PXVXL sequence and interactions at the flanking residues tune selectivity and stability of HP1a complexes.
FIGURE 8.
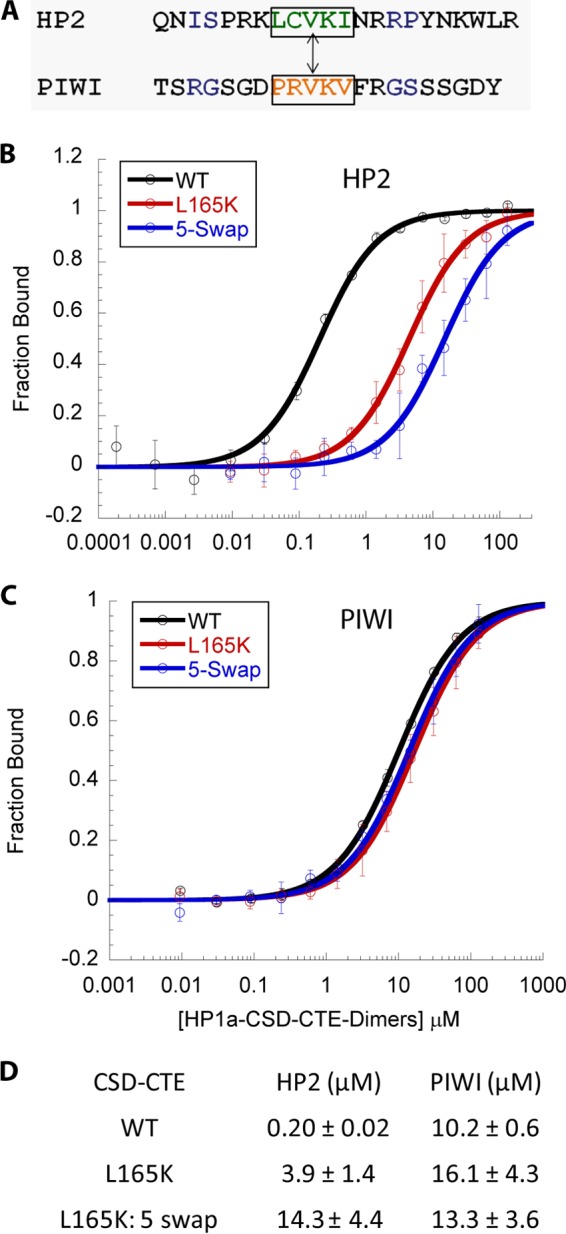
The HP1a-CSD-CTE-L165K peptide in conjunction with a pentamer swap between HP2 and PIWI imparts equivalent binding of HP2 and PIWI. A, pentamer exchange between HP2 and PIWI. B and C, fluorescence polarization binding curves for HP1a-CSD-CTE WT and the L165K mutated peptide with HP2 (B) and PIWI (C) using either the WT or pentamer-swapped counterpart in the latter case. D, table of the dissociation constant values demonstrating the equivalent binding of the pentamer-swap peptides to the L165K mutant CDS.
DISCUSSION
In this study we explore how HP1 paralogues achieve unique functions in the genome by examining their ability to bind to unique partners. To help elucidate their ability to discriminate among partners, we have examined the binding surface of HP1a in greater detail. This binding surface is created by homodimerization. We identified two of nine residues in the dimerization interface that could be involved in differential regulation of dimerization, residues that are not conserved in HP1a in comparison with Mm/Hs HP1β and Dm HP1b: Arg-188 and Ser-199. Residue 188 is a conserved glutamine in HP1b, HP1c and Mm/Hs HP1β while residue 199 is more variable among the Dm HP1a family. Consequently we focused on Arg-188, and found that the R188Q substitution does improve HP1a dimer stability (Fig. 1). However, this change does not impact partner binding (Fig. 2). Because the dimerization parameters did not provide much insight into the mechanism for CSD differential partner binding, we next looked at the binding surface of the CSD.
Our previous study showed that the CTE was important for HP1a tight binding to both of the tested peptides, HP2 and PIWI (12). This indicated that the CTE might be part of the HP1a binding surface. HP1a and HP1b differ in the length and sequence of their CTEs. HP1a has a short, negatively charged CTE of just five residues whereas HP1b has a CTE of ∼80 residues, with neutral residues in positions where the HP1a CTE has charged residues. Mutations that replace the HP1a CTE charged residues with neutral residues result in a loss of partner binding affinity (Fig. 5), confirming the importance of the charged CTE for HP1a function. In contrast, the addition of the neutral CTE to the HP1b CSD weakens dimerization (Fig. 3); this change results in similar or decreased peptide binding affinity (Fig. 4). Thus the characteristics of the CTE contribute to the ability of HP1a to bind to HP2 (an essential heterochromatin protein) with high affinity compared with the binding by HP1b (Figs. 2, 4, 5).
