

Background: Green tea polyphenol (EGCG) has beneficial effects on cardiovascular dysfunction.
Results: EGCG stimulates autophagy through a CaMKKβ-mediated mechanism, which contributes to degradation of lipid droplets.
Conclusion: Regulation of autophagic flux by EGCG plays a role in intracellular lipid accumulation.
Significance: Findings show a novel mechanism for beneficial effects of EGCG in cardiovascular complications.
Keywords: AMP-activated Kinase (AMPK), Autophagy, Calcium, Lipid Droplet, Polyphenols, CaMKKβ, EGCG, Lipophagy
Abstract
Epigallocatechin gallate (EGCG) is a major polyphenol in green tea that has beneficial effects in the prevention of cardiovascular disease. Autophagy is a cellular process that protects cells from stressful conditions. To determine whether the beneficial effect of EGCG is mediated by a mechanism involving autophagy, the roles of the EGCG-stimulated autophagy in the context of ectopic lipid accumulation were investigated. Treatment with EGCG increased formation of LC3-II and autophagosomes in primary bovine aortic endothelial cells (BAEC). Activation of calmodulin-dependent protein kinase kinase β was required for EGCG-induced LC3-II formation, as evidenced by the fact that EGCG-induced LC3-II formation was significantly impaired by knockdown of calmodulin-dependent protein kinase kinase β. This effect is most likely due to cytosolic Ca2+ load. To determine whether EGCG affects palmitate-induced lipid accumulation, the effects of EGCG on autophagic flux and co-localization of lipid droplets and autophagolysosomes were examined. EGCG normalized the palmitate-induced impairment of autophagic flux. Accumulation of lipid droplets by palmitate was markedly reduced by EGCG. Blocking autophagosomal degradation opposed the effect of EGCG in ectopic lipid accumulation, suggesting the action of EGCG is through autophagosomal degradation. The mechanism for this could be due to the increased co-localization of lipid droplets and autophagolysosomes. Co-localization of lipid droplets with LC3 and lysosome was dramatically increased when the cells were treated with EGCG and palmitate compared with the cells treated with palmitate alone. Collectively, these findings suggest that EGCG regulates ectopic lipid accumulation through a facilitated autophagic flux and further imply that EGCG may be a potential therapeutic reagent to prevent cardiovascular complications.
Introduction
Ectopic accumulation of lipids, including neutral lipid and cholesterol esters, contributes to inflammatory status and endoplasmic reticulum (ER)2 stress in vascular endothelium that is associated with endothelial dysfunction and atherosclerosis (1). Degradation of lipid droplets by stimulation of autophagy (lipophagy) reduces ER stress and inflammation (2). Autophagy is a catabolic process that plays pivotal roles in metabolism, cell death, and differentiation (3, 4). An excess amount of lipids, aggregated proteins, and organelles is degraded through the autophagic process, which is one of the protective mechanisms used to remove unused cellular materials (5, 6). Macroautophagy (hereafter autophagy) occurs through a series of events forming membrane-like structure, compartmentalization, and fusion of vesicles that generate autophagolysosome (5). Impairment of the lysosomal degradation process causes reduced autophagic flux leading to serious disorders in cardiovascular and metabolic tissues (7–9).
An 11-year follow-up study shows that green tea consumption is associated with reduced mortality due to cardiovascular diseases but not with mortality due to cancer (10). We and others have shown that the most abundant green tea polyphenol, epigallocatechin-3-gallate (EGCG), has beneficial health effects in various pathophysiological conditions, including insulin resistance, endothelial dysfunction, and ischemia-reperfusion injuries (11–13). One of the molecular mechanisms for EGCG-mediated protective effects is through activation of adenosine monophosphate-activated protein kinase (AMPK) (14, 15). However, the molecular mechanisms for linking EGCG-stimulated AMPK and autophagy with regard to lipid metabolism are not known. Furthermore, although polyphenols, including EGCG and resveratrol, have effects on lipolysis, it is not known whether the lipolysis is associated with lipophagy (16, 17).
In this study, we investigated the mechanism of EGCG-induced autophagy and its role in accumulation of intracellular lipid accumulation. Here, we show that EGCG-stimulated autophagy is (at least in part) through a CaMKKβ/AMPK-mediated mechanism and has a role in degradation of lipid droplets in vascular endothelial cells. Our data demonstrate a novel mechanism for polyphenols to regulate lipid metabolism in vascular endothelial cells.
EXPERIMENTAL PROCEDURES
Materials
Anti-LC3 (4599), anti-ATG5 (8540), anti-pAMPK (2531), anti-AMPK (2532), anti-pAkt (9271), anti-pULK1 (5869), anti-ULK (4773), anti-pmTOR (2971), and anti-mTOR (2972) antibodies were obtained from Cell Signaling Technology. Anti-CaMKKβ (sc-50341) and anti-LAMP-1 (sc-17768) antibodies were obtained from Santa Cruz Biotechnology. EGCG (Sigma, E4143) and anti-β-actin (Sigma, A5316) antibody were obtained from Sigma. Anti-SQSTM1(p62) was purchased from BD Biosciences (610832). Dicer siRNA for bovine ATG5 and CaMKKβ and scrambled dicer siRNA were purchased from Integrated DNA Technologies. 3-Methyladenine (3977), PD98059 (1213), SB203580 (1202), compound C (3093), and STO-609 (1551) were obtained from Tocris Biosciences (Minneapolis, MN). Some of the inhibitors were dissolved in dimethyl sulfoxide (DMSO), and we confirmed that the vehicle alone did not affect our results.
Cell Culture and Transfection
Bovine aortic endothelial cells (BAEC) in primary culture were obtained from Cell Applications (San Diego) and maintained in F-12K media containing 5% fetal bovine serum (FBS), endothelial cell growth supplement (15 μmol/ml, CB40006B, BD Biosciences), heparin sulfate (50 μg/ml, Sigma, H3393), penicillin (100 units/ml), and streptomycin (100 μg/ml). All experiments were conducted on BAEC between three and six passages. BAEC were transfected with dicer siRNA using INTERFERin® reagents (Polyplus Transfection, 409-50) according to the manufacturer's instructions. One day after transfection, cells were serum-starved for 2 h and then treated with EGCG or inhibitors as indicated in the legends to figures. The sequences are as follows: dicer siRNA for bovine atg5, 5′-rArArUrCrUrCrUrCArCrUrGrUrUrCrArUrUrArUrCrArArArGrUrU-3′ and 5′-rCrUrUrUrGrArUrArArUrGrArArCrArGrUrGrArGrArGrATT-3′; bovine CaMKKβ, 5′-rGrCrUrUrCrUrUrGrArGrGrArUrGrGrCrArArUrUrUrCrCrUrGrGrU-3′ and 5′-rCrArGrGrArArUrUrGrCrCrArUrCrCrUrCrArArGrArAGC-3′; and dsi scrambled RNA, 5′-rArUrArCrGrCrGrUrArUrUrArUrArCrGrCrGrArUrUrArArCrGrArC-3′ and 5′-rCrGrUrUrArArUrCrGrCrGrUrArUrArArUrArCrGrCrGrUrAT-3′.
Preparation of Cell Lysate and Immunoblotting
Cells were briefly washed with ice-cold PBS after the indicated treatments. Cells were then scraped in lysis buffer containing 50 mm Tris (pH 7.2), 125 mm NaCl, 1% Triton X-100, 0.5% Nonidet P-40, 1 mm EDTA, 1 mm Na3VO4, 20 mm NaF, 1 mm sodium pyrophosphate, and complete protease inhibitor mixture (Roche Applied Science, 05056489001). Cell debris was pelleted by centrifugation at 17,000 × g for 10 min at 4 °C. Supernatants were then boiled with Laemmli sample buffer for 5 min, and proteins were resolved by 12% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with primary antibodies, and peroxidase-conjugated secondary antibodies were incubated. The bands were visualized by using SuperSignal chemiluminescent substrate (Pierce, 34078). Immunoblots were quantified by Image analyzer (Vision Works LS) and UVP Bioimaging Systems.
