

Background: Catalytic activity of prokaryotic and eukaryotic arylamine N-acetyltransferases (NATs) relies on a strictly conserved catalytic Cys-His-Asp triad.
Results: Structural and biochemical studies identified a functional NAT with a Cys-His-Glu catalytic triad.
Conclusion: The catalytic triad of these acetyltransferases is more plastic than previously believed.
Significance: (BACCR)NAT3 represents the first known functional acetyltransferase with a non-canonical Cys-His-Glu catalytic triad.
Keywords: Acetyl Coenzyme A, Crystal Structure, Enzyme Mechanisms, Enzyme Mutation, Enzyme Structure
Abstract
Arylamine N-acetyltransferases (NATs), a class of xenobiotic-metabolizing enzymes, catalyze the acetylation of aromatic amine compounds through a strictly conserved Cys-His-Asp catalytic triad. Each residue is essential for catalysis in both prokaryotic and eukaryotic NATs. Indeed, in (HUMAN)NAT2 variants, mutation of the Asp residue to Asn, Gln, or Glu dramatically impairs enzyme activity. However, a putative atypical NAT harboring a catalytic triad Glu residue was recently identified in Bacillus cereus ((BACCR)NAT3) but has not yet been characterized. We report here the crystal structure and functional characterization of this atypical NAT. The overall fold of (BACCR)NAT3 and the geometry of its Cys-His-Glu catalytic triad are similar to those present in functional NATs. Importantly, the enzyme was found to be active and to acetylate prototypic arylamine NAT substrates. In contrast to (HUMAN) NAT2, the presence of a Glu or Asp in the triad of (BACCR)NAT3 did not significantly affect enzyme structure or function. Computational analysis identified differences in residue packing and steric constraints in the active site of (BACCR)NAT3 that allow it to accommodate a Cys-His-Glu triad. These findings overturn the conventional view, demonstrating that the catalytic triad of this family of acetyltransferases is plastic. Moreover, they highlight the need for further study of the evolutionary history of NATs and the functional significance of the predominant Cys-His-Asp triad in both prokaryotic and eukaryotic forms.
Introduction
Arylamine N-acetyltransferases (NATs4; EC. 2.3.1.5) are phase II xenobiotic-metabolizing enzymes. These xenobiotic-metabolizing enzymes play an important role in the biotransformation and biological fates of aromatic amine compounds like drugs, carcinogens, and toxins (1, 2). NATs are structurally related to transglutaminases and cysteine proteases and possess a strictly conserved Cys-His-Asp catalytic triad (3–5). In functional NATs, this catalytic triad allows the N-acetylation of arylamines and/or O-acetylation of arylhydroxylamines through a Ping Pong Bi Bi mechanism (6, 7).
Site-directed mutagenesis experiments have shown that the NAT catalytic reaction unequivocally requires each of the Cys, His, and Asp catalytic residues (8–10). Accordingly, all known functional prokaryotic and eukaryotic NATs harbor a Cys-His-Asp catalytic triad (11, 12). Natural variations in the catalytic triad residues are poorly documented among the NAT family. So far, one unique natural modification of a catalytic triad residue has been reported and characterized. A single nucleotide polymorphism (364G→A) in a (HUMAN)NAT2 isozyme (NAT2*12D) leads to an inactive NAT2 variant in which the catalytic triad Asp residue is replaced by Asn (10). Surprisingly, mutation of the catalytic Asp to the chemically analogous Glu in (HUMAN)NAT2 produced a variant with very low enzyme activity (2% of the wild-type activity). This Asp-to-Glu variant is also expressed at very low levels, suggesting that the substitution alters the structure of the enzyme (10).
Analyses of functional prokaryotic and eukaryotic NATs and those with triad variants prompted the hypothesis that a strict Cys-His-Asp catalytic triad is required for catalysis by this family of acetyltransferases (10, 11). Interestingly, however, among the three nat genes present in Bacillus cereus, one encodes for a putative NAT isoenzyme, arylamine N-acetyltransferase 3 ((BACCR)NAT3), with an atypical Cys-His-Glu catalytic triad (9). The catalytic activity and structure of this atypical NAT have not been studied presumably because it was assumed to be inactive.
Here, x-ray crystallography, site-directed mutagenesis, biophysical, and computational approaches were combined to characterize this atypical NAT protein. We show that, despite the presence of a Glu residue in its catalytic triad, this atypical NAT is functional and able to acetylate prototypic NAT substrates. In contrast to (HUMAN)NAT2, (BACCR)NAT3 can support either a Glu or Asp residue in its catalytic triad with no significant structural or catalytic impact. Our data show that, contrary to current knowledge, the plasticity of the active site of this family of acetyltransferases also extends to their catalytic triad residues. More broadly, it raises the question of the evolutionary history of NAT enzymes and in particular of the functional significance of the predominance of the Cys-His-Asp triad in both prokaryotic and eukaryotic isozymes.
EXPERIMENTAL PROCEDURES
Chemical Compounds, Strains, and Culture Conditions
4-Aminosalicylic acid, 2-aminofluorene, isoniazid, acetyl coenzyme A (AcCoA), para-nitrophenyl acetate (PNPA), iodoacetamide (IAA), isopropyl β-d-1-thiogalactopyranoside, imidazole, lysozyme, kanamycin, and guanidine hydrochloride (GdmCl) were purchased from Sigma. Substrates were prepared in Milli-Q water or dimethyl sulfoxide (DMSO; Euromedex) at 100 mm and stored at −20 °C until use. Protease inhibitor mixture (100×) was prepared with 100 mm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride and 2 mm leupeptin (Euromedex). Other compounds were provided by Sigma unless specified. Escherichia coli BL21 strains were cultured in Luria broth (LB) Miller medium at 37 °C. B. cereus strain ATCC 14579 was kindly provided by Pr. I. Martin-Verstraete (Institut Pasteur, Paris, France).
Amino Acid Sequence Analysis of B. cereus NAT3
The nat3 open reading frame (ORF) coding for the (BACCR)NAT3 was identified using BLASTp. Multiple sequence alignments with other NATs were achieved with Muscle at the European Bioinformatics Institute. All parameters were set to defaults. Alignment outputs were generated with ESPript V2.2.
Cloning of B. cereus nat3 ORF and Construction of an E123D Mutant
Cloning of the ORF coding for (BACCR)NAT3 wild type (WT) was reported in Kubiak et al. (13). The (BACCR)NAT3 E123D mutant was generated by site-directed mutagenesis. Two independent PCRs were conducted on genomic DNA, one using primers (BACCR)NAT3_for (5′-CCGGATCCATGACCGACTTTCAAAAACA-3′) and E123D_rev (5′-CGAATCCTACGTCAATTAAAT-3′) and the other using primers E123_for (5′-ATTTAATTGACGTAGGATTCG-3′) and (BACCR)NAT3_rev (5′-GGGAGCTCTTATAATGTAATTCCGTT-3′). A third PCR using primers (BACAN)NAT3_for and (BACAN)NAT3_rev was used to obtain a single amplicon containing the E123D mutation. PCR products were double digested using BamHI and XhoI (New England Biolabs) and cloned into a pET28(a) plasmid for further expression of His6-tagged fusion recombinant proteins. Cloning was verified by DNA sequencing.
