

Background: 6′-Guanidinonaltrindole (6′-GNTI) activates G protein coupling to κ-opioid receptors (KOR) without β-arrestin2 recruitment in transfected cells.
Results: In striatal neurons, 6′-GNTI activates Akt but not ERK1/2; U69,593 activates both kinases.
Conclusion: In neurons, U69,593-induced activation of ERK1/2 is β-arrestin2-dependent, whereas activation of Akt is G protein-mediated.
Significance: Identification of KOR signaling pathways in endogenous systems will inform the development of KOR-directed medications.
Keywords: Arrestin, Brain, Drug Screening, G Protein-coupled Receptors (GPCR), Gene Knockout, MAP Kinases (MAPKs), Neurons, Opiate Opioid, Receptor Endocytosis, κ-Opioid Receptor
Abstract
There is considerable evidence to suggest that drug actions at the κ-opioid receptor (KOR) may represent a means to control pain perception and modulate reward thresholds. As a G protein-coupled receptor (GPCR), the activation of KOR promotes Gαi/o protein coupling and the recruitment of β-arrestins. It has become increasingly evident that GPCRs can transduce signals that originate independently via G protein pathways and β-arrestin pathways; the ligand-dependent bifurcation of such signaling is referred to as “functional selectivity” or “signaling bias.” Recently, a KOR agonist, 6′-guanidinonaltrindole (6′-GNTI), was shown to display bias toward the activation of G protein-mediated signaling over β-arrestin2 recruitment. Therefore, we investigated whether such ligand bias was preserved in striatal neurons. Although the reference KOR agonist U69,593 induces the phosphorylation of ERK1/2 and Akt, 6′-GNTI only activates the Akt pathway in striatal neurons. Using pharmacological tools and β-arrestin2 knock-out mice, we show that KOR-mediated ERK1/2 phosphorylation in striatal neurons requires β-arrestin2, whereas Akt activation depends upon G protein signaling. These findings reveal a point of KOR signal bifurcation that can be observed in an endogenous neuronal setting and may prove to be an important indicator when developing biased agonists at the KOR.
Introduction
Activation of the G protein-coupled κ-opioid receptor (KOR)5 by its endogenous ligand, dynorphin, impacts diverse physiological functions. In particular, the dynorphin/KOR system has been shown to modulate dopaminergic and serotonergic tone in the CNS and therefore has become a favorable target for drug discovery efforts aimed at treating addiction and mood disorders. Current KOR ligands are limited by unwanted side effects and unusual pharmacodynamics. KOR agonists, for example, have been shown to decrease the reinforcing and rewarding effects of abused drugs (1–5) and reduce the symptoms of mania (6, 7). However, they also induce sedation, dissociation, and dysphoria (8). KOR antagonists have antidepressant and anxiolytic effects in rodent models (9–11) and may provide a therapeutic means of treating relapse to addictive drugs (12, 13); yet the currently available, selective KOR antagonists are extremely long acting, which may limit their clinical utility (14, 15). Therefore, the question remains as to how this receptor should be acted upon to mediate the desired effects while limiting undesired consequences, whether that be through the use of partial agonists or functionally selective agonists or antagonists.
Functional selectivity, biased agonism, and ligand-directed signaling conceptually describe the condition wherein ligand binding to G protein-coupled receptors (GPCR) promotes receptor coupling to certain signaling cascades preferentially over others (16–20), and evidence of ligand-directed signaling at the KOR has been reported. For example, U69,593 and MOM-salvinorin B differ in their ability to induce astrocyte proliferation and in the temporal activation of ERK1/2 observed in astrocytes following drug treatment. In these cells, U69,593 activates ERK1/2 via both early phase G protein-dependent and later β-arrestin2-dependent mechanisms, the latter of which was correlated with astrocyte proliferation. In contrast, MOM-salvinorin B only activates the early G protein-dependent ERK1/2 pathway and does not induce astrocyte proliferation (21). In another demonstration of functional selectivity at KOR, the classic KOR antagonists norbinaltorphimine (nor-BNI) and JDTic were shown to antagonize G protein coupling but activate the MAP kinase JNK in a KOR-mediated manner (22). It is currently hypothesized that functional selectivity results from ligands directly influencing the conformation of the receptor and thereby directing selective interactions with particular second messenger systems. When coupled with molecular modeling studies and the introduction of the recent crystal structure of KOR, the characterization of KOR ligands that promote distinct pharmacological profiles may lead to structure-function-inspired drug design (23–25).
6′-Guanidinonaltrindole (6′-GNTI) was developed by Portoghese and colleagues (26) as a guanidino derivative of the selective δ-opioid receptor (DOR) antagonist naltrindole. Interestingly, the addition of a guanidino group to naltrindole converts it to a KOR-selective compound, and moving the guanidino group from the 5′ to 6′ position changes the ligand from a KOR antagonist (5′-GNTI) to a KOR agonist (6′-GNTI). Initially, 6′-GNTI agonist activity at KOR was demonstrated by showing that 6′-GNTI-induced inhibition of smooth muscle contraction in the guinea pig ileum could be selectively blocked by nor-BNI (26). Recently, 6′-GNTI was reported to partially activate G protein-mediated signaling pathways without inducing β-arrestin2 recruitment in HEK-293 cells (27). These findings make 6′-GNTI of great interest for dissecting the roles of different KOR signaling cascades as well as KOR drug development for the treatment of pain and addiction. In the present study, we sought to further characterize the pharmacological properties of 6′-GNTI in multiple signaling pathways in both KOR-expressing Chinese hamster ovary (CHO) cells and primary neuronal cultures where the KOR is expressed endogenously. We confirm that in CHO cells, 6′-GNTI is a potent partial agonist in G protein signaling and that it antagonizes β-arrestin2 recruitment to the KOR and receptor internalization (27). Although 6′-GNTI does not recruit β-arrestins, it does induce the phosphorylation of ERK1/2 and Akt, suggesting that these kinases are activated independent of KOR/β-arrestin2 interactions. However, when we assessed kinase activation profiles in primary striatal neurons, we found that only U69,593 is able to induce ERK1/2 activation and it does so in a β-arrestin2-dependent and pertussis toxin-insensitive manner. Conversely, both U69,593 and 6′-GNTI induce Akt phosphorylation in striatal neurons, activation that is blocked by pretreatment with pertussis toxin. These results indicate that 6′-GNTI is a functionally selective ligand at the endogenously expressed KOR.
EXPERIMENTAL PROCEDURES
Cell Culture and Cell Line Construction
CHO-K1 cells were transfected with an N-terminal hemagglutinin (HA)-tagged human KOR in pcDNA3.1(+) (Gene ID 4986, Missouri S&T cDNA Resource Center, Rolla, MO) by electroporation (GenePulser XCell, Bio-Rad). Transfected cells were selected via HA-antibody cell sorting by flow cytometry (FACSAria and LSRII, BD Biosciences), and the top 3% expression level cell population was used for all experiments. CHO-KOR cells were characterized for KOR membrane expression via confocal microscopy and total receptor number determined by [3H]U69,593 saturation binding (KD ∼ 0.6 nm, Bmax ∼ 1800 fmol/mg protein) (28). For all experiments with the CHO-KOR line, untransfected CHO cells served as negative controls (data not shown).
Drugs
U69,593 was obtained from Sigma, and nor-BNI dihydrochloride, 5′-GNTI ditrifluoroacetate, and 6′-GNTI ditrifluoroacetate were all obtained from Tocris Bioscience (Ellisville, MO). U69,593 was prepared in ethanol, 6′-GNTI and 5′-GNTI were prepared in DMSO, and nor-BNI was prepared in dH2O. In all cases, vehicle contained 1% DMSO and 1% ethanol in PBS (unless stated otherwise in the figure legends). Pertussis toxin from Bordetella pertussis was obtained from Sigma.
Mice
β-Arrestin2 knock-out (βarr2-KO) and wild-type (WT) littermates were used to assess [35S]GTPγS coupling in mouse brain and for the culturing of primary neurons (28). All experiments were performed with the approval of the Institutional Animal Care and Use Committee of The Scripps Research Institute.
[35S]GTPγS Coupling
CHO-KOR cells were collected, and membrane pellets were prepared by homogenization with a polytronic Tissue-Tearor in buffer (10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EDTA) followed by 20,000 × g centrifugation at 4 °C for 30 min. The resultant membrane pellet was resuspended in assay buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2, 1 mm EDTA, 40 μm GDP) via Teflon-on-glass Dounce homogenizer. Fifteen μg of membrane protein was incubated with increasing concentrations of drug and 0.1 nm [35S]GTPγS (PerkinElmer Life Sciences) for 1 h at 30 °C in a total reaction volume of 200 μl. For antagonist experiments, protein was preincubated with test compounds for 15 min prior to the addition of 100 nm U69,593 and [35S]GTPγS. Reactions were terminated by rapid filtration using a 96-well plate Brandel cell harvester (Brandel, Gaithersburg, MD) followed by washes with ice cold water. Microscint-20 (PerkinElmer Life Sciences) was added to the plates after drying, and radioactivity was read with a TopCount NXT HTS microplate scintillation and luminescence counter (PerkinElmer Life Sciences). All compounds were run in parallel assays in duplicate for comparison.