Transmission Electron Microscopy
Bovine aortic endothelial cells were serum-starved for 2 h and then treated with EGCG (10 μm) for 2 h. The cells were fixed with 2.5% glutaraldehyde in 0.1 mm cacodylate buffer (pH 7.4) for 1 h at 4 °C and post-fixed, dehydrated, and then embedded in epoxy resin. Ultra-thin sections (70–90 nm) were observed with a transmission electron microscope (FEI Tecnai T12 120 kV (Hillsboro, OR)). Images were captured on an AMT XR 60B CCD (Danvers, MA). Quantification was performed in individual frames using ImageJ software (National Institutes of Health, Bethesda). The data were shown by the ratio of the total area of autophagosomes and autolysosomes to the total cytoplasmic area.
Immunocytochemistry
To visualize lipid droplets, cells grown on coverslips were fixed with 4% formaldehyde in PBS and then stained with BODIPY 493/503 (Invitrogen, D3922) for 30 min at room temperature. For immunofluorescent staining of cells, cells were treated as described in the figure legends. After stimulation, cells were fixed in 4% paraformaldehyde/PBS for 10 min, and the cells were washed with PBS. Cells were then permeabilized with 0.1% Triton X-100/PBS for 5 min and washed with PBS. Cells were blocked with 5% BSA/PBS for 1 h and then incubated with anti-LC3 or anti-LAMP-1 antibody in 5% BSA/PBS at 4 °C overnight. Cells were washed with PBS three times (5 min each) and then incubated with Alexa 555-conjugated goat anti-rabbit IgG (Invitrogen, A21422) for 1 h at room temperature. Images were acquired with an Axiovert fluorescence microscope (Carl Zeiss Ltd., Thornwood, NY). 10–15 cells were randomly selected from each treatment to calculate the average number of lipid droplets and the percentage of co-localization per cell. Quantification was performed using ImageJ software (National Institutes of Health), and the percentage of co-localization was calculated by JACoP plugin of ImageJ. The data shown were from one representative experiment of three independent repeats.
Calcium Measurements
The intracellular Ca2+ levels in BAEC were recorded using the Ca2+ indicator fluo-3 (Invitrogen, F-14218) and a modification of previously described procedures (18, 19). Cell suspension was applied onto polyethyleneimine (1 mg/ml)-coated coverslips. Cells were allowed to adhere for 1 h, upon which floating cells were washed away. Coverslips containing adhered cells were placed overnight in culturing medium in a 5% CO2, 95% air atmosphere incubator at 37 °C. The following day, after 2 h of serum starvation at 37 °C, BAEC were loaded with the acetoxymethyl (AM) ester of fluo-3 (10 μg/ml) for 20 min at room temperature (20–24 °C) in external solution containing (in mm) the following: 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.4); dispersion of the ester was aided by 0.025% Pluronic F-127 (Invitrogen, P3000MP). After washing, the indicator was permitted to de-esterify for 20 min in BAEC at room temperature in external solution. Coverslips containing fluo-3-loaded BAEC were then mounted onto a recording chamber and visualized. Images were acquired every 5 s, the first 10 s being used as a base line to establish F0 (see below). After image 10, we bath applied EGCG (10 μm, 200 s) to test its effects on intracellular Ca2+ levels. Ca2+ responsiveness was confirmed in cells at the end of each experiment by using the Ca2+ ionophore 4-bromo-A23187 (10 μm, 200 s, B1494, Invitrogen) as described previously (20).
To assess contribution of the ER Ca2+ store to EGCG-induced Ca2+ dynamics in BAEC, we employed a commonly used procedure described elsewhere (21). Briefly, BAEC were preincubated in external solution supplemented with cyclopiazonic acid (CPA, 20 μm, 30 min; Sigma, C1530), a blocker of ER Ca2+-ATPase. After allowing the ER store to deplete for 30 min, we bath applied EGCG in the presence of CPA.
All experiments were done at room temperature. We used an inverted microscope (TE 300; Nikon, Melville, NY) equipped with wide field epifluorescence.
For visualization of fluo-3, we used a standard fluorescein/FITC filter set (Chroma, Rockingham, VT). Images were captured through a ×60 PlanApo oil-immersion objective (numerical aperture (NA), 1.4; Nikon) using a CoolSNAP-HQ-cooled charge-coupled device camera (Roper Scientific, Tucson, AZ) driven by V++ imaging software (Digital Optics, Auckland, New Zealand). For time-lapse image acquisition, a camera and an electronic shutter (Vincent Associates, Rochester, NY) inserted in the excitation pathway were controlled by software. A xenon arc lamp (100 watts) was used as a light source. All images shown in figures and supplemental movies represent raw data with their pixel intensities within the camera's (CoolSNAP-HQ) dynamic range (0–4095). In analysis, all imaging data were background subtracted using regions of the coverslip field containing no cells. Data are expressed as dF/F0 (%), where dF represents the change of fluorescence, and F0 represents the base-line fluorescence of cells (10 consecutive images). Ca2+ transients from background subtracted fluo-3 emission traces were declared Ca2+ peaks if over two consecutive images their dF values exceeded the F0 ± S.D. Oscillatory events were counted for each cell, and their peaks (amplitudes) dF/F0 were determined. Cumulative Ca2+ responses were calculated by summing dF/F0 for individual time points during the entire EGCG challenge of BAECs.
All data on Ca2+ dynamics fulfill normality as established using the Shapiro-Wilk test. The effects of CPA on EGCG-induced Ca2+ dynamics (number of oscillatory events, peak and cumulative dF/F0) were tested using t tests. Data are expressed as means ± S.E.
RT-PCR
The cells were treated as described in the figure legends. Total RNA was prepared by using TRIzol (Invitrogen, 15596018) according to the manufacturer's instructions. One microgram of total RNA was used for cDNA synthesis by using the Omniscript RT kit (Qiagen, 205113). Then the cDNA was subjected to semi-quantitative PCR analysis by using Hot Star Taq Master Mix kit (Qiagen, 203445). PCR product was visualized with fluorescent dye (Helix Technologies, HDS001), and the image was analyzed and quantified by Image analyzer (Vision Works LS) and UVP. The primers for CaMKKβ are forward, TGAAGACCAGGCCCGTTTCTACTT, and reverse, TCACACCAAAGTCCGCGATCTTGA; and for β-actin are forward, CTGGCACCCAGCACAATGAAG, and reverse, TAGAAGCATTGCGGTGGACG.