Expression and Purification of Recombinant (BACCR)NAT3 WT and E123D Enzymes
E. coli BL21 strains containing the (BACCR)NAT3 WT or E123D constructions were grown at 37 °C until absorbance at 600 nm reached 0.7. Addition of 500 μm isopropyl β-d-1-thiogalactopyranoside for 5 h at 25 °C started the induction of NAT protein overexpression. Bacteria were then resuspended in phosphate-buffered saline (PBS), pH 7.4, 10 mg of lysozyme, 0.1% Triton X-100, and 1× protease inhibitor mixture prior to sonication at 4 °C (3 min; 8-s pulse on, 30-s pulse off; Branson Digital Sonicator model 450). Bacterial debris were pelleted (30 min, 12,000 × g, 4 °C), and supernatants were incubated with Chromatrix nickel-nitrilotriacetic acid beads (Jena Bioscience) for a minimum of 2 h at 4 °C. Extensive washes were achieved on columns successively with PBS with 0, 20, and 30 mm imidazole before elution of His6-tagged NAT enzymes with PBS with 300 mm imidazole. Finally, proteins were reduced with 10 mm dithiothreitol (DTT) and dialyzed overnight against 25 mm Tris-HCl, pH 7.5. Proteins were digested with thrombin and further purified by ion exchange chromatography (Mono Q column, ÄKTA system, GE Healthcare) as described previously (13). Purification yield was estimated by SDS-PAGE, and protein concentration was determined by absorbance at 280 nm.
Crystal Structure Determination
Crystallization and data collection of the (BACCR)NAT3 wild-type protein were reported previously (13). Briefly, crystals were obtained with the hanging drop-vapor diffusion method with a 1:1 mixture of 30 mg·ml−1 (BACCR)NAT3 protein with 1.6 m sodium citrate, pH 6.5, 0.28 m NDSB-221 as additive (Hampton Research) at 18 °C. The best crystals were flash cooled in liquid nitrogen with a 1:1 volume ratio of Paratone and paraffin oil as cryoprotectant before diffraction data collection at the Swiss Light Source synchrotron (Beamline PX3, Paul Scherrer Institute, Switzerland). The crystallographic parameters and data statistics were reported in Kubiak et al. (13). Briefly, data collection was achieved at 0.984 Å with a 1° oscillation per image on a charge-coupled device mar225 detector. XDS was used for data integration (14). The (BACCR)NAT3 structure was resolved by molecular replacement using the program Phaser (15) implemented in the CCP4 program suite (16). A model of (BACCR)NAT3 was built using CHAINSAW (17) with (BACAN)NAT1 structure (Protein Data Bank code 3LNB) as the search model. The protein structure was obtained by iterative manual rebuild using Coot (18) and refined by Twin Lattice Symmetry and restrained refinement using REFMAC5 (19). After addition of water molecules, a final refinement step was achieved using BUSTER 2.0 software (20). The stereochemical quality of the model was followed during the refinement process using the program MolProbity (21). Water molecules were added with a 3.0σ cutoff and manual verification before final validation was performed with PROCHECK (22). The final refinement statistics and model parameters are shown in Table 1. All structural representations were generated with PyMOL (23). Atomic coordinates and structure factors of the (BACCR)NAT3 protein have been deposited in the Protein Data Bank under accession code 4DMO.
TABLE 1.
Refinement statistics and model parameters of (BACCR)NAT3 WT
SLS, Swiss Light Source.
| Data collection | |
| Beam line | X06DA, SLS |
| Wavelength (Å) | 0.984 |
| Space group | C121 |
| Cell dimensions | |
| a, b, c (Å) | 90.43, 44.52, 132.97 |
| α, β, γ (°) | 90.0, 103.8, 90.0 |
| Resolution (Å) | 20.0-2.14 (2.27-2.14)a |
| Rmeas (%)b | 10.9 (103.1) |
| I/σ(I) | 14.1 (2.0) |
| Completeness (%) | 99.2 (96.0) |
| Multiplicity | 7.4 (7.3) |
| No. of reflections | 208,589 (29,418) |
| No. of unique reflections | 28,411 (4,348) |
| Wilson B-factor (Å2) | 37.8 |
| Refinement | |
| Resolution (Å) | 20.0-2.14 |
| R/Rfree (%) | 20.4/25.5 |
| No. of non-hydrogen atoms | |
| Protein | 4,064 |
| Solvent | 128 |
| Ligands | 42 |
| B-factor (Å2) | |
| Protein | 27.8 |
| Water molecules | 24.1 |
| Ligands | 60.4 |
| r.m.s.d. bond lengths (Å) | 0.012 |
| r.m.s.d. bond angle (°) | 1.2 |
| Ramachandran plot (%) | |
| Favored | 97.0 |
| Allowed | 3.0 |
| Disallowed | 0.0 |
a Numbers in parentheses are for the highest resolution shell.
b Rmeas = Σh√(n/n − 1)Σi|Ihi − Ih|/ΣhΣiIhi where Ihi is the ith observation of the reflection h, Ih is the mean intensity of reflection h, and n is the multiplicity of reflection h.
Determination of Apparent Kinetic Parameters of (BACCR)NAT3 and the E123D Variant Enzymes
Steady-state kinetic experiments were conducted using the 5,5′-dithiobis(nitrobenzoic acid) assay (24) to determine the apparent kinetics of (BACCR)NAT3 and E123D variant enzymes parameters Kmapp and kcat toward 4-aminosalicylic acid, 2-aminofluorene, and isoniazid. Initial velocities (Vi) were determined in the presence of arylamine substrate at various concentrations, 400 μm AcCoA, and 0.5 μg·ml−1 enzyme in 25 mm Tris-HCl, pH 7.5. Alternatively, various concentrations of acetyl donors were used with 500 μm 4-aminosalicylic acid to determine the apparent kinetic parameters toward AcCoA. The PNPA assay described by Cleland and Hengge (25) was used to obtain kinetic parameters toward the acetyl donor PNPA. Control experiments were conducted in the absence of arylamine substrate. Vi values were plotted against substrate concentration and fitted to the Michaelis-Menten equation using Kaleidagraph 3.5 (Synergy Software). Results of triplicate experiments are shown including standard deviations.
Circular Dichroism (CD) Spectra and Chemical Unfolding Followed by CD
Recombinant enzyme samples were extensively dialyzed against 10 mm sodium phosphate, pH 7.5 buffer before recording CD spectra in an Aviv215 spectropolarimeter. Far-UV CD spectra (180–260 nm) were acquired with 505 and 463 μg·ml−1 final concentrations of (BACCR)NAT3 and E123D variant, respectively, through a cylindrical cell with a 0.02-cm path length. The ellipticity signal was recorded each 0.5 nm (1-nm bandwidth) with an integration time of 1 s per step. The average spectra of four successive scans was normalized to the protein concentration, and ellipticity was converted to differential molar extinction coefficient Δϵ per residue. Secondary structure quantitative composition was deduced from the decomposition of the spectra using the CONTIN analysis included in the CDPro package with the 29-protein basis of Johnson and co-worker (26). Near-UV CD spectra (250–350 nm) were obtained from an average of five successive scans between 250 and 350 nm through a 1-cm path length rectangular cell with 2.016 and 1.852 mg·ml−1 final concentration of (BACCR)NAT3 and E123D variant, respectively. Spectra were normalized to the protein concentration as Δϵ per chain.
Native and 5 m GdmCl totally unfolded proteins both at 0.1 mg·ml−1 were prepared in 20 mm Tris-HCl, pH 7.8. Various amounts of native and unfolded proteins were mixed in a 1-ml final volume to obtain GdmCl concentrations ranging from 0 to 5 m with a 0.1 m steps. The samples were held for 24 h at 25 °C before recording ellipticity at 222 nm using a 0.5-cm path length rectangular cell (10-s integration time, 2-nm bandwidth). The ellipticity at 222 nm was expressed as a function of GdmCl concentration and then fitted to the following two-state transition model equation.