We also investigated the residues in HP1a outside of the dimerization interface that might allow for the observed differential affinity for HP2 and PIWI. Leu-165 proved to be a critical player, impacting binding of the HP2 peptide but not the PIWI peptide (Figs. 6 and 7). The results with Leu-165 establish the principle of differential contact points in the HP1a CSD dimer that impart differential affinity among the partner proteins.
Finally, the beta strands of the partner proteins that interact with the dimerization interface of the CSD also have a highly variable sequence, and the PXVXL motif itself is variable, both of which provide another potential means of regulating partner affinity. Swapping the PXVXL motif between the HP2 (LSVKI) and PIWI (PRVKV) peptides, and measuring binding to the L165K mutant HP1a CSD-CTE eliminates the remaining differential affinity observed between HP2 and PIWI (Fig. 8). Thus the differential roles of the HP1 paralogs can in part be traced to their binding surface.
The work presented herein focuses on the binding surface for proteins generated by the CSD and the region C-terminal to it. At the N-terminal end of the protein, the CD has been implicated in H3K9me2/3 binding. While interaction with H3K9me2/3 clearly stabilizes HP1a at the pericentric heterochromatin and other domains where this mark is enriched, it is not the only determinant of HP1a distribution (6). Further, HP1a, HP1b, and HP1c do not distribute coincidently (5). The chromo shadow domain could play a role by interacting with DNA-binding proteins that localize to specific regions in the DNA. For example HP1c binds to WOC, which is a DNA binding zinc finger protein (23, 24). HP1a binds to Su(var)3–7, which also has DNA binding activity (26). Thus the specificity of the CSD in partner selection may play an important role in determining the distribution of the paralogs. More work is needed understand the significance of those interactions.
Although we are learning more about CSD function, there are still many open questions. By understanding the mechanisms in place to discriminate between different partners, including the CSD binding partners that have DNA binding activity as well as those with other functions, we will be able to gain a fuller understanding of the roles that the HP1 paralogs, and HP1a in particular, are playing in the genome.
Acknowledgments
We thank Washington University Bio 4522 students for generating mutant forms of HP1a for this study: Philip Chen, Degian Ghebermicael, Vincent Huang, Noliyanda James, Sung Yeop Jeong, Yamini Krishnamurthy, Sujay Kulshrestha, Jennifer Lapp, Benjamin Marks, Suzanne Mazhuvanchery, Sheri McClerklin, Chantel Miller, Akhila Narla, David Nathin, Neil Savalia, Nancy Shen, Mai Phuong Tran, Joy Wang, Vivian Wang, Yijun Yang. We thank the Elgin laboratory for critical review of the manuscript.
This work was supported, in whole or in part, by the National Institutes of Health Grant GM068388 (to S. C. R. E.), the National Science Foundation under Grant No. 0852037-Cellular and Developmental Biology Research Apprenticeship Program (CD-BioRAP) at Washington University, St. Louis, and by Washington University, St Louis.
N. C. Riddle and I. Umana, Washington University, personal communication.
- HP1a
- heterochromatin protein 1a
- CSD
- chromo shadow domain
- CTE
- C-terminal extension
- CD
- chromodomain
- WOC
- without children.