RESULTS
EGCG Stimulates Autophagy
Because formation of LC3-II by cleavage and lipidation is an indication of autophagy (22–24), we examined whether EGCG stimulates LC3-II formation. Treatment of BAEC with EGCG increased LC3-II formation in a time-dependent (Fig. 1, A and B) and dose-dependent (Fig. 1, C and D) manner. To confirm that EGCG-stimulated LC3-II formation is through an autophagy-dependent mechanism, we transiently transfected BAEC with siRNA for atg5 and then treated BAEC without or with EGCG. Knockdown of atg5 (one of the genes required for autophagy, 40% reduction in ATG5 protein expression) reduced EGCG-stimulated LC3-II formation (Fig. 1, E and F). The data suggest that EGCG-stimulated LC3-II formation is through an autophagy-dependent mechanism. Next, we also observed the autophagosome formation by electron microscopy after treating the BAEC without or with EGCG (10 μm) (Fig. 2). EGCG stimulated formation of autophagolysosome-like structure and made compartments that have less dense areas compared with the normal cellular density (white triangles) and autophagosomes (Fig. 2A, solid line arrow). Phagophore and double membrane structures indicate that active autophagy occurs with EGCG treatment (Fig. 2B). The autophagosome area (Fig. 2C) and autophagolysosomal (Fig. 2D) area were increased 3- and 7-fold, respectively, in the EGCG-treated cells when compared with untreated cells. The rates of autophagosome formation and degradation are steady state in normal conditions, which is altered in various stressful conditions (25). We examined whether EGCG stimulates autophagic flux by comparing accumulation of LC3-II with and without inhibition of lysosomal degradation. We blocked lysosomal degradation by using ammonium chloride/leupeptin (NH4Cl/Leu) as reported previously (26). Treatment with NH4Cl/Leu alone causes accumulation of LC3-II, and treatment with EGCG and NH4Cl/Leu further enhanced the accumulation of LC3-II (Fig. 3, A and B). The subtracted values (the difference in the amount of LC3-II without blocking lysosomal degradation from the value with lysosomal degradation) indicate that EGCG stimulates autophagic flux (Fig. 3C). Sequestosome 1 (SQSTM1, p62), a ubiquitin-binding protein, is involved in autophagy and is known as an indication of autophagic degradation (27–29). We examined whether EGCG facilitates degradation of SQSTM1. Treatment of EGCG significantly decreased the SQSTM1 level (Fig. 3, D and E). This suggests that EGCG enhances autophagic degradation.
FIGURE 1.

EGCG stimulates autophagy. BAEC were treated with EGCG for the indicated times. Cell lysates were analyzed by Western blot using antibodies against the indicated proteins. A and B, time course of EGCG (10 μm) effect on LC3-II formation. Data are mean ± S.E. *, p < 0.05; **, p < 0.01 versus control (0 time point). C and D, dose-dependent response to EGCG on LC-3-II formation. Data are mean ± S.E. **, p < 0.01; ***, p < 0.001 versus control (no treatment). E and F, BAEC were transfected with control scrambled or ATG5 siRNA and incubated for 48 h. The cells were serum-starved for 2 h and then treated without or with EGCG (10 μm) for 4 h. LC3-II bands from three independent experiments were quantified and normalized for β-actin. Data are mean ± S.E. (**, p < 0.05, versus scrambled siRNA treated).
FIGURE 2.

Electron microscopic images of BAEC treated without or with EGCG. A and B, BAEC were treated without (upper panels) or with (lower panels) 10 μm EGCG for 2 h. Autophagosomes (thin solid arrows), autophagolysosomes (white triangle), and phagophore (black arrow) are indicated. C and D, areas of autophagosome and autophagolysosome were calculated as described under “Experimental Procedures.” Data are mean ± S.E. (***, p < 0.001, versus nontreated (NT) cells).
FIGURE 3.

EGCG enhances autophagic flux. A and B, BAEC were treated without or with EGCG (10 μm, 4 h). The lysosomal inhibitor (NH4Cl (20 mm), Leu (200 μm)) was treated 1 h prior to cell harvest, and cell lysate was collected and analyzed by immunoblotting for LC3-II formation. Three independent experiments were performed, and the density of LC3-II/LC3-I was quantified. Data are mean ± S.E. (***, p < 0.001). C, differences between the absence and presence of NH4Cl/Leu were calculated for the indication of autophagic flux. Three independent experiments were quantified and calculated. Data are mean ± S.E. (*, p < 0.05). D and E, BAEC were treated without or with EGCG (10 μm) for the indicated time points. Cell lysate was harvested and analyzed by immunoblot with anti-SQSTM1(p62) antibody. SQSTM1 was significantly degraded by treatment with EGCG, which indicates autophagic degradation was enhanced. Three independent experiments were performed, and data are mean ± S.E. (***, p < 0.001). NT, not treated.
EGCG-stimulated LC3-II Formation Is through a CaMKKβ/AMPK-mediated Mechanism
We previously reported that EGCG stimulates AMPK (30). Because AMPK directly phosphorylates uncoordinated-51-like kinase (ATG1/ULK1) (31, 32), we examined whether EGCG stimulates phosphorylation of AMPK and ULK1. Treatment of BAEC with EGCG stimulated the phosphorylation of AMPK and ULK1 in a time-dependent manner (Fig. 4A). To identify signal transduction pathways for EGCG-stimulated autophagy, we treated BAEC with various inhibitors. EGCG-stimulated LC3-II formation was inhibited by 3-methyladenine (PI3K inhibitor), compound C (AMPK inhibitor), and STO-609 (CaMKKβ inhibitor) but not by PD98059 (MEK inhibitor) and SB203580 (p38 MAPK inhibitor) (Fig. 4, B and C). We previously demonstrated that EGCG stimulates H2O2 production, which is known to stimulate CaMKKβ and AMPK in vascular endothelial cells (33). Thus, we examined whether H2O2 is involved in LC3-II formation. However, pretreatment of cells with N-acetylcysteine, an antioxidant, was not able to inhibit the EGCG-stimulated LC3-II formation (Fig. 4, D and E). In contrast, the same amount of N-acetylcysteine was able to inhibit EGCG-stimulated phosphorylation of Akt (Fig. 4, F and G). Inhibition of CaMKKβ by STO-609 inhibited EGCG-stimulated phosphorylation of AMPK and ULK1 (Fig. 5, A and B). mTOR plays an important role in autophagy, and inhibition of mTOR stimulates autophagy in response to various stimuli (34, 35). We examined whether EGCG inhibits phosphorylation of mTOR. Surprisingly, EGCG did not inhibit the phosphorylation of mTOR, and pretreatment with STO-609 did not affect the EGCG-stimulated phosphorylation of mTOR (Fig. 5C). These data suggest that the EGCG-stimulated autophagy is independent of mTOR activity. To confirm that CaMKKβ is involved in EGCG-stimulated LC3-II formation, we transiently transfected BAEC with siRNA for CaMKKβ, and we then treated with EGCG. Knockdown of CaMKKβ by siRNA was able to reduce the mRNA expression of CaMKKβ by 48%. Reduction of CaMKKβ inhibited EGCG-stimulated LC3-II formation (Fig. 5D). Thus, the data suggest that EGCG stimulates autophagy through a CaMKKβ-mediated mechanism.
FIGURE 4.

EGCG-stimulated autophagy is through AMPK and CaMKKβ but not through reactive oxygen species production. A, BAEC were treated with EGCG (10 μm) for the indicated times, and then cell extracts were analyzed by immunoblotting with the indicated antibodies. Three independent experiments were performed and quantified. B and C, BAEC were pretreated with inhibitors for 30 min; 3-methyladenine (3-MA, 5 mm), PD98059 (PD, 25 μm), SB203508 (SB, 10 μm), compound C (CC, 10 μm), or STO-609 (STO, 10 μm), and then treated with EGCG (10 μm, 4 h). Cell extracts were analyzed by Western blot by using the indicated antibodies. Data are mean ± S.E. (*, p < 0.05; **, p < 0.01 versus EGCG alone). D–G, BAEC were pretreated with the indicated dose of N-acetylcysteine and then treated with EGCG (10 μm, 4 h, D and E, or 3 h, F and G). Cell extracts were analyzed by Western blot using the indicated antibodies. Experiments were repeated three times and quantified by densitometry. Data are mean ± S.E., ***, p < 0.001.
FIGURE 5.