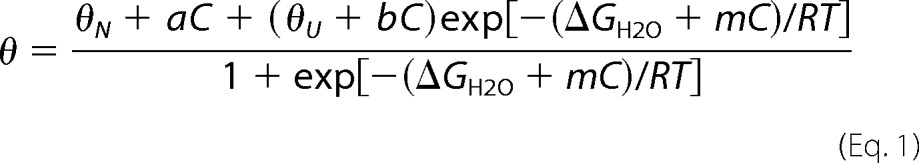 |
where C is the concentration of GdmCl (M), ΔGH2O is the unfolding free energy in the absence of denaturant, and m is the dependence of ΔG on denaturant concentration. a and b are the slopes of the ellipticity base line (supposedly linear) of the native and unfolded states, respectively. Finally, the ellipticity signal was normalized to the total amplitude difference between the fully unfolded protein and native protein (27).
pH-dependent Inactivation by Iodoacetamide
Recombinant enzyme was mixed with either 50 mm MES, pH 6.0–6.5; 50 mm Tris-HCl, pH 7–9; or 50 mm CAPS, pH 9.5–10. Addition of 1 mm IAA to a solution containing 15 μm enzyme started the inactivation reaction at 25 °C. The percentage of residual activity was measured at different incubation times by adding 5-μl aliquots to a solution containing 25 mm Tris-HCl, pH 7.5, 2 mm PNPA, and 500 μm 4-aminosalicylic acid as described above. The percentage of residual activity fitted a single exponential curve allowing us to determine the first-order inactivation constant kinact, which was corrected by the kinact determined in the absence of IAA. ln(kinact) was plotted as a function of pH. The catalytic cysteine pKa value was determined using the Henderson-Hasselbalch equation (28).
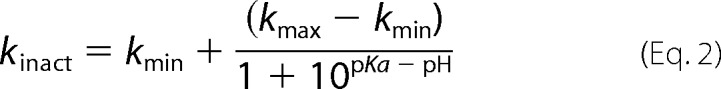 |
where kmax is the highest and kmin is the lowest measured kinact value, respectively. Standard deviations were calculated from three independent experiments.
Phylogenetic Analysis of NAT Sequences
Amino acid sequences were retrieved with BLASTp on the non-redundant sequence databases using the (BACCR)NAT3 sequence as a query. The 250 protein sequences with the highest scores were aligned. Of these, one strain representative of each species with different NAT isoform sequences was selected. Sequences with lower scores but presenting a putative Cys-His-Glu catalytic triad were added to this selection. All sequences were aligned using Muscle with the (BACCR)NAT1, (BACCR)NAT2, and (BACCR)NAT3 sequences and sequences of previously characterized NATs. The phylogenetic tree was constructed using a distance method implemented in MEGA (29) through a neighbor-joining analysis computed using the Dayhoff model. A total of 500 bootstrap replications were performed to determine statistical support for clades.
RESULTS AND DISCUSSION
Analysis of B. cereus Strain ATCC 14579 NAT Sequences Reveals an Isoform with a Cys-His-Glu Catalytic Triad
B. cereus strain ATCC14579 possesses three NAT homologue sequences (BACCR)NAT1, (BACCR)NAT2, and (BACCR)NAT3, which share 94, 83, and 49% sequence identity with the characterized Bacillus anthracis NAT1, NAT2, and NAT3, respectively, indicating that they are orthologous (5, 9, 30). We aligned the sequences of these three NATs with characterized eukaryotic and prokaryotic NATs (Fig. 1); all three B. cereus NAT sequences possess the characteristic NAT functional motifs, suggesting that these proteins belong to the NAT family of acetyltransferases (11, 31). Importantly, as reported previously, one of the B. cereus NAT sequences, (BACCR)NAT3, contains Glu in place of the canonical catalytic triad Asp residue (5, 9).
FIGURE 1.

Alignment of B. cereus NAT sequences with sequences of functionally characterized NATs. All three NAT sequence homologues from B. cereus ((BACCR)NAT1, (BACCR)NAT2, and (BACCR)NAT3) possess the two conserved NAT functional motifs (black bars). The catalytic Cys and His residues (★) are fully conserved throughout all B. cereus NAT homologues. However, (BACCR)NAT3 and (BACAN)NAT3 harbor a Glu residue instead of the canonical Asp residue at catalytic position 123 (▴). The alignment suggests a three-domain (α-helical bundle (1–85), β-barrel (86–192), and C-terminal α/β lid (193–263)) organization of the B. cereus NAT homologues by comparison with the secondary structure of (SALTY)NAT1 indicated on top of the alignment. The region corresponding to a mammalian-like insertion loop is underlined in gray. The alignment was performed by the Muscle algorithm, and the output was achieved with ESPript 2.0. Numbers indicate amino acid position in (SALTY)NAT1.
Expression of (BACCR)NAT3 and an E123D (BACCR)NAT3 Variant
The only characterized NAT enzyme harboring a putative Cys-His-Glu catalytic triad is (BACCAN)NAT3, a short NAT homologue devoid of NAT activity (5, 30). Previous evidence suggests that NAT sequences harboring a Cys-His-Glu catalytic triad are devoid of NAT activity (9, 10, 30). To characterize the (BACCR)NAT3, we cloned and expressed the protein in E. coli and purified it to homogeneity. In parallel, a variant of (BACCR)NAT3 in which the catalytic Glu was replaced by an Asp residue (E123D variant) was also cloned, expressed, and purified. Both proteins were soluble and had identical purification yields (5 mg/liter of culture). SDS-PAGE revealed a unique band of ∼34 kDa consistent with the predicted (ProtParam) molecular mass of 33,465 and 33,450 Da for the wild-type and mutant proteins, respectively (Fig. 2A). Altogether, these data suggest that, in contrast to (HUMAN)NAT2 isozyme, the presence of a Glu or Asp residue in the catalytic triad of (BACCR)NAT3 does not impact protein stability and does not lead to aggregation during expression (10).
FIGURE 2.

Expression yields and structural similarities between (BACCR)NAT3 wild-type and E123D enzymes. A, separation profile of purified recombinant (BACCR)NAT3 wild-type and E123D proteins (1 μg) on a 12.5% SDS-PAGE gel stained with Coomassie Blue. Lane M, molecular mass markers. B, far-UV CD spectra. Decomposition of the spectra (in box) reveals identical fraction of secondary structure content (expressed in percent) between the wild-type (black line) and the E123D mutant enzymes (gray line). H, helix; S, strand; Unrd, unordered. C, near-UV CD spectra (black line, wild type; gray line, E123D mutant). D, guanidinium chloride-induced unfolding of (BACCR)NAT3 wild-type and E123D enzymes followed at 222 nm. The solid lines correspond to the simulated transition curved built from the fit of the data to a two-state transition between 1.2 and 5 m GdmCl (black boxes and line, wild type; open circles and gray line, E123D mutant). The experimental conditions are detailed under “Experimental Procedures.”
Further analysis of (BACCR)NAT3 and its E123D variant by circular dichroism revealed that their secondary and tertiary structures were very similar to one another (Fig. 2, B and C). The unfolding profiles of both proteins showed an unusual transient dramatic ellipticity decrease centered on 0.6–0.7 m GdmCl (Fig. 2D). At higher denaturant concentration, a regain in ellipticity was observed, and at GdmCl concentrations above 1.1 m, the signal change appears as a classical unfolding transition. Both proteins showed very similar unfolding profiles (including the large transient 0.1–1 m GdmCl ellipticity loss). Fitting the ellipticity change between 1.2 and 5 m denaturant to the classical two-state model led to very similar stability characteristics with a midpoint at 1.83 m (ΔGH2O = 6377.0; m = 3486.3) and 1.72 m (ΔGH2O = 8043.5; m = 4683.0) GdmCl for the wild-type and E123D isozymes, respectively. The large ellipticity loss preceding the standard unfolding transition coincided with a very significant transient increase of light scattering intensity reflected by the dynode high voltage during CD measurement. The marginal loss in ellipticity at small denaturant concentration may denote the transient accumulation of ordered or unordered soluble aggregates (fibrils?) induced by a helix → coil or helix → β-structure conformational transition. This atypical phenomenon could be related to the “denaturation trough” initially reported by Goldberg and co-workers (32) and then by other authors (33–35). Although the (HUMAN)NAT2 D122N protein has been suggested to retain its global folding over molecular dynamics experiments, mutagenesis studies showed that the (HUMAN)NAT2 D122E variant was unstable and prematurely degraded (10, 36). In contrast, our data demonstrate that the presence of either an Asp or a Glu residue in the catalytic triad appears not to affect the folding properties of (BACCR)NAT3.
