

Background: Complement is implicated in obesity and insulin resistance; however, the specific complement activation pathway involved is not known.
Results: C1q in the classical pathway is required for activation of complement in response to high fat diets.
Conclusion: C1q is an important contributor to high fat diet-induced insulin resistance.
Significance: C1q may be an important therapeutic target for treating the derangements in metabolism associated with obesity.
Keywords: Adipose Tissue, Apoptosis, Complement, Inflammation, Insulin Resistance, Obesity
Abstract
Complement activation is implicated in the development of obesity and insulin resistance, and loss of signaling by the anaphylatoxin C3a prevents obesity-induced insulin resistance in mice. Here we have identified C1q in the classical pathway as required for activation of complement in response to high fat diets. After 8 weeks of high fat diet, wild-type mice became obese and developed glucose intolerance. This was associated with increased apoptotic cell death and accumulation of complement activation products (C3b/iC3b/C3c) in liver and adipose tissue. Previous studies have shown that high fat diet-induced apoptosis is dependent on Bid; here we report that Bid-mediated apoptosis was required for complement activation in adipose and liver. Although C1qa deficiency had no effect on high fat diet-induced apoptosis, accumulation of complement activation products and the metabolic complications of high fat diet-induced obesity were dependent on C1q. When wild-type mice were fed a high fat diet for only 3 days, hepatic insulin resistance was associated with the accumulation of C3b/iC3b/C3c in the liver. Mice deficient in C3a receptor were protected against this early high fat diet-induced hepatic insulin resistance, whereas mice deficient in the negative complement regulator CD55/DAF were more sensitive to the high fat diet. C1qa−/− mice were also protected from high fat diet-induced hepatic insulin resistance and complement activation. Evidence of complement activation was also detected in adipose tissue of obese women compared with lean women. Together, these studies reveal an important role for C1q in the classical pathway of complement activation in the development of high fat diet-induced insulin resistance.
Introduction
Obesity is a strong risk factor for the development of metabolic syndrome; collectively, metabolic syndrome is characterized by an increased risk for chronic disease, including insulin resistance and type 2 diabetes, dyslipidemia, and cardiovascular disease as well as non-alcoholic liver disease (nonalcoholic fatty liver disease/non-alcoholic steatohepatitis) (1). The innate immune system is critical to the development of obesity-induced metabolic syndrome, including the development of insulin resistance (2). Obesity, insulin resistance, and type 2 diabetes are associated with an inflammatory response in adipose tissue, characterized by macrophage accumulation and the production of inflammatory mediators, that contributes to the development of peripheral insulin resistance (3–6). More recent data suggest that innate immunity and resident macrophages are also involved in the development of hepatic insulin resistance. For example, Lanthier et al. (4) identified Kupffer cells, the resident macrophage in the liver, as critical to the development of hepatic insulin resistance in response to high fat diets.
Although it is clear that innate immune responses and resident tissue macrophages are critical to the development of insulin resistance in both adipose tissue and liver, the mechanisms for the activation of resident macrophages and the innate immune response are not completely understood. The role of TLR4 in activation of tissue macrophages in models of obesity has been well explored. For example, free fatty acids interact with TLR4 to activate hepatic macrophages (7), and cholesterol may interfere with signaling processes regulating TLR signaling and innate immune function (7, 8). TLR4 signaling can also be activated in response to increased endotoxin/lipopolysaccharide (LPS) in the circulation, the result of impaired barrier function in the intestine in response to high fat diets and/or obesity (9).
Complement is another component of the innate immune system that may contribute to the activation of resident tissue macrophages and chronic inflammation in obesity. The complement cascade is a phylogenetically ancient part of the immune system critical to an organism's ability to ward off infection (10). Activation of the complement pathway can occur via the classical, lectin, or alternative pathway, all three pathways culminate in the activation of C3. These pathways leading to C3 cleavage are “triggered enzyme cascades,” analogous to the regulated activation of the coagulation pathway (11). The cleavage products C3a and C5a, termed the anaphylatoxins, are important regulators of the inflammatory response. C3a and C5a stimulate the production of cytokines in a number of cell types (12, 13), either alone or in the presence of other inflammatory mediators, such as LPS (13). C5a also has chemoattractant properties, recruiting neutrophils to the site of infection/injury by regulating the expression of chemokines and adhesion molecules (14, 15).
Although complement is classically associated with the protection from invading organisms, a growing body of evidence implicates complement in chronic inflammatory diseases, such as alcoholic (16–18) and non-alcoholic liver disease (19). A recent study identified C3a receptor as a key determinant of insulin resistance and adipose tissue inflammation in diet-induced obesity in mice (20), and complement activation products are detected in the liver of patients with non-alcoholic steatohepatitis (19).
Although these studies indicate a role of complement in the innate immune response to high fat diets and obesity, the specific pathways of complement activation in response to high fat diets/obesity have not been investigated. Recent data in models of alcoholic liver disease have implicated C1q in the classical pathway as a critical mediator of both adipose and liver inflammation in response to alcohol exposure (16, 17, 21). The classical pathway of complement is activated upon the binding of C1q, the recognition subunit of first component (C1) in the classical pathway of activation, to immune complexes. The interaction of C1q with cell surface markers on apoptotic cells, including phosphatidylserine (22), surface blebs (23), or nucleic acids on the cell surface (24), is an important mechanism for the activation of the classical pathway as well as clearance of apoptotic cells (25). Because adipocyte death via Bid-dependent apoptotic pathways is an important link between high fat diet-induced obesity and insulin resistance (26), here we tested the hypothesis that Bid-mediated apoptosis would contribute to the activation of C1q in response to high fat diet-induced obesity and that C1q and complement are critical contributors in the development of adipose tissue inflammation and impaired glucose tolerance/insulin sensitivity in response to high fat diet-induced obesity.