For coupling in mouse brain, striata were isolated from 4–5-month-old male WT mice. Tissue was homogenized, and membranes were prepared as described above. Coupling reactions, containing 2.5 μg of protein, 20 μm GDP, and 0.1 nm [35S]GTPγS, were incubated at room temperature for 2 h prior to harvesting, as described above.
β-Arrestin2 Translocation
To visually assess β-arrestin2 translocation to the KOR, CHO-KOR cells were transiently transfected with βarr2-GFP (5 μg) by electroporation and plated on collagen-coated glass-bottom confocal dishes (MatTek, Ashland, MA). After incubation at 37 °C for 24 h, the cells were serum-starved for 1 h in serum-free MEM without phenol red (Invitrogen). Drugs were added, and live cell images were obtained at the time points indicated using an Olympus FluoView IX81 confocal microscope (Olympus, Center Valley, PA) as described previously (29).
β-Arrestin2 translocation was also studied using the PathHunter® β-arrestin assay in CHO-K1 cells expressing the KOR (DiscoveRx, Fremont, CA) according to the manufacturer's protocol. Cells were treated with agonist for 90 min prior to assessment of enzyme complementation. For antagonist experiments, the cells were incubated with the antagonist for 60 min prior to agonist addition. Luminescence values were obtained using a Synergy HT luminometer (BioTek, Winooski, VT). All compounds were run in parallel experiments in duplicate.
KOR Internalization
To qualitatively determine agonist-induced receptor internalization, CHO-KOR cells were plated on collagen-coated glass-bottom confocal dishes and serum-starved for 30 min prior to the experiment. The KOR was labeled in live cells by incubation with an anti-HA-AlexaFluor 488 antibody (1:100, Invitrogen) for 15 min at 37 °C. Following washes, drug (10 μm) was added, and the cells were monitored by confocal imaging for HA-KOR internalization over the times indicated (29, 30).
Agonist-induced KOR internalization was also assessed in CHO-KOR cells using a cell surface biotinylation assay. This assay was performed as described previously (30). The cells were serum-starved for 30 min, and surface receptors were biotinylated prior to drug treatment. Drug was added at 10 μm, and cells were treated for 30 min or 1 h (30 min for the antagonist and then 30 min for the agonist, where applicable). The remaining surface biotinylation was removed by glutathione-stripping buffer, and the protected biotinylation was immunoprecipitated by streptavidin-coated beads. Western blots were performed as described previously with 10% bis-Tris NuPage gels (Invitrogen) and transferred to nitrocellulose membranes (18, 30). The receptor was detected using a KOR antibody (Neuromics, Edina, MN). Blots were imaged using the Odyssey infrared imager, and densitometry of bands was measured using Odyssey 2.1 software (Li-Cor, Lincoln, NE). The -fold stimulation over vehicle was calculated by dividing the densitometry values of the treated groups by the average value of the vehicle-treated group.
In-cell Western
To assess agonist-induced ERK1/2 phosphorylation via In-cell Western assay (Li-Cor), CHO-KOR cells were plated into 384-well clear-bottom, black-walled plates (BD Biosciences) at a density of 15,000 cells/well. The cells were serum-starved for 1 h prior to treatment with increasing drug concentrations for 10 min. The cells were fixed (4% paraformaldehyde in PBS), washed in 0.1 m glycine, permeabilized (0.2% Triton X-100), and blocked with Li-Cor blocking buffer in PBS (Li-Cor). Cells were then incubated with both phosphorylated and total ERK1/2 primary antibodies (p-ERK1/2, catalog No. 4370, at 1:300, and t-ERK1/2, catalog No. 4696, at 1:200; Cell Signaling Technology, Beverly, MA) overnight in Li-Cor blocking buffer. Secondary antibodies (anti-rabbit IRDye800 and anti-mouse IRDye680, both at 1:500; Li-Cor) were added in Li-Cor blocking buffer + 0.025% Tween 20. Fluorescence was determined using the Odyssey infrared imager. The -fold ERK1/2 phosphorylation over vehicle levels was determined by first normalizing phosphorylated ERK1/2 levels to total ERK1/2 levels within each well and then normalizing to the average of the vehicle treated cells within each experiment. All compounds were run in parallel with 6 replicate wells per point.
Signaling by Western Blot
CHO-KOR cells were plated in 6-well dishes and serum-starved for 1 h in serum-free medium prior to treatment. Concentrations and durations of treatment are indicated in the legends for Figs. 4 and 5. For antagonist studies, nor-BNI was added during the last 15 min of the serum starvation. For pertussis toxin pretreatment studies, 100 ng/ml pertussis toxin was added to the complete medium at ∼24 h prior to the assay. Following treatment, cells were washed once in PBS, and lysates were prepared in lysis buffer (20 mm Tris, 150 mm NaCl, 2 mm EDTA, 0.1% SDS, 1% Nonidet P-40, 0.25% deoxycholate, 1 mm sodium orthovanadate, 1 mm PMSF, 1 mm NaF, and protease inhibitors). Western blots were performed to assess phospho-ERK1/2 (1:1000, Cell Signaling Technology, catalog No. 4370), total ERK1/2 (1:1000, Cell Signaling Technology, catalog No. 4696), phospho-Akt (1:1000, Cell Signaling Technology, catalog No. 2965), and total Akt (1:1000, Cell Signaling Technology, catalog No. 2920) levels as described above.
FIGURE 4.

6′-GNTI-stimulated ERK1/2 phosphorylation in CHO-KOR cells is pertussis toxin-sensitive. CHO-KOR cells were serum-starved for 1 h prior to drug treatment and the determination of ERK1/2 phosphorylation. Representative images of plates or Western blots and densitometric analyses are provided. For each sample, phosphorylated ERK1/2 (p-ERK1/2) was first normalized to total ERK1/2 (t-ERK1/2) and the -fold stimulation over vehicle or basal is provided (mean ± S.E.). A, following treatment with increasing concentrations of drug for 10 min, -fold stimulation was determined by immunocytochemistry in a 384-well plate format (In-cell Western). 6′-GNTI induces ERK1/2 phosphorylation to a similar extent as U69,593 (two-way ANOVA for drug: F(1,108) = 0.07, p = 0.7919; for concentration: F(7,108) = 79.85, p < 0.0001). 5′-GNTI does not induce ERK1/2 phosphorylation (n = 9 (U69,593 and 6′-GNTI) and n = 5 (5′-GNTI) performed in replicates of six). B, both U69,593-induced (U69, 10 μm for 10 min) and 6′-GNTI-induced (6′, 10 μm for 10 min) ERK1/2 phosphorylation are sensitive to pretreatment with nor-BNI (Nor; 10 μm for 15 min) as determined by Western blot analysis (one-way ANOVA: F(5,41) = 79.49, p < 0.0001; Bonferroni's post hoc test: vehicle versus drug treatment, ***, p < 0.001, U69,593 versus nor-BNI+U69,593, ^^^, p < 0.001, 6′-GNTI versus nor-BNI+6′-GNTI, ###, p < 0.001; n = 4 independent experiments). C, time course analysis of the 10 μm concentrations of U69,593 and 6′-GNTI reveals that ERK1/2 phosphorylation levels remain elevated for up to 2 h post-treatment with either ligand (one-way ANOVA: for U69,593, F(5,27) = 7.886, p < 0.0001; for 6′-GNTI, F(5,27) = 9.619, p < 0.0001; Bonferroni's post hoc test: basal versus U69,593, *, p < 0.05, **, p < 0.01, ***, p < 0.001, basal versus 6′-GNTI, ^, p < 0.05, ^^^, p < 0.001). U69,593 and 6′-GNTI do not induce any significant differences between their time courses of activation (two-way ANOVA for drug: F(1,54) = 3.17, p = 0.0805; for time: F(5,54) = 16.65, p < 0.0001; n = 4 independent experiments). D, U69,593- and 6′-GNTI-induced (10 μm for 10 min) ERK1/2 phosphorylation can be blocked by an overnight pretreatment (100 ng/ml) with pertussis toxin (PTX) (Student's t test: vehicle (veh) versus drug treatment, ***, p < 0.001; n = 3 independent experiments).
FIGURE 5.