CaMKKβ is involved in EGCG-stimulated AMPK, ULK1, and autophagy but not mTOR. A–C, BAEC were pretreated with STO-609 (10 μm) for 30 min and then treated with EGCG (10 μm) for 2 min. Cell lysates were analyzed by immunoblotting with the indicated antibody. The increased fold was calculated by LC3-II/β-actin. Experiments were repeated three times and quantified by densitometry. Data are mean ± S.E.; ns > 0.05, **, p < 0.01, and ***, p < 0.001. D, BAEC were transfected with control scrambled or CaMKKβ siRNA and incubated for 48 h. The cells were serum-starved for 2 h and then treated without or with EGCG (10 μm) for 4 h. LC3-II bands from three independent experiments were quantified and normalized for β-actin. Reduction of CaMKKβ was examined by RT-PCR. Data are mean ± S.E. (**, p < 0.01, and ***, p < 0.001).
EGCG Stimulates Cytosolic Ca2+ Levels That Are Required for LC3-II Formation
Because CaMKKβ requires intracellular calcium signaling to be activated (36), we evaluated if BAEC have the ability to increase cytosolic Ca2+ (Ca2+cyt) in response to EGCG. Serum-starved BAEC were loaded with the Ca2+ dye fluo-3 to monitor their Ca2+cyt levels. Bath applied EGCG (10 μm, 200 s) caused an increase in Ca2+cyt levels, which displayed an oscillatory pattern (n = 44) (supplemental Movie 1). It has been established that in other cells, e.g. astrocytes, this oscillatory pattern resembles calcium release from internal stores, mainly ER (19, 37). To test whether internal Ca2+ stores, in particular the ER store, supply Ca2+ during EGCG stimulation of BAEC, we pretreated serum-starved BAEC with CPA (20 μm, 30 min), a widely used blocker of ER Ca2+-ATPase. During the course of this pretreatment with CPA, the depletion of the ER store in the presence of extracellular Ca2+ was evident as a slow increase in Ca2+cyt levels (n = 36; dF/F0 = 47 ± 3%; p < 0.01, paired t test), which reached a new base line (Fig. 6, compare images a in A and B). Because we were interested in the ability of BAEC to handle Ca2+cyt once the ER store had been depleted, we used Ca2+cyt levels after the CPA pretreatment as a base line (F0) for further analysis. Indeed, CPA was kept throughout the remainder of the experimental paradigm. Hence, bath application of EGCG to CPA-treated cells (n = 36) caused attenuated Ca2+cyt dynamics (Fig. 6B and supplemental Movie 2); the average number of oscillatory events was reduced when compared with those recorded from the control cells (n = 44) exposed to EGCG in the absence of CPA (Fig. 6C; 3.3 ± 0.6 and 5.7 ± 0.9, respectively). Furthermore, peaks/amplitudes of oscillatory events were decreased in CPA-treated cells in comparison with control cells (peak dF/F0 = 24 ± 7 and 55 ± 16%, respectively) (Fig. 6D). These data point toward BAEC utilization of the ER store for supply of cytosolic Ca2+ during the EGCG challenge.
FIGURE 6.

Elevation of intracellular calcium is required for EGCG-stimulated autophagy. A, serum-starved BAEC were pre-loaded with fluo-3 (10 μg/ml) and then stimulated with EGCG (10 μm). Cytosolic fluorescence was observed and quantified. The pseudocolor scale is a linear representation of the fluorescence intensity ranging from 200 to 1200 intensity units. Oscillation of calcium signaling was observed in EGCG-treated cells. B, BAEC were pretreated with CPA (20 μm) 30 min prior to EGCG treatment. C–E, pretreatment of BAEC with CPA reduced calcium signaling. C, number of events; D, peak of signaling; and E, cumulative fluorescence were quantified as described under “Experimental Procedures”; F, bovine aortic endothelial cells were pretreated with Ca2+ chelators for 30 min, EGTA (1 mm), or BAPTA-AM (10 μm), and then treated with EGCG (10 μm, 4 h). **, p < 0.01. Cell lysates were harvested and analyzed by immunoblotting. LC3-II bands from three independent experiments were quantified and normalized for β-actin. Data are mean ± S.E. (*, p < 0.05).
To critically evaluate the amount of Ca2+ supplied from the ER store to cytosol during the entire EGCG stimulus, we obtained cumulative Ca2+ responses. CPA-treated cells in comparison with control cells showed grossly reduced EGCG-induced cumulative Ca2+ responses (cumulative dF/F0 = 108 ± 18 and 654 ± 157%, respectively; t test, p < 0.01) (Fig. 6E). Therefore, BAEC respond to EGCG by an increase in Ca2+cyt showing an oscillatory pattern, and this Ca2+ excitability requires the supply of Ca2+ from the ER store. It should be noted, however, that the cumulative response is not completely blocked by CPA, which may indicate that other sources of Ca2+, such as the entry from the external space and mitochondria, might also contribute to EGCG-induced Ca2+ excitability; for discussion of Ca2+ sources, see Ref. 20. This intracellular Ca2+ contributed to EGCG-stimulated LC3-II formation because chelating intracellular Ca2+ by BAPTA-AM (10 μm, 30 min) or by the presence of extracellular EGTA (1 mm, 30 min) inhibited LC3-II formation (Fig. 6F).
EGCG Facilitates Autophagic Flux and Lipophagy
It has been demonstrated that treatment with palmitate inhibits autophagic flux in pancreatic beta cells (38). So, we examined whether EGCG affects autophagic flux that was inhibited by palmitate. We treated BAEC with palmitate (200 μm, 4 h) in the presence or absence of lysosomal inhibitors (NH4Cl/Leu) (Fig. 7). As expected, the ratio of LC3-II/LC3-I was not further increased in the presence of the lysosomal inhibitor complex (NH4Cl/Leu) compared with the cells incubated with palmitate in the absence of NH4Cl/Leu. In contrast, treatment of BAEC with EGCG and palmitate increased the ratio of LC3-II/LC3-I in the presence of NH4Cl/Leu compared with the cells treated without NH4Cl/Leu. The results suggest that palmitate inhibits lysosomal degradation of LC3-II, which is opposed by treatment with EGCG. Excess intake of lipid causes obesity and ectopic lipid accumulation, which is implicated as one of the causes for cardiometabolic syndrome (39–41). Fatty acid overload increases intracellular lipid droplets, and the presence of lipid droplets in non-adipose tissue plays a role in various pathophysiologies (42, 43). We examined whether EGCG-stimulated autophagic flux contributes to the reduction of intracellular lipid droplets. Treatment with palmitate caused accumulation of lipid droplets (Fig. 8, A–D, the 3rd rows, and E) that are decreased by EGCG (Fig. 8, A and C, the 4th rows, and E). This EGCG-stimulated reduction of lipid droplets was diminished by treatment with NH4Cl/Leu (Fig. 8, B and D, the 4th rows, and E). The accumulation of lipid droplets was observed only in the cells treated with palmitate and not in the cells treated with BSA or BSA plus EGCG (Fig. 8, A–D, the 1st and the 2nd rows). To test whether lipid droplets are associated with lipophagy, we examined whether LC3 (Fig. 8, A and B) or LAMP-1 (Fig. 8, C and D) co-localize with lipid droplets. In the cells treated with palmitate, only a small number of co-localizations was observed both in the presence and absence of NH4Cl/Leu (Fig. 8, A–D, the 3rd rows). When the cells were treated with palmitate and EGCG, the number of co-localizations between lipid droplets with LC3 or LAMP-1 was slightly increased in the absence of NH4Cl/Leu, probably due to the rapid degradation (Fig. 8, A and C, the 4th rows), and significantly increased in the presence of NH4Cl/Leu (Fig. 8, B and D, the 4th rows). To exclude the possibility that EGCG inhibits formation of lipid droplets, we treated BAEC with EGCG after incubation with palmitate (Fig. 8H, right column) and compared the results with co-treatment with palmitate and EGCG (Fig. 8H, left column). The number of lipid droplets was almost identical whether EGCG was treated together with or after palmitate (Fig. 8I). This suggests that the effect of EGCG in reduction of lipid droplets is mainly dependent on degradation but not inhibition of formation. These results suggest that EGCG decreases accumulation of lipid droplets through facilitation of lysosomal degradation, which may contribute to prevention of lipotoxicity in vascular endothelial cells.