MATERIALS AND METHODS
Animals and Care
5–8-week-old male mice of each of the following genotypes were used in the described studies. Wild-type mice (C57BL/6J) were purchased from Jackson Laboratory (Bar Harbor, ME). C3aR−/− mice were generated on a mixed genetic background by R. Wetsel (27) and back-crossed into the C57BL/6 background by M. E. Medof (Case Western Reserve University, Cleveland, OH). CD55−/− mice (also known as Daf1−/−) were generated by M. E. Medof (28). C1qa−/− (29) mice were provided by M. Carroll (Harvard Medical School, Boston, MA). C57BL/6 Bid−/− mice were provided by Dr. Xiao-Ming Yin (University of Pittsburgh) (30). Animals were housed in pairs in standard microisolator cages and maintained on a 12-h/12-h light/dark cycle. All animals received humane care, and all procedures were approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
C57BL/6 and the different strains of complement deficient mice used in the 8-week-diet studies were fed a high fat diet (D12331; 58% of calories from fat, 25% of calories from maltose dextrins/sucrose) or a micronutrient-matched low fat diet (D12329; 11% of calories from fat, 72% of calories from maltose dextrins/sucrose) (Research Diets, Brunswick, NJ). Mice were weighed weekly, and food intake per cage was measured twice weekly. C57BL/6 and Bid−/− were fed a high fat diet or low fat diet exactly as described previously (26). Mice used in 3-day diet studies were fed the high fat diet or standard Purina laboratory chow. Mice and food were weighed at the beginning and end of the 3-day protocol. All mice were fasted for 6 h prior to euthanasia. Portions of liver or epididymal adipose were flash frozen in liquid nitrogen and stored at −80 °C until Western blot analysis, preserved in RNAlater (Qiagen, Valencia, CA), and stored at −20 °C until RNA isolation or fixed in 10% formalin or frozen in optimal cutting temperature compound (OCT)2 (Sakura Finetek U.S.A., Inc., Torrance, CA) for histology.
Portal Insulin Injections
Mice were anesthetized, blood was collected, and then mice were injected with insulin (I6634 or I9278; Sigma-Aldrich) via the portal vein (1 μg/g of body weight) or an equal volume of 0.09% saline. Liver and epididymal adipose were harvested 2 min after injection. Tissues were immediately frozen in liquid nitrogen.
Clodronate Injections
Mice were injected with clodronate-containing liposomes (purchased from ClodronateLiposomes.org; 5 mg/ml) or an equal volume of PBS-containing liposomes via tail vein (200 μl/mouse) 24 h prior to beginning the high fat diet for 3 days.
Western Blot Analysis and Quantitative Real-time PCR
Frozen liver or epididymal adipose was homogenized in radioimmune precipitation assay lysis buffer, and lysates were used for Western blot analysis as described previously (21). RNA was isolated from liver and adipose, cDNA was prepared, and quantitative RT-PCR was carried out as described previously (21).
Metabolic Analyses
Fasting blood glucose was measured from a tail nick using the OneTouch Ultra Blood Glucose Meter and test strips (Lifescan, Milpitas, CA) prior to administration of anesthetic, if applicable. Glucose tolerance tests were performed on fasted mice after 7 weeks of high fat diet feeding. For glucose tolerance tests, mice received 20% d-glucose solution (2 mg/kg of body weight) or an equal volume of saline by intraperitoneal injection; blood glucose was measured at base line and at 15, 30, 60, and 120 min. Fasting insulin was measured in plasma samples using a commercially available ELISA kit (Mercodia, Uppsala, Sweden). HOMA-IR was calculated using the formula, (fasting glucose (mg/dl) × fasting insulin (ng/ml))/405.
Plasma ALT and Liver Triglycerides
Plasma samples were assayed for ALT using commercially available enzymatic assay kits (Diagnostic Chemicals, Ltd., Oxford, CT) according to the manufacturer's instructions. Total liver triglycerides were measured using the Triglyceride Reagent Kit from Pointe Scientific Inc. (Lincoln Park, MI) according to the manufacturer's instructions.
Immunohistochemistry
For C3b/iC3b/C3c (C3b), tumor necrosis factor-α (TNFα), and TUNEL staining in liver, frozen sections were mounted on glass slides, fixed with 4% paraformaldehyde, and washed three times in PBS. For C3b and TUNEL staining in epididymal adipose, paraffin-embedded tissue was deparaffinized, and then slides were processed for staining. Sections were blocked with 2% bovine serum albumin (diluted in PBS) or 10% normal goat serum containing 0.1% sodium azide and 0.1% Triton-X-100 for 1 h, followed by overnight incubation with a 1:50 dilution of primary antibody against TNFα (R&D Systems, Minneapolis, MN) or C3b primary antibody (HM1065, Hycult Biotech, Plymouth Meeting, PA) at 4 °C. The C3b antibody recognized a neoepitope revealed only after cleavage of C3. Sections were washed in PBS, incubated with the fluorochrome-conjugated secondary antibody (Alexa Fluor 488 conjugates goat anti-rabbit IgG, 1:250 diluted in blocking buffer) for 2 h in the dark at room temperature, washed again in PBS, and mounted with VECTASHIELD with DAPI containing anti-fade reagent (Vector Laboratories, Inc., Burlingame, CA). Fluorescent images were acquired using a LEICA confocal microscope. No specific immunostaining was seen in sections incubated without primary antibody. TUNEL was visualized using the ApopTag Plus In Situ Apoptosis Detection kit (S7165 for rhodamine staining in adipose and S7111 for fluoroscein staining in liver; Millipore, Billerica, MA) following the manufacturer's instructions.
Omental Adipose Tissue from Obese and Lean Women
Human omental fat samples were obtained, as previously described, from women undergoing elective abdominal surgery (31). This study was approved by the Institutional Review Board at the Cleveland Clinic. Samples were taken from the omental fat depot adjacent to the greater curvature of the stomach and were immediately placed in saline prior to lysis. Tissues were lysed in 20 mm Tris, 1% Np-40, 137 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 10% (v/v) glycerol, 1 mm DTT, 1 mm PMSF, 2 mm Na3VO4 and immediately stored at −80 °C. For Western analysis, 25 μg of total protein was loaded onto an 8% SDS-polyacrylamide gel and transferred to PVDF membranes via a semidry transfer technique. Membranes were blocked with 3% bovine serum albumin, fraction V in Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated overnight with a goat polyclonal antibody against human C3 (1:1000 in TBST) from MP Biomedicals (catalog no. 55033). The C3 cleavage product, C3d, identified based on apparent molecular weight, was detected using chemiluminescence, and images were captured with an Eastman Kodak Co. Image Station 4000R. Membranes were reprobed overnight with a mouse monoclonal antibody against human HSC70 (1:16,000 in TBST) from Santa Cruz Biotechnology, Inc. (catalog no. sc-7298) and analyzed as above. Plasma from a control subject was incubated with or without zymosan (2 mg/ml) for 30 min at 36 °C and used as a positive control in the Western blot analysis.