6′-GNTI-stimulated Akt phosphorylation in CHO-KOR cells is pertussis toxin-sensitive. CHO-KOR cells were serum-starved for 1 h prior to drug treatment and the determination of Akt phosphorylation. Representative images of plates or Western blots and densitometric analyses are provided. For each sample, phosphorylated Akt (p-Akt) was first normalized to total Akt (t-Akt), and the -fold stimulation over vehicle or basal is provided (mean ± S.E.). A, following treatment with increasing concentrations of drug for 10 min, -fold stimulation was determined by Western blot analysis. 6′-GNTI partially induces Akt phosphorylation when compared with U69,593 (one-way ANOVA: for U69,593, F(5,15) = 97.69, p < 0.0001; for 6′-GNTI, F(5,15) = 7.57, p < 0.0001; Bonferroni's post hoc test: vehicle versus U69,593, ***, p < 0.001, basal versus 6′-GNTI, ^, p < 0.05, ^^, p < 0.01, ^^^, p < 0.001; two-way ANOVA for drug × concentration: U69,593 versus 6′-GNTI, F(5,30) = 16.98, p < 0.0001; n = 3 independent experiments). B, both U69,593-stimulated (U69, 10 μm for 10 min) and 6′-GNTI-stimulated (6′, 10 μm for 10 min) Akt phosphorylation are sensitive to pretreatment with nor-BNI (Nor; 10 μm for 15 min) as determined by Western blot analysis (one-way ANOVA: F(5,41) = 34.81, p < 0.0001; Bonferroni's post hoc test: vehicle versus drug treatment, **, p < 0.01, ***, p < 0.001; U69,593 versus nor-BNI+U69,593, ^^^, p < 0.001; 6′-GNTI versus nor-BNI+6′-GNTI, ###, p < 0.001; n = 4 independent experiments). C, time course analysis of the 10 μm concentrations of U69,593 and 6′-GNTI reveals that Akt phosphorylation levels peak at 10 min post-treatment with either drug and quickly return to baseline (one-way ANOVA: for U69,593, F(5,27) = 3.934, p < 0.0001; for 6′-GNTI, F(5,27) = 3.352, p = 0.0001; Bonferroni's post hoc test: basal versus U69,593, *, p < 0.05, basal versus 6′-GNTI, ^, p < 0.05). U69,593 and 6′-GNTI do not induce any significant differences between their time courses of activation (two-way ANOVA for drug: F(1,54) = 0.05, p = 0.8238; for time: F(5,54) = 7.23, p < 0.0001; n = 4 independent experiments). D, U69,593- and 6′-GNTI-induced (10 μm for 10 min) Akt phosphorylation can be blocked by an overnight pretreatment (100 ng/ml) with pertussis toxin (PTX) (Student's t test: vehicle (Veh) versus drug treatment, **, p < 0,01; ***, p < 0.001; n = 3 independent experiments).
Striatal Neurons
Primary striatal neuronal cultures were generated from postnatal day 1 mouse neonates obtained from homozygous breeding of βarr2-KO and WT mice. Striata were isolated and digested in 0.0375% papain (Sigma-Aldrich) followed by trituration to generate a single cell suspension as described previously (18, 31). Neurons were plated in Neurobasal-A medium (Invitrogen) supplemented with 2% B-27 serum supplement, 0.5 mm l-glutamine, 1.675 μg/liter sodium selenite, 2.5 mg/liter insulin, 1.375 mg/liter transferrin, and 50 μg/ml gentamycin onto 12-well tissue culture dishes coated with poly-l-lysine (Sigma-Aldrich). Cytosine β-d-arabinofuranoside (Sigma-Aldrich) was added to the culture medium 4 days after plating, and the medium was replaced with fresh culture medium at 5 days post-plating.
On the tenth day after plating, kinase activation was determined. Neurons were incubated in serum-free MEM for 1 h prior to drug treatment. For agonist studies, neurons were treated with 10 μm drug for 10 min. For nor-BNI pretreatment studies, nor-BNI was added to the medium to a final concentration of 1 μm during the last 15 min of the serum starvation period. For pertussis toxin pretreatment studies, pertussis toxin was added to the medium 24 h prior to drug treatment at a final concentration of 100 ng/ml. Following drug treatment, neurons were washed once in PBS; lysates were prepared, and Western blots were performed as described above.
Data Analysis and Statistics
All statistical comparisons were made using GraphPad Prism 6.01 software (GraphPad, La Jolla, CA). To generate concentration response curves, vehicle values were subtracted from the raw data, and three-parameter nonlinear regression curve-fit analysis was performed. For determination of agonist stimulation, the percentage of maximal U69,593 was calculated by normalizing all values to the top of the U69,593 curve. For antagonist studies, values were normalized to the average of 100 nm or 1 μm U69,593 alone, and the percentage is provided (Figs. 1B and 2C). The potency and efficacy values, obtained from the averages of the nonlinear regression analysis performed on each individual curve, are reported as the mean ± S.E. in Tables 1 and 2. To determine ligand differences within and between assays, curves were fit to the operational model (32), and the ratio of τ/KA was calculated (GraphPad Prism). The within assay differences were determined by subtracting the log(τ/KA) for U69,593 (the reference ligand) from the values obtained for 6′-GNTI, which generated the “transduction efficiency” for 6′-GNTI, or the Δlog(τ/KA). To determine bias between assays, relative bias factors were calculated by subtracting the Δlog(τ/KA) for each assay, presented as ΔΔ(τ/KA)path1-path2 (Table 1) (33–35). Student's t test indicates an unpaired, two-tailed analysis. In all cases, significance was considered to be p ≤ 0.05. All studies were performed n ≥ 3 independent experiments performed in multiple replicates, with the specific number of individual experiments for each assay noted in the figure legends.
FIGURE 1.
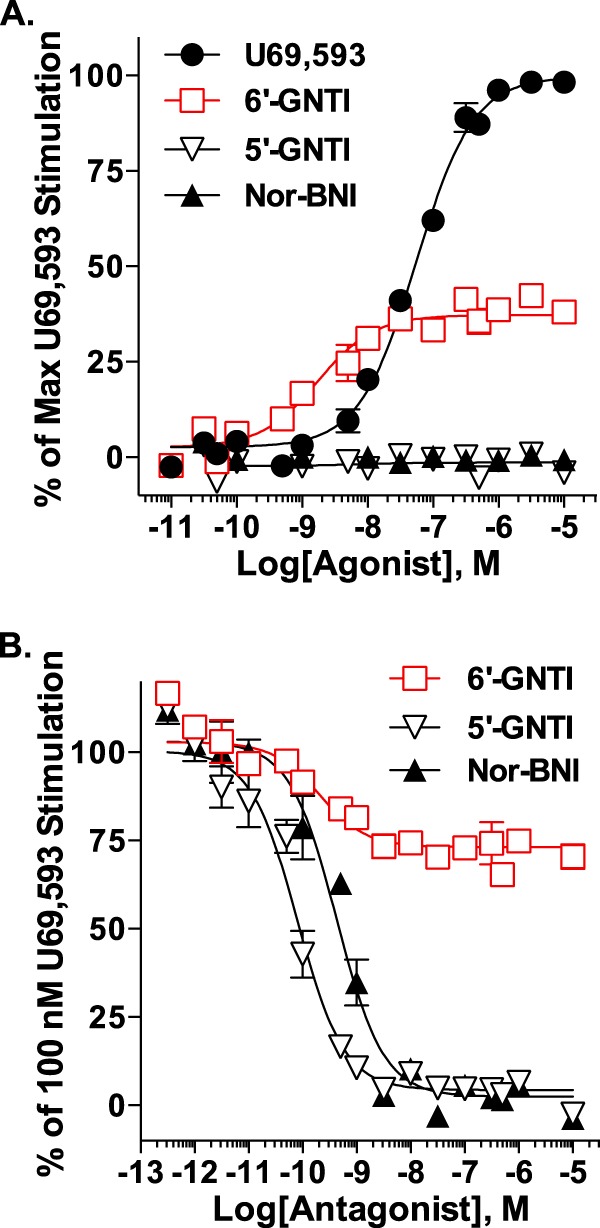
6′-GNTI is a potent partial agonist for G protein coupling in CHO-KOR cells. [35S]GTPγS incorporation was determined for CHO-KOR membranes following drug treatment. Values are reported as the mean ± S.E. Data were fit to nonlinear regression curves. A, agonist mode. Membranes were treated with increasing concentrations of drug or vehicle for 1 h. Relative to U69,593, 6′-GNTI potently, although less efficaciously, stimulates [35S]GTPγS binding (two-way ANOVA for interaction of drug × concentration: U69,593 versus 6′-GNTI, F(14,168) = 70.75, p < 0.0001). Neither 5′-GNTI nor nor-BNI stimulates [35S]GTPγS binding over vehicle levels (non-convergence, R2 < 0.6). The percent maximal U69,593-induced coupling is provided (n = 13 (U69,593), n = 12 (6′-GNTI), n = 7 (5′-GNTI), and n = 4 (nor-BNI) performed in duplicate). B, antagonist mode. Membranes were preincubated for 15 min with vehicle (0.1% DMSO) or increasing concentrations of nor-BNI, 6′-GNTI or 5′-GNTI prior to the addition of 100 nm U69,593 for 1 h. 6′-GNTI partially blocks U69,593-stimulated [35S]GTPγS coupling, whereas nor-BNI and 5′-GNTI completely block coupling (two-way ANOVA for interaction of drug × concentration: 6′-GNTI versus nor-BNI, F(15,116) = 21.61, p < 0.0001; 6′-GNTI versus 5′-GNTI, F(15,120) = 18.59, p < 0.0001). The percent coupling observed for 100 nm U69,593 is provided (n = 9 (6′-GNTI) and n = 8 (5′-GNTI, nor-BNI) in duplicate).
FIGURE 2.