A Cys-His-Glu Catalytic Triad Is Consistent with NAT Activity in (BACCR)NAT3 Wild Type: a Kinetic Comparison with the E123D Mutant
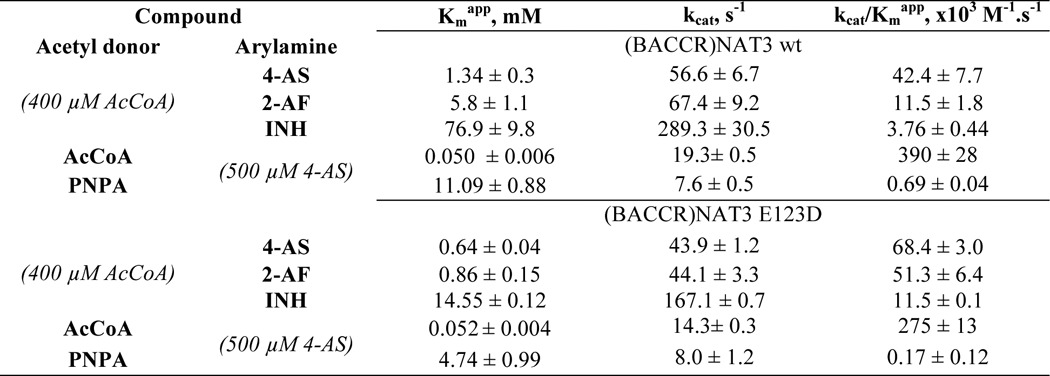
As both the (BACCR)NAT3 and (BACCR)NAT3 E123D proteins appeared properly folded, we investigated their potential to N-acetylate three model aromatic amine substrates (4-aminosalicylic acid, 2-aminofluorene, and isoniazid). (BACCR)NAT3 displayed significant N-acetylation activity toward these three NAT substrates (Table 2). Restoring the Asp residue at position 123 slightly decreased the turnover of the enzyme (kcat) by 1.3–1.7 times and the affinity constant (Kmapp) by 2.1–6.8 times. (BACCR)NAT3 wild-type catalytic efficiency (kcat/Kmapp) was 1.6–4.4 times lower than that of the E123D mutant, but both enzymes were in the range of mean catalytic efficiency values observed for other bacterial NATs (30, 37). Similarly, the Kmapp value of (BACCR)NAT3 for the acetyl donor PNPA was 2 times higher than that of the mutant, and the catalytic efficiency was 2.5 times higher. However, only small differences were observed between the two enzymes in kinetic parameters for the physiological acetyl donor AcCoA (Table 2).
TABLE 2.
Kinetic parameters of (BACCR)NAT3 WT and E123D enzymes for prototypic arylamines and acetyl donors
4-AS, 4-aminosalicylic acid; 2-AF, 2-aminofluorene; INH, isoniazid.
Consistent with previous reports for other NATs, PNPA was a less efficient acetyl donor than AcCoA for both NATs we studied (37). AcCoA has been shown to possess numerous stabilizing interactions all over the active site cleft in contrast to aromatic amines that interact deeply in the active site to reach the acetyl-cysteine intermediate for acetylation (38–41). Thus, the differences in kinetic parameters observed for aromatic amine substrates and the aromatic amine structural analogue PNPA suggest that the shape of the active site proximal to the catalytic triad might be affected; a similar mechanism has been proposed for the (HUMAN)NAT2 D122N mutant (36). Glu and Asp residues share very similar chemical properties and have similar side chain pKa values of 4.15 and 3.71, respectively. The third catalytic residue of the triad has been suggested to act as a base assisting the deprotonation of the two other catalytic residues; a change in the catalytic mechanism would therefore be expected to modify the reactivity of the catalytic cysteine (42, 43). Hence, we conducted a pH-dependent inactivation experiment by IAA to determine the reactivity/pKa of the catalytic Cys69 in both (BACCR)NAT3 and the E123D variant. The pKa values of the catalytic cysteine of the wild-type and E123D mutant enzymes were found to be similar (8.26 ± 0.11 and 7.80 ± 0.14, respectively) (Fig. 3). This suggests that the presence of a catalytic Asp or Glu residue has no significant impact on the reactivity of the catalytic cysteine of (BACCR)NAT3. Moreover, the IAA/pH inactivation profile is more similar to that of Mycobacterium tuberculosis NAT (whose catalytic cysteine pKa lies above 10) than to that of hamster NAT2 for which the catalytic cysteine pKa is 5.23. Therefore, the catalytic mechanism of (BACCR)NAT3 is unlikely to rely on a thiolate/imidazolium ion pair as shown for hamster NAT2 (42) but rather on a general base catalysis mechanism in which the catalytic Cys69 is deprotonated in the presence of substrate at physiological pH as proposed for M. tuberculosis NAT (Fig. 4) (43).
FIGURE 3.
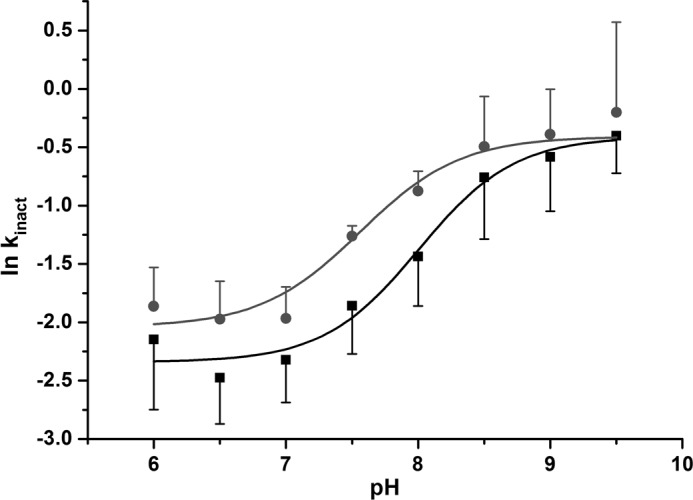
pH-dependent inactivation of (BACCR)NAT3 by IAA. The first-order inactivation constant (kinact) was determined between pH 6 and 9.5 by measuring the ln of residual activity after 1 mm IAA inactivation of the enzyme. The non-linear fit of data to the Henderson-Hasselbalch equation (Equation 2) indicates pKa values of 8.26 and 7.80 for the Cys69 catalytic residue of the wild type (black curve) and E123D mutant (red curve), respectively. Standard error bars are shown for three independent experiments.
FIGURE 4.

