

Background: The x-ray structure and mechanism of activation of prothrombin remain elusive.
Results: X-ray and solution studies document conformation flexibility of prothrombin. The two sites of cleavage at Arg-271 and Arg-320 have distinct solvent accessibility.
Conclusion: Burial of Arg-320 prevents prothrombin autoactivation and directs prothrombinase to cleave at Arg-271 first.
Significance: A structure-based mechanism of prothrombin activation emerges.
Keywords: Coagulation factors, Enzyme mutation, Enzyme structure, Structural biology, Thrombin, Zymogen
Abstract
The zymogen prothrombin is composed of fragment 1 containing a Gla domain and kringle-1, fragment 2 containing kringle-2, and a protease domain containing A and B chains. The prothrombinase complex assembled on the surface of platelets converts prothrombin to thrombin by cleaving at Arg-271 and Arg-320. The three-dimensional architecture of prothrombin and the molecular basis of its activation remain elusive. Here we report the first x-ray crystal structure of prothrombin as a Gla-domainless construct carrying an Ala replacement of the catalytic Ser-525. Prothrombin features a conformation 80 Å long, with fragment 1 positioned at a 36° angle relative to the main axis of fragment 2 coaxial to the protease domain. High flexibility of the linker connecting the two kringles suggests multiple arrangements for kringle-1 relative to the rest of the prothrombin molecule. Luminescence resonance energy transfer measurements detect two distinct conformations of prothrombin in solution, in a 3:2 ratio, with the distance between the two kringles either fully extended (54 ± 2 Å) or partially collapsed (≤34 Å) as seen in the crystal structure. A molecular mechanism of prothrombin activation emerges from the structure. Of the two sites of cleavage, Arg-271 is located in a disordered region connecting kringle-2 to the A chain, but Arg-320 is well defined within the activation domain and is not accessible to proteolysis in solution. Burial of Arg-320 prevents prothrombin autoactivation and directs prothrombinase to cleave at Arg-271 first. Reversal of the local electrostatic potential then redirects prothrombinase toward Arg-320, leading to thrombin generation via the prethrombin-2 intermediate.
Introduction
Prothrombin, or coagulation factor II, was discovered in 1872 as the causative agent for the conversion of fibrinogen into fibrin (1) and is abundantly present in the blood where it circulates at a concentration of 0.1 mg/ml (2). In the penultimate step of the coagulation cascade, prothrombin is proteolytically converted to the active protease thrombin by the prothrombinase complex comprising the protease factor Xa and the cofactor Va assembled on the surface of platelets in the presence of Ca2+ (3). Prothrombin (see Fig. 1) is a vitamin K-dependent zymogen composed of fragment 1 (residues 1–155), fragment 2 (residues 156–271), and a protease domain (residues 272–579). Fragment 1 contains a Gla domain (residues 1–46) and a kringle (residues 65–143), fragment 2 contains a second kringle (residues 170–248), and the protease domain contains the A chain (residues 272–320) and the catalytic B chain (residues 321–579). Three linker domains connect kringle-1 to the Gla domain (residues 47–64), the two kringles (residues 144–169), and kringle-2 to the A chain (residues 249–284), respectively. Neither kringle is involved in the binding of prothrombin to the platelet membrane (4) required for efficient thrombin generation; that role is played by the Gla domain in the presence of Ca2+ (5, 6). Kringle-1 and kringle-2 interact with cofactor Va (7), and so do residues within the catalytic B chain scattered between the autolysis loop (8) and exosite I (9). Factor Xa interacts with kringle-2 (10) and residues near exosite II of the B chain (11). The prothrombinase complex converts prothrombin to thrombin by cleaving at Arg-271 and Arg-320 (see Fig. 1). Cleavage at Arg-271 sheds the auxiliary domains, fragment 1 and fragment 2, and generates the inactive precursor prethrombin-2. The alternative cleavage at Arg-320 separates the A and B chains, which remain connected through a disulfide bond (Cys-293–Cys-439), and yields the active intermediate meizothrombin. Under physiological conditions on the surface of platelets, activation of prothrombin proceeds via the Arg-271 cleavage (12) in a concerted manner that precludes accumulation of either meizothrombin or prethrombin-2 (13). The molecular basis of this preferred pathway of activation is unknown.
FIGURE 1.