6′-GNTI recruits little or no β-arrestin2 to the KOR and antagonizes U69,593-stimulated KOR/β-arrestin2 coupling. A, the DiscoveRx PathHunter® enzyme complementation assay was utilized to quantify β-arrestin2 recruitment to the KOR in CHO cells exposed to drug for 90 min. Although it still has efficacy over vehicle levels, 6′-GNTI induces weak recruitment of β-arrestin2 to the KOR in comparison with U69,593 (two-way ANOVA for interaction of drug × concentration: U69,593 versus 6′-GNTI, F(6,24) = 114.28, p < 0.0001). Neither 5′-GNTI nor nor-BNI stimulates β-arrestin2 interactions with the KOR. All responses were normalized to vehicle and are presented as the percent maximal stimulation induced by U69,593 ± S.E. (n = 3 performed in duplicate or triplicate). B, β-arrestin2 recruitment in CHO-KOR cells was visualized by confocal microscopy of fluorescently tagged β-arrestin2 (βarr2-GFP). In vehicle-treated cells (15 min), βarr2-GFP is localized to the cytosol. Upon treatment with U69,593 (10 μm), βarr2-GFP translocation is visible, as evident by puncta at the plasma membrane (arrow). 6′-GNTI (10 μm) does not induce any detectable βarr2-GFP translocation at 15 min post-treatment. C, pretreatment with 6′-GNTI for 1 h partially inhibits U69,593-induced (1 μm, 90 min) recruitment in the KOR DiscoveRx PathHunter® CHO cells compared with nor-BNI and 5′-GNTI, both of which fully antagonize U69,593-mediated interactions. However, 5′-GNTI is slightly more potent than nor-BNI in this assay (two-way ANOVA for interaction of drug × concentration: nor-BNI versus 6′-GNTI, F(5,24) = 11.58, p < 0.0001; 5′-GNTI versus 6′-GNTI, F(5,24) = 8.68, p < 0.0001; nor-BNI versus 5′-GNTI, F(5,24) = 4.31, p = 0.0061). The data were normalized to the percent stimulation caused by 1 μm U69,593 and presented as the mean ± S.E. (n = 3 performed in duplicate). D, confocal microscopy reveals that a 10-min pretreatment with nor-BNI (10 μm) or 6′-GNTI (10 μm) effectively prevents βarr2-GFP translocation to the plasma membrane of CHO-KOR cells treated with U69,593 (1 μm) for 10 min in comparison with cells pretreated with vehicle (0.1% DMSO) alone (arrow). E, U69,593-induced (1 μm, 10 min) βarr2-GFP translocation in CHO-KOR cells is reversed by a 10-min incubation with nor-BNI or 6′-GNTI (10 μm).
TABLE 1.
Agonist activity at KOR in the CHO-KOR cell line
Emax is expressed relative to maximal U69,593-induced activation. The EC50 and Emax values (mean ± S.E., n > 3) were calculated by nonlinear regression analysis compared with the reference ligand, U69,593. The change in transduction efficiency (ΔTE) within each assay was calculated based on the equation ΔTE = 10 × [log(τ/KA)6′-GNTI-U69]. Individual n values are presented in the figure legends. Student's t test analysis, U69,593 vs. 6′-GNTI: *, p < 0.05; ***, p < 0.001. 5′-GNTI and Nor-BNI failed to converge to stimulatory nonlinear regression curve-fitting analysis. The transduction ratio between assays ΔΔlog(τ/KA)G protein-βarr2 was calculated; the divergence from 1 indicates the degree of bias for G protein coupling over β-arrestin2 recruitment for 6′-GNTI relative to the performance of the reference ligand, U69,593, in each assay (33, 34).
| Ligand | [35S]GTPγSa |
β-Arrestin2a |
ERK1/2 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 | Emax | ΔTE | EC50 | Emax | ΔTE | EC50 | Emax | ΔTE | |
| nm | % | nm | % | nm | % | ||||
| U69,593 | 68.7 ± 9.7 | 100 | 59.3 ± 4.4 | 100 | 6.7 ± 2.3 | 100 | |||
| 6′-GNTI | 2.1 ± 0.5*** | 37 ± 2*** | 9.8 | 5.9 ± 3.3*** | 12 ± 3*** | 1.7 | 0.4 ± 0.1* | 75 ± 5*** | 7.2 |
a ΔΔ(τ/KA)G protein-βarr2 = 5.82.
TABLE 2.
Antagonist activity at KOR in CHO-KOR cell line
% Inhibition represents the relative inhibition of the stimulation induced by U69,593 in the [35S]GTPγS (100 nm) and β-arrestin2 (1 μm) assays. IC50 and % Inhibition values were calculated by nonlinear regression analysis compared with the reference antagonist nor-BNI and are presented as mean ± S.E. (n ≥ 3). Individual n values are presented in the figure legends. Student's t test analysis, nor-BNI vs. either 6′-GNTI or 5′-GNTI: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
| Ligand | [35S]-GTPγS |
β-Arrestin2 |
||
|---|---|---|---|---|
| IC50 | % Inhibition | IC50 | % Inhibition | |
| nm | nm | |||
| Nor-BNI | 0.46 ± 0.12 | 100 ± 0.01 | 2.1 ± 0.2 | 100 ± 4 |
| 6′-GNTI | 0.44 ± 0.15 | 31.7 ± 1.5*** | 10.1 ± 1.8* | 69 ± 2*** |
| 5′-GNTI | 0.07 ± 0.02** | 99.8 ± 4.3 | 0.8 ± 0.1** | 101 ± 3 |
RESULTS
G protein coupling assays were used to compare 6′-GNTI and the reference agonist, U69,593. 6′-GNTI proved to be more potent that U69,593, although it behaved as a partial agonist (Fig. 1A and Table 1). As expected, the antagonists had no effect. The classification of 6′-GNTI as a partial agonist was further confirmed by its partial inhibition of U69,593-stimulated G protein coupling, as compared with 5′-GNTI and nor-BNI (Fig. 1B and Table 2).
6′-GNTI was tested for its ability to promote KOR/β-arrestin2 interactions using both a commercial enzyme complementation assay (DiscoveRx PathHunter®) and confocal microscopy. In the DiscoveRx PathHunter® assay, U69,593 robustly induced interactions between β-arrestin2 and the KOR. In contrast to U69,593, 6′-GNTI induced very little recruitment of β-arrestin2 to the KOR (Fig. 2A and Table 1). A bias factor was calculated using the operational model (34), and the divergence from unity confirmed the apparent bias of 6′-GNTI for G protein coupling over β-arrestin2 recruitment (Table 1). Confocal microscopy was also used to assess the translocation of βarr2-GFP to the plasma membrane of CHO-KOR cells. In the basal state, βarr2-GFP could be seen distributed throughout the cell; however, upon stimulation with U69,593, βarr2-GFP robustly translocated to the plasma membrane, as evidenced by the formation of puncta near the surface of the cell within 10 min of drug treatment. As expected, neither 6′-GNTI nor the antagonists induced recruitment (Fig. 2B). In the DiscoveRx PathHunter® assay, 6′-GNTI potently, but not fully, blocked U69,593-induced recruitment of β-arrestin2, which supports its role as a partial agonist in this system (Fig. 2C and Table 2). When antagonism of β-arrestin2 recruitment was determined by confocal microscopy, 6′-GNTI, as well as nor-BNI, fully prevented and reversed U69,593-induced βarr2-GFP translocation (Fig. 2, D and E).
6′-GNTI was also tested for its ability to induce KOR internalization, which has been shown to be a β-arrestin-dependent process (36). Confocal images reveal that the N-terminal HA-tagged KOR translocates from the surface of CHO cells into intracellular vesicles following U69,593, but not 6′-GNTI, treatment (Fig. 3A). U69,593-induced internalization of the KOR can also be blocked by pretreating the cells with either 6′-GNTI or 5′-GNTI (Fig. 3B). Results from a cell surface biotinylation assay support the findings of the confocal studies, wherein treatment with U69,593, but not 6′-GNTI or 5′-GNTI, induces KOR internalization, and both 6′-GNTI and 5′-GNTI fully block U69,593-induced internalization (Fig. 3C). These findings demonstrate, in both a different cellular background and in different signaling assays than those previously reported (27), that 6′-GNTI maintains its bias toward G protein-mediated signaling over β-arrestin2 recruitment.
FIGURE 3.

6′-GNTI does not promote KOR internalization and prevents U69,593-stimulated KOR internalization. A and B, agonist-induced (10 μm) internalization of KOR in CHO-KOR cells was determined by live cell confocal microscopy. Receptors were live cell-labeled with anti-HA-tagged antibody coupled to AlexaFluor 488 prior to drug treatment. Representative images from three independent experiments are shown. A, exposure to U69,593 for 1 h causes pronounced KOR internalization, as evidenced by the marked increase in intracellular vesicles (arrow) compared with those in vehicle-treated cells. One-hour treatments with either 6′-GNTI or 5′-GNTI do not induce receptor internalization. B, pretreating the cells with either 6′-GNTI or 5′-GNTI for 30 min prevents U69,593-induced internalization (arrow) (30 min). C, agonist-induced (10 μm, 30 min) receptor internalization was quantitated in CHO-KOR cells by a cell surface biotinylation assay. For antagonist studies, cells were pretreated with either 6′-GNTI or 5′-GNTI for 30 min prior to the addition of U69,593 (U69) (one-way ANOVA: F(5,22) = 25.57, p < 0.0001; Bonferroni's post hoc test: vehicle versus drug, ***, p < 0.001; U69,593 versus 6′-GNTI or 5′-GNTI + U69,593, ^^^, p < 0.001). A representative Western blot and densitometric analysis, normalized to vehicle-treated controls and presented as the mean ± S.E., are provided. Experimental controls: 100%, no glutathione stripping; Strip, cells stripped directly after biotin labeling with no drug treatment (n = 5).