Putative catalytic mechanism for the Glu- and Asp-containing (BACCR)NAT3 enzymes. The pKa values of the catalytic cysteine residue suggest that it exists mostly as a thiol at physiological pH (<8), which would be inconsistent with the nucleophilic attack on the AcCoA molecule (first reaction step). Thus, as suggested by Sikora et al. (43) for the M. tuberculosis NAT enzyme, we propose that the Glu/Asp123 and His108 dyad act as general base that facilitates the formation of a thiolate. In the absence of AcCoA in the active site, the thiol proton is shared between Cys60 and His108 (dashed line). The AcCoA binding in the active site could displace this equilibrium and help in the deprotonation of the thiol, leading to the nucleophilic attack of the thiolate on the AcCoA. The following catalysis steps are likely to involve the canonical tetrahedral intermediate states (in brackets) that are known to occur in the Ping Pong Bi Bi mechanism of NAT enzymes. The active site is represented by a gray dashed curve, the His108 and Glu/Asp123 residues are buried, and the Cys69 is solvent-accessible.
Mutation of individual catalytic residues led to a dramatic loss of activity in NATs (10, 38). For the first time, we demonstrate the existence of a non-canonical but functional Cys-His-Glu catalytic triad in an NAT. Our data suggest that this non-canonical triad has no significant impact on activity or substrate-specific catalytic mechanisms when compared with other characterized prokaryotic NATs. This is an important finding for documenting the catalytic diversity of NAT-related enzymes. Indeed, only two catalytic triad variants of the papain-like cysteine peptidases cathepsins L have been reported and are associated with normal activity levels but greatly altered substrate specificity (44). The catalytic triads of transglutaminases and deubiquitinating enzymes are strictly conserved (45). Nevertheless, two esterases (serine proteases) have been reported to have normal activity levels and substrate specificity despite an Asp-to-Glu mutation in the Cys-His-Asp catalytic triad (46). Alternatively, normal activity levels have been reported in particular inteins with a modified catalytic residue but with an accompanying change in the catalytic mechanism (47, 48).
Crystal Structure of (BACCR)NAT3 Wild-type Enzyme: Overall Structure and Catalytic Triad Geometry
Although mutagenesis and computational studies have been conducted on catalytic triad variants of NAT enzymes, no structural determination studies have been reported. Thus, we have solved the crystal structure of (BACCR)NAT3 at 2.14 Å. The Matthews coefficient value of 2.16 Å3·Da−1 and solvent content of 43.03% are consistent with the presence of two molecules in the asymmetric unit after molecular replacement. According to PROCHECK, 97% of the residues are in favored regions of the Ramachandran plot, and 3% are in additional allowed regions, and the R-factor and Rfree values are 20.4 and 25.5%, respectively (Table 1). Three molecules of NDSB-221 additive have been identified in the asymmetric unit with no biological significance. No electron density was observed for the two residues Gly−1 and Ser0 that remained after the His6 tag thrombin cleavage. Clear electron density was observed for all residues except for side chains of surfaced-distributed residues 6, 15–18, 23, 54, 88, 230, 241, and 249. The enzyme has a typical three-domain fold consisting of an α-helical bundle (1–85), a β-barrel (86–192), and a C-terminal α/β lid (193–263) (Fig. 5A) (11). Interestingly, no electron density was observed for residues Asn168–Ala177, which align with the “mammalian insertion” present in human NATs and inferred in (BACAN)NAT1 (Fig. 1) (40, 41). This suggests the presence of a similar highly flexible loop in (BACCR)NAT3 and reinforces the idea that some Bacillus NAT isoforms may have undergone a particular evolutionary history (e.g. horizontal gene transfer from a eukaryotic host, giving rise to eukaryotic features in these bacterial NATs). The crystal structure of (BACCR)NAT3 shows unambiguous and strong electron densities for residues Cys69, His108, and more importantly the non-canonical Glu123, shaping a catalytic triad (Fig. 5B). Furthermore, this catalytic triad is highly superimposable with the canonical Cys-His-Asp triad of other NATs as shown by r.m.s.d. values between 0.627 and 0.869 Å. These values are ∼4-fold lower compared with r.m.s.d. values between the catalytic triads of canonical NATs likely due to the presence of a larger Glu residue in (BACCR)NAT3 rather than to a different triad conformation as all catalytic triads are similarly oriented (Fig. 5C). The side chains of Cys69, His108, and Glu123 are similarly oriented compared with the Asp and Cys residues of canonical NATs, and the Cys69-His108 and His108-Glu123 in (BACCR)NAT3 are also comparable with those found in other NATs. Altogether, these observations depict clear structural bases for the catalytic activity measured for (BACCR)NAT3 and demonstrate that the geometry of the non-canonical Cys-His-Glu catalytic triad fulfills the structural and chemical requirements of NAT catalysis in the (BACCR)NAT3 enzyme.
FIGURE 5.

Three-dimensional structure of the (BACCR)NAT3 wild-type enzyme. A, schematic and surface representation of the two (BACCR)NAT3 molecules in the asymmetric unit, solved at 2.14 Å. Each enzyme chain has a typical three-domain fold including an α-helical bundle (red), a β-barrel (yellow), and a C-terminal α/β lid (blue). The structure confirms that residues Cys69, His108, and Glu123 form a catalytic triad (green ball and stick). No electron density was attributed to the region spanning from Lys167 and Asp178 in both molecules, indicating the presence of a flexible loop corresponding to the mammalian-like insertion found in human NAT enzymes. B, 2Fo − Fc density map at 1.5σ for (BACCR)NAT3 catalytic residues Cys69, His108, and Glu123. Hydrogen bonds are shown as black dashes, and distances (molecule A in the asymmetric unit) are expressed in Å. Distances in molecule B are shown in parentheses. C, superimposition of (BACCR)NAT3 (red) with (SALTY)NAT1 (yellow), (PSEAE)NAT1 (blue), (MYCMR)NAT1 (orange), and (RHILO)NAT1 (green) catalytic triads with r.m.s.d. values of 0.869, 0.672, 0.825, and 0.812 Å, respectively. It reveals a conserved geometry for the non-canonical Cys-His-Glu catalytic triad in (BACCR)NAT3 compared with other canonical NAT catalytic triads. The (BACCR)NAT3 backbone is shown in orange.
Protein Packing and Catalytic Residue Environment Likely Explain the Accommodation of a Glu in the Catalytic Triad of (BACCR)NAT3
As previously reported by Zang et al. (10), the loss of activity in (HUMAN)NAT2 enzyme likely results from disruption of the protein structure integrity. The structures of (BACCR)NAT3 and (HUMAN)NAT2 are highly superimposable with r.m.s.d. values of 1.483 and 0.539 Å for the whole structure and the catalytic triad, respectively. Both enzymes have a similar catalytic triad geometry, although His108 appears slightly remote in (BACCR)NAT3 compared with its position in (HUMAN)NAT2 (Fig. 6A). In (BACCR)NAT3, the catalytic Glu123 residue is stabilized by a complex network of hydrogen bonds established with residues Asn73, His108, Gly125, and Tyr184 and unexpectedly with two water molecules that are further stabilized through three hydrogen bonds to residues Leu121, Leu132, and Ala133 (Fig. 6B, upper panel). In (HUMAN)NAT2, the catalytic Asp122 interacts with the same residues (except for Ser125), but the main chain is stabilized by a single hydrogen bond with Gln130 rather than with water molecules (Fig. 6B, lower panel). The topology of (BACCR)NAT3 is similar to that of (HUMAN)NAT2. Despite 27 additional residues in the (HUMAN)NAT2 enzyme compared with (BACCR)NAT3, the solvent-accessible surface of both proteins is very similar with values of 13,552 and 12,485 Å2, respectively (Fig. 6C). In addition, (BACCR)NAT3 has nine unresolved/absent surfaced-exposed residues in its inferred mammalian-like loop. Fifteen of the additional residues in (HUMAN)NAT2 compose a C-terminal extension that is stabilized by a 2.8-Å hydrogen bond between Thr289 and Ser127 (Fig. 7A) (40). In contrast, the C-terminal amino acid of (BACCR)NAT3 is 19 Å far from the corresponding Ser128 position. In both enzymes, this position is located on the turn of the loop carrying the third catalytic residue. Ten of the additional residues are located in the “mammalian-like insertion,” which is stabilized by several interactions with residues on domain III and hence is much more rigid than the unresolved flexible loop in (BACCR)NAT3 (Fig. 6B). This suggests a higher packing of the (HUMAN)NAT2 enzyme that could prevent structural rearrangements of the enzyme and thus preclude the accommodation of the larger Glu residue in the catalytic triad. Additionally, several differences are found in the vicinity (<5 Å) of the third catalytic residue. In particular, the large residues Gln130 and Met131 in (HUMAN)NAT2 are replaced by the smaller Leu130 and Pro131 residues, respectively, in (BACCR)NAT3 (Fig. 7A). These amino acid differences lead to a larger space available around the Glu residue in (BACCR)NAT3 (167 Å3) compared with the Asp residue in (HUMAN)NAT2 (101 Å3). It is likely that the interaction between the Asp122 main chain and Gln130 in (HUMAN)NAT2 allows fewer structural rearrangement of the two residues compared with the conformations available for the Glu123 main chain and the water molecules in (BACCR)NAT3. Thus, considering the similar chemistry of the Asp and Glu residues and the results of the kinetic study of (BACCR)NAT3 enzymes, we propose that the accommodation of a Glu as a catalytic residue in NAT enzymes relies on a combination of low steric constraints, favorable protein packing, and amino acid environment.