Statistical Analysis
Values shown in all figures represent means ± S.E. Data were analyzed by analysis of variance using the general linear model procedure (SAS, Carey, IN). Data were log-transformed, as needed, to obtain a normal distribution. Follow-up comparisons were made by least square means testing. Values labeled with different lowercase letters (see Figs. 1–3, 5, and 6 and Tables 1 and 3) are significantly different from one another (p < 0.05).
FIGURE 1.

C1qa deficiency prevents C3b deposition and inflammation in epididymal adipose after 8 weeks of high fat diet; obesity is associated with complement activation in human adipose tissue. C57BL/6J and C1qa−/− mice were allowed free access to a high fat diet (HFD) or low fat diet (LFD) for 8 weeks. A, paraffin-embedded sections of epididymal adipose tissue were stained with hematoxylin and eosin (H&E). Images are representative of 4–6 mice/group and are shown at ×20. B, TUNEL-positive nuclei (red) were detected in adipose tissue from wild-type and C1qa−/− mice. Nuclei were labeled with DAPI (blue). TUNEL-positive nuclei were counted and expressed as a percentage of DAPI-positive nuclei. C, immunoreactive C3b was visualized by immunohistochemistry in paraffin-embedded sections from epididymal adipose and semiquantified. Images are shown at ×40 magnification. B and C, values represent means ± S.E. (error bars). Results labeled with different lowercase letters are significantly different from one another (p < 0.05), n = 4–6 mice. D, relative expression of TNFα and MCP-1 mRNA was measured by RT-PCR in epididymal adipose and normalized to 18 S. Values represent means ± S.E. Results labeled with different lowercase letters are significantly different from one another (p < 0.05), n = 10–12 mice. E, relative protein expression of C3d was assessed in the omental adipose of lean and obese women by Western blotting and normalized to HSC70. Values represent means ± S.E. Results labeled with different lowercase letters are significantly different from one another (p < 0.05), n = 8 for lean and 16 for obese females.
FIGURE 2.

C1qa deficiency prevents altered glucose homeostasis after 8 weeks of high fat diet. C57BL/6J and C1qa−/− mice were allowed free access to a high fat diet (HFD) or low fat diet (LFD) for 8 weeks. Mice were fasted for 6 h prior to blood collection and euthanasia. A, fasting blood glucose was measured from a tail nick using a standard glucometer. B, fasting insulin was measured in plasma samples using a commercially available ELISA kit. C, HOMA-IR was calculated using the formula, (fasting glucose (mg/dl) × fasting insulin (ng/ml))/405. D, for glucose tolerance tests, mice received 20% d-glucose solution (2 mg/kg of bodyweight) by intraperitoneal injection. Blood glucose was measured at base line and at 15, 30, 60, and 120 min. For all graphs, values represent means ± S.E. (error bars). A–C, results labeled with different lowercase letters are significantly different from one another (p < 0.05), n = 4–6. D, *, p < 0.05 compared with C1qa−/− mice.
FIGURE 3.

C1qa−/− mice are protected from 8-week high fat diet-induced hepatic C3b deposition and hepatic steatosis. C57BL/6J and C1qa−/− mice were allowed free access to a high fat diet (HFD) or the low fat diet (LFD) for 8 weeks. A, TUNEL-positive nuclei (green) were detected in liver from wild-type and C1qa−/− mice. Nuclei were labeled with DAPI (blue). TUNEL-positive nuclei were counted and expressed as a percentage of DAPI-positive nuclei. B, hepatic triglycerides were measured by a biochemical assay. C, plasma ALT was assayed using commercially available enzymatic assay kit. D, immunoreactive C3b was visualized by immunohistochemistry in OCT-frozen liver sections. Total fluorescence was determined using Image Pro software. Images are shown at ×40 magnification E, relative expression of TNFα and MCP-1 mRNA was measured by qRT-PCR in liver and normalized to 18 S. Values represent means ± S.E. (error bars) Results labeled with different lowercase letters are significantly different from one another (p < 0.05), n = 4–8. *, p < 0.05 compared with C1qa−/− mice.
FIGURE 5.

Three-day high fat diet induces hepatic insulin resistance and complement activation. Male C57BL/6J mice were allowed free access to a high fat diet (HFD; 58% of calories from fat) or standard laboratory chow (Chow) for 3 days. A, immunoreactive C3b was visualized by immunohistochemistry in OCT-frozen liver sections. The total number of fluorescent foci per field was determined using Image Pro. B–D, to assess insulin sensitivity, mice were injected with insulin (I) or an equal volume of saline (S) via the portal vein. Relative quantity of phospho-AKT was assessed in adipose (B) and liver (C and D) by Western blotting and normalized to HSC70. D and E, Kupffer cells were depleted in C57BL/6J mice by injection with clodronate- or PBS-containing liposomes via tail vein 24 h prior to starting a 3-day high fat (HFD) diet. Expression of F4/80 mRNA was reduced to less than 16% of saline-treated mice (data not shown). E, immunoreactive C3b was visualized by immunohistochemistry in OCT-frozen liver sections. Values represent mean ± S.E., n = 3–6 mice/group. Values labeled with different lowercase letters are significantly different from each other (p < 0.05).
FIGURE 6.

Role of C3aR, CD55, or C1qa in high fat diet-induced hepatic insulin resistance. C57BL/6J, C3aR−/−, CD55−/−, or C1qa−/− mice were allowed free access to a high fat diet (HFD) or chow (Ch) diet for 3 days (2 days for CD55−/−). Mice were injected with insulin (I) or saline (S) via the portal vein. Relative quantity of phospho-AKT was assessed in the liver of C3aR−/− (A), CD55−/− (B), or C1qa−/− (C) by Western blotting and normalized to HSC70. Values below the images represent mean ± S.E. Values with an asterisk are significantly different from those for saline-injected chow-fed mice within each genotype (p < 0.05), n = 3–8. D, immunoreactive C3b was visualized by immunohistochemistry in OCT-frozen liver sections. The total number of fluorescent foci per field was determined using Image Pro. Images are representative of 3–4 mice/group and are shown at 40× magnification. Values labeled with different lowercase letters are significantly different from each other (p < 0.05). E, immunoreactive TNFα was visualized by immunohistochemistry of OCT-frozen liver sections. Representative images are shown at ×40 magnification.
TABLE 1.