REFERENCES
- 1. James T. C., Eissenberg J. C., Craig C., Dietrich V., Hobson A., Elgin S. C. (1989) Distribution patterns of HP1, a heterochromatin-associated nonhistone chromosomal protein of Drosophila. Eur. J. Cell Biol. 50, 170–180 [PubMed] [Google Scholar]
- 2. James T. C., Elgin S. C. (1986) Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell. Biol. 6, 3862–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eissenberg J. C., James T. C., Foster-Hartnett D. M., Hartnett T., Ngan V., Elgin S. C. (1990) Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 87, 9923–9927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vermaak D., Henikoff S., Malik H. S. (2005) Positive selection drives the evolution of rhino, a member of the heterochromatin protein 1 family in Drosophila. PLoS genetics 1, 96–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vermaak D., Malik H. S. (2009) Multiple roles for heterochromatin protein 1 genes in Drosophila. Annu. Rev. Genet. 43, 467–492 [DOI] [PubMed] [Google Scholar]
- 6. Smothers J. F., Henikoff S. (2001) The hinge and chromo shadow domain impart distinct targeting of HP1-like proteins. Mol. Cell. Biol. 21, 2555–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riddle N. C., Minoda A., Kharchenko P. V., Alekseyenko A. A., Schwartz Y. B., Tolstorukov M. Y., Gorchakov A. A., Jaffe J. D., Kennedy C., Linder-Basso D., Peach S. E., Shanower G., Zheng H., Kuroda M. I., Pirrotta V., Park P. J., Elgin S. C., Karpen G. H. (2011) Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 21, 147–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y., Kirschmann D. A., Wallrath L. L. (2002) Does heterochromatin protein 1 always follow code? Proc. Natl. Acad. Sci. U.S.A. 99, 16462–16469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thiru A., Nietlispach D., Mott H. R., Okuwaki M., Lyon D., Nielsen P. R., Hirshberg M., Verreault A., Murzina N. V., Laue E. D. (2004) Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 23, 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jacobs S. A., Taverna S. D., Zhang Y., Briggs S. D., Li J., Eissenberg J. C., Allis C. D., Khorasanizadeh S. (2001) Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 20, 5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu B. Y., Emtage P. C., Duyf B. J., Hilliker A. J., Eissenberg J. C. (2000) Heterochromatin protein 1 is required for the normal expression of two heterochromatin genes in Drosophila. Genetics 155, 699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mendez D. L., Kim D., Chruszcz M., Stephens G. E., Minor W., Khorasanizadeh S., Elgin S. C. (2011) The HP1a disordered C terminus and chromo shadow domain cooperate to select target peptide partners. Chembiochem 12, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Badugu R., Yoo Y., Singh P. B., Kellum R. (2005) Mutations in the heterochromatin protein 1 (HP1) hinge domain affect HP1 protein interactions and chromosomal distribution. Chromosoma 113, 370–384 [DOI] [PubMed] [Google Scholar]
- 14. Brasher S. V., Smith B. O., Fogh R. H., Nietlispach D., Thiru A., Nielsen P. R., Broadhurst R. W., Ball L. J., Murzina N. V., Laue E. D. (2000) The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 19, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Canzio D., Chang E. Y., Shankar S., Kuchenbecker K. M., Simon M. D., Madhani H. D., Narlikar G. J., Al-Sady B. (2011) Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 41, 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaffer C. D., Stephens G. E., Thompson B. A., Funches L., Bernat J. A., Craig C. A., Elgin S. C. (2002) Heterochromatin protein 2 (HP2), a partner of HP1 in Drosophila heterochromatin. Proc. Natl. Acad. Sci. U.S.A. 99, 14332–14337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang S. H., Elgin S. C. (2011) Drosophila Piwi functions downstream of piRNA production mediating a chromatin-based transposon silencing mechanism in female germ line. Proc. Natl. Acad. Sci. U.S.A. 108, 21164–21169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smothers J. F., Henikoff S. (2000) The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr. Biol. 10, 27–30 [DOI] [PubMed] [Google Scholar]
- 19. Wang W., Malcolm B. A. (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. BioTechniques 26, 680–682 [DOI] [PubMed] [Google Scholar]
- 20. Jacobs S. A., Fischle W., Khorasanizadeh S. (2004) Assays for the determination of structure and dynamics of the interaction of the chromodomain with histone peptides. Methods Enzymol. 376, 131–148 [DOI] [PubMed] [Google Scholar]
- 21. Demeler B. (2005) Ultrascan A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments, Royal Society of Chemistry, UK [Google Scholar]
- 22. Huang Y., Myers M. P., Xu R. M. (2006) Crystal structure of the HP1-EMSY complex reveals an unusual mode of HP1 binding. Structure 14, 703–712 [DOI] [PubMed] [Google Scholar]
- 23. Abel J., Eskeland R., Raffa G. D., Kremmer E., Imhof A. (2009) Drosophila HP1c is regulated by an auto-regulatory feedback loop through its binding partner Woc. PloS one 4, e5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Font-Burgada J., Rossell D., Auer H., Azorín F. (2008) Drosophila HP1c isoform interacts with the zinc-finger proteins WOC and Relative-of-WOC to regulate gene expression. Genes Dev. 22, 3007–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mendez D. L. (2012) Key Factors in Drosophila melanogaster HP1a Partner Binding, Washington University in Saint Louis [Google Scholar]
- 26. Cléard F., Delattre M., Spierer P. (1997) SU(VAR)3–7, a Drosophila heterochromatin-associated protein and companion of HP1 in the genomic silencing of position-effect variegation. EMBO J. 16, 5280–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shaffer C. D., Cenci G., Thompson B., Stephens G. E., Slawson E. E., Adu-Wusu K., Gatti M., Elgin S. C. (2006) The large isoform of Drosophila melanogaster heterochromatin protein 2 plays a critical role in gene silencing and chromosome structure. Genetics 174, 1189–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]