

Background: hnRNP L is a multifunctional RNA-binding protein implicated in alternative splicing regulation, etc.
Results: RRM1 and RRM34 of hnRNP L adopt nearly typical RRM topology and use β-sheets to bind RNA.
Conclusion: hnRNP L RRM34 facilitates the target RNA with appropriate in-between distance to form a loop.
Significance: RNA looping suggests a general mechanism for alternative splicing regulators possessing position-dependent dual roles.
Keywords: Crystal Structure, Protein Structure, RNA Splicing, RNA-binding Proteins, RNA-Protein Interaction, RNA Binding, RNA Looping, RRM Domain, hnRNP-L
Abstract
Heterogeneous nuclear ribonucleoprotein L (hnRNP L) is an abundant RNA-binding protein implicated in many bioprocesses, including pre-mRNA processing, mRNA export of intronless genes, internal ribosomal entry site-mediated translation, and chromatin modification. It contains four RNA recognition motifs (RRMs) that bind with CA repeats or CA-rich elements. In this study, surface plasmon resonance spectroscopy assays revealed that all four RRM domains contribute to RNA binding. Furthermore, we elucidated the crystal structures of hnRNP L RRM1 and RRM34 at 2.0 and 1.8 Å, respectively. These RRMs all adopt the typical β1α1β2β3α2β4 topology, except for an unusual fifth β-strand in RRM3. RRM3 and RRM4 interact intimately with each other mainly through helical surfaces, leading the two β-sheets to face opposite directions. Structure-based mutations and surface plasmon resonance assay results suggested that the β-sheets of RRM1 and RRM34 are accessible for RNA binding. FRET-based gel shift assays (FRET-EMSA) and steady-state FRET assays, together with cross-linking and dynamic light scattering assays, demonstrated that hnRNP L RRM34 facilitates RNA looping when binding to two appropriately separated binding sites within the same target pre-mRNA. EMSA and isothermal titration calorimetry binding studies with in vivo target RNA suggested that hnRNP L-mediated RNA looping may occur in vivo. Our study provides a mechanistic explanation for the dual functions of hnRNP L in alternative splicing regulation as an activator or repressor.
Introduction
Heterogeneous nuclear ribonucleoproteins (hnRNPs)3 are RNA-binding proteins that are involved in a variety of cellular functions, including nascent transcript packaging, alternative splicing, and translation regulation (1). The importance of RNA processing steps to human diseases, especially cancer, is becoming clearer (2). hnRNP L is an abundant and multifunctional protein that shuttles between the nucleus and cytoplasm. It mediates selective exon inclusion during alternative splicing (3), facilitates polyadenylation (4), assists export of intronless gene mRNA (5), promotes internal ribosomal entry site-dependent translation (6, 7), and regulates mRNA stability (8, 9). hnRNP L interacts with other hnRNPs, including hnRNP A2 (10), E2, I, and K (11). hnRNP L also associates with lysine methyltransferase-3 during chromatin modification (12), the MED23 protein of Mediator during transcriptional regulation (13) and apurinic/apyrimidinic endonuclease 1 during transcriptional regulation (14).
CA-repetitive sequences are the most common simple sequence repeat in the human genome (15). CA-repeat and CA-rich elements are a widespread class of regulators that function in mammalian alternative splicing (16). hnRNP L binds to these elements specifically and acts as an activator or repressor, depending on the context (3). hnRNP L binds to various types of genes and regulates the alternative splicing and expression of different isoforms, for example, pro- and anti-apoptotic isoforms of caspase-9 in non-small cell lung cancer (2), multiple isoforms of CD45 during an immune response (17, 18), and the isoforms of carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) implicated in carcinogenesis (19). Many recent studies have focused on hnRNP L-mediated splicing regulation. Heiner et al. (20) showed that hnRNP L represses exon inclusion by binding to intronic high score binding motifs located close to splice sites of tight junction protein 1 (TJP1) and solute carrier family 2 (facilitated glucose transporter) member 2 (SLC2A2) genes, which sterically prevent splice site recognition by snRNPs. House and Lynch (21) demonstrated that hnRNP L binds to an exonic splicing silencer in CD45 exon 4 and forms a ternary complex with the U1 and U2 snRNPs flanking the exon, which interferes with cross-intron snRNP pairing and blocks the transition from an A or A-like complex to a B complex in the developing spliceosome. Motta-Mena et al. (3) proposed that hnRNP L represses CD45 exon 5 usage in T cells by inhibiting the binding of the exonic splicing enhancer (ESE) to the enhancer complex that recruits the U2 snRNP. However, how hnRNP L binds RNA to regulate alternative pre-mRNA splicing remains unknown.
hnRNP L contains four RNA recognition motifs (RRMs), also known as RNA-binding domains. The RRM domain is ∼90 amino acids long and contains conserved eight- and six-residue motifs named RNP-1 and RNP-2, respectively. It usually folds into a typical β1α1β2β3α2β4 structure, including a four-stranded antiparallel β-sheet as the primary RNA-binding surface. Three aromatic residues at key positions of the canonical RNP-1 and RNP-2 motifs on the central strands are responsible for nonspecific contacts with RNA (22). Surprisingly, no conserved aromatic residues at key positions in RNP-1 and RNP-2 are found in any of the four RRM domains of hnRNP L (Fig. 1A). Several structures of RRM domains without conserved aromatic residues in RNPs have been determined (23, 24). However, no structure of the RRM domains of human hnRNP L has been reported, and how they individually or together bind to RNA is unknown.
FIGURE 1.

RRM domains and domain organization of human hnRNP L. A, sequence comparison of RNP-2 and RNP-1 motifs in hnRNP L RRMs with the RRM family consensus. Nonconserved residues from the consensus are shaded gray. Conserved aromatic positions important for RNA binding are indicated by black triangles. B, schematic representation of the domain organization of hnRNP L and the constructs used in this study.
In this paper, we investigated the RNA-binding affinities of different constructs of hnRNP L and determined the crystal structures of RRM1 and RRM34. We used site-directed mutagenesis and SPR assays to map the binding surface between hnRNP L and CA repeats. In vitro studies using different RNAs suggested an RNA-looping mechanism for hnRNP L splicing regulation. In addition, we propose models for how hnRNP L functions as an alternative splicing regulator with dual roles and suggest the possibility of hnRNP L-mediated recruitment of other factors without direct interactions.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
cDNAs encoding different fragments of hnRNP L FLΔN(32–589), RRMN1(32–180), RRM1(90–180), RRM2(177–295), RRM12(32–295), and RRM34(380–589) were PCR-amplified from the pET-30a/hnRNP L(32–589) plasmid, a gift from Dr. Zhu Bing (12). PCR products were digested with restriction endonucleases NdeI and XhoI and ligated into a modified pET-28a(+) plasmid lacking the thrombin cleavage site. Mutations were generated with a QuikChange site-directed mutagenesis kit (Stratagene) using the manufacturer's protocol. Recombinant plasmids were confirmed by DNA sequencing (Invitrogen) and transformed into Escherichia coli BL21 (DE3) (Novagen) to produce target proteins as N-terminal His6 fusions. E. coli cells were cultured in LB medium at 310 K with 50 mg/liter kanamycin until the A600 reached 0.8–1.2, after which bacteria were induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside at 289 K for 20 h. Bacteria were collected by centrifugation, resuspended in buffer A (50 mm Tris-HCl, 500 mm NaCl, pH 7.5), and lysed by ultrasonication. Cell extracts were centrifuged at 14,000 × g for 30 min at 277 K. Supernatants were purified with Ni2+-nitrilotriacetate affinity resin (Qiagen) pre-equilibrated with buffer A. Eluted proteins were concentrated by centrifugal ultrafiltration (Amicon Ultra-15, 3-kDa molecular mass cutoff, Millipore), loaded onto a pre-equilibrated HiLoadTM 16/60 SuperdexTM 75-pg column or a HiLoad 16/60 Superdex 200 column (GE Healthcare) in an Äkta-System (GE Healthcare), eluted at a flow rate of 1 ml/min with the same buffer, and collected in 1.2-ml fractions. Peak fractions were analyzed by Tricine/SDS-PAGE (15%, w/v) and stained with Coomassie Brilliant Blue R-250. Purified fractions were pooled together and concentrated by centrifugal ultrafiltration. The concentration was determined by A280. For crystallization, RRM1(90–180) was dialyzed against buffer B (20 mm Tris-HCl, 50 mm NaCl, 5 mm β-mercaptoethanol, pH 7.5) and concentrated to 40 mg/ml. RRM34(380–589) was further purified by HiTrap Q FF (5 ml) ion exchange chromatography (GE Healthcare). The final protein in buffer C (20 mm Tris-HCl, 50 mm NaCl, pH 8.0) was concentrated to 39 mg/ml for crystallization trials.