Metabolic characteristics of wild-type and C1qa−/− mice on low fat and high fat diets for 8 weeks
Values represent means ± S.E., n = 10–12. HFD, high fat diet; LFD, low fat diet.
| C57BL/6J LFD | C57BL/6J HFD | C1qa−/− LFD | C1qa−/− HFD | |
|---|---|---|---|---|
| Initial body weight (g) | 20.0 ± 0.2a | 19.9 ± 0.4a | 21.3 ± 0.7a | 20.8 ± 0.6a |
| Final body weight (g) | 25.4 ± 0.5a | 33.8 ± 0.8b | 28.6 ± 0.9a | 34.3 ± 1.3b |
| Epididymal adipose (g)/body weight (g) | 1.8 ± 0.2a | 4.9 ± 0.3b | 2.0 ± 0.3a | 3.6 ± 0.5b |
a,b Values with different superscript letters are significantly different from each other, p < 0.05.
TABLE 3.
Metabolic characteristics of wild-type and C1qa−/− mice on chow or high fat diets for 3 days
Values represent means ± S.E., n = 10–12. HFD, high fat diet.
| C57BL/6J chow | C57BL/6J HFD | C1qa−/− chow | C1qa−/− HFD | |
|---|---|---|---|---|
| Weight gain (3 days) | 1.7 ± 0.2a | 1.7 ± 0.3a | 2.7 ± 0.6a | 2.7 ± 0.5a |
| Hepatic triglycerides (mg/g) | 17.9 ± 0.7a | 27.2 ± 2.0b | 21.5 ± 2.1a | 31.5 ± 3.4b |
| Plasma ALT (units/liter) | 14.7 ± 0.5a | 19.5 ± 2.2a | 23.3 ± 2.4b | 25.7 ± 3.1b |
a,b Values with different superscript letters are significantly different from each other, p < 0.05.
RESULTS
Mice Deficient in C1q Become Obese on High Fat Diets but Are Protected from Activation of Complement in Adipose Tissue and Expression of Inflammatory Cytokines
Complement has been implicated in the development of insulin resistance because C3a receptor-deficient mice are protected from the metabolic effects of high fat diets in adipose tissue (20). However, the pathway of complement activation in response to high fat diet-induced obesity has not yet been identified. The classical pathway has been implicated in both alcoholic liver disease (17, 21) and non-alcoholic liver disease (19). Therefore, to establish if this pathway contributes to the metabolic effects of high fat diet-induced obesity, wild-type mice and mice deficient in C1qa, a required element in the classical pathway of complement activation, were fed the high fat or low fat diets for 8 weeks. Initial body weights were similar among all groups; after 8 weeks, mice fed the high fat diet had greater final bodyweight compared with mice on the low fat diet (Table 1). Weight gain in response to high fat diet feeding was equivalent in wild-type and C1qa−/− mice (Table 1). Epididymal adipose weight, relative to total body weight, was increased in both wild-type and C1qa−/− mice after high fat diet compared with mice on the low fat diet; there was no effect of genotype on this response (Table 1).
High fat diets increased epididymal adipocyte size in both wild-type and C1qa−/− mice (Fig. 1A) as well as increasing the number of TUNEL-positive nuclei (Fig. 1B). High fat diets were also associated with evidence of complement activation in adipose tissue. Accumulation of C3b, a cleavage product of C3, was increased in the adipose of wild-type mice after 8 weeks of high fat diet feeding (Fig. 1C). In contrast, there was no evidence of complement activation in the adipose of C1qa−/− mice, suggesting a critical role for the classical pathway in complement activation in adipose tissue. Similarly, the relative expression of TNFα and monocyte chemotactic protein-1 (MCP-1) mRNA, indicators of adipose inflammation, were increased in the epididymal adipose of wild-type but not C1qa−/− mice on the high fat diet (Fig. 1D).
Complement Activation in Adipose Tissue in Obese Insulin-resistant Women
Because high fat diet-induced obesity activated complement in adipose tissue of mice, we hypothesized that complement would also be activated in adipose tissue from obese insulin-resistant humans. C3d, another C3 cleavage product, was assessed by Western blotting in omental adipose tissue obtained from lean and obese women while undergoing elective abdominal surgery (Table 2). The relative quantity of immunoreactive C3d was increased in the omental adipose from obese insulin-resistant women compared with lean controls (Fig. 1E). Taken together, these data indicate that adipose inflammation is associated with complement activation in both mouse models of obesity and in obese insulin-resistant women.
TABLE 2.
Demographics of lean and obese women
Values are means ± S.E., n = 8 for lean and 16 for obese. BMI, body mass index; ND, not determined.
| Lean women | Obese women | |
|---|---|---|
| Age | 39 ± 7 | 46 ± 3 |
| BMI | 22.3 ± 0.9 | 46.1 ± 1.4 |
| Fasting blood glucose | 81.3 ± 3.3 | 121.9 ± 12.4 |
| Triglycerides | ND | 177.6 ± 21.5 |
| Cholesterol | ND | 203.9 ± 9.1 |
| LDL | ND | 104.9 ± 8.8 |
| HDL | ND | 52.2 ± 2.9 |
C1qa Deficiency Prevents Altered Glucose Homeostasis after 8 Weeks of High Fat Diet
Because C1q-dependent complement activation was associated with adipose inflammation following high fat diet feeding, we hypothesized that C1qa would also mediate impaired glucose homeostasis after feeding high fat diets. Following 8 weeks of high fat diet feeding, fasting blood glucose, fasting plasma insulin, and HOMA-IR (an indicator of insulin resistance) were increased in wild-type mice compared with mice on the low fat diet (Fig. 2, A–C), whereas C1qa−/− mice were resistant to these effects of high fat diets. C1qa−/− mice exhibited a lower blood glucose excursion compared with wild-type mice after high fat diet feeding in a glucose tolerance test (Fig. 2D).
C1q Deficiency Prevents Hepatic Steatosis and C3b Deposition after 8 Weeks of High Fat Diet
High fat feeding for 8 weeks to C57BL/6 mice resulted in a phenotype typical of high fat diet-induced steatosis, including increased TUNEL-positive nuclei in the liver (Fig. 3A) as well as increases in hepatic triglycerides (Fig. 3B), circulating ALT concentrations (Fig. 3C), and expression of inflammatory cytokines (Fig. 3E). Complement activation products are evident in the liver of obese patients with non-alcoholic steatohepatitis; importantly, complement activation correlates with disease severity (19). Wild-type mice fed high fat diets for 8 weeks also exhibit evidence of complement activation, evidenced by increased accumulation of C3b (Fig. 3D); C3b accumulation was not observed in C1qa−/− mice (Fig. 3A). C1qa deficiency did not prevent high fat diet-induced increases in TUNEL-positive nuclei in the liver but did protect mice from hepatic steatosis, inflammatory cytokine expression, and also partially reduced plasma ALT concentrations compared with wild-type mice (Fig. 3).