To assess the impact of the 6′-GNTI bias toward the activation of G protein coupling over β-arrestin2 recruitment on the activation of downstream signaling, ERK1/2 and Akt phosphorylation profiles were assessed in the CHO-KOR cell line. Both 6′-GNTI and U69,593 robustly induce ERK1/2 phosphorylation, whereas 5′-GNTI has no effect when assessed in an immunocytochemistry-based assay. In this assay, 6′-GNTI was again more potent than U69,593 and yet only slightly less efficacious (Fig. 4A and Table 1). ERK1/2 activation was confirmed by Western blot analysis, and the effects of both drugs can be antagonized by nor-BNI (Fig. 4B). As KOR-induced ERK1/2 phosphorylation can be biphasic, with an early and late phase of activation (21), we compared the time courses of kinase activation for the 10 μm concentration of U69,593 and 6′-GNTI by Western blot analysis. U69,593 and 6′-GNTI induced similar temporal profiles of ERK1/2 activation, with phosphorylation levels peaking by 10 min and persisting over basal levels for up to 2 h post-drug treatment (Fig. 4C). Pertussis toxin pretreatment completely abolished both U69,593- and 6′-GNTI-mediated ERK1/2 activation, indicating a Gαi/o-dependent signaling mechanism (Fig. 4D). 6′-GNTI and U69,593 stimulated Akt phosphorylation in a very similar manner to that observed for ERK1/2 (Fig. 5, A–C). Again, Akt phosphorylation in CHO-KOR cells can be blocked by pertussis toxin pretreatment (Fig. 5D).
Both the ligand and the complement of intracellular proteins available to interact with a receptor can dictate the pathway by which a receptor signals, and therefore ligand signaling profiles can vary in different cell types (19, 37). To assess whether the ligand bias we observed between U69,593 and 6′-GNTI is maintained in a more physiologically relevant system, we assessed the ability of 6′-GNTI to activate KOR-mediated signaling pathways in striatum and in primary striatal neurons from mice. In striatal membranes prepared from adult mice, U69,593 induced stimulation of G protein coupling, whereas 6′-GNTI had no effect (Fig. 6). It is common to encounter small assay windows when performing G protein coupling in brain tissue, as evidenced by the 40% stimulation achieved by the full agonist, U69,593. Therefore, it is not surprising that 6′-GNTI, which could only stimulate 35% of the maximal coupling achieved with U69,593 in the overexpression system, did not produce detectable stimulation of G protein coupling in this system. To overcome this limitation of the system, we tested whether 6′-GNTI would maintain its role as a partial agonist by determining whether it could displace U69,593-stimulated coupling. In accordance with its role as a partial agonist at KOR, 6′-GNTI, when included at a constant concentration, suppressed the efficacy of U69,593, demonstrating that 6′-GNTI has a direct impact on KOR-mediated G protein coupling in striatum.
FIGURE 6.
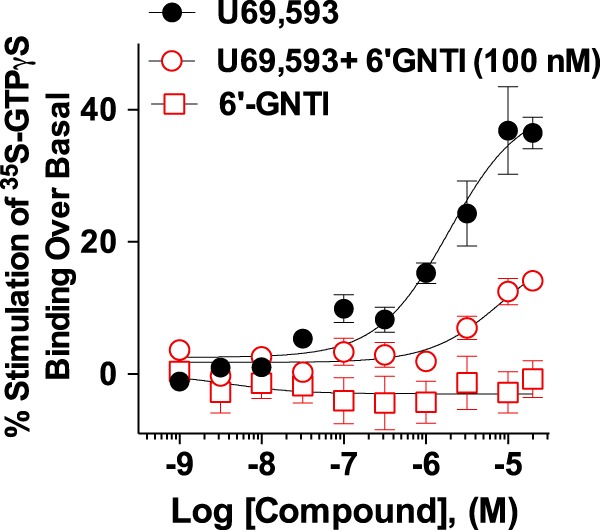
6′-GNTI modulates U69,593-mediated G protein coupling in mouse striatum. [35S]GTPγS incorporation was determined for striatal membranes prepared from WT mice following drug treatment (2 h). U69,593 stimulates [35S]GTPγS binding over vehicle levels (EC50 = 25,680 nm ± 15,590 nm; Emax = 1.752 ± 0.203), whereas 6′-GNTI has no effect (non-convergence, R2 < 0.6). 6′-GNTI (100 nm) reduces U69,593-induced G protein coupling (EC50 = 14,210 nm ± 3,715 nm; Emax = 1.244 ± 0.036) (two-way ANOVA for interaction of drug × concentration: U69,593 versus U69,593 + 6′-GNTI, F(9,96) = 8.66, p < 0.0001). Values are reported as the mean ± S.E. Data were fit to nonlinear regression curves. The -fold stimulation over vehicle treatment is provided (n = 6; performed in duplicate).
In striatal neurons cultured from WT neonates, U69,593 induced the phosphorylation of ERK1/2, which was blocked by pretreatment with nor-BNI (Fig. 7A). In contrast to U69,593, exposure to 6′-GNTI caused a decrease in ERK1/2 phosphorylation to a level below that observed in vehicle treated neurons. The 6′-GNTI-induced repression of ERK1/2 was not sensitive to nor-BNI pretreatment; however, nor-BNI alone suppressed ERK1/2 phosphorylation below vehicle levels, suggesting that 6′-GNTI may act through a mechanism similar to nor-BNI to dampen the basal activation of this pathway. The time course of 6′-GNTI-induced ERK1/2 phosphorylation was not shifted, as treatment with 6′-GNTI for 2 min also revealed no ERK1/2 phosphorylation, demonstrating that the kinase activation window was not simply overshot by the 10-min incubation period.
FIGURE 7.
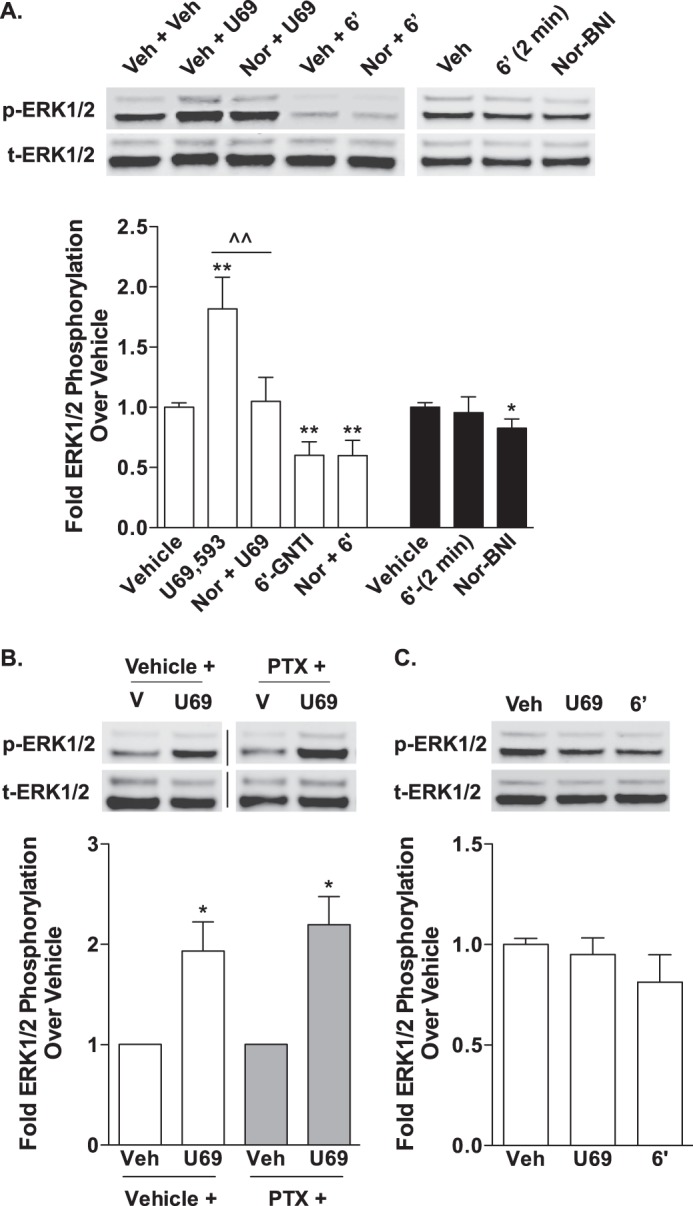
U69,593 and 6′-GNTI have divergent signaling effects on ERK1/2 in mouse primary striatal neurons. Primary striatal neurons were serum-starved for 1 h prior to the determination of ERK1/2 phosphorylation (10 μm for 10 min). Representative images of Western blots and densitometric analyses are provided. For each sample, phosphorylated ERK1/2 (p-ERK1/2) was first normalized to total ERK1/2 (t-ERK1/2), and the -fold stimulation over vehicle is provided (mean ± S.E.). A, WT neurons were pretreated with either vehicle (Veh, 0.9% saline) or nor-BNI (Nor, 1 μm) during the last 15 min of the serum starvation. U69,593 (U69) activates ERK1/2 in striatal neurons in a nor-BNI-sensitive manner (one-way ANOVA: for U69,593, F(2,18) = 9.550, p = 0.0015; Bonferroni's post hoc test: vehicle versus U69,593, **, p < 0.01, U69,593 versus nor-BNI + U69,593, ^^, p < 0.01). In contrast, 6′-GNTI (6′) represses ERK1/2 activation in striatal neurons, and this repression is not sensitive to nor-BNI (one-way ANOVA: for 6′-GNTI, F(2,19) = 9.527, p = 0.0014; Bonferroni's post hoc test: vehicle versus drug treatment groups, **, p < 0.01; n = 5–6 independent neuronal preparations). In a separate set of experiments, the neurons were treated with 6′-GNTI for 2 min or nor-BNI (1 μm) alone for 10 min. 6′-GNTI-mediated activation of ERK1/2 is not merely shifted, as a 2-min treatment does not shift phosphorylation levels away from basal (Student's t test: p > 0.05). Nor-BNI treatment alone also represses ERK1/2 phosphorylation levels (Student's t test: *, p < 0.05; n = 4 independent neuronal preparations). B, U69,593-mediated ERK1/2 activation in WT striatal neurons is not sensitive to overnight pretreatment with pertussis toxin (PTX; 100 ng/ml) (Student's t test: vehicle versus U69,593 within each pretreatment group, *, p < 0.05; n = 3 independent neuronal preparations). Representative images are from different parts of the same gel. C, neither U69,593 nor 6′-GNTI induces an increase in ERK1/2 phosphorylation levels in striatal neurons cultured from βarr2-KO mice (Student's t test: p > 0.05; n = 4 independent neuronal preparations).