FIGURE 6.

Comparison of structural properties between (BACCR)NAT3 and (HUMAN)NAT2. A, structural alignment of (BACCR)NAT3 (orange) and (HUMAN)NAT2 (cyan) catalytic triads reveals a very similar geometry. Electrostatic interactions, hydrogen bonds, and distances are represented as indicated in Fig. 5 legend. B, amino acid network interactions of the third catalytic residues in (BACCR)NAT3 (upper panel) and (HUMAN)NAT2 (lower panel). In contrast to Asp122 in (HUMAN)NAT2, the Glu123 main chain interacts with two water molecules (red spheres) that are further stabilized through hydrogen bonds with residues Leu121, Leu132, and Ala133. C, structural alignment representing the main differences between (BACCR)NAT3 (light orange) and (HUMAN)NAT2 (light blue) conformations. The two structures were aligned using PyMOL (The PyMOL Molecular Graphics System, Version 0.99, Schrödinger, LLC.); additional residues found in one enzyme but not the other are colored in red and blue for amino acid extensions in (BACCR)NAT3 and (HUMAN)NAT2, respectively. The structural alignment was further supported by structural alignment of the two sequences using DaliLite at the European Bioinformatics Institute.
FIGURE 7.
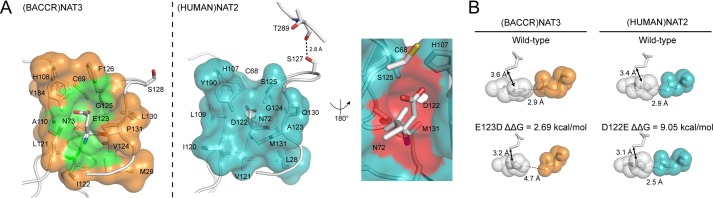
A model proposed for the differential accommodation of Glu as a catalytic residue in (BACCR)NAT3 and (HUMAN)NAT2. A, the cavity formed by residues surrounding (<5 Å) the Glu123 or Asp122 catalytic residue shows a smaller size with values of 167 and 101 Å3 (CastP calculation (53)) for (BACCR)NAT3 (green pocket) and (HUMAN)NAT2 (red pocket), respectively. This difference is likely due to the presence of the Pro131 residue in (BACCR)NAT3 instead of Met131 residue in (HUMAN)NAT2. Both enzymes are represented in the same orientation, and a vertical 180° rotation view of the cavity in (HUMAN)NAT2 is shown in the right panel. The surface of Asn72 has been subtracted in (HUMAN)NAT2 for increased visibility of the cavity, but it was included for the cavity volume calculation. The hydrogen bond in (HUMAN)NAT2 between the C-terminal residue Thr289 and Ser127 is represented in dashes (distances in Å); the corresponding Ser128 does not interact with the C-terminal region in (BACCR)NAT3. B, FoldX energy minimization models of the E123D and D122E mutants for (BACCR)NAT3 and (HUMAN)NAT2, respectively. The difference in free energy variation (ΔΔG) generated upon mutation was calculated by FoldX. The wild-type and mutant catalytic triads are represented in sticks, and the catalytic His are represented as Van der Waals spheres (1-Å radius; white). The Asp or Glu residue surface is also represented in Van der Waals spheres (1-Å radius) for (BACCR)NAT3 (orange) and (HUMAN)NAT2 (cyan) models. These models show that a steric clash occurs between His and Glu upon D122E mutation in (HUMAN)NAT2.
Energy calculations and energy minimization models of the (BACCR)NAT3 E123D and (HUMAN)NAT2 D122E mutants using FoldX 3.0 and Popmusic 2.1 support this model (49, 50). The ΔΔG energy values obtained using the FoldX and Popmusic algorithms are +2.69 and 0.31 kcal/mol upon E123D mutation in (BACCR)NAT3 and +9.05 and +2.51 kcal/mol upon D122E mutation in (HUMAN)NAT2, respectively. This clearly indicates that the presence of a glutamate is energetically favorable in (BACCR)NAT3 in contrast to its destabilizing effect in (HUMAN)NAT2. Energy minimization models suggest that such an increase in ΔG is concomitant with a steric clash between the His107 and Glu122 catalytic residues in (HUMAN)NAT2 (Fig. 7B). Although the model seems robust, the absence of protein backbone rearrangements upon the mutation leads to a His108-Asp123 distance of 4.7 Å, which is incompatible with the catalytic activity observed experimentally in the (BACCR)NAT3 E123D mutant enzyme (Fig. 7B).
The geometry of the His108, Phe125, and Tyr184 residues suggests the existence of a strong network of π-π interactions (Fig. 6B). In hamster NAT2, residue Tyr190 has been suggested to be important for maintaining the catalytic triad conformation by interacting with the catalytic Asp122 and His107 residues through hydrogen bonding and π-π interactions, respectively (51). The presence of a Ser instead of a Phe residue at position 125 in (HUMAN)NAT2 is likely to weaken this network. One can only speculate that local structural rearrangements upon insertion of a larger Glu residue may trigger the disruption of this network. Coupled with a lower degree of freedom of the Asp122-containing loop (Ser127-Thr287 interaction) and a greater compaction, this could nevertheless decrease the ability of the protein to “find” an energetically favorable conformation, leading to the disruption of the protein structure integrity and its premature degradation. Molecular dynamics simulations have only been performed on the (HUMAN)NAT2 D122N mutant, but these suggest that the enzyme possesses fewer hydrogen bonds, which could decrease its overall stability (36).