Complement Activation in Adipose and Liver Occurs in Response to Bid-dependent Apoptosis
High fat diets and obesity are linked with increased apoptosis in adipose tissue and liver (26). Our group has previously found that adipocyte apoptosis is dependent on Bid, a proapoptotic protein that links the extrinsic and intrinsic cell death pathways, and that Bid−/− mice are protected from high fat diet-induced insulin resistance and hepatic steatosis (26). Because one important mechanism for the activation of the classical complement pathway is via the binding of C1q to apoptotic cells, we hypothesized that C1q provides the link between apoptosis and complement activation. If this hypothesis were true, then complement activation in response to high fat diet-induced obesity should be reduced in Bid−/− mice. Indeed, C3b deposition in both adipose and liver of was not detected in Bid−/− mice after high fat diet feeding (Fig. 4). Taken together, these data suggest that high fat diet-induced obesity is associated with a C1q-dependent activation of complement in both adipose and liver and that complement activation is downstream of high fat diet-induced apoptosis and then contributes to multiple aspects of the metabolic syndrome.
FIGURE 4.
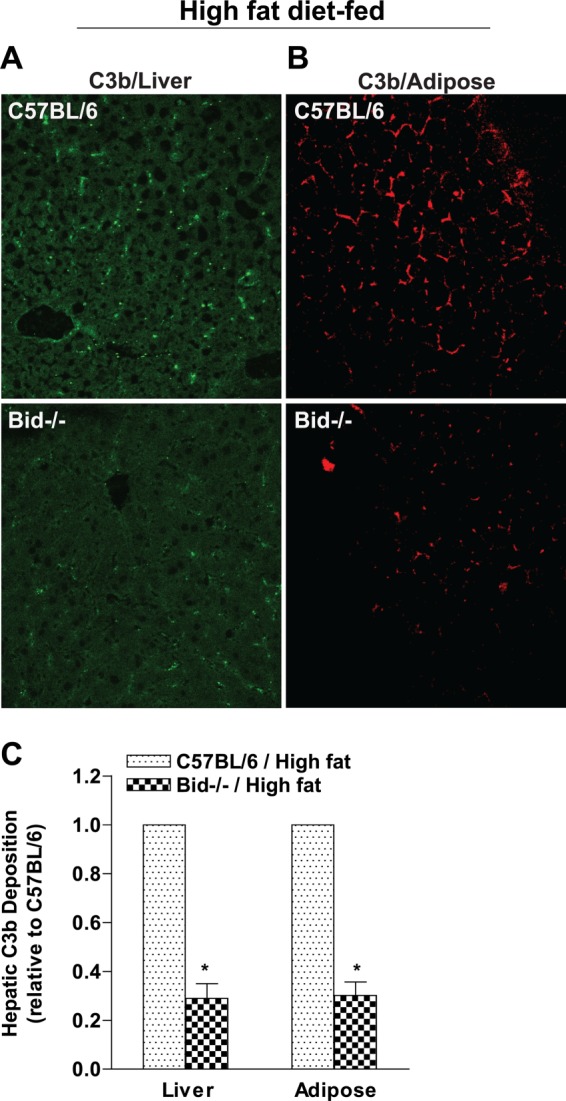
Bid−/− mice are protected from activation of complement in response to high fat diet feeding. C57BL/6 and Bid−/− mice were provided with high fat diet, and immunoreactive C3b was visualized by immunohistochemistry in paraffin-embedded sections from epididymal adipose (A) and liver (B). Minimal C3b staining was observed in mice fed low fat diets (data not shown). Images were semiquantified (C) as described under “Materials and Methods.” Images are shown at ×40 magnification. Values represent means ± S.E. (error bars), n = 4–6. *, p < 0.05 compared with Bid−/− mice.
Three-day High Fat Diet Feeding Results in Hepatic Insulin Resistance in Wild-type Mice
Inflammatory mediators contribute to hepatic insulin resistance induced by short term high fat diet feeding (4) as well as impaired glucose homeostasis that develops after long term feeding. The development of hepatic insulin resistance is considered pivotal in the eventual progression of systemic insulin resistance and type 2 diabetes (5). Because complement was activated after long term high fat diet feeding in both adipose and liver, we investigated whether complement was also involved in the early hepatic responses to high fat diets. Even short courses of high fat diet feeding result in hepatic steatosis and insulin resistance (32). Wild-type mice were fed a high fat diet or a standard laboratory chow diet for 3 days. Total weight gain was equal in high fat- and chow-fed mice (Table 3). High fat diet feeding for 3 days slightly increased hepatic triglycerides in both genotypes (Table 3). The short exposure to high fat diets had no effect on plasma ALT, but ALT concentrations were slightly higher in C1qa−/− mice compared with wild type on both the chow and high fat diets (Table 3). Complement activation in the liver, assessed by the accumulation of the C3 degradation product C3b, was increased in wild-type mice after 3 days of high fat diet with a predominately sinusoidal deposition (Fig. 5A). To assess insulin sensitivity, mice were injected with insulin or vehicle (saline) via the portal vein. Following challenge with insulin, phosphorylated AKT increased in adipose (Fig. 5B) and liver (Fig. 5C) of chow-fed mice. Feeding of high fat diets for 3 days decreased insulin stimulation of phospho-AKT in liver but not adipose tissue of wild-type mice, indicating a rapid development of hepatic insulin resistance prior to insulin resistance in adipose tissue (Fig. 5, B and C). Consistent with previous reports (4), early hepatic insulin resistance was mediated via activation of hepatic macrophages. Depletion of hepatic macrophages with clodronate prevented the high fat diet-induced complement activation (Fig. 5E) as well as the loss of hepatic insulin sensitivity, evidenced by a normal insulin-stimulated phosphorylation of Akt in clodronate-treated mice, after high fat diet feeding for 3 days (Fig. 5D).
Complement Activation Contributes to the Early Effects of High Fat Diet-induced Hepatic Insulin Resistance
Mamane et al. (20) reported that C3aR is required for impaired insulin sensitivity after high fat diet-induced obesity. Here we tested whether C3aR was also required for the development of hepatic insulin resistance after 3 days of high fat diet feeding. Although high fat diet reduced insulin-stimulated phospho-AKT in the liver of wild-type mice, C3aR−/− mice were protected from this loss of insulin sensitivity (Fig. 6A). CD55/DAF is a negative regulator of complement, and thus CD55/DAF deficiency would be hypothesized to exacerbate the effects of complement activation in the development of short term high fat diet-induced hepatic insulin resistance. In order to test this hypothesis, insulin sensitivity was measured in wild-type and CD55/DAF−/− mice after only 2 days of high fat diet. Whereas wild-type mice were still insulin-sensitive after only 2 days of high fat diet feeding, mice deficient in CD55/DAF already exhibited decreased phospho-AKT after challenge with insulin (Fig. 6B). Taken together, these data suggest that complement activation contributes to hepatic insulin resistance induced by short term high fat diet.