In CHO-KOR cells, ERK1/2 phosphorylation was found to be pertussis toxin-sensitive. Therefore, we next determined whether U69,593-mediated ERK1/2 phosphorylation in striatal neurons is G protein-dependent. Unlike in the CHO-KOR cells, pertussis toxin pretreatment had no effect on U69,593-induced ERK1/2 activation (Fig. 7B), suggesting that U69,593 induces ERK1/2 phosphorylation independent of Gαi/o protein coupling. Because GPCRs can activate ERK1/2 by both G protein- and β-arrestin-dependent mechanisms, striatal neurons cultured from neonates lacking β-arrestin2 (βarr2-KO) were used to assess the contributions of β-arrestin2 to ERK1/2 activation. Similar to 6′-GNTI in WT neurons, treatment with U69,593 did not induce ERK1/2 phosphorylation above vehicle levels in the βarr2-KO neurons (Fig. 7C). Together, these studies indicate that U69,593 induces ERK1/2 phosphorylation through a KOR-mediated, β-arrestin2-dependent, pertussis toxin-insensitive mechanism.
Interestingly, when we assessed Akt phosphorylation in WT striatal neurons following treatment with U69,593 and 6′-GNTI, we found that both agonists induced Akt phosphorylation to similar levels (Fig. 8A). Pretreatment with nor-BNI blocked the Akt phosphorylation observed with either compound, suggesting that 6′-GNTI has the potential to fully activate this KOR-mediated signaling event in striatal neurons. In contrast to the ERK1/2 studies in striatal neurons, pretreatment with pertussis toxin fully inhibited U69,593 and 6′-GNTI-stimulated Akt phosphorylation (Fig. 8B), whereas both agonists were able to activate Akt in the absence of β-arrestin2 (Fig. 8C).
FIGURE 8.
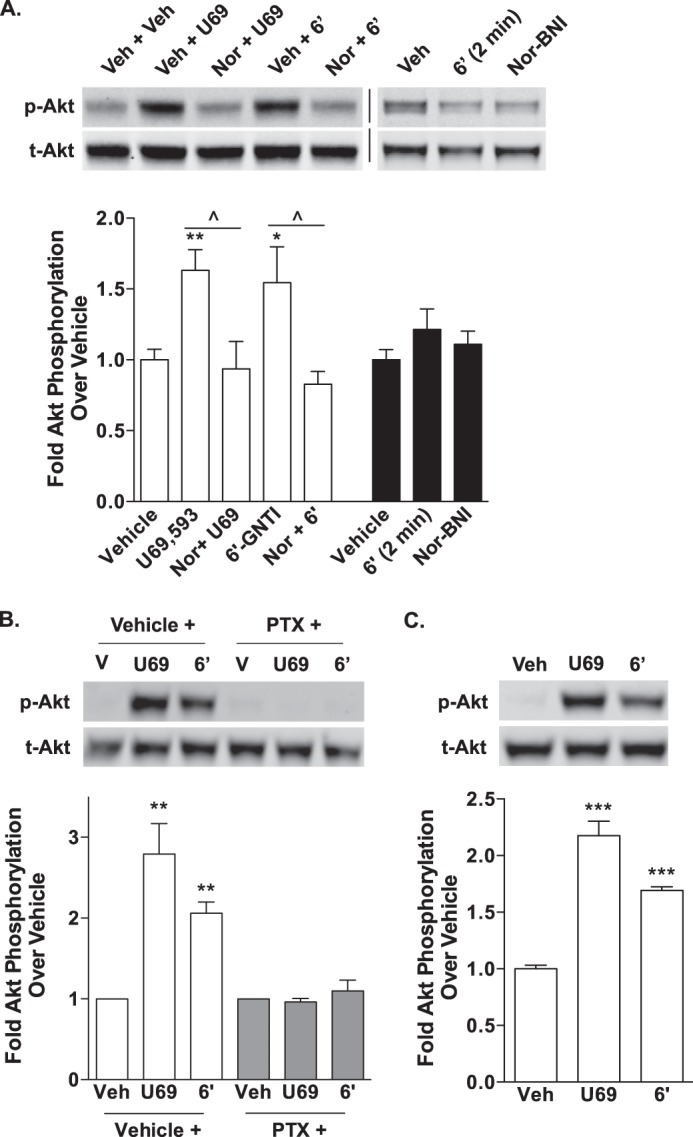
U69,593- and 6′-GNTI-mediated Akt phosphorylation in mouse primary striatal neurons is pertussis toxin-sensitive and β-arrestin2-independent. Primary striatal neurons were serum-starved for 1 h prior to the determination of Akt phosphorylation (10 μm for 10 min). Representative images of Western blots and densitometric analyses are provided. For each sample, phosphorylated Akt (p-Akt) was first normalized to total Akt (t-Akt), and the -fold stimulation over vehicle is provided (mean ± S.E.). A, WT neurons were pretreated with either vehicle (Veh, 0.9% saline) or nor-BNI (Nor, 1 μm) during the last 15 min of the serum starvation. Akt is activated by both U69,593 (U69) and 6′-GNTI (6′) in a nor-BNI-sensitive manner (one-way ANOVA: for U69,593, F(2,13) = 8.247, p = 0.0049; for 6′-GNTI, F(2,14) = 5.662, p = 0.0158; Bonferroni's post hoc test: vehicle versus drug treatment groups, *, p < 0.05, **, p < 0.01; drug versus nor-BNI + drug, ^, p < 0.05; n = 5–6 independent neuronal preparations). Nor-BNI treatment alone does not alter Akt phosphorylation from vehicle levels (Student's t test: p > 0.05; n = 4 independent neuronal preparations). B, U69,593- and 6′-GNTI-induced Akt activation in WT striatal neurons is sensitive to overnight pretreatment with pertussis toxin (PTX; 100 ng/ml) (Student's t test: vehicle (V) versus drug treatment within each pretreatment group, **, p < 0.01; n = 3 independent neuronal preparations). C, U69,593 and 6′-GNTI induce an increase in Akt phosphorylation levels in striatal neurons cultured from βarr2-KO mice (Student's t test: ***, p < 0.001; n = 4 independent neuronal preparations).
DISCUSSION
In comparison with U69,593, 6′-GNTI is a highly potent, partial agonist in G protein coupling (Fig. 1) as well as in ERK1/2 (Fig. 4) and Akt (Fig. 5) phosphorylation assays when assessed in KOR-expressing CHO cell lines. We confirmed here that 6′-GNTI promotes only marginal β-arrestin2 recruitment in the commercial DiscoveRx PathHunterTM cell line and no detectable recruitment of a GFP-tagged β-arrestin2 to KOR, as assessed by confocal microscopy (Fig. 2). Consistent with its weak ability to recruit β-arrestin2, 6′-GNTI did not promote subsequent receptor internalization (Fig. 3). Overall, 6′-GNTI displayed signaling bias toward G protein coupling over β-arrestin2 recruitment in the cell lines (Table 1).
When the two agonists are compared for induction of KOR signaling in neuronal cultures, it was found that U69,593 activates ERK1/2, whereas 6′-GNTI does not. Conversely, 6′-GNTI decreases the overall level of ERK1/2 activation, an effect that is also observed with the KOR antagonist nor-BNI (Fig. 7). It is attractive to hypothesize that the lack of ERK1/2 activation is due to the inability of 6′-GNTI to recruit β-arrestin2 and that β-arrestin2 may mediate KOR signaling to ERK1/2. This idea is supported by the following observations: 1) U69,593-induced ERK1/2 activation is not sensitive to pertussis toxin, suggesting that KOR to ERK1/2 signaling is G protein-independent; and 2) U69,593-induced ERK1/2 signaling does not ensue in the absence of β-arrestin2, as demonstrated in striatal neurons isolated from βarr2-KO mice.