Crystallization and Data Collection
hnRNP L RRM1(90–180) was crystallized using the hanging drop vapor diffusion method by mixing 1 μl of protein solution and 1 μl of reservoir solution at 287 K. The crystal suitable for x-ray diffraction was grown in reservoir solution consisting of 0.1 m HEPES and 25% (w/v) polyethylene glycol 3,350, pH 7.5 (Hampton Research). Data collection was performed at 100 K with cryoprotectant solution (reservoir solution supplemented with an additional 20% (v/v) glycerol). Diffraction data were collected in-house to 2.0-Å resolution using a Rigaku RU-H3R generator and a Mar345 detector. For hnRNP L RRM34(380–589), the crystal suitable for x-ray diffraction was grown in reservoir solution containing 30% (v/v) pentaerythritol ethoxylate (15:4 EO/OH), 50 mm ammonium sulfate, and 50 mm BisTris, pH 6.5 (Hampton Research). Diffraction data were collected at beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF) at 100 K with cryoprotectant solution (reservoir solution with 20% (v/v) glycerol).
Structure Determination and Refinement
For hnRNP L RRM1(90–180), the diffraction data set was processed using iMOSFLM (25) and scaled using the Scala program in the CCP4 suite (26). The phase was determined by molecular replacement using the program Phaser (27) with the structure of hnRNP L-like (hnRNP LL) RRM1 (PDB code 1WEX) as the search model. Cycles of refinement and model building were carried out using REFMAC5 (28) and COOT (29) until the crystallography R-factor and free R-factor converged to 20.3 and 25.6%, respectively. TLS refinement (30) was executed in REFMAC5 at the last stage. Ramachandran analysis showed that 95.1% of the residues were in the most favored region, with 4.9% in the additionally allowed region. For hnRNP L RRM34(380–589), the diffraction data set was processed and scaled using the HKL2000 package (31). The phase was determined by molecular replacement using the program Molrep (32) and Phaser (27) with the isolated RRM3 and RRM4 of PTB (PDB code 2ADC) as the search model. Cycles of refinement and model building were carried out using REFMAC5 (28) and COOT (29) until the crystallography R-factor and free R-factor converged to 19.0 and 24.3%, respectively. TLS refinement (30) was executed in REFMAC5 at the last stage. Ramachandran analysis showed that, similarly to hnRNP L RRM1, 95.1% of the residues were in the most favored region, with 4.9% in the additionally allowed region. The qualities of these structures were checked using the program MolProbity (33). The details of data collection and processing are presented in Table 1. All structure figures were prepared with PyMOL (DeLano Scientific).
TABLE 1.
Binding affinity of various constructs of hnRNP L with 5′(CA)213′ RNA by SPR assays
| hnRNP L constructs | KDa |
|---|---|
| μm | |
| FLΔN | 1.2 ± 0.1 × 10−3 |
| RRMN12 | 0.14 ± 0.01 |
| RRM34 | 2.2 ± 0.1 × 10−2 |
| RRM1 | — |
| RRMN1 | 10 ± 1 |
| RRM2 | 0.50 ± 0.06 |
| RRMN12-Linker-1 (K178A) | 0.17 ± 0.01 |
| RRMN12-Linker-2 (D184A/D186A/D187A/R188A/N192A) | 0.10 ± 0.01 |
a Values are based on three independent experiments and the mean ± S.E. is indicated.
SPR Spectroscopy
SPR spectroscopy was carried out using a BIAcore 3000 (GE Healthcare). RNA segments of 21 (CA) repeats biotinylated at the 3′-end were immobilized on an SA chip. For kinetic analysis, proteins were passed over the chip at different concentrations in RNase-free running buffer (20 mm Tris-HCl, 500 mm NaCl, pH 7.5) and washed with 0.17% SDS (w/v). SPR assays were performed at 298 K. Data were analyzed with BIAevaluation version 4.1 software (Biacore Life Sciences).
Gel Electrophoretic Mobility Shift Binding Assays (EMSA)
Protein-RNA interactions were evaluated by EMSA. For FRET-EMSA, 12 pmol of doubly labeled RNAs (Takara) were mixed with 48 pmol or decreased amounts of hnRNP L RRM34 and incubated on ice for 30 min. Complexes were resolved on a 9% native polyacrylamide gel at a constant voltage of 6 V/cm at 277 K in 50 mm Tris acetate, 50 mm potassium acetate, pH 8.0. After electrophoresis, the gel was scanned with an LAS-4000 (GE Healthcare) excited with a 490-nm laser. The donor emission was defined as green and the acceptor emission as red (34). For EMSA of hnRNP L RRM34 with 34-nt RNA, the concentration of 34-nt was 40 μm, whereas concentrations of hnRNP L RRM34 ranged from 0 to 160 μm. The gel was visualized with toluidine.
Steady-state FRET Assays
Steady-state FRET measurements of doubly labeled RNAs were carried out in a spectrofluorometer. The excitation wavelength was 490 nm (5-nm bandwidth). For quantitative measurement of the FRET efficiency, the donor and acceptor emissions were measured at 520 and 580 nm, respectively. The FRET efficiency was calculated as I580/(I580 + I520), where I represents the intensity. The global dissociation constant (KD) was obtained by plotting FRET efficiency as a function of [hnRNP L RRM34], and fitting the curve to the modified Hill equation: FRET = FRET0 + (FRETmax − FRET0) [protein]n/(KDn + [protein]n).
Cross-linking Assays
Cross-linking assays were carried out using disuccinimidyl suberate (Sigma). The reaction buffer contained 50 mm HEPES, 300 mm NaCl, and 10% glycerol, pH 7.5. hnRNP L RRM34 (∼1 mg/ml) was added with an equimolar amount of double-labeled RNA and incubated on ice for 30 min. A reaction containing protein only without RNA was carried out for comparison. Disuccinimidyl suberate dissolved in DMSO was added to reactions at the molar ratio of 1 hnRNP L RRM34, 20 disuccinimidyl suberate. Mixtures were incubated on ice for another 30 min before quenching with 1 m Tris, pH 8.0. Reactions were directly analyzed by SDS-PAGE after quenching for 15 min at room temperature. Protein bands were visualized with Coomassie Brilliant Blue (Bio-Rad). RNA bands were visualized using LAS-4000 (GE Healthcare).