C1qa Is Required for Hepatic C3b Deposition after a 3-Day High Fat Diet
Because C1q-dependent activation of complement was critical for the metabolic effects of high fat diet-induced obesity, we next investigated whether C1qa also mediated the early hepatic response to high fat diets. In contrast to wild-type mice (Figs. 5C and 6C), mice deficient in C1qa−/− maintained insulin-stimulated phosphorylation of AKT in the liver after high fat diet feeding for 3 days (Fig. 6C). Consistent with this protective effect on insulin sensitivity, hepatic C3b deposition was not increased in C1qa−/− mice after 3 days of high fat diet feeding (Fig. 6D). Finally, both C1qa−/− and C3aR−/− mice express less TNFα after high fat diet feeding compared with wild type mice (Fig. 6E). Taken together, these data indicate a critical role for C1q-dependent complement activation in the development of early hepatic insulin resistance in response to high fat diet feeding.
DISCUSSION
Although it is well appreciated that the innate immune system is critical to the development of insulin resistance and the complications of obesity, very little is known about the role of complement, a key effector in the innate immune response, in high fat diet/obesity-induced insulin resistance metabolic syndrome. Here we provide evidence of complement activation in adipose tissue in mouse models of high fat diet-induced obesity as well as in the omental adipose tissue of obese women. Importantly, mice deficient in C1qa, a key component in complement activation via the classical pathway, were protected from high fat diet-induced adipose inflammation and systemic glucose intolerance. Even short term feeding of high fat diets, prior to the onset of obesity, increased complement activation in the liver. Early complement activation in the liver was C1qa-dependent and was required for the development of hepatic insulin resistance. Taken together, these data suggest that the classical pathway is a key effector of the metabolic abnormalities associated with high fat diets and obesity.
Previous correlative studies have implicated complement in the development of diabetes and metabolic syndrome. For example, increased plasma C3a is associated with obesity and type 2 diabetes (33, 34). Indeed, serum C3 is a more robust marker of insulin resistance than other markers, such as C-reactive protein and leukocyte count (35). Complement is also activated in plasma of type 1 diabetics (36). Complement activation is implicated in the micro- and macrovascular complications in diabetics, associated with cardiovascular disease and renal disease (37, 38). Interestingly, genome-wide association studies between single nucleotide polymorphisms (SNPs) and expression of genes in metabolically active tissues, including liver and adipose tissue, have identified the complement cascade as an important candidate pathway in type 2 diabetes (39). CR1, C4A/B, and C5 genes were highly significant in this analysis (39). The most well studied aspect of the relationship between adipose tissue and complement stems from the identification of C3adesArg, a C3 degradation product that does not interact with the C3a receptor, as acylation-stimulating protein, a peptide involved in triglyceride homeostasis (40). Adipose tissue also produces C3, factor B, and factor D, essential components for the alternative pathway of activation, as well as properdin (40). C3adesArg/acylation-stimulating protein is detected locally in adipose tissue in response to meals, associated with increased storage of triglycerides (41).
More recent mechanistic studies have identified an important role for C3aR and C5aR in mediating adipose inflammation and insulin resistance in mouse and rat models of high fat diet-induced obesity (20, 42). Here, making use of C1qa−/−-deficient mice, we have identified the classical pathway of complement activation as required for activation of complement in both mouse and human obesity. In adipose tissue, C1q-dependent complement activation was evident after 8 weeks of high fat diet feeding (Fig. 1) but not after 3 days (data not shown). Complement proteins C1q and C3b act as opsonins to facilitate the clearance of apoptotic cell bodies (25, 43, 43). High fat diet-induced obesity is associated with apoptosis in both adipose and liver. Apoptosis may be due to toxic effects of lipids and/or due to the overexpansion of adipocyte size (44, 45). The current data are consistent with the involvement of C1q in the clearance of apoptotic cells within the inflamed adipose tissue; when apoptosis in adipose is prevented by Bid deficiency, complement was not activated, and inflammation was reduced. This role may be similar to the critical link between apoptosis in adipose of alcohol-fed mice and complement activation via C1q (21) and neuronal apoptosis and C1q activation in some murine models of Alzheimer disease (46). Alternatively, C1q may be binding to oxidized phospholipids in the inflamed adipose tissue (47).
Hepatic insulin resistance occurs early in response to high fat diets, prior to systemic glucose intolerance (48–50) and is mediated, at least in part, via increased expression of inflammatory mediators, such as TNFα. Inflammatory cytokines induce insulin resistance through phosphorylation of negative regulatory sites in the insulin signaling cascade (2, 51, 52). Expression of inflammatory cytokines in the liver is mediated via the activation of Kupffer cells, the resident macrophage in the liver, in a number of acute and chronic inflammatory conditions (9, 18). Kupffer cells also contribute to the development of hepatic insulin resistance, even after short term feeding of high fat diets (Fig. 4) (9, 50, 53). The mechanisms for Kupffer cell activation in response to the early phases of high fat diet feeding are not yet understood. Activation of TLR4 by LPS and/or fatty acids has been implicated in Kupffer cell activation during high fat diet feeding (9). Oxidized LDL can also activate Kupffer cells during high fat diet feeding to LDL receptor-deficient mice (54). Here we find that high fat diets increased complement activation in the liver within 3 days of feeding; this activation was dependent on C1q. Evidence of complement activation in the liver of both male and female patients with non-alcoholic steatohepatitis also suggests the involvement of the classical pathway in obese humans (19). Because Kupffer cells express both C3aR and C5aR receptors (13), our data suggest that complement activation in the liver contributes to activation of Kupffer cells and expression of inflammatory cytokines in the liver during high fat diet feeding and obesity.