FIGURE 7.
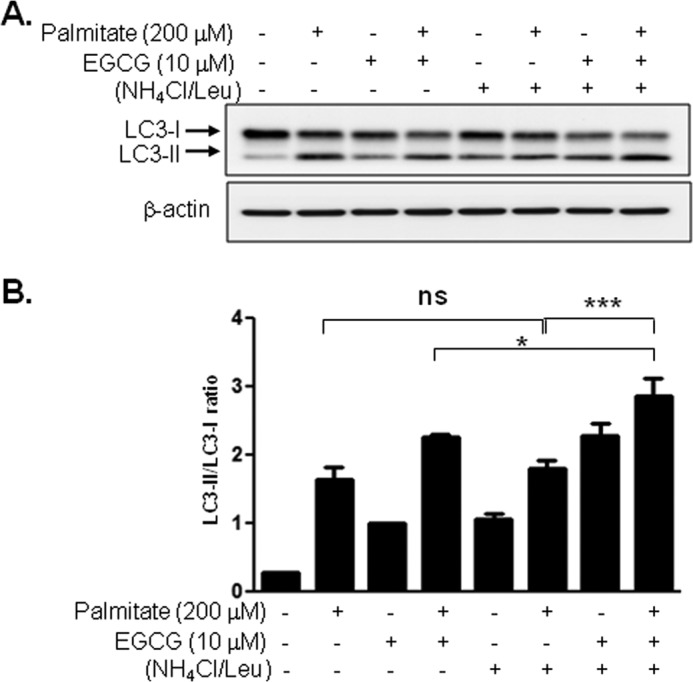
Palmitate-induced inhibition of autophagic flux, which was opposed by co-treatment with EGCG. BAEC were treated with BSA (0.1%) or palmitate (200 μm)/BSA conjugate in the presence or absence of EGCG (10 μm) for 4 h. Lysosomal inhibitor (20 mm NH4Cl, 200 μm leupeptin) was treated 1 h prior to cell harvest. Cell lysate was analyzed by immunoblotting for LC3-II formation. Three independent experiments were performed, and density of LC3-II/LC3-I was quantified. Data are mean ± S.E. (*, p < 0.05; ***, p < 0.001). ns, not significant.
FIGURE 8.

EGCG enhances lipophagy. BAEC were seeded on glass coverslips. Cells were treated with BSA (0.1%) (the top two rows in A–D) or palmitate (200 μm)/BSA (0.1%)) conjugate (the bottom two rows in A–D) in the absence or presence of EGCG (10 μm) for 4 h without (A and C) or with (B and D) lysosomal inhibitors (20 mm NH4Cl, 200 μm leupeptin). The accumulation of lipid droplets was stained with BODIPY 493/503 (green). The co-localization of lipid droplets with LC3 (A and B, red) or LAMP-1 (C and D, red) was by immunocytochemistry using anti-LC3 and anti-LAMP-1 antibodies as described under “Experimental Procedures.” Insets show the higher magnification of the areas in the squares. Arrows indicate co-localization events. Nucleus was stained with Hoechst 33342 (blue). E, number of lipid droplets (LDs) was counted, and the average number of lipid droplets per cell was calculated. Data are mean ± S.E. (*, p < 0.05; **, p < 0.01, and ***, p < 0.001, n = 12). The percentage of lipid droplet co-localization with LC3 (F) or LAMP-1 (G) in cells treated with palmitate in the absence or presence of EGCG without or with NH4Cl/leupeptin was quantified. Data are mean ± S.E. (*, p < 0.05, n = 6). H and I, BAEC were incubated with palmitate (200 μm)/BSA (0.1%) conjugate without or with EGCG (10 μm) for 4 h (left column) or preincubated with palmitate for 4 h and then treated without or with EGCG (10 μm) for another 4 h (right column). Cells were fixed with paraformaldehyde and then stained with BODIPY 493/503 (green). Nuclei were stained with Hoechst 33342 (blue). The number of lipid droplets (LDs) was counted, and the average number of lipid droplets per cell was calculated. Data are mean ± S.E. (***, p < 0.001, n = 10).
DISCUSSION
This study demonstrates that EGCG stimulates autophagy through a CaMKKβ/AMPK-dependent mechanism and facilitates autophagic flux (Fig. 9). Furthermore, EGCG-stimulated lysosomal degradation leads to reduced accumulation of intracellular lipid droplets in vascular endothelial cells. These EGCG effects in vascular endothelium may contribute to protection from lipid-mediated endothelial dysfunction and cardiovascular complications.
FIGURE 9.
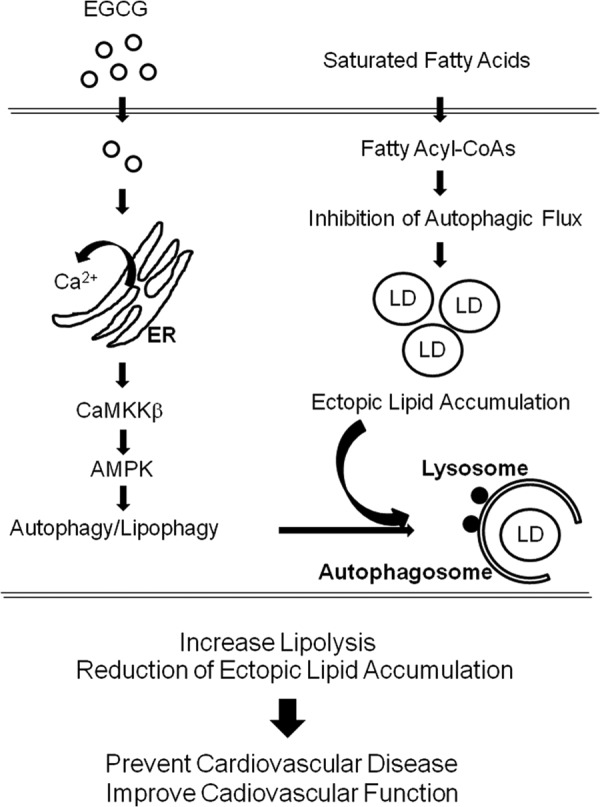
Schematic diagram of proposed EGCG-stimulated signaling pathway to activate lipophagy. EGCG-induced oscillation of cytosolic Ca2+ levels by stimulating Ca2+ store in ER that activates CaMKKβ/AMPK pathways and facilitation of autophagic flux. Saturated fatty acid (palmitate) causes impairment of autophagic flux, which leads to accumulation of ectopic lipid accumulation. The facilitated autophagic flux by EGCG affects degradation of accumulated lipid droplets in vascular endothelial cells, which may contribute to improvement of cardiovascular function and prevention of cardiovascular disease. This EGCG-stimulated lipophagy may explain the beneficial health effect of green tea consumption.
EGCG Induces Autophagy through AMPK and CaMKKβ
In this study, we demonstrate that EGCG induces autophagosome and autophagolysosome formation (Fig. 2) through a CaMKKβ/AMPK signaling pathway leading to facilitation of autophagic flux (Figs. 3 and 5).