Dynamic Light Scattering Assays
Dynamic light scattering assays were performed in a DynaPro-MS800 (ATC) at 298 K with buffer (20 mm Tris, 100 mm NaCl, pH 7.5). hnRNP L RRM34 (∼0.5 mg/ml) was added with an equimolar amount of RNA and incubated for 30 min before measurements. Protein without RNA was also measured for comparison.
Isothermal Titration Calorimetry (ITC) Measurements
ITC measurements were performed with an iTC200 (Microcal) calorimeter at 298 K. Protein samples were dialyzed in buffer (25 mm HEPES, 300 mm NaCl, pH 7.5). The concentration of 34-nt RNA in the syringe was 300 μm. Protein concentration in the cell (200 μl volume) was 15 μm. Data were analyzed according to a single-site binding model with Microcal ORIGIN software.
RESULTS
Determination of the RNA Binding Activity of hnRNP L Constructs
Previous studies indicated that hnRNP L binds specifically to both CA-repeat and CA-rich elements (16). However, the RNA-binding property of hnRNP L has not been addressed. To elucidate the RNA-binding property of individual and multiple RRMs of hnRNP L, we purified various constructs of hnRNP L for SPR studies (Fig. 1B). Also, we attempted to generate individual RRM3 and RRM4, but these constructs were insoluble. Notably, the construct FLΔN (32–589 amino acids), lacking the N-terminal 31 amino acids, is usually regarded as full-length hnRNP L (4, 12, 13). hnRNP L specifically activates eNOS pre-mRNA splicing by binding to intronic variable-length CA repeats, and it binds to 20-repeat CA RNA moderately (35). For these reasons, a 21-copy CA repeat construct was chosen as the immobilized RNA substrate. As shown in Table 1, FLΔN showed the strongest RNA-binding ability (KD = 1.2 ± 0.1 nm). The C-terminal tandem domains RRM34 showed strong RNA-binding ability (KD = 22 ± 1 nm), although it was 17-fold weaker than that of FLΔN. The individual RRM, especially RRM1, showed almost no RNA binding activity. Interestingly, with the addition of 58 N-terminal amino acids, RRMN1(32–180) showed weak RNA-binding ability (KD = 10 ± 1 μm), which was 19-fold weaker than that of RRM2 (KD = 0.50 ± 0.06 μm). RRMN1 and RRM2 were arranged in cis as RRMN12, which showed moderately strong RNA-binding ability (KD = 0.14 ± 0.01 μm). These results indicated that both the C- and N-terminal parts of hnRNP L contribute to RNA binding, but RRM34 showed six times the binding capability of RRMN12. Furthermore, when the individual RRM (RRMN1 and RRM2) or tandem RRM domains (RRMN12 and RRM34) were tethered together on a single polypeptide, the affinities for RNA increased extensively. Although these values were 1,000-fold less than the product of the affinities of the component RRMs (36), this result suggested that RRMs cooperatively bind RNA with higher affinity. Because the linker between RRM1 and RRM2 is rich in hydrophilic residues (178KISRPGDSDDSRSVNSVL195), it is most likely solvent-exposed and nonstructured. When Lys-178 in the linker was substituted with alanine (Linker-1), the RNA-binding affinity was similar to wild-type hnRNP L RRMN12. When the other charged or polar residues in the linker were simultaneously replaced by alanines (Linker-2), the RNA-binding affinity did not change. Thus, the linker between the two N-terminal RRMs did not contribute to RNA binding.
Crystal Structure of hnRNP L RRM1
We determined the crystal structure of RRM1 at 2.0 Å. Details about data collection and refinement are summarized in Table 2. Although possessing atypical ribonucleoprotein motifs, hnRNP L RRM1 adopts the typical β1α1β2β3α2β4 topology, consisting of an antiparallel four-stranded β-sheet adjacent to two α-helices on the opposite side (Fig. 2A). The C-terminal loop of RRM1 stretches across the β-sheet in a conformation stabilized by interactions with residues in the N-terminal loop and β-sheet. Interestingly, the N-terminal loop is oriented toward the β-sheet through a hydrogen bond formed between Thr-98 and Asn-172, resulting in a conformation more like that in the structure of RNA-bound PTB RRM1 (23) than that in the free PTB RRM1 (37) (Fig. 2B). When a longer N-terminal loop is present, it may interact with RNA and cooperate with the C-terminal loop and β-sheet to bind RNA like PTB RRM1 in an RNA complex. This structure may explain why the RNA-binding affinity of hnRNP L RRMN1 is much stronger than RRM1. Notably, residues involved in the interactions mentioned above are highly conserved across species (Fig. 2D).
TABLE 2.
Data collection and refinement statistics for hnRNP L RRM1 and RRM34
| Data collection | RRM1(90–180) | RRM34(380–589) |
|---|---|---|
| Resolution | 39.25 to 2.03 Å (2.14 to 2.03 Å)a | 50.0 to 1.82 Å (1.85 to 1.82 Å)a |
| Wavelength | 1.5418 Å | 0.97907 Å |
| Oscillation width | 1° | 1° |
| Exposure time | 294 s | 1 s |
| Space group | P31 | P212121 |
| Unit cell parameters | a = b = 43.17, c = 78.53 Å | a = 36.57, b = 56.55, c = 88.74 Å |
| No. of unique reflections | 10,541 (1521)a | 17,147 (830) a |
| Redundancy | 3.5 (3.5)a | 6.5 (6.6)a |
| Completeness | 99.6% (97.6%)a | 100% (100%)a |
| Average I/σ (I) | 14.0 (3.7)a | 23.8 (4.8)a |
| Rmergeb | 7.1% (34.1%)a | 7.4% (43.8%)a |
| Refinement | ||
| Resolution | 39.25 to 2.04 Å | 47.69 to 1.82 Å |
| No. of reflections for refinement/test | 9988/502 | 16,234/865 |
| Rworkc/Rfreed | 20.4%/25.5% | 19.0%/24.3% |
| r.m.s.d. for bonds | 0.009 Å | 0.010 Å |
| r.m.s.d. for angles | 1.112° | 1.241° |
| Mean B factor | 20.02 Å2 | 21.02 Å2 |
| No. of water oxygen atoms | 82 | 71 |
| Ramachandran plot | ||
| Most favored regions | 95.1% | 95.1% |
| Additional allowed regions | 4.9% | 4.9% |
a Values in parentheses are for the highest resolution shell.
b Rmerge = Σ|Ii − 〈I〉|/Σ|I|, where Ii is the intensity of an individual reflection and 〈I〉 is the average intensity of that reflection.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors for reflections, respectively.
d Rfree was calculated as Rwork using the 5% of reflections that were selected randomly and omitted from refinement.
FIGURE 2.

Structure and RNA-binding surface mapping of hnRNP L RRM1. A, ribbon representation of hnRNP L RRM1 structure. ribonucleoprotein motifs are colored orange. Residues at key aromatic positions are color-coded by atom type (cyan, carbon; red, oxygen; blue, nitrogen). B, structure comparison of hnRNP L RRM1 (green) and PTB RRM1 in both RNA-free (blue, PDB code 1SJQ) and RNA-bound (red, PDB code 2AD9) states. RNA in the complex structure of PTB RRM1-CUCUCU is shown in magenta. Major structural differences are highlighted. The hydrogen bond between Thr-98 and Asn-172 in hnRNP L RRM1 is indicated. C, residues selected for mapping the RNA-binding surface of hnRNP L RRM1. Mutations of residues that reduced, increased, and had no effect on RNA binding are indicated in red, green, and yellow, respectively. D, sequence alignment of hnRNP L RRM1. Residues related to the spatial direction of the N-terminal loop (Thr-98 and Asn-172) are indicated by black triangles. Residues involved in RNA binding are designated with black stars. Different substitutions in hnRNP L RRM1 at the positions important for U2 and U4 recognition of PTB RRM1 are indicated by dots.