An alternative hypothesis is that 6′-GNTI, as a partial agonist, is not efficacious enough to reveal a response in the endogenous setting where receptor numbers are relatively low compared with the overexpressing cell line (CHO-KOR cell line, ∼1800 fmol/mg membrane protein; mouse striatum, ∼40 fmol/mg membrane protein (5)). The fact that 6′-GNTI-induced G protein coupling could not be observed in striatal membranes supports this idea. However, treatment of striatal neurons with 6′-GNTI stimulated Akt phosphorylation to the same extent as treatment with U69,593, suggesting that the agonist is capable of fully activating KOR in this setting (Fig. 8). Moreover, the activation of Akt is pertussis toxin-sensitive, suggesting that 6′-GNTI, as well as U69,593, utilizes a Gαi/o protein signaling pathway to induce this response in striatal neurons. It is not clear, however, why 6′-GNTI behaves as a full agonist for activating AKT in the neurons when it only partially activates Gαi/o coupling in brain. This could be due to the differences in brain tissue (whole striatum) versus neuronal culture preparations. Alternatively, it could reflect differences in assessing the proximal G protein coupling in the isolated membranes versus measuring the accumulation of the downstream amplified AKT phosphorylation that is measured after treating live, intact neurons. Another potential explanation is that the G protein-mediated signal to Akt is less hampered when the β-arrestin2 is antagonized by 6′-GNTI, as suggested in the cell culture assays.
Whereas KOR-mediated activation of ERK1/2 requires β-arrestin2 in striatal neurons, ERK1/2 phosphorylation in CHO cells appears to be completely dependent upon pertussis toxin-sensitive G protein signaling. It is not clear from this study why the neurons and the CHO cells differ regarding their utilization of β-arrestin2 in this signaling cascade; however, such observations have precedence. For example, serotonin, N-methyltryptamine, and 2,5-dimethoxy-4-iodoamphetamine induce Akt phosphorylation downstream of the serotonin 2A receptor in C6 glioma and PC12 cells (38),6 but only serotonin induces Akt phosphorylation in mouse cortical neurons, a process that has been shown to require β-arrestin2 to mediate serotonin-induced activation of Akt in mouse cortical neurons (18). It is attractive to speculate that there are components of the immediate environment of KOR endogenously expressed in neurons, that are not present in the transfected Chinese hamster ovary cells and that provide an assembly of a functional “receptorsome” unique to the striatal neurons. These observations suggest that KOR expressed in striatum require β-arrestin2 to activate ERK1/2. The nature of these neuronal culture-specific complements remains to be determined, as well as the physiological and behavioral consequences of such signaling. The development and evaluation of agonists that selectively activate one pathway over another, much like 6′-GNTI, will allow for such elucidations. Overall, the findings described here underscore the importance of evaluating compound functional selectivity in an endogenous setting.
In the current study, 6′-GNTI was shown to be very potent at activating KOR-mediated G protein coupling, which is in agreement with a study showing that 6′-GNTI potently inhibits cAMP accumulation (27). However, in HEK-293 cells expressing a modified Gα protein (Δ6-Gqi4-myr) coupled to a calcium channel reporter assay, the potency of 6′-GNTI was substantially lower (∼224 nm) than the potency observed for U69,593 (∼0.8 nm) (39). The co-expression of the DOR along with the KOR improved the potency of 6′-GNTI (∼40 nm) but did not affect the potency for U69,593, which led us to conclude that 6′-GNTI is a weak agonist at KOR but a more potent agonist at the KOR/DOR heteromer. This observation was not recapitulated in studies assessing cAMP accumulation, where DOR co-expression had no effect on the potency of 6′-GNTI (27). In the present study, we also found that 6′-GNTI is a potent, partial agonist at the KOR using a standard [35S]GTPγS binding assay in CHO-KOR cellular membranes, and as only KOR is overexpressed in the cell line, presumably the actions due to activation of KOR. One potential explanation for the discrepancies is the use of different cell lines. Another possible explanation may be found in the nature of the assays themselves. In contrast to the [35S]GTPγS binding assay, which assesses interactions between the receptor and its endogenous G proteins, the Δ6-Gqi4-myr-expressing cell line measures changes in calcium fluorophore release due to receptor activation of a chimeric Gαq/i protein (40). Therefore, these differences may not be reflective of the low capacity of 6′-GNTI to activate the KOR monomer/homomer but rather its relative low potency in promoting KOR coupling to the modified, chimeric G proteins. Collectively, these findings emphasize the importance of the cellular context in receptor and drug function, as the complement of intracellular proteins, which differ between cell types (i.e. the presence of homomers or heteromers, G protein-coupled receptor kinases, and other scaffolding or regulatory proteins), will determine the signaling and regulatory events that occur downstream of receptor activation.
Discrepancies between pharmacological characterizations made across cell-based assays and between those made in endogenous cell types underscore the need to be cautious in assigning classifications of ligands (such as agonist, biased agonist, partial agonist, etc.). Cell-based assays, such as DiscoveRx PathHunter® or those employing the Δ6-Gqi4-myr protein, are optimized to facilitate high-throughput screening of drug properties. However, as these assays are increasingly utilized for pharmacological assessment, care must be taken in determining whether the differential properties of the ligand detected (such as functional selectivity) can be extrapolated to a physiological context or whether they are artifacts of the cell-based assay utilized in the initial characterization (41). The differences in the results between the two assays used to determine β-arrestin2 translocation to the KOR demonstrate how the use of highly modified cellular systems can pose a challenge in determining the properties of a drug. Consistent with the published finding utilizing bioluminescence resonance energy transfer (BRET) (27), the confocal studies presented in this current study reveal that 6′-GNTI does not induce any detectable translocation of β-arrestin2 to the plasma membrane (Fig. 2B). In contrast, in the KOR-expressing CHO-K1 DiscoveRx PathHunter® cell-based assay, 6′-GNTI induces low efficacy but high potency recruitment of β-arrestin2 to the KOR (Fig. 2A). These differences may be due to intrinsic differences in receptor numbers, assay sensitivity, cell type, time scales, or competition with endogenously expressed, untagged β-arrestin2. Furthermore, the differences could be artifacts of the modified β-arrestin2 or KOR constructs used in each assay, encompassing GFP, Renilla luciferase 8, and modified LacZ holoenzyme fusion proteins, which may obscure or distort subtle variations in drug-induced β-arrestin2 recruitment. However, 6′-GNTI does not induce receptor internalization in either transiently transfected HEK-293 cells (data not shown) or stably transfected CHO-KOR cells, as determined by confocal microscopy, biotinylation, and FACS analysis (Fig. 3) (27). Moreover, 6′-GNTI does not promote KOR-mediated ERK1/2 activation in striatal neurons, a signaling pathway that we show here requires β-arrestin2, yet 6′-GNTI maintains full efficacy for activating G protein-dependent signaling to Akt in the same neuronal population. Together these observations suggest that the bias for G protein- over β-arrestin2-mediated signaling induced by 6′-GNTI actions at KOR are preserved in an endogenous setting.
Functionally biased compounds may prove to be useful tools in delineating the relationship between the activation of particular pathways and specific effects at the cellular level in a manner that may also be translated and studied at the behavioral level in animal models, particularly in determining the involvement of KOR interactions with β-arrestin2 in the biological effects of KOR ligands. As our data demonstrate, drug screening in more physiologically relevant systems is necessary to truly ascertain the pharmacological profile of a drug. Although the studies in the KOR-expressing cell lines would suggest that KOR-induced activation of ERK1/2 is mediated entirely via pertussis toxin-sensitive G proteins, the studies in striatal neurons demonstrate that KOR signals to ERK1/2 by way of a G protein-independent, β-arrestin2-dependent pathway. This finding may be particularly useful in determining whether newly developed agonists that are biased against β-arrestin2 recruitment maintain that bias in the endogenous setting, as the activation of ERK1/2 in neurons can be readily measured. The elucidation of basic drug properties at the isolated receptor in simplified culture systems is still an important part of the drug discovery process. These systems provide the basic understanding of drug/receptor function and capabilities, which can then be modified by the study of different cellular contexts with different complements of receptor-binding proteins and other components of the signaling cascade. However, care must be taken when trying to attribute the signaling responses observed in cell-based assays to the pharmacological profiles obtained in vivo.
Acknowledgments
We thank Drs. Cécile Béguin and Bruce Cohen (McLean Hospital/Harvard Medical School) for providing the library of KOR ligands used in the initial screen. We thank Dr. Arthur Christopoulos of (Monash University, Melbourne, Australia) for thoughtful discussions on bias factor determinations.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DA031927 (to L. M. B.).
C. L. Schmid and L. M. Bohn, unpublished observations.
- KOR
- κ-opioid receptor
- ANOVA
- analysis of variance
- βarr2
- β-arrestin2
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DOR
- δ-opioid receptor
- GNTI
- guanidinonaltrindole
- GPCR
- G protein-coupled receptor
- nor-BNI
- norbinaltorphimine.