Previously, it has been shown that a high concentration (50–100 μm) of EGCG stimulates autophagy leading to cell death in cancer cells (44, 45). Another study reported that EGCG stimulates autophagy, which leads to inhibition of endotoxin-induced septic shock through EGCG-induced degradation of HMGB1, a late lethal inflammatory factor (46). However, a molecular mechanism for the EGCG-stimulated autophagy with regard to Ca2+/CaMKKβ and lipid droplet is unknown. In this study, we demonstrate a novel mechanism for EGCG-stimulated autophagy and its functional consequence in degradation of lipid droplets.
AMPK is a key mediator for the initial process of autophagy by stimulating the phosphorylation of ULK and formation of its protein complex with multiple autophagic proteins (47). Thus, activation of AMPK is crucial for initiation of autophagy. Because AMPK is an energy-sensing enzyme recognizing the AMP/ATP ratio, starvation conditions activate autophagy through an AMPK-dependent mechanism (48). Thus, EGCG may be mimicking starvation or caloric restriction conditions, which are consistent with the beneficial health effects of polyphenols, including EGCG and resveratrol (49–52). EGCG also has an anti-diabetic effect that is similar to metformin, an anti-diabetic drug that activates AMPK (53). This suggests that EGCG and metformin may have a common mechanism to ameliorate metabolic and cardiovascular disorders. Knockdown of ATG5 or CaMKKβ was significantly but not completely able to block the LC3-II formation (Figs. 1E and 5D). This may be due to the incomplete removal of ATG5 or CaMKKβ, because siRNAs were able to knock down only 40 and 48% of ATG5 and CaMKKβ, respectively. However, it is possible that the remaining LC3-II after knockdown of ATG5 or CaMKKβ could be due to the CaMKKβ- or ATG5-independent mechanism. Using primary cells from knock-out mice may help to understand the precise mechanism. In this study, our results suggest that EGCG stimulates LC3-II formation at least in part through an ATG5- and CaMKKβ-mediated mechanism.
We observed that EGCG activates AMPK, whereas EGCG did not stimulate the phosphorylation of mTOR (Fig. 5C). Activation of AMPK inhibits mTOR in starvation-induced autophagy (54, 55). However, we were not able to observe that mTOR is inhibited by EGCG, which may be a cell type-specific or a stimulus-specific response. Interestingly, we observed that EGCG was not able to induce LC3-II formation in mouse embryonic fibroblasts (data not shown). In addition, other studies have shown that palmitate induces autophagy through an mTOR-independent mechanism (56). This unexpected result suggests that EGCG-stimulated accumulation of LC3-II is independent of the mTOR pathway.
EGCG Induces Cytosolic Ca2+ Dynamics Implicated in Autophagy
EGCG induces intracellular Ca2+ dynamics, and chelating cytosolic calcium by BAPTA-AM or reducing availability of extracellular Ca2+ by EGTA suppressed autophagy (Fig. 6F). This suggests that cytosolic Ca2+ load is necessary for EGCG-induced autophagy. Consistent with our data, previous reports show that the heightened free cytosolic calcium induces autophagy through activation of CaMKKβ and AMPK (57–59). EGCG stimulates Ca2+ release from the ER store, because treatment with CPA, a blocker of ER Ca2+-ATPase, reduced cytosolic Ca2+ load. It has been shown that Ca2+ is required for CaMKKβ to activate AMPK, although CaMKKβ can autonomously activate other substrates, including CaMKI and CaMKIV, without Ca2+/CaM binding (60). Our data suggest that the elevated cytosolic Ca2+ dynamics are necessary for EGCG-stimulated autophagy. Resveratrol, a major polyphenol in red wine, activates CaMKKβ/AMPK through inhibition of cAMP-degrading phosphodiesterase and PKCϵ that leads to the activation of ryanodine receptor to increase intracellular calcium levels (52). However, the link between autophagy and polyphenol-induced intracellular calcium is not known. Here, we show for the first time that EGCG elicits an increase in cytosolic Ca2+, which contributes to autophagy. In addition to the signaling molecules to stimulate the CaMKKβ/AMPK axis, increased cytosolic Ca2+ may contribute to autophagosomal membrane formation from the ER membrane. Previously, it has been shown that ER membranes co-localize with calcium phosphate precipitate-induced LC3-positive autophagosome (61). This suggests that calcium-induced autophagosome formation may be attributable to interaction with the ER membrane. Further studies are necessary to reveal the role of cytosolic Ca2+ and CaMKKβ in autophagy.
EGCG Facilitates Lipophagy
In this study, we demonstrate that EGCG facilitates autophagic flux, which contributes to oppose palmitate-induced accumulation of lipid droplets in endothelial cells (Figs. 7 and 8). Reduction of lipid droplets in endothelial cells through autophagic flux suggests a novel mechanism for EGCG-mediated beneficial health effects. Autophagy (or lipophagy) plays a role in lipid metabolism both in adipose tissue and in non-adipose tissue (42, 43, 62). Reduction of lipid accumulation in adipose tissue leads to weight loss and improvement of whole body metabolism. In contrast, reduced lipid accumulation in vascular endothelium may contribute to cardiovascular function, because triglyceride and cholesterols are the major components of lipid droplets that are associated with atherosclerosis and coronary heart disease. Chronic high fat diet and acute high cholesterol diet lead to impaired lysosomal degradation (63), and fatty acid inhibits autophagic flux due to the failure of lysosomal degradation in beta cells (38). Consistent with these reports, our data show that palmitate impairs autophagic flux in primary aortic endothelial cells, and EGCG enhances degradation of lipid droplets through facilitation of autophagic flux.
Despite the reduced number of lipid droplets in the EGCG-treated cells, a larger number of lipid droplets co-localize with LC3 and LAMP-1 in the presence of NH4Cl/Leu (Fig. 8). The reason for the smaller number of lipid droplets and the fewer co-localizations of lipid droplets and LC3 or LAMP-1 in the presence of EGCG without NH4Cl/Leu seems to be due to the rapid degradation of lipid droplets.
The accumulation of lipid droplets is much more prominent in the cells treated with palmitate alone than the cells treated with palmitate along with EGCG. One possibility is that triglyceride synthesis could be slower in EGCG-treated cells. However, we observed that reduction of the lipid droplet was almost identical whether the cells were co-treated or post-treated with EGCG with palmitate (Fig. 8, H and I). Moreover, co-localization of lipid droplets with autophagosomes was markedly increased in the EGCG-treated cells when lysosomal degradation was blocked (Fig. 8). These suggest that treatment with palmitate seems to inhibit the fusion process of autophagosome and lysosome, and the fusion is facilitated by EGCG.
Autophagic flux can be divided into three steps as follows: (i) formation of autophagosome; (ii) formation of autolysosome by fusion of lysosome and autophagosome; and (iii) lysosomal degradation. Formation of autophagosome was dramatically increased by EGCG as shown the samples with lysosomal inhibitors (Fig. 3, A and B, 2nd and 4th lanes), and lysosomal degradation was increased (Fig. 3, C–E). Nonetheless, our results suggest that EGCG stimulates lipophagy through facilitation of autophagosome formation, lysosomal fusion, and degradation. This does not exclude the possibility that other lipases, including hormone-sensitive lipase and endothelial lipase, may contribute to reduction of lipid accumulation. In fact, EGCG stimulates hormone-sensitive lipase in adipocytes and pancreatic lipases in the serum that are associated with weight loss in adipose tissue (64, 65). In contrast, our data present for the first time that EGCG reduces endothelial ectopic lipid accumulation. We previously reported that EGCG intake protects from insulin resistance, hypertension, and ischemia-reperfusion injury in the heart in spontaneously hypertensive rats (12). These beneficial health effects of EGCG in cardiovascular tissues may be associated with the EGCG-induced facilitation of autophagy. Further studies are required to understand the more detailed regulatory mechanisms for EGCG-stimulated autophagic flux.