Phylogenetic Analysis of Glu Residue Distribution in the Catalytic Triads of NAT Enzymes
To date, only the (BACCR)NAT3 and (BACAN)NAT3 orthologous enzymes and the (HUMAN)NAT2 D122N variant have been shown to naturally harbor a mutation in a catalytic residue. A closer investigation of the diversity of the NAT catalytic triad was achieved by BLASTing the non-redundant protein with (BACCR)NAT3 as query. The 250 highest scoring sequences retrieved all correspond to NAT homologues belonging to Bacillus species (supplemental Fig. S1). These sequences cluster into three main clades because of sequence identity with B. cereus NAT1, NAT2, and NAT3 enzymes (Fig. 8). Interestingly, every strain possessing several NAT sequences systematically harbors one with a Glu residue instead of an Asp at the catalytic position 123. More strikingly, seven NAT homologue sequences are present in the low score range and also present a putative Cys-His-Glu catalytic triad, including three sequences from Dictyostelium species and four from other bacteria (Fig. 8). Glenn et al. (5) showed that NAT sequences from B. anthracis and B. cereus nat genes were closely related to NATs from slime molds, suggesting the possible involvement of horizontal nat gene transfer events. Thus, the presence of a non-canonical NAT enzyme in the majority of Bacillus strains might stem from early horizontal gene transfer events from a eukaryotic host. Still, other unrelated bacteria harbor such atypical NAT homologues. Considering the environmental adaptation role proposed for prokaryotic NATs, such findings suggest that other non-canonical NAT enzymes may be functional and that such enzymes might have particular properties, e.g. substrate specificity, conferring specific features to these organisms. The characterization of the different B. cereus NAT isoforms and other non-canonical enzymes will help in answering this question.
FIGURE 8.

Distribution of the non-canonical Cys-His-Glu catalytic triad throughout species. NAT homologue sequences from Bacillus species cluster differently from canonical bacterial NATs and are closely related to lower eukaryotes as reported previously (5). The Bacillus sequences cluster in three main clades corresponding to (BACCR)NAT1, (BACCR)NAT2, and (BACCR)NAT3 orthologous sequences. All NAT homologues present in the (BACCR)NAT3-containing clade have a Glu residue instead of the Asp at catalytic position 123 (in green). Three sequences from lower eukaryotes and four bacterial sequences also possess a putative Glu as the third catalytic residue (in red). Characterized NAT sequences are indicated according to the NAT nomenclature; other sequences are indicated as genus species strain followed by the UniProt identifier in brackets.
Concluding Remarks
Although the activity of the prokaryotic and eukaryotic NAT enzymes was known to rely on the presence of a strictly conserved Cys-His-Asp catalytic triad, we report here the characterization of a fully functional NAT enzyme possessing a non-canonical Cys-His-Glu catalytic triad. The crystal structure of this NAT enzyme from B. cereus ((BACCR)NAT3) suggests that the accommodation of a Glu residue might be achieved only in certain NAT enzymes that possess favorable packing and steric constraints around the catalytic position rather than depending on chemical properties of the third catalytic residue, although it remains unclear which regions or particular residues are involved. The identification of both prokaryotic and eukaryotic NAT sequences adds clues in this direction. However, it raises the question of the evolutionary history of the catalytic triad of NAT enzymes and the significance of the predominance of Cys-His-Asp catalytic triads over the non-canonical Cys-His-Glu catalytic triad, which can be catalytically active as shown in this study. A similar question has been proposed for the conservation of an Asp residue instead of the equivalent Glu residue in the catalytic triad of acetylcholine esterase (52). Finally, our work reinforces recent studies demonstrating greater structural and functional diversity than expected in the NAT enzyme family of acetyltransferases. The systematic functional and structural characterization of NATs harboring atypical features, in particular the homologues carrying the non-canonical Cys-His-Glu catalytic triad, will be necessary to better understand the basis of this emerging diversity.
Acknowledgments
We thank Dr. Karine Moncoq and Ximing Xu for helpful discussions on crystallogenesis assays. We acknowledge the core platform “Biophysique des macromolécules et de leurs intéractions” (Institut Pasteur) for provision of CD facilities. We also acknowledge Synchrotron Swiss Light Source (Villigen, Switzerland) for the provision of synchrotron radiation facilities, and we thank Meitian Wang for assistance with the use of the Beamline X06DA.
This work was supported in part by the Université Paris Diderot-Paris 7, Délégation Générale de l'Armement (DGA), the Institut Pasteur Paris, the CNRS and French Infrastructure for Integrated Structural Biology (FRISBI) Grant ANR-10-INSB-05-01.

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (code 4DMO) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NAT
- arylamine N-acetyltransferase (unless specified, all NAT enzymes referred to in the text are named according to the current official NAT nomenclature)
- (BACCR)NAT3
- B. cereus NAT3
- AcCoA
- acetyl coenzyme A
- PNPA
- para-nitrophenyl acetate
- IAA
- iodoacetamide
- GdmCl
- guanidine hydrochloride
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Hein D. W. (2002) Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat. Res. 506–507, 65–77 [DOI] [PubMed] [Google Scholar]
- 2. Sim E., Fakis G., Laurieri N., Boukouvala S. (2012) Arylamine N-acetyltransferases—from drug metabolism and pharmacogenetics to identification of novel targets for pharmacological intervention. Adv. Pharmacol. 63, 169–205 [DOI] [PubMed] [Google Scholar]
- 3. Sinclair J. C., Sandy J., Delgoda R., Sim E., Noble M. E. (2000) Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat. Struct. Biol. 7, 560–564 [DOI] [PubMed] [Google Scholar]
- 4. Rodrigues-Lima F., Deloménie C., Goodfellow G. H., Grant D. M., Dupret J. M. (2001) Homology modelling and structural analysis of human arylamine N-acetyltransferase NAT1: evidence for the conservation of a cysteine protease catalytic domain and an active-site loop. Biochem. J. 356, 327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glenn A. E., Karagianni E. P., Ulndreaj A., Boukouvala S. (2010) Comparative genomic and phylogenetic investigation of the xenobiotic metabolizing arylamine N-acetyltransferase enzyme family. FEBS Lett. 584, 3158–3164 [DOI] [PubMed] [Google Scholar]
- 6. Riddle B., Jencks W. P. (1971) Acetyl-coenzyme A:arylamine N-acetyltransferase. Role of the acetyl-enzyme intermediate and the effects of substituents on the rate. J. Biol. Chem. 246, 3250–3258 [PubMed] [Google Scholar]
- 7. Weber W. W., Hein D. W. (1985) N-Acetylation pharmacogenetics. Pharmacol. Rev. 37, 25–79 [PubMed] [Google Scholar]
- 8. Dupret J. M., Grant D. M. (1992) Site-directed mutagenesis of recombinant human arylamine N-acetyltransferase expressed in Escherichia coli. Evidence for direct involvement of Cys68 in the catalytic mechanism of polymorphic human NAT2. J. Biol. Chem. 267, 7381–7385 [PubMed] [Google Scholar]
- 9. Sandy J., Mushtaq A., Holton S. J., Schartau P., Noble M. E., Sim E. (2005) Investigation of the catalytic triad of arylamine N-acetyltransferases: essential residues required for acetyl transfer to arylamines. Biochem. J. 390, 115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zang Y., Zhao S., Doll M. A., Christopher States J., Hein D. W. (2007) Functional characterization of the A411T (L137F) and G364A (D122N) genetic polymorphisms in human N-acetyltransferase 2. Pharmacogenet. Genomics 17, 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sim E., Lack N., Wang C.-J., Long H., Westwood I., Fullam E., Kawamura A. (2008) Arylamine N-acetyltransferases: structural and functional implications of polymorphisms. Toxicology 254, 170–183 [DOI] [PubMed] [Google Scholar]