Importantly, C1q-dependent complement activation was also required for the development of the early phase of hepatic insulin resistance, evidenced by impaired phosphorylation of Akt in response to a challenge with insulin. After longer term feeding of high fat diets and the development of obesity, systemic glucose intolerance was also found to be dependent on C1q. Previous work in mouse and rat models of high fat diet-induced obesity have demonstrated a critical role for C3aR and C5aR in the impairment of glucose homeostasis (20, 42). Taken together with the current data, these studies identify C1q-mediated activation of complement as an essential component in the development of both early hepatic insulin resistance and later systemic glucose intolerance. Evidence of complement activation in both adipose and liver suggest that complement is involved in multiple aspects of the tissue pathophysiology leading to insulin resistance and may be an important therapeutic target for preventing and/or reducing the derangements in metabolism associated with obesity.
Acknowledgments
We thank M. Edward Medof, Marina Botta, Xia-MingYin, and John Carroll for providing genetically modified mice for this study.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grants 5R01AA016399 and 5R37 AA011876 (to L. E. N.), RO1DK089547 (to J. P. K.), RO1 DK076853 and RO1DK082451 (to A. E. F.), support from Grant T32DK07319 (to A. D. H.), and NIH, NCRR, Grant CTSA UL1RR024989 (Case Western Reserve University/Cleveland Clinic).
- OCT
- optimal cutting temperature compound
- HOMA-IR
- homeostatic model assessment-insulin resistance
- ALT
- alanine transaminase
- C3b
- C3b/iC3b/C3c.
REFERENCES
- 1. Khashab M. A., Liangpunsakul S., Chalasani N. (2008) Nonalcoholic fatty liver disease as a component of the metabolic syndrome. Curr. Gastroenterol. Rep. 10, 73–80 [DOI] [PubMed] [Google Scholar]
- 2. Gregor M. F., Hotamisligil G. S. (2011) Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445 [DOI] [PubMed] [Google Scholar]
- 3. Musso G., Gambino R., Cassader M. (2010) Non-alcoholic fatty liver disease from pathogenesis to management. An update. Obes. Rev. 11, 430–445 [DOI] [PubMed] [Google Scholar]
- 4. Boden G., Shulman G. I. (2002) Free fatty acids in obesity and type 2 diabetes. Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Invest. 32, 14–23 [DOI] [PubMed] [Google Scholar]
- 5. Savage D. B., Petersen K. F., Shulman G. I. (2007) Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 87, 507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baffy G. (2009) Kupffer cells in non-alcoholic fatty liver disease. The emerging view. J. Hepatol. 51, 212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wouters K., van Gorp P. J., Bieghs V., Gijbels M. J., Duimel H., Lütjohann D., Kerksiek A., van Kruchten R., Maeda N., Staels B., van Bilsen M., Shiri-Sverdlov R., Hofker M. H. (2008) Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48, 474–86 [DOI] [PubMed] [Google Scholar]
- 9. Valenti L., Fracanzani A. L., Fargion S. (2009) The immunopathogenesis of alcoholic and nonalcoholic steatohepatitis. Two triggers for one disease? Semin. Immunopathol. 31, 359–369 [DOI] [PubMed] [Google Scholar]
- 10. Gasque P. (2004) Complement. A unique innate immune sensor for danger signals. Mol. Immunol. 41, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 11. Walport M. J. (2001) Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 12. Monsinjon T., Gasque P., Chan P., Ischenko A., Brady J. J., Fontaine M. C. (2003) Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 17, 1003–1014 [DOI] [PubMed] [Google Scholar]
- 13. Schieferdecker H. L., Schlaf G., Jungermann K., Götze O. (2001) Functions of anaphylatoxin C5a in rat liver. Direct and indirect actions on nonparenchymal and parenchymal cells. Int. Immunopharmacol. 1, 469–481 [DOI] [PubMed] [Google Scholar]
- 14. Jauneau A. C., Ischenko A., Chan P., Fontaine M. (2003) Complement component anaphylatoxins upregulate chemokine expression by human astrocytes. FEBS Lett. 537, 17–22 [DOI] [PubMed] [Google Scholar]
- 15. DiScipio R. G., Daffern P. J., Jagels M. A., Broide D. H., Sriramarao P. (1999) A comparison of C3a and C5a-mediated stable adhesion of rolling eosinophils in postcapillary venules and transendothelial migration in vitro and in vivo. J. Immunol. 162, 1127–1136 [PubMed] [Google Scholar]
- 16. Pritchard M. T., McMullen M. R., Stavitsky A. B., Cohen J. I., Lin F., Medof M. E., Nagy L. E. (2007) Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology 132, 1117–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cohen J. I., Roychowdhury S., McMullen M. R., Stavitsky A. B., Nagy L. E. (2010) Complement and alcoholic liver disease. Role of C1q in the pathogenesis of ethanol-induced liver injury in mice. Gastroenterology 139, 664–674, 674.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H. J., Gao B., Zakhari S., Nagy L. E. (2012) Inflammation in alcoholic liver disease. Annu. Rev. Nutr. 32, 343–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rensen S. S., Slaats Y., Driessen A., Peutz-Kootstra C. J., Nijhuis J., Steffensen R., Greve J. W., Buurman W. A. (2009) Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 20. Mamane Y., Chung Chan C., Lavallee G., Morin N., Xu L. J., Huang J., Gordon R., Thomas W., Lamb J., Schadt E. E., Kennedy B. P., Mancini J. A. (2009) The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes 58, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sebastian B. M., Roychowdhury S., Tang H., Hillian A. D., Feldstein A. E., Stahl G. L., Takahashi K., Nagy L. E. (2011) Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J. Biol. Chem. 286, 35989–35997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Païdassi H., Tacnet-Delorme P., Garlatti V., Darnault C., Ghebrehiwet B., Gaboriaud C., Arlaud G. J., Frachet P. (2008) C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J. Immunol. 180, 2329–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Korb L. C., Ahearn J. M. (1997) C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes. Complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 158, 4525–4528 [PubMed] [Google Scholar]
- 24. Elward K., Griffiths M., Mizuno M., Harris C. L., Neal J. W., Morgan B. P., Gasque P. (2005) CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J. Biol. Chem. 280, 36342–36354 [DOI] [PubMed] [Google Scholar]