In summary, EGCG induces autophagy through a Ca2+/CaMKKβ/AMPK-mediated mechanism, which contributes to reduction in the palmitate-induced accumulation of lipid droplets in endothelial cells. These findings suggest the following: 1) heightened intracellular calcium dynamics activating CaMKKβ/AMPK may play an important role in the beneficial health effect of green tea; 2) EGCG stimulates autophagic flux, a key step for autophagic degradation, which may help reduce the accumulation of lipid; 3) supplementation of green tea may have the a beneficial effect in endothelial function through facilitation of lipophagy. These effects of green tea polyphenol may help prevent metabolic and cardiovascular disorders.
This work was supported, in whole or in part, by National Institutes of Health Grant P60 DK-079626 (to University of Alabama at Birmingham Diabetes Research Training Center-sponsored Pilot and Feasibility Program). This work was also supported by American Diabetes Association Grants 1-09-JF-33 and 1-12-BS-99 (to J. K.), American Heart Association Grant 13GRNT17220057 (to J. K.), and National Science Foundation Grant CBET 0943343 (to V. P.).
This article was selected as a Paper of the Week.

This article contains supplemental Movies 1 and 2.
- ER
- endoplasmic reticulum
- EGCG
- epigallocatechin-gallate
- CaMKKβ
- calmodulin-dependent protein kinase kinase β
- BAEC
- bovine aortic endothelial cell
- AMPK
- AMP-activated protein kinase
- ULK
- uncoordinated-51-like kinase
- LAMP-1
- lysosomal associated membrane protein1
- CPA
- cyclopiazonic acid
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- NH4Cl/Leu
- ammonium chloride/leupeptin
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1. Kolattukudy P. E., Niu J. (2012) Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ. Res. 110, 174–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Younce C., Kolattukudy P. (2012) MCP-1-induced protein promotes adipogenesis via oxidative stress, endoplasmic reticulum stress, and autophagy. Cell. Physiol. Biochem. 30, 307–320 [DOI] [PubMed] [Google Scholar]
- 3. Chimal-Monroy J., Abarca-Buis R. F., Cuervo R., Diaz-Hernandez M., Bustamante M., Rios-Flores J. A., Romero-Suarez S., Farrera-Hernandez A. (2011) Molecular control of cell differentiation and programmed cell death during digit development. IUBMB Life 63, 899–906 [DOI] [PubMed] [Google Scholar]
- 4. Sridhar S., Botbol Y., Macian F., Cuervo A. M. (2012) Autophagy and disease: always two sides to a problem. J. Pathol. 226, 255–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cuervo A. M. (2004) Autophagy: in sickness and in health. Trends Cell Biol. 14, 70–77 [DOI] [PubMed] [Google Scholar]
- 6. Singh R. (2010) Autophagy and regulation of lipid metabolism. Results Probl. Cell Differ. 52, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh R., Cuervo A. M. (2011) Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gustafsson A. B., Gottlieb R. A. (2009) Autophagy in ischemic heart disease. Circ. Res. 104, 150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Meyer G. R., Martinet W. (2009) Autophagy in the cardiovascular system. Biochim. Biophys. Acta 1793, 1485–1495 [DOI] [PubMed] [Google Scholar]
- 10. Kuriyama S., Shimazu T., Ohmori K., Kikuchi N., Nakaya N., Nishino Y., Tsubono Y., Tsuji I. (2006) Green tea consumption and mortality due to cardiovascular disease, cancer, and all causes in Japan: the Ohsaki study. JAMA 296, 1255–1265 [DOI] [PubMed] [Google Scholar]
- 11. Bose M., Lambert J. D., Ju J., Reuhl K. R., Shapses S. A., Yang C. S. (2008) The major green tea polyphenol, (−)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice. J. Nutr. 138, 1677–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Potenza M. A., Marasciulo F. L., Tarquinio M., Tiravanti E., Colantuono G., Federici A., Kim J. A., Quon M. J., Montagnani M. (2007) EGCG, a green tea polyphenol, improves endothelial function and insulin sensitivity, reduces blood pressure, and protects against myocardial I/R injury in SHR. Am. J. Physiol. Endocrinol. Metab. 292, E1378–E1387 [DOI] [PubMed] [Google Scholar]
- 13. Kim J. A., Formosa G., Li Y., Potenza M. A., Marasciulo F. L., Montagnani M., Quon M. J. (2007) Epigallocatechin gallate, a green tea polyphenol, mediates NO-dependent vasodilation using signaling pathways in vascular endothelium requiring reactive oxygen species and Fyn. J. Biol. Chem. 282, 13736–13745 [DOI] [PubMed] [Google Scholar]
- 14. Hwang J. T., Park I. J., Shin J. I., Lee Y. K., Lee S. K., Baik H. W., Ha J., Park O. J. (2005) Genistein, EGCG, and capsaicin inhibit adipocyte differentiation process via activating AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 338, 694–699 [DOI] [PubMed] [Google Scholar]
- 15. Moon H. S., Chung C. S., Lee H. G., Kim T. G., Choi Y. J., Cho C. S. (2007) Inhibitory effect of (−)-epigallocatechin-3-gallate on lipid accumulation of 3T3-L1 cells. Obesity 15, 2571–2582 [DOI] [PubMed] [Google Scholar]
- 16. Ahn J., Cho I., Kim S., Kwon D., Ha T. (2008) Dietary resveratrol alters lipid metabolism-related gene expression of mice on an atherogenic diet. J. Hepatol. 49, 1019–1028 [DOI] [PubMed] [Google Scholar]
- 17. Sohle J., Knott A., Holtzmann U., Siegner R., Gronniger E., Schepky A., Gallinat S., Wenck H., Stab F., Winnefeld M. (2009) White tea extract induces lipolytic activity and inhibits adipogenesis in human subcutaneous (pre)-adipocytes. Nutr. Metab. 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Montana V., Ni Y., Sunjara V., Hua X., Parpura V. (2004) Vesicular glutamate transporter-dependent glutamate release from astrocytes. J. Neurosci. 24, 2633–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee W., Malarkey E. B., Reyes R. C., Parpura V. (2008) Micropit: A new cell culturing approach for characterization of solitary astrocytes and small networks of these glial cells. Front. Neuroeng. 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stout R. F., Jr., Parpura V. (2011) Voltage-gated calcium channel types in cultured C. elegans CEPsh glial cells. Cell Calcium 50, 98–108 [DOI] [PubMed] [Google Scholar]
- 21. Malarkey E. B., Ni Y., Parpura V. (2008) Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia 56, 821–835 [DOI] [PubMed] [Google Scholar]
- 22. Fujita N., Itoh T., Omori H., Fukuda M., Noda T., Yoshimori T. (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanida I., Sou Y. S., Ezaki J., Minematsu-Ikeguchi N., Ueno T., Kominami E. (2004) HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 279, 36268–36276 [DOI] [PubMed] [Google Scholar]
- 24. Tanida I., Ueno T., Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mizushima N., Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy 3, 542–545 [DOI] [PubMed] [Google Scholar]
- 26. Hubbard V. M., Valdor R., Patel B., Singh R., Cuervo A. M., Macian F. (2010) Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol. 185, 7349–7357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bjørkøy G., Lamark T., Johansen T. (2006) p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2, 138–139 [DOI] [PubMed] [Google Scholar]