- 12. Kubiak X., Dairou J., Dupret J.-M., Rodrigues-Lima F. (2013) Crystal structure of arylamine N-acetyltransferases: insights into the mechanisms of action and substrate selectivity. Expert Opin. Drug Metab. Toxicol. 9, 349–362 [DOI] [PubMed] [Google Scholar]
- 13. Kubiak X., Pluvinage B., Li de la Sierra-Gallay I., Weber P., Haouz A., Dupret J.-M., Rodrigues-Lima F. (2012) Purification, crystallization and preliminary x-ray characterization of Bacillus cereus arylamine N-acetyltransferase 3 [(BACCR)NAT3]. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 196–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP 4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 18. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bricogne G., Blanc E., Brnadl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER, Version 2.10.0, Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 21. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 23. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 0.99rc6, Schrödinger, LLC, New York [Google Scholar]
- 24. Brooke E. W., Davies S. G., Mulvaney A. W., Pompeo F., Sim E., Vickers R. J. (2003) An approach to identifying novel substrates of bacterial arylamine N-acetyltransferases. Bioorg. Med. Chem. 11, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 25. Cleland W. W., Hengge A. C. (1995) Mechanisms of phosphoryl and acyl transfer. FASEB J. 9, 1585–1594 [DOI] [PubMed] [Google Scholar]
- 26. King S. M., Johnson W. C. (1999) Assigning secondary structure from protein coordinate data. Proteins 35, 313–320 [PubMed] [Google Scholar]
- 27. Le H.-T., Chaffotte A. F., Demey-Thomas E., Vinh J., Friguet B., Mary J. (2009) Impact of hydrogen peroxide on the activity, structure, and conformational stability of the oxidized protein repair enzyme methionine sulfoxide reductase A. J. Mol. Biol. 393, 58–66 [DOI] [PubMed] [Google Scholar]
- 28. Krekel F., Samland A. K., Macheroux P., Amrhein N., Evans J. N. (2000) Determination of the pKa value of C115 in MurA (UDP-N-acetylglucosamine enolpyruvyltransferase) from Enterobacter cloacae. Biochemistry 39, 12671–12677 [DOI] [PubMed] [Google Scholar]
- 29. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pluvinage B., Dairou J., Possot O. M., Martins M., Fouet A., Dupret J.-M., Rodrigues-Lima F. (2007) Cloning and molecular characterization of three arylamine N-acetyltransferase genes from Bacillus anthracis: identification of unusual enzymatic properties and their contribution to sulfamethoxazole resistance. Biochemistry 46, 7069–7078 [DOI] [PubMed] [Google Scholar]
- 31. Rodrigues-Lima F., Dupret J.-M. (2002) In silico sequence analysis of arylamine N-acetyltransferases: evidence for an absence of lateral gene transfer from bacteria to vertebrates and first description of paralogs in bacteria. Biochem. Biophys. Res. Commun. 293, 783–792 [DOI] [PubMed] [Google Scholar]
- 32. London J., Skrzynia C., Goldberg M. E. (1974) Renaturation of Escherichia coli tryptophanase after exposure to 8 M urea. Evidence for the existence of nucleation centers. Eur. J. Biochem. 47, 409–415 [DOI] [PubMed] [Google Scholar]
- 33. Mitraki A., Betton J.-M., Desmadril M., Yon J. M. (1987) Quasi-irreversibility in the unfolding-refolding transition of phosphoglycerate kinase induced by guanidine hydrochloride. Eur. J. Biochem. 163, 29–34 [DOI] [PubMed] [Google Scholar]
- 34. Hammarström P., Persson M., Freskgård P.-O., Mårtensson L.-G., Andersson D., Jonsson B.-H., Carlsson U. (1999) Structural mapping of an aggregation nucleation site in a molten globule intermediate. J. Biol. Chem. 274, 32897–32903 [DOI] [PubMed] [Google Scholar]
- 35. Xu Q., Xie Z., Ding J., Lin S.-X., Xu G. (2004) Monoclonal antibodies assisting refolding of firefly luciferase. Protein Sci. 13, 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rajasekaran M., Abirami S., Chen C. (2011) Effects of single nucleotide polymorphisms on human N-acetyltransferase 2 structure and dynamics by molecular dynamics simulation. PLoS One 6, e25801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Westwood I. M., Sim E. (2007) Kinetic characterisation of arylamine N-acetyltransferase from Pseudomonas aeruginosa. BMC Biochem. 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sandy J., Holton S., Fullam E., Sim E., Noble M. (2005) Binding of the anti-tubercular drug isoniazid to the arylamine N-acetyltransferase protein from Mycobacterium smegmatis. Protein Sci. 14, 775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fullam E., Westwood I. M., Anderton M. C., Lowe E. D., Sim E., Noble M. E. (2008) Divergence of cofactor recognition across evolution: coenzyme A binding in a prokaryotic arylamine N-acetyltransferase. J. Mol. Biol. 375, 178–191 [DOI] [PubMed] [Google Scholar]
- 40. Wu H., Dombrovsky L., Tempel W., Martin F., Loppnau P., Goodfellow G. H., Grant D. M., Plotnikov A. N. (2007) Structural basis of substrate-binding specificity of human arylamine N-acetyltransferases. J. Biol. Chem. 282, 30189–30197 [DOI] [PubMed] [Google Scholar]
- 41. Pluvinage B., Li de la Sierra-Gallay I., Kubiak X., Xu X., Dairou J., Dupret J.-M., Rodrigues-Lima F. (2011) The Bacillus anthracis arylamine N-acetyltransferase ((BACAN)NAT1) that inactivates sulfamethoxazole, reveals unusual structural features compared with the other NAT isoenzymes. FEBS Lett. 585, 3947–3952 [DOI] [PubMed] [Google Scholar]
- 42. Wang H., Vath G. M., Gleason K. J., Hanna P. E., Wagner C. R. (2004) Probing the mechanism of hamster arylamine N-acetyltransferase 2 acetylation by active site modification, site-directed mutagenesis, and pre-steady state and steady state kinetic studies. Biochemistry 43, 8234–8246 [DOI] [PubMed] [Google Scholar]
- 43. Sikora A. L., Frankel B. A., Blanchard J. S. (2008) Kinetic and chemical mechanism of arylamine N-acetyltransferase from Mycobacterium tuberculosis. Biochemistry 47, 10781–10789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pillay D., Boulangé A. F., Coetzer T. H. (2010) Expression, purification and characterisation of two variant cysteine peptidases from Trypanosoma congolense with active site substitutions. Protein Expr. Purif. 74, 264–271 [DOI] [PubMed] [Google Scholar]
- 45. Mehta D. K., Eckert R. L. (2005) Transglutaminases: Family of Enzymes with Diverse Functions, Karger Publishers, Basel, Switzerland [Google Scholar]
- 46. Chu X., He H., Guo C., Sun B. (2008) Identification of two novel esterases from a marine metagenomic library derived from South China Sea. Appl. Microbiol. Biotechnol. 80, 615–625 [DOI] [PubMed] [Google Scholar]
- 47. Southworth M. W., Benner J., Perler F. B. (2000) An alternative protein splicing mechanism for inteins lacking an N-terminal nucleophile. EMBO J. 19, 5019–5026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saleh L., Southworth M. W., Considine N., O'Neill C., Benner J., Bollinger J. M., Jr., Perler F. B. (2011) Branched intermediate formation is the slowest step in the protein splicing reaction of the Ala1 KlbA intein from Methanococcus jannaschii. Biochemistry 50, 10576–10589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guerois R., Nielsen J. E., Serrano L. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387 [DOI] [PubMed] [Google Scholar]
- 50. Gonnelli G., Rooman M., Dehouck Y. (2012) Structure-based mutant stability predictions on proteins of unknown structure. J. Biotechnol. 161, 287–293 [DOI] [PubMed] [Google Scholar]
- 51. Zhou X., Zhang N., Liu L., Walters K. J., Hanna P. E., Wagner C. R. (2009) Probing the catalytic potential of the hamster arylamine N-acetyltransferase 2 catalytic triad by site-directed mutagenesis of the proximal conserved residue, Tyr190. FEBS J. 276, 6928–6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soreq H., Seidman S. (2001) Acetylcholinesterase—new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302 [DOI] [PubMed] [Google Scholar]
- 53. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]