Identifying the RNA-binding Surface of hnRNP L RRM1
We further mapped the RNA-binding surface on hnRNP L RRM1. Some residues on the surface of the structure were selected for alanine substitution (Fig. 2C), and their RNA-binding properties were analyzed by SPR (Table 3). The mutants exhibited similar CD spectra to that of wild-type protein, indicating that the mutations did not affect protein folding (data not shown). Simultaneous substitution of Leu-141 and Ser-174 with alanines (L141A/S174A) remarkably decreased the RNA-binding affinity by 15-fold compared with the wild-type protein. Mutant H105A had moderately reduced RNA-binding affinity (6-fold reduction). Both R107A and Q139A had almost unchanged RNA-binding affinity. All of these residues are located on the central β-sheets, β1 and β3. Unexpectedly, V132A moderately increased RNA-binding affinity (4-fold), whereas that of N172A was increased by 1-fold. There may be reduced steric hindrance, which is important for hnRNP L in binding a large nucleotide, such as adenine. In particular, Val-132 and Asn-172 are located on the lateral β-strands β2 and β4, respectively. However, when we simultaneously mutated Lys-137 and Arg-138 on loop3 to alanines (K137A/R138A), there was no obvious difference in RNA-binding affinity compared with wild-type protein, indicating that they were nonessential for RNA binding. Therefore, like most RRM domains, the β-sheet of hnRNP L RRM1 is responsible for RNA binding. Instead of the characteristic aromatic side-chain residues (Phe/Tyr at RNP2 site 2, RNP1 site 3, and Phe at RNP1 site 5) that usually engage in stacking interactions with nucleotides (38), His-105, Gln-139, and Leu-141 occupy these positions in hnRNP L RRM1 (Figs. 2A and 1A). His-105 and Leu-141 are important for RNA binding, yet Gln-139 may not directly contact RNA (Table 3). Potentially, hnRNP L RRM1 binds RNA via a β-sheet as usual, although the related residues and the detailed binding mode may be different. The residues of hnRNP L RRM1 involved in RNA binding are highly conserved across species (Fig. 2D), indicating that the mode of RNA binding is evolutionarily conserved.
TABLE 3.
Binding affinity of wild-type and variants of hnRNP L RRMN12 with 5′(CA)213′ RNA by SPR assays
| KDa | KD relative to WT | |
|---|---|---|
| μm | ||
| Wild type RRMN12 | 0.14 ± 0.01 | 1 |
| H105A | 0.91 ± 0.02 | 7 |
| R107A | 0.20 ± 0.01 | 1 |
| V132A | 0.024 ± 0.001 | 0.2 |
| Q139A | 0.18 ± 0.01 | 1 |
| N172A | 0.083 ± 0.004 | 0.6 |
| L141A/S174A | 2.3 ± 0.1 | 16 |
| K137A/R138A | 0.17 ± 0.01 | 1 |
a Values are based on three independent experiments, and the mean ± S.E. is indicated.
Crystal Structure of hnRNP L RRM34
We determined the three-dimensional structure of hnRNP L RRM34 at 1.8 Å (Table 2). Four residues in loop3 of RRM4 (537GKSE540) were not observed in the electron density map, most likely because they are disordered. Each RRM adopted the classical β1α1β2β3α2β4 structure, except for an unusual fifth β-strand that was antiparallel to β2 in RRM3 (Fig. 3A). β5 is connected to β4 by an extended linker loop 6, which is stretched over the β-sheet and stabilized by close interactions with residues located on the β-sheet. The mutations that destroy the stability of loop 6 (K413A and I459A) moderately reduced RNA-binding affinity, implying its role in RNA binding (Table 4).
FIGURE 3.

Structure and RNA-binding surface mapping of hnRNP L RRM34. A, ribbon representation of hnRNP L RRM34 structure. Secondary structure elements of hnRNP L RRM34 are labeled. The structure is colored by domains as follows: RRM3 (residues 380–480), green; interdomain linker (residues 481–500), red; RRM4 (residues 501–588), blue. Disordered residues between β2 and β3 of RRM4 are modeled as dashed lines. B, direct interactions between RRM3 and RRM4. C, indirect interdomain interactions mediated by the linker. Residues involved in B and C are represented by sticks, and the dotted surfaces represent hydrophobic interaction surfaces. D, sequence alignment of human hnRNP L RRM34 with its homologues. Amino acids involved in the interdomain interactions are designated with black triangles below; residues involved in RNA binding are designated with black stars. Different substitutions in hnRNP L RRM34 at the positions important for U2 and U4 recognition of PTB RRM34 are indicated by dots. E and F, residues selected for mapping the RNA binding surface of hnRNP L RRM3 (E) and RRM4 (F). Residues whose mutations reduced and had no effect on RNA binding are indicated in red and yellow, respectively.
TABLE 4.
Binding affinity of wild-type and variants of hnRNP L RRM34 with 5′(CA)213′ RNA by SPR assays
| KDa | KD relative to WT | |
|---|---|---|
| nm | ||
| Wild type RRM34 | 22 ± 1 | 1 |
| Y387A | 48 ± 1 | 2 |
| K413A | 60 ± 6 | 3 |
| S454A | 30 ± 1 | 1 |
| I459A | 43 ± 3 | 2 |
| P461A | 18 ± 3 | 1 |
| R495A | 43 ± 1 | 2 |
| H504A | 3.1 ± 0.1 × 102 | 14 |
| F506A | 3.8 ± 0.1 × 102 | 17 |
| K533A | 15 ± 1 | 1 |
| F535A | 40 ± 2 | 2 |
| K579A | 83 ± 4 | 4 |
a Values are based on two or three independent experiments, and the mean ± S.E. is indicated.
The two RRMs, connected by a 20-residue peptide linker containing a short α-helix, interact intimately with each other, burying a solvent-accessible surface area of ∼1400 Å2. The interaction through their helical surfaces aligns their antiparallel β-sheets facing solvent in opposite directions. The hydrophobic and hydrogen bond interactions occur via direct inter-RRM contacts (Fig. 3B) and indirect contacts mediated by the interdomain linker (Fig. 3C). The packed crystal structure of RRM34 is consistent with a previous observation, by NMR spectroscopy, of the RRM3 and RRM4 interaction (39). Interestingly, the amino acids involved in interdomain contacts are highly conserved across species (Fig. 3D), suggesting that RRM34 in all homologous proteins is in the same spatial arrangement and that this topology may be functionally significant.
The compact structure of hnRNP L RRM34 is different from all reported tandem RRM structures except PTB RRM34 (40). hnRNP L RRM34 has the most sequence identity (30%) with PTB RRM34, and the structure of hnRNP L RRM34 is similar to that of PTB in both the free (40) and RNA-bound (23) states with the root mean square deviations for 177 Cα positions 2.38 and 2.46 Å, respectively.