REFERENCES
- 1. Glick S. D., Maisonneuve I. M., Raucci J., Archer S. (1995) Kappa opioid inhibition of morphine and cocaine self-administration in rats. Brain Res. 681, 147–152 [DOI] [PubMed] [Google Scholar]
- 2. Negus S. S., Mello N. K., Portoghese P. S., Lin C. E. (1997) Effects of kappa opioids on cocaine self-administration by rhesus monkeys. J. Pharmacol. Exp. Ther. 282, 44–55 [PubMed] [Google Scholar]
- 3. Schenk S., Partridge B., Shippenberg T. S. (1999) U69593, a kappa-opioid agonist, decreases cocaine self-administration and decreases cocaine-produced drug seeking. Psychopharmacology (Berl.) 144, 339–346 [DOI] [PubMed] [Google Scholar]
- 4. Potter D. N., Damez-Werno D., Carlezon W. A., Jr., Cohen B. M., Chartoff E. H. (2011) Repeated exposure to the kappa-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol. Psychiatry 70, 744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van't Veer A., Bechtholt A. J., Onvani S., Potter D., Wang Y., Liu-Chen L. Y., Schütz G., Chartoff E. H., Rudolph U., Cohen B. M., Carlezon W. A., Jr. (2013) Ablation of kappa-opioid receptors from brain dopamine neurons has anxiolytic-like effects and enhances cocaine-induced plasticity. Neuropsychopharmacology 38, 1585–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carlezon W. A., Jr., Béguin C., DiNieri J. A., Baumann M. H., Richards M. R., Todtenkopf M. S., Rothman R. B., Ma Z., Lee D. Y., Cohen B. M. (2006) Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J. Pharmacol. Exp. Ther. 316, 440–447 [DOI] [PubMed] [Google Scholar]
- 7. Cohen B. M., Murphy B. (2008) The effects of pentazocine, a kappa agonist, in patients with mania. Int. J. Neuropsychopharmacol. 11, 243–247 [DOI] [PubMed] [Google Scholar]
- 8. Walsh S. L., Geter-Douglas B., Strain E. C., Bigelow G. E. (2001) Enadoline and butorphanol: evaluation of kappa-agonists on cocaine pharmacodynamics and cocaine self-administration in humans. J. Pharmacol. Exp. Ther. 299, 147–158 [PubMed] [Google Scholar]
- 9. Bodkin J. A., Zornberg G. L., Lukas S. E., Cole J. O. (1995) Buprenorphine treatment of refractory depression. J. Clin. Psychopharmacol. 15, 49–57 [DOI] [PubMed] [Google Scholar]
- 10. Mague S. D., Pliakas A. M., Todtenkopf M. S., Tomasiewicz H. C., Zhang Y., Stevens W. C., Jr., Jones R. M., Portoghese P. S., Carlezon W. A., Jr. (2003) Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J. Pharmacol. Exp. Ther. 305, 323–330 [DOI] [PubMed] [Google Scholar]
- 11. Knoll A. T., Muschamp J. W., Sillivan S. E., Ferguson D., Dietz D. M., Meloni E. G., Carroll F. I., Nestler E. J., Konradi C., Carlezon W. A., Jr. (2011) Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol. Psychiatry 70, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Land B. B., Bruchas M. R., Schattauer S., Giardino W. J., Aita M., Messinger D., Hnasko T. S., Palmiter R. D., Chavkin C. (2009) Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc. Natl. Acad. Sci. U.S.A. 106, 19168–19173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aldrich J. V., Patkar K. A., McLaughlin J. P. (2009) Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc. Natl. Acad. Sci. U.S.A. 106, 18396–18401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Broadbear J. H., Negus S. S., Butelman E. R., de Costa B. R., Woods J. H. (1994) Differential effects of systemically administered nor-binaltorphimine (nor-BNI) on kappa-opioid agonists in the mouse writhing assay. Psychopharmacology (Berl.) 115, 311–319 [DOI] [PubMed] [Google Scholar]
- 15. Ko M. C., Willmont K. J., Lee H., Flory G. S., Woods J. H. (2003) Ultra-long antagonism of kappa opioid agonist-induced diuresis by intracisternal norbinaltorphimine in monkeys. Brain Res. 982, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kenakin T. (2007) Functional selectivity through protean and biased agonism: who steers the ship? Mol. Pharmacol. 72, 1393–1401 [DOI] [PubMed] [Google Scholar]
- 17. Urban J. D., Clarke W. P., von Zastrow M., Nichols D. E., Kobilka B., Weinstein H., Javitch J. A., Roth B. L., Christopoulos A., Sexton P. M., Miller K. J., Spedding M., Mailman R. B. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1–13 [DOI] [PubMed] [Google Scholar]
- 18. Schmid C. L., Bohn L. M. (2010) Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ss-arrestin2/Src/Akt signaling complex in vivo. J. Neurosci. 30, 13513–13524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raehal K. M., Schmid C. L., Groer C. E., Bohn L. M. (2011) Functional selectivity at the mu-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 63, 1001–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mailman R. B. (2007) GPCR functional selectivity has therapeutic impact. Trends Pharmacol. Sci. 28, 390–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McLennan G. P., Kiss A., Miyatake M., Belcheva M. M., Chambers K. T., Pozek J. J., Mohabbat Y., Moyer R. A., Bohn L. M., Coscia C. J. (2008) Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J. Neurochem. 107, 1753–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bruchas M. R., Yang T., Schreiber S., Defino M., Kwan S. C., Li S., Chavkin C. (2007) Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J. Biol. Chem. 282, 29803–29811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Negri A., Rives M. L., Caspers M. J., Prisinzano T. E., Javitch J. A., Filizola M. (2013) Discovery of a novel selective kappa-opioid receptor agonist using crystal structure-based virtual screening. J. Chem. Inf. Model. 53, 521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spetea M., Asim M. F., Noha S., Gerhard G., Schmidhammer H. (2013) Current kappa opioid receptor ligands and discovery of a new molecular scaffold as a kappa opioid receptor antagonist using pharmacophore-based virtual screening. Curr. Pharm. Des., in press [DOI] [PubMed] [Google Scholar]
- 25. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human kappa-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sharma S. K., Jones R. M., Metzger T. G., Ferguson D. M., Portoghese P. S. (2001) Transformation of a kappa-opioid receptor antagonist to a kappa-agonist by transfer of a guanidinium group from the 5′- to 6′-position of naltrindole. J. Med. Chem. 44, 2073–2079 [DOI] [PubMed] [Google Scholar]
- 27. Rives M. L., Rossillo M., Liu-Chen L. Y., Javitch J. A. (2012) 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment. J. Biol. Chem. 287, 27050–27054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., Lin F. T. (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 29. Bohn L. M., Dykstra L. A., Lefkowitz R. J., Caron M. G., Barak L. S. (2004) Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol. Pharmacol. 66, 106–112 [DOI] [PubMed] [Google Scholar]
- 30. Groer C. E., Tidgewell K., Moyer R. A., Harding W. W., Rothman R. B., Prisinzano T. E., Bohn L. M. (2007) An opioid agonist that does not induce mu-opioid receptor-arrestin interactions or receptor internalization. Mol. Pharmacol. 71, 549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmid C. L., Raehal K. M., Bohn L. M. (2008) Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 1079–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Black J. W., Leff P. (1983) Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 220, 141–162 [DOI] [PubMed] [Google Scholar]
- 33. Kenakin T., Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216 [DOI] [PubMed] [Google Scholar]
- 34. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Evans B. A., Broxton N., Merlin J., Sato M., Hutchinson D. S., Christopoulos A., Summers R. J. (2011) Quantification of functional selectivity at the human alpha(1A)-adrenoceptor. Mol. Pharmacol. 79, 298–307 [DOI] [PubMed] [Google Scholar]
- 36. Li J. G., Luo L. Y., Krupnick J. G., Benovic J. L., Liu-Chen L. Y. (1999) U50,488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J. Biol. Chem. 274, 12087–12094 [DOI] [PubMed] [Google Scholar]
- 37. Bohn L. M. (2009) Selectivity for G protein- or arrestin-mediated signaling, in Functional Selectivity of G Protein-Coupled Receptor Ligands (Neve K., ed) 1st Ed., pp. 71–85, Humana Press, Totowa, NJ [Google Scholar]
- 38. Cowen D. S., Johnson-Farley N. N., Travkina T. (2005) 5-HT receptors couple to activation of Akt, but not extracellular-regulated kinase (ERK), in cultured hippocampal neurons. J. Neurochem. 93, 910–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Waldhoer M., Fong J., Jones R. M., Lunzer M. M., Sharma S. K., Kostenis E., Portoghese P. S., Whistler J. L. (2005) A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. U.S.A. 102, 9050–9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kostenis E. (2001) Is Galpha16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors? Trends Pharmacol. Sci. 22, 560–564 [DOI] [PubMed] [Google Scholar]
- 41. Bohn L. M., McDonald P. H. (2010) Seeking ligand bias: assessing GPCR coupling to beta-arrestins for drug discovery. Drug Discov. Today Technol. 7, e37–e42 [DOI] [PMC free article] [PubMed] [Google Scholar]