- 25. Mevorach D., Mascarenhas J. O., Gershov D., Elkon K. B. (1998) Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med. 188, 2313–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alkhouri N., Gornicka A., Berk M. P., Thapaliya S., Dixon L. J., Kashyap S., Schauer P. R., Feldstein A. E. (2010) Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 285, 3428–3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kildsgaard J., Hollmann T. J., Matthews K. W., Bian K., Murad F., Wetsel R. A. (2000) Cutting edge. Targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J. Immunol. 165, 5406–5409 [DOI] [PubMed] [Google Scholar]
- 28. Lin F., Fukuoka Y., Spicer A., Ohta R., Okada N., Harris C. L., Emancipator S. N., Medof M. E. (2001) Tissue distribution of products of the mouse decay-accelerating factor (DAF) genes. Exploitation of a Daf1 knock-out mouse and site-specific monoclonal antibodies. Immunology 104, 215–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Botto M., Dell'Agnola C., Bygrave A. E., Thompson E. M., Cook H. T., Petry F., Loos M., Pandolfi P. P., Walport M. J. (1998) Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19, 56–59 [DOI] [PubMed] [Google Scholar]
- 30. Yin X. M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K. A., Korsmeyer S. J. (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400, 886–891 [DOI] [PubMed] [Google Scholar]
- 31. Kelly K. R., Kashyap S. R., O'Leary V. B., Major J., Schauer P. R., Kirwan J. P. (2010) Retinol-binding protein 4 (RBP4) protein expression is increased in omental adipose tissue of severely obese patients. Obesity 18, 663–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maher J. J., Leon P., Ryan J. C. (2008) Beyond insulin resistance. Innate immunity in nonalcoholic steatohepatitis. Hepatology 48, 670–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y., Lu H. L., Zhang J., Yu H. Y., Wang H. W., Zhang M. X., Cianflone K. (2006) Relationships among acylation stimulating protein, adiponectin and complement C3 in lean vs. obese type 2 diabetes. Int. J. Obes. (Lond.) 30, 439–446 [DOI] [PubMed] [Google Scholar]
- 34. Weyer C., Tataranni P. A., Pratley R. E. (2000) Insulin action and insulinemia are closely related to the fasting complement C3, but not acylation stimulating protein concentration. Diabetes Care 23, 779–785 [DOI] [PubMed] [Google Scholar]
- 35. Muscari A., Antonelli S., Bianchi G., Cavrini G., Dapporto S., Ligabue A., Ludovico C., Magalotti D., Poggiopollini G., Zoli M. (2007) Serum C3 is a stronger inflammatory marker of insulin resistance than C-reactive protein, leukocyte count, and erythrocyte sedimentation rate. Comparison study in an elderly population. Diabetes Care 30, 2362–2368 [DOI] [PubMed] [Google Scholar]
- 36. Sundsmo J. S., Papin R. A., Wood L., Hirani S., Waldeck N., Buckingham B., Kershnar A., Ascher M., Charles M. A. (1985) Complement activation in type 1 human diabetes. Clin. Immunol. Immunopathol. 35, 211–225 [DOI] [PubMed] [Google Scholar]
- 37. Østergaard J., Hansen T. K., Thiel S., Flyvbjerg A. (2005) Complement activation and diabetic vascular complications. Clin. Chim. Acta 361, 10–19 [DOI] [PubMed] [Google Scholar]
- 38. Flyvbjerg A. (2010) Diabetic angiopathy, the complement system and the tumor necrosis factor superfamily. Nat. Rev. Endocrinol. 6, 94–101 [DOI] [PubMed] [Google Scholar]
- 39. Zhong H., Beaulaurier J., Lum P. Y., Molony C., Yang X., Macneil D. J., Weingarth D. T., Zhang B., Greenawalt D., Dobrin R., Hao K., Woo S., Fabre-Suver C., Qian S., Tota M. R., Keller M. P., Kendziorski C. M., Yandell B. S., Castro V., Attie A. D., Kaplan L. M., Schadt E. E. (2010) Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 6, e1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pattrick M., Luckett J., Yue L., Stover C. (2009) Dual role of complement in adipose tissue. Mol. Immunol. 46, 755–760 [DOI] [PubMed] [Google Scholar]
- 41. MacLaren R. E., Cui W., Lu H., Simard S., Cianflone K. (2010) Association of adipocyte genes with ASP expression. A microarray analysis of subcutaneous and omental adipose tissue in morbidly obese subjects. BMC Med. Genomics 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lim J., Iyer A., Suen J. Y., Seow V., Reid R. C., Brown L., Fairlie D. P. (2013) C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. 27, 822–831 [DOI] [PubMed] [Google Scholar]
- 43. Trouw L. A., Blom A. M., Gasque P. (2008) Role of complement and complement regulators in the removal of apoptotic cells. Mol. Immunol. 45, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 44. Gornicka A., Fettig J., Eguchi A., Berk M. P., Thapaliya S., Dixon L. J., Feldstein A. E. (2012) Adipocyte hypertrophy is associated with lysosomal permeability both in vivo and in vitro. Role in adipose tissue inflammation. Am. J. Physiol. Endocrinol. Metab. 303, E597–E606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lumeng C. N., DelProposto J. B., Westcott D. J., Saltiel A. R. (2008) Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rivera C. A., Gaskin L., Allman M., Pang J., Brady K., Adegboyega P., Pruitt K. (2010) Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol. 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Galvan M. D., Foreman D. B., Zeng E., Tan J. C., Bohlson S. S. (2012) Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J. Immunol. 188, 3716–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kraegen E. W., Clark P. W., Jenkins A. B., Daley E. A., Chisholm D. J., Storlien L. H. (1991) Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 40, 1397–1403 [DOI] [PubMed] [Google Scholar]
- 49. Samuel V. T., Liu Z. X., Qu X., Elder B. D., Bilz S., Befroy D., Romanelli A. J., Shulman G. I. (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353 [DOI] [PubMed] [Google Scholar]
- 50. Lanthier N., Molendi-Coste O., Horsmans Y., van Rooijen N., Cani P. D., Leclercq I. A. (2010) Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G107–G116 [DOI] [PubMed] [Google Scholar]
- 51. Tilg H. (2010) The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis. 28, 179–185 [DOI] [PubMed] [Google Scholar]
- 52. Vallerie S. N., Hotamisligil G. S. (2010) The role of JNK proteins in metabolism. Sci. Transl. Med. 2, 60rv5. [DOI] [PubMed] [Google Scholar]
- 53. Trauner M., Arrese M., Wagner M. (2010) Fatty liver and lipotoxicity. Biochim. Biophys. Acta 1801, 299–310 [DOI] [PubMed] [Google Scholar]
- 54. Bieghs V., van Gorp P. J., Walenbergh S. M., Gijbels M. J., Verheyen F., Buurman W. A., Briles D. E., Hofker M. H., Binder C. J., Shiri-Sverdlov R. (2012) Specific immunization strategies against oxidized low-density lipoprotein. A novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology 56, 894–903 [DOI] [PMC free article] [PubMed] [Google Scholar]