Identifying the RNA-binding Surface of hnRNP L RRM34
We investigated the surface and critical residues for RNA binding in hnRNP L RRM34 through mutations and SPR assays. The selected surface residues of RRM34 are shown in Fig. 3, E and F, and the SPR results are summarized in Table 4. The mutations did not affect protein folding, as assessed by CD spectra (data not shown). Although three mutants, S454A, Y461A, and K533A, had almost no effect on binding, the rest showed moderately or remarkably reduced binding ability. Some mutations in RRM3 of hnRNP L (Y387A, K413A, and I459A) showed very moderately reduced binding ability, suggesting their moderate contribution to RNA binding. Whereas Ile-459 resides on loop 6, Lys-413, and Tyr-387 are located on the β2 and β1 strands of RRM3, respectively. Additionally, both Lys-413 and Ile-459 stabilize loop 6 as described above, so their impact on RNA binding may be partially due to more flexibility in loop 6, which could prevent the β-sheet from binding RNA. The binding abilities of several mutants in RRM4 (R495A, F535A, K579A, and especially H504A and F506A) were reduced by varying degrees, indicating that these residues are directly involved in RNA binding. The mutations H504A and F506A showed the most reduced affinity, suggesting that these residues are critical for RNA binding. Phe-506 and His-504 are located at positions 4 and 2 of the RNP2 sequence, respectively. Phe-535 and Lys-579 reside on β2 and β4, respectively. Arg-495 is in the interdomain linker. Therefore, hnRNP L RRM34 binds RNA mainly through the β-sheets of the two RRMs, with the assistance of some residues in loop 6 of RRM3 and the interdomain linker. The residues involved in RNA binding are well conserved (Fig. 3D).
Given the similarity in the structures and RNA-binding surfaces, hnRNP L RRM1 and RRM34 may adopt a similar mode of protein-RNA interaction as PTB RRM1 and RRM34 (23). However, their binding specificity is different. The preferred sequence for hnRNP L is a CA-repeat; for PTB, it is poly(CU) (23). In the reported complex structures, PTB RRM1 and RRM34 specifically recognize U2C3U4 (23). Through structure-based, optimized sequence alignments between hnRNP L and PTB, we found that for both RRM1 and RRM34, the residues corresponding to those participating in recognition of C3 in PTB are conserved in hnRNP L, whereas those involved in recognition of U2 and U4 are less conserved in hnRNP L (Fig. 4, A and B). The residues forming hydrogen bonds to U2 and U4 are extremely different. For example, Gln-129, Lys-137, Thr-407, Ser-525, and His-411 in PTB are replaced by Asn-172, Ser-180, Cys-452, Cys-581, and Gln-456, respectively, in hnRNP L. These changes may directly affect recognition specificity. For those residues forming a hydrophobic pocket that contacts U2 and U4, the substitutions may possibly change the size or shape of the spatial cavity. Consistent with the larger size of adenine compared with uracil, the cavities of hnRNP L are larger and deeper than the corresponding cavities of PTB RRM1 and RRM3 for U4 (Fig. 4C). Furthermore, the substituted residues in hnRNP L display a high degree of conservation across species (Figs. 2D and 3D), suggesting their conserved roles in RNA recognition. Overall, these results provide a preliminary view of how hnRNP L interacts with RNA and how it preserves its binding specificity.
FIGURE 4.

Residues affecting the binding specificity of hnRNP L RRM34 in comparison with PTB RRM34. A, structure-based sequence alignment of hnRNP L RRM1 and PTB RRM1. Residues in PTB RRM1 involved in the recognition of C3 are indicated by triangles and those of U2 and U4 by stars, colored red for the hydrophobic interactions and black for the hydrogen bond interactions. Different substitutions in hnRNP L RRM1 at the positions important for U2 and U4 recognition of PTB RRM1 are indicated by dots. B, structure-based sequence alignment of hnRNP L RRM34 and PTB RRM34. Residues in PTB RRM34 involved in the special recognition of C3 are indicated by triangles and those of U2 and U4 by stars, colored red for the hydrophobic interactions and black for the hydrogen bond interactions. Different substitutions in hnRNP L RRM34 at the positions important for U2 and U4 recognition of PTB RRM34 are indicated by dots. C, comparison of the cavities of hnRNP L (cyan) with the corresponding binding cavities of PTB RRM1 (PDB code 2AD9) and RRM3 (PDB code 2ADC) (violet) for U4.
RNA Looping Induced by hnRNP L RRM34
Because we found that the two β-sheets of RRM3 and RRM4 align in an antiparallel fashion, facing opposite directions, and that both are involved in RNA binding, which would require the RNA backbone to bend to interact with both RRM domains, we speculated that hnRNP L RRM34 may bind to two separate binding sites within the same RNA by inducing RNA looping. To test this hypothesis, we performed binding studies of hnRNP L RRM34 with three different RNAs. These RNAs contained two CACACA hexamers at the two ends separated by 5 (referred to as “U5”), 15 (“U15”), and 21 (“U21”) uracils (Fig. 5A). RNA 5′- and 3′-ends were labeled with carboxyfluorescein and tetramethylrhodamine, respectively. Carboxyfluorescein and tetramethylrhodamine act as donor and acceptor, respectively, in a FRET pair (41). The donor emission is defined as green and the acceptor emission as red (34). If the two RNA ends are placed in close proximity to each other, FRET increases; this property was used to monitor RNA conformational changes during binding reactions (Fig. 5B).
FIGURE 5.

FRET-based gel shift and steady-state FRET assays of hnRNP L RRM34 binding to three different RNAs. A, sequences of three different RNAs with the CA repeats underlined. B, schematic representation of the RNA looping mechanism of hnRNP L RRM34. C, FRET-based gel shift assays of the fluorophore-labeled RNAs in the absence and presence of hnRNP L RRM34. Donor emission in the figure is shown in green and acceptor emission in red. The figure was generated by superposition using Photoshop 11.0 (Adobe). D, EMSA for binding of hnRNP L RRM34 to U21, establishing a 1:1 stoichiometry of the complex. E, fluorescence emission spectra of labeled U21 in the absence (dashed line) and presence (solid line) of 750 nm hnRNP L RRM34. F, FRET efficiencies of labeled U5, U15, and U21 in the absence (black) and presence (gray) of 750 nm hnRNP L RRM34. G, FRET efficiency of U21 as a function of [hnRNP L RRM34]. The data were fitted with a modified Hill equation (see “Experimental Procedures”). Error bars were derived from three independent assays.
First, we tested the binding of hnRNP L RRM34 to the three different RNAs using a FRET-based gel shift assay (Fig. 5C). As expected, free RNAs shifted more slowly and showed less red as the RNA size increased. U5 produced a more intense red band than free U15 and U21 because of the smaller distance between RNA ends. When hnRNP L RRM34 was assessed, the protein·RNA complexes shifted more slowly and with more intense red bands than the corresponding free RNA bands. This result suggested that the binding of hnRNP L RRM34 brings the two ends of RNAs into close proximity, which is consistent with the expected RNA looping mode. The diffuse bands of protein·U5 complexes suggested that the binding affinity of hnRNP L RRM34 to U5 is weak, such that the complex dissociated during electrophoresis. However, hnRNP L RRM34 complexed with U15 or U21 shifted as a clean band, suggesting a strong binding affinity. Given that the three RNAs have the same binding sites and that the poly(U) was observed to not bind to hnRNP L RRM34 (data not shown), the lower binding affinity of U5 is possibly due to the short distance between the two binding sites; the loop required for the two binding sites to simultaneously bind to hnRNP L RRM34 could not form as in U15 and U21. In EMSAs using different protein, U21 molar ratios, we found that hnRNP L RRM34 binds to U21 with 1:1 stoichiometry (Fig. 5D), and a similar result was obtained for U15 (data not shown). Thus, both binding sites within U21 and U15 RNA are bound by each RRM of hnRNP L RRM34 to form a complex, which may induce RNA looping.