- 29. Ichimura Y., Kominami E., Tanaka K., Komatsu M. (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 30. Reiter C. E., Kim J. A., Quon M. J. (2010) Green tea polyphenol epigallocatechin gallate reduces endothelin-1 expression and secretion in vascular endothelial cells: roles for AMP-activated protein kinase, Akt, and FOXO1. Endocrinology 151, 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Egan D. F., Shackelford D. B., Mihaylova M. M., Gelino S., Kohnz R. A., Mair W., Vasquez D. S., Joshi A., Gwinn D. M., Taylor R., Asara J. M., Fitzpatrick J., Dillin A., Viollet B., Kundu M., Hansen M., Shaw R. J. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee J. W., Park S., Takahashi Y., Wang H. G. (2010) The association of AMPK with ULK1 regulates autophagy. PLoS One 5, e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin B. Y., Sartoretto J. L., Gladyshev V. N., Michel T. (2009) Endothelial nitric oxide synthase negatively regulates hydrogen peroxide-stimulated AMP-activated protein kinase in endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 106, 17343–17348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shang L., Wang X. (2011) AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 7, 924–926 [DOI] [PubMed] [Google Scholar]
- 35. Ravikumar B., Vacher C., Berger Z., Davies J. E., Luo S., Oroz L. G., Scaravilli F., Easton D. F., Duden R., O'Kane C. J., Rubinsztein D. C. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 [DOI] [PubMed] [Google Scholar]
- 36. Hawley S. A., Pan D. A., Mustard K. J., Ross L., Bain J., Edelman A. M., Frenguelli B. G., Hardie D. G. (2005) Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19 [DOI] [PubMed] [Google Scholar]
- 37. Reyes R. C., Parpura V. (2009) The trinity of Ca2+ sources for the exocytotic glutamate release from astrocytes. Neurochem. Int. 55, 2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Las G., Serada S. B., Wikstrom J. D., Twig G., Shirihai O. S. (2011) Fatty acids suppress autophagic turnover in β-cells. J. Biol. Chem. 286, 42534–42544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lara-Castro C., Garvey W. T. (2008) Intracellular lipid accumulation in liver and muscle and the insulin resistance syndrome. Endocrinol. Metab. Clin. North Am. 37, 841–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suganami T., Tanaka M., Ogawa Y. (2012) Adipose tissue inflammation and ectopic lipid accumulation. Endocr. J. 59, 849–857 [DOI] [PubMed] [Google Scholar]
- 41. Gustafson B. (2010) Adipose tissue, inflammation, and atherosclerosis. J. Atheroscler. Thromb. 17, 332–341 [DOI] [PubMed] [Google Scholar]
- 42. Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., Czaja M. J. (2009) Autophagy regulates lipid metabolism. Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weidberg H., Shvets E., Elazar Z. (2009) Lipophagy: selective catabolism designed for lipids. Dev. Cell 16, 628–630 [DOI] [PubMed] [Google Scholar]
- 44. Satoh M., Takemura Y., Hamada H., Sekido Y., Kubota S. (2013) EGCG induces human mesothelioma cell death by inducing reactive oxygen species and autophagy. Cancer Cell Int. 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y., Yang N. D., Zhou F., Shen T., Duan T., Zhou J., Shi Y., Zhu X. Q., Shen H. M. (2012) (−)-Epigallocatechin-3-gallate induces non-apoptotic cell death in human cancer cells via ROS-mediated lysosomal membrane permeabilization. PLoS One 7, e46749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li W., Zhu S., Li J., Assa A., Jundoria A., Xu J., Fan S., Eissa N. T., Tracey K. J., Sama A. E., Wang H. (2011) EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem. Pharmacol. 81, 1152–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao M., Klionsky D. J. (2011) AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. 13, 119–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Birkenfeld A. L., Lee H. Y., Guebre-Egziabher F., Alves T. C., Jurczak M. J., Jornayvaz F. R., Zhang D., Hsiao J. J., Martin-Montalvo A., Fischer-Rosinsky A., Spranger J., Pfeiffer A. F., Jordan J., Fromm M. F., König J., Lieske S., Carmean C. M., Frederick D. W., Weismann D., Knauf F., Irusta P. M., De Cabo R., Helfand S. L., Samuel V. T., Shulman G. I. (2011) Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14, 184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Boer V. C., de Goffau M. C., Arts I. C., Hollman P. C., Keijer J. (2006) SIRT1 stimulation by polyphenols is affected by their stability and metabolism. Mech. Ageing Dev. 127, 618–627 [DOI] [PubMed] [Google Scholar]
- 50. Huang C. H., Tsai S. J., Wang Y. J., Pan M. H., Kao J. Y., Way T. D. (2009) EGCG inhibits protein synthesis, lipogenesis, and cell cycle progression through activation of AMPK in p53 positive and negative human hepatoma cells. Mol. Nutr. Food Res. 53, 1156–1165 [DOI] [PubMed] [Google Scholar]
- 51. Baur J. A., Pearson K. J., Price N. L., Jamieson H. A., Lerin C., Kalra A., Prabhu V. V., Allard J. S., Lopez-Lluch G., Lewis K., Pistell P. J., Poosala S., Becker K. G., Boss O., Gwinn D., Wang M., Ramaswamy S., Fishbein K. W., Spencer R. G., Lakatta E. G., Le Couteur D., Shaw R. J., Navas P., Puigserver P., Ingram D. K., de Cabo R., Sinclair D. A. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Park S. J., Ahmad F., Philp A., Baar K., Williams T., Luo H., Ke H., Rehmann H., Taussig R., Brown A. L., Kim M. K., Beaven M. A., Burgin A. B., Manganiello V., Chung J. H. (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen D., Pamu S., Cui Q., Chan T. H., Dou Q. P. (2012) Novel epigallocatechin gallate (EGCG) analogs activate AMP-activated protein kinase pathway and target cancer stem cells. Bioorg. Med. Chem. 20, 3031–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alers S., Löffler A. S., Wesselborg S., Stork B. (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross-talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim J., Kundu M., Viollet B., Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tan S. H., Shui G., Zhou J., Li J. J., Bay B. H., Wenk M. R., Shen H. M. (2012) Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J. Biol. Chem. 287, 14364–14376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Abbott M. J., Edelman A. M., Turcotte L. P. (2009) CaMKK is an upstream signal of AMP-activated protein kinase in regulation of substrate metabolism in contracting skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1724–R1732 [DOI] [PubMed] [Google Scholar]
- 58. Witczak C. A., Fujii N., Hirshman M. F., Goodyear L. J. (2007) Ca2+/calmodulin-dependent protein kinase kinase-alpha regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes 56, 1403–1409 [DOI] [PubMed] [Google Scholar]
- 59. Hurley R. L., Anderson K. A., Franzone J. M., Kemp B. E., Means A. R., Witters L. A. (2005) The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280, 29060–29066 [DOI] [PubMed] [Google Scholar]
- 60. Racioppi L., Means A. R. (2012) Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J. Biol. Chem. 287, 31658–31665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen X., Li M., Chen D., Gao W., Guan J. L., Komatsu M., Yin X. M. (2012) Autophagy induced by calcium phosphate precipitates involves endoplasmic reticulum membranes in autophagosome biogenesis. PLoS One 7, e52347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh R. (2011) Hypothalamic lipophagy and energetic balance. Aging 3, 934–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rodriguez-Navarro J. A., Kaushik S., Koga H., Dall'Armi C., Shui G., Wenk M. R., Di Paolo G., Cuervo A. M. (2012) Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U.S.A. 109, E705–E714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Grove K. A., Sae-tan S., Kennett M. J., Lambert J. D. (2012) (−)-Epigallocatechin-3-gallate inhibits pancreatic lipase and reduces body weight gain in high fat-fed obese mice. Obesity 20, 2311–2313 [DOI] [PubMed] [Google Scholar]
- 65. Lee M. S., Kim C. T., Kim I. H., Kim Y. (2009) Inhibitory effects of green tea catechin on the lipid accumulation in 3T3-L1 adipocytes. Phytother. Res. 23, 1088–1091 [DOI] [PubMed] [Google Scholar]