We also confirmed the binding of hnRNP L RRM34 to the three RNAs in solution through steady-state FRET (42). Fig. 5E shows the emission spectrum of U21 in the absence (dashed line) and presence (solid line) of hnRNP L RRM34. For free RNA, the intensity of the donor fluorophore (520 nm) is larger than that of the acceptor fluorophore (580 nm), indicating that the two ends of U21 are distant. When bound to hnRNP L RRM34, the donor intensity of U21 decreased, whereas the acceptor intensity increased, indicating that the two ends of U21 were brought into close proximity by hnRNP L RRM34. The calculated FRET efficiency for U21 increased from 0.20 to 0.59. A similar result was observed for U15 but not U5 (Fig. 5F). We used the observed FRET efficiency in increasing protein concentrations to quantify the binding affinity of hnRNP L RRM34 to RNA (Fig. 5G). The binding curves of hnRNP L RRM34 to U21 and U15 were fitted to a modified Hill equation, yielding similar dissociation constants (KD) of 0.23 ± 0.01 and 0.25 ± 0.01 μm, respectively. In agreement with the FRET-EMSA, the binding affinity of hnRNP L RRM34 to U5 was too low to be measured accurately.
Theoretically, two possible models of protein·RNA complex formation are suggested by the results above. hnRNP L could be present in a looped monomeric complex (Fig. 6A) or in a dimeric complex composed of two hnRNP L RRM34 and two unlooped RNAs (Fig. 6B). To determine the complex structure, we investigated protein·RNA complexes in solution using chemical cross-linking assays. Compared with the protein-only cross-linking system, addition of U21 RNA produced an extra band with an apparent molecular mass of ∼40 kDa in SDS-PAGE. This band was determined to be a monomeric complex containing one molecule of hnRNP L RRM34 (∼25 kDa) and one molecule of looped U21 RNA (∼11 kDa) (Fig. 6C). As expected, RNA was observed in the complex band by fluorescence emission (Fig. 6C). We further analyzed the complex by dynamic light scattering (Fig. 6D). In the absence of U21 RNA, the protein radius indicated that its apparent molecular mass is ∼25 kDa. In the presence of U21 RNA, the protein and RNA formed a complex with an apparent molecular mass of 35 kDa (Fig. 6D), which was consistent with the cross-linking results. Thus, the protein and RNA formed an RNA-looped monomeric complex but not a dimeric complex. In sum, hnRNP L RRM34 facilitated RNA looping, which required an appropriate distance of more than five nucleotides between two binding sites within the same RNA molecule. Because the distance between the two β-sheets in hnRNP L RRM34 (∼40 Å) corresponded to the length of about seven nucleotides, a seven-nucleotide distance may be required for RNA looping.
FIGURE 6.

hnRNP L RRM34 binds to U21 RNA in RNA-looped monomers but not dimers. A, schematic representation of the RNA-looped monomer. B, schematic representation of the dimer containing two hnRNP L RRM34 and two RNAs. C, cross-linking SDS-PAGE analysis of the compound form of hnRNP L RRM34 with RNA. The bands for protein·RNA complex are indicated. D, dynamic light scattering assays of hnRNP L RRM34 in the absence (black) and presence (red) of U21 RNA.
To investigate whether hnRNP L RRM34 could facilitate RNA looping in vivo, we detected binding between hnRNP L RRM34 and its in vivo target (designated as 34-nt RNA; Fig. 7A, upper panel), a sequence important for regulation of CD45 exon 5 (3, 43). Two exonic activation-responsive sequence motifs, each binding specifically to hnRNP L and containing a short conserved element (MCYYGCA in which M represents cytosine or adenine and Y is any pyrimidine (44)), are separated by a 21-nt ESE. hnRNP L RRM34 bound moderately to the 34-nt RNA, as determined by EMSA (Fig. 7A, lower panel). Bands of the protein·34-nt RNA complex were dispersed along the lanes, suggesting weaker binding affinity compared with that of U21 or U15 RNA, which is possibly due to the much weaker binding sites. For a quantitative estimation of binding efficiency, we monitored the binding affinities by ITC measurements, and the data were fitted to a single-site binding model (Fig. 7B). Consistently, the KD value of the hnRNP-L RRM34·34-nt RNA complex is 8.9 ± 1.0 μm, which is higher than that of the hnRNP-L RRM34·U21 or ·U15 complex. Moreover, the estimated n value (the number of binding sites per protein monomer) is 1.03 ± 0.06 for the complex, indicating that hnRNP L RRM34 binds to the 34-nt RNA at 1:1 stoichiometry as expected. These results suggested that the binding of hnRNP L RRM34 could facilitate formation of a loop by the target RNA in vivo.
FIGURE 7.

Binding of hnRNP L RRM34 to its in vivo target RNA. A, electrophoretic mobility gel shift data for binding of 34-nt RNA to hnRNP L RRM34. The RNA sequence is shown above. Activation-responsive sequence motifs (underlined and in bold) of CD45 exon 5, which are separated by a 21-nt spacer, represent binding sites for individual hnRNP L RRM domains. B, ITC measuring binding of hnRNP L RRM34 to 34-nt RNA (top, raw titration data; bottom, integrated heat measurements). The curve was fitted using a single-site binding model with KD and n indicated.
DISCUSSION
Our studies revealed that all four RRMs in hnRNP L cooperate to bind with RNA. RRM34 functioned as a unit that is more critical for RNA binding than RRMN12. RRM2 showed moderate RNA-binding affinity. The extra N-terminal 58 amino acids are required for RRM1 to bind RNA. The linker between RRM1 and RRM2 is not involved in RNA binding. Furthermore, we obtained the crystal structures of RRM1 and RRM34 of hnRNP L. Structure-based mutations combined with RNA-binding tests showed that RRM1 and RRM34 of hnRNP L bind to RNA mainly through the β-sheets of RRMs. Interestingly, the crystal structure of RRM34 indicated that the β-sheets of RRM3 and RRM4 point away from each other on opposite surfaces. A series of binding studies demonstrated that hnRNP L RRM34 is sufficient to bind two appropriately separate binding sites within the same RNA by inducing RNA looping. The presence of the first two RRMs may cooperate to enhance the binding ability of hnRNP L to the target gene. In this way, hnRNP L can simultaneously bind to multiple separate binding motifs present in many regulated genes. Alternatively, the first two RRMs may play other roles, such as interactions with itself or other proteins at the splice sites, as reported (11).
Its capacity for RNA looping helps to explain why hnRNP L possesses location-dependent dual functions, either as a repressor or an activator in alternative splicing. If it loops out an alternative exon or an exonic ESE to make the exon less accessible for recognition, the exon is excluded, as in the case of CD45 exon 5 (Fig. 8A) (3). hnRNP L simultaneously binds the flanking silencers (S1 and S2 in Fig. 8A) on both sides of the ESE, loops out the ESE sequence, blocks the recruitment of a splicing enhancer factor SF2/ASF, and prevents recruitment of the U2 snRNP to the 3′-splicing site region upstream of exon 5, which altogether results in exclusion of exon 5. Looping may be required for repression by hnRNP L; in the absence of the ESE, the silencers (S1 and S2) have no independent silencer activity (3). If, however, hnRNP L loops out the intron, it might bring the 5′ and 3′ splice sites into close proximity to stimulate splicing, as in the case of removal of DAF (CD55) intron 7 by simultaneous hnRNP L binding to two distant CA clusters (Fig. 8B) (4). Therefore, the location of the binding sites relative to the regulated splice site is important for determining the alternative splicing outcome, in agreement with the previous report of the position- and context-dependent dual functions of hnRNP L (3, 16). Other splicing regulatory proteins also perform dual functions depending on the context, such as neuro-oncological ventral antigen 1 (Nova-1) (45) and muscleblind-like splicing regulator 1 (MBNL1) (46), which mediate RNA looping. Thus, RNA looping may be a general mechanism for this kind of protein to regulate alternative splicing.
FIGURE 8.
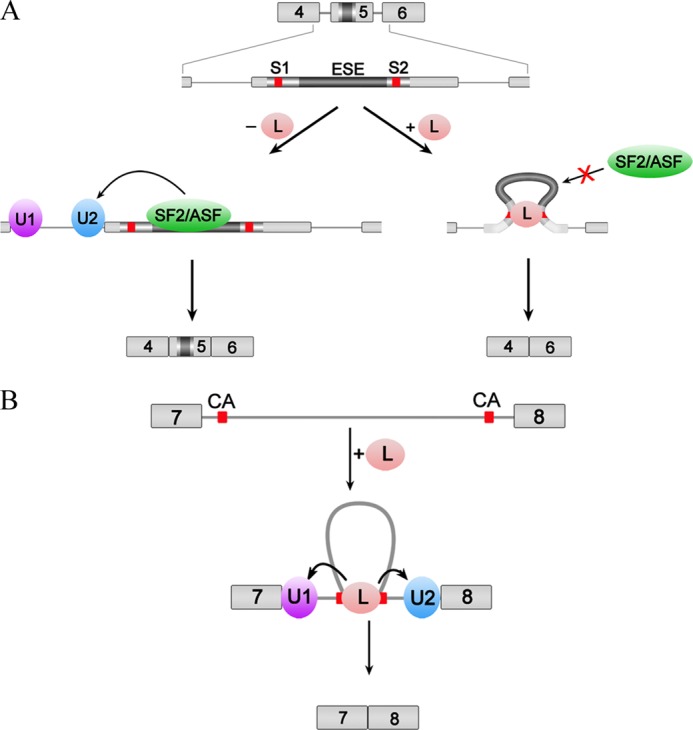
Models for hnRNP L regulation of alternative splicing. A, hnRNP L represses inclusion of an alternative exon in the case of the splicing regulation of exon 5 of CD45. hnRNP L binds two motifs flanking the ESE in exon 5 and changes the conformation of the ESE, consequently repressing its binding to the enhancer complex and blocking the activity of the splicing enhancer. B, hnRNP L promotes removal of a specific intron in the case of the splicing regulation of DAF. hnRNP L binds two distant CA clusters in intron 7, loops out the long in-between sequence, brings the splice sites into close proximity, and recruits U1 and U2 snRNP to recognize the 5′ and 3′ splice sites. Red boxes indicate hnRNP L-binding motifs.
In light of RNA looping, it can be interpreted that hnRNP L autoregulates its own expression by binding to two clusters of CA-rich motifs separated by an ∼215-nt sequence (47). The two distant clusters of CA-rich motifs, each containing 11 and 14 CA-rich motifs, are brought in close proximity by looping. This RNA conformation favors binding of a second molecule of hnRNP L to the remaining CA motifs and then a third one, etc. Multiple bound hnRNP L molecules may interact with each other. Therefore, a subtle change in protein concentration can be augmented to cause a large change in conformation, generating a concentration-dependent splice-regulatory signal for autoregulation. Additionally, our data support the proposed model in which hnRNP L/hnRNP LL mediates cross-exon 4–6 interaction causing exon 5 to be looped out to generate the three-exon skipped form of CD45, R0 (18). The looping conformation of RNA, caused by simultaneous hnRNP L binding on CA motifs in CD45 exons 4 and 6, may promote the binding of hnRNP LL. After assembly, hnRNP L and hnRNP LL may interact with each other due to their close proximity. This proximity-mediated interaction helps to explain why the L/LL interaction is exon 4- and 6-dependent and CD45-specific in human B cells (18). Thus, by introducing RNA looping, hnRNP L may nucleate the assembly of additional proteins to RNA without direct protein-protein interactions.
These structural and biochemical analyses have shed light on the location-dependent dual functions of hnRNP L in alternative splicing. We propose that hnRNP L facilitates RNA looping by binding RNA. hnRNP L is the fifth example of a protein that loops out RNA through intramolecular interactions; the others are Mbnl1 (46), Nova-1 (45), KSRP (48), and PTB (42). In addition, hnRNP A/B and hnRNP F/H facilitate RNA looping through intermolecular interactions (49). Thus, RNA looping may be a widespread mechanism for RNA-binding proteins to change RNA secondary structure for special functions. However, further in vitro and in vivo investigations will be required to uncover the details of this looping mechanism.
Acknowledgment
We thank the staff at SSRF beamline BL17U for assistance with synchrotron data collection.
This work was supported by Chinese Ministry of Science and Technology Grants 2012CB917200 and 2009CB825500, Chinese National Natural Science Foundation Grants 31270014, 31130018, and 30900224, and Science and Technological Fund of Anhui Province for Outstanding Youth Grant 10040606Y11.
The atomic coordinates and structure factors (codes 3R27 and 3TO8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- RRM
- RNA recognition motif
- SPR
- surface plasmon resonance
- ITC
- isothermal titration calorimetry
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- nt
- nucleotide
- snRNP
- small nuclear ribonucleoprotein
- ESE
- exonic splicing enhancer
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- PDB
- Protein Data Bank.
REFERENCES
- 1. Han S. P., Tang Y. H., Smith R. (2010) Functional diversity of the hnRNPs: past, present and perspectives. Biochem. J. 430, 379–392 [DOI] [PubMed] [Google Scholar]
- 2. Shankarling G., Lynch K. W. (2010) Living or dying by RNA processing: caspase expression in NSCLC. J. Clin. Invest. 120, 3798–3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Motta-Mena L. B., Heyd F., Lynch K. W. (2010) Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol. Cell 37, 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hung L. H., Heiner M., Hui J., Schreiner S., Benes V., Bindereif A. (2008) Diverse roles of hnRNP L in mammalian mRNA processing: a combined microarray and RNAi analysis. RNA 14, 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guang S., Felthauser A. M., Mertz J. E. (2005) Binding of hnRNP L to the pre-mRNA processing enhancer of the herpes simplex virus thymidine kinase gene enhances both polyadenylation and nucleocytoplasmic export of intronless mRNAs. Mol. Cell. Biol. 25, 6303–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang B., Lim J. H., Hahm B., Jang S. K., Lee S. W. (2009) hnRNP L is required for the translation mediated by HCV IRES. Biochem. Biophys. Res. Commun. 378, 584–588 [DOI] [PubMed] [Google Scholar]
- 7. Majumder M., Yaman I., Gaccioli F., Zeenko V. V., Wang C., Caprara M. G., Venema R. C., Komar A. A., Snider M. D., Hatzoglou M. (2009) The hnRNA-binding proteins hnRNP L and PTB are required for efficient translation of the Cat-1 arginine/lysine transporter mRNA during amino acid starvation. Mol. Cell. Biol. 29, 2899–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shih S. C., Claffey K. P. (1999) Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J. Biol. Chem. 274, 1359–1365 [DOI] [PubMed] [Google Scholar]
- 9. Söderberg M., Raffalli-Mathieu F., Lang M. A. (2007) Identification of a regulatory cis-element within the 3′-untranslated region of the murine inducible nitric oxide synthase (iNOS) mRNA; interaction with heterogeneous nuclear ribonucleoproteins I and L and role in the iNOS gene expression. Mol. Immunol. 44, 434–442 [DOI] [PubMed] [Google Scholar]
- 10. Hamilton B. J., Nichols R. C., Tsukamoto H., Boado R. J., Pardridge W. M., Rigby W. F. (1999) hnRNP A2 and hnRNP L bind the 3′ UTR of glucose transporter 1 mRNA and exist as a complex in vivo. Biochem. Biophys. Res. Commun. 261, 646–651 [DOI] [PubMed] [Google Scholar]
- 11. Kim J. H., Hahm B., Kim Y. K., Choi M., Jang S. K. (2000) Protein-protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J. Mol. Biol. 298, 395–405 [DOI] [PubMed] [Google Scholar]
- 12. Yuan W., Xie J., Long C., Erdjument-Bromage H., Ding X., Zheng Y., Tempst P., Chen S., Zhu B., Reinberg D. (2009) Heterogeneous nuclear ribonucleoprotein L is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J. Biol. Chem. 284, 15701–15707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang Y., Li W., Yao X., Lin Q. J., Yin J. W., Liang Y., Heiner M., Tian B., Hui J., Wang G. (2012) Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol. Cell 45, 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuninger D. T., Izumi T., Papaconstantinou J., Mitra S. (2002) Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res. 30, 823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lander E. S. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 [DOI] [PubMed] [Google Scholar]
- 16. Hui J., Hung L. H., Heiner M., Schreiner S., Neumüller N., Reither G., Haas S. A., Bindereif A. (2005) Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J. 24, 1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rothrock C. R., House A. E., Lynch K. W. (2005) hnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J. 24, 2792–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Preussner M., Schreiner S., Hung L. H., Porstner M., Jack H. M., Benes V., Ratsch G., Bindereif A. (2012) hnRNP L and L-like cooperate in multiple-exon regulation of CD45 alternative splicing. Nucleic Acids Res. 40, 5666–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dery K. J., Gaur S., Gencheva M., Yen Y., Shively J. E., Gaur R. K. (2011) Mechanistic control of carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) splice isoforms by the heterogeneous nuclear ribonuclear proteins hnRNP L, hnRNP A1, and hnRNP M. J. Biol. Chem. 286, 16039–16051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heiner M., Hui J., Schreiner S., Hung L. H., Bindereif A. (2010) hnRNP L-mediated regulation of mammalian alternative splicing by interference with splice site recognition. RNA Biol. 7, 56–64 [DOI] [PubMed] [Google Scholar]
- 21. House A. E., Lynch K. W. (2006) An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat. Struct. Mol. Biol. 13, 937–944 [DOI] [PubMed] [Google Scholar]
- 22. Maris C., Dominguez C., Allain F. H. (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272, 2118–2131 [DOI] [PubMed] [Google Scholar]
- 23. Oberstrass F. C., Auweter S. D., Erat M., Hargous Y., Henning A., Wenter P., Reymond L., Amir-Ahmady B., Pitsch S., Black D. L., Allain F. H. (2005) Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309, 2054–2057 [DOI] [PubMed] [Google Scholar]
- 24. Liker E., Fernandez E., Izaurralde E., Conti E. (2000) The structure of the mRNA export factor TAP reveals a cis arrangement of a non-canonical RNP domain and an LRR domain. EMBO J. 19, 5587–5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leslie A. G. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 26. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 29. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 31. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 32. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 33. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rueda D., Wick K., McDowell S. E., Walter N. G. (2003) Diffusely bound Mg2+ ions slightly reorient stems I and II of the hammerhead ribozyme to increase the probability of formation of the catalytic core. Biochemistry 42, 9924–9936 [DOI] [PubMed] [Google Scholar]
- 35. Hui J., Stangl K., Lane W. S., Bindereif A. (2003) HnRNP L stimulates splicing of the eNOS gene by binding to variable-length CA repeats. Nat. Struct. Biol. 10, 33–37 [DOI] [PubMed] [Google Scholar]
- 36. Shamoo Y., Abdul-Manan N., Williams K. R. (1995) Multiple RNA binding domains (RBDs) just don't add up. Nucleic Acids Res. 23, 725–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simpson P. J., Monie T. P., Szendröi A., Davydova N., Tyzack J. K., Conte M. R., Read C. M., Cary P. D., Svergun D. I., Konarev P. V., Curry S., Matthews S. (2004) Structure and RNA interactions of the N-terminal RRM domains of PTB. Structure 12, 1631–1643 [DOI] [PubMed] [Google Scholar]
- 38. Auweter S. D., Oberstrass F. C., Allain F. H. (2006) Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 34, 4943–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skrisovska L., Allain F. H. (2008) Improved segmental isotope labeling methods for the NMR study of multidomain or large proteins: application to the RRMs of Npl3p and hnRNP L. J. Mol. Biol. 375, 151–164 [DOI] [PubMed] [Google Scholar]
- 40. Vitali F., Henning A., Oberstrass F. C., Hargous Y., Auweter S. D., Erat M., Allain F. H. (2006) Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. EMBO J. 25, 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rueda D., Walter N. G. (2006) Fluorescent energy transfer readout of an aptazyme-based biosensor. Methods Mol. Biol. 335, 289–310 [DOI] [PubMed] [Google Scholar]
- 42. Lamichhane R., Daubner G. M., Thomas-Crusells J., Auweter S. D., Manatschal C., Austin K. S., Valniuk O., Allain F. H., Rueda D. (2010) RNA looping by PTB: Evidence using FRET and NMR spectroscopy for a role in splicing repression. Proc. Natl. Acad. Sci. U.S.A. 107, 4105–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tong A., Nguyen J., Lynch K. W. (2005) Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J. Biol. Chem. 280, 38297–38304 [DOI] [PubMed] [Google Scholar]
- 44. Rothrock C., Cannon B., Hahm B., Lynch K. W. (2003) A conserved signal-responsive sequence mediates activation-induced alternative splicing of CD45. Mol. Cell 12, 1317–1324 [DOI] [PubMed] [Google Scholar]
- 45. Teplova M., Malinina L., Darnell J. C., Song J., Lu M., Abagyan R., Musunuru K., Teplov A., Burley S. K., Darnell R. B., Patel D. J. (2011) Protein-RNA and protein-protein recognition by dual KH1/2 domains of the neuronal splicing factor Nova-1. Structure 19, 930–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teplova M., Patel D. J. (2008) Structural insights into RNA recognition by the alternative splicing regulator muscleblind-like MBNL1. Nat. Struct. Mol. Biol. 15, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rossbach O., Hung L. H., Schreiner S., Grishina I., Heiner M., Hui J., Bindereif A. (2009) Auto- and cross-regulation of the hnRNP L proteins by alternative splicing. Mol. Cell. Biol. 29, 1442–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Díaz-Moreno I., Hollingworth D., Kelly G., Martin S., García-Mayoral M., Briata P., Gherzi R., Ramos A. (2010) Orientation of the central domains of KSRP and its implications for the interaction with the RNA targets. Nucleic Acids Res. 38, 5193–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martinez-Contreras R., Fisette J. F., Nasim F. H., Madden R., Cordeau M., Chabot B. (2006) Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 4, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]