Top, schematic representation of prothrombin composed of fragment 1 (residues 1–155), fragment 2 (residues 156–271), and a protease domain (residues 272–579). Fragment-1 contains a Gla domain (residues 1–44) and a kringle (residues 65–143), fragment-2 contains a second kringle (residues 170–248), and the protease domain contains the A (residues 272–320) and B (residues 321–579) chains. Thrombin is generated by two cleavages at Arg-271 and Arg-320, producing the inactive precursor prethrombin-2 and the active intermediate meizothrombin, respectively. The A and B chain remain covalently attached after activation through the disulfide bond Cys-293–Cys-439 (Cys-1–Cys-122). Cleavage at Arg-284 by thrombin itself reduces the length of the A chain to its final 36 amino acids composition. Bottom, x-ray crystal structure of Gla-domainless prothrombin with kringle-1 (red) positioned at an angle of 36° relative to kringle 2 (green) that is coaxial to the protease domain (B chain in yellow and A chain in orange). The active site region is indicated by a circle, and the termini for each domain visible in the orientation are noted. Of the two sites of cleavage, Arg-320 (Arg-15) in the activation domain is visible, but Arg-271 is part of a 20-residue segment missing in the electron density map because of disorder.
The structure of prothrombin is currently unknown, and so is the solvent accessibility of the two sites of cleavage at Arg-271 and Arg-320. A model of prothrombin bound to the prothrombinase complex has recently been proposed (14) based on the structure of prethrombin-1 (15) that differs from prothrombin for the absence of fragment 1. Of the two intermediates along the prothrombin activation pathway, prethrombin-2 has been crystallized free (16) and bound to hirugen (17) or staphylocoagulase (18), and the active intermediate meizothrombin has been crystallized without fragment 1 and in the bound form (19, 20). These previous crystal structures offer no insight on the arrangement of fragment 1 relative to the rest of the prothrombin molecule. Here we report the first x-ray crystal structure of prothrombin, obtained from a Gla-domainless construct carrying an Ala replacement of the catalytic Ser-525 to prevent autoactivation (21). The structure reveals an intrinsic conformational flexibility that is confirmed by studies in solution and suggests a molecular mechanism of activation.
EXPERIMENTAL PROCEDURES
Numbering
To facilitate comparison among different trypsin-like proteases, it is customary to number residues in the protease domain based on the sequence of chymotrypsinogen (22). Insertions relative to chymotrypsinogen (e.g. residues of the A chain in the activation domain) are labeled with a lowercase suffix. The chymotrypsinogen numbering becomes impractical for a zymogen-like prothrombin carrying >200 residues in auxiliary domains such as fragments 1 and 2 not present in the mature enzyme or chymotrypsinogen itself. To avoid possible duplications in numbering between residues in auxiliary domains and the protease domain, all prothrombin residues in this study are numbered sequentially based on the sequence of the zymogen. The corresponding number according to chymotrypsinogen for residues in the protease domain (A and B chains) is noted by parentheses, e.g. the catalytic Ser is identified as Ser-525 (Ser-195).
Materials
Gla-domainless prothrombin (residues 45–579) mutant S525A and the quadruple Ala mutant E311A/D318A/E323A/S525A of residues Glu-311 (Glu-14e), Asp-318 (Asp-14l), Glu-323 (Glu-18), and Ser-525 (Ser-195) were expressed in baby hamster kidney cells after introduction of an ApaI/BamHI restriction site in the human prothrombin cDNA (21). Recombinant full-length prothrombin (residues 1–579) wild-type and mutant S101C/S210C were expressed in baby hamster kidney cells after introduction of a SmaI/BamHI restriction site in the human prothrombin cDNA. In all cases, the resulting cDNA was ligated in the pNUT-prethrombin-1 vector. The nucleotide sequence of the constructs was confirmed by sequencing. Constructs were transfected into baby hamster kidney cells by lipofection, and selection was obtained by methotrexate. Expression levels of the secreted protein were analyzed by Western blot using an anti-human prothrombin antibody. The addition of vitamin K to the full-length prothrombin media ensured correct γ-carboxylation of the Gla domain. Purification of the recombinant proteins was carried out by affinity chromatography, ion exchange chromatography, and size exclusion chromatography, as described previously (15, 20, 23). Homogeneity and chemical identity of final preparations were verified by SDS-PAGE and by reverse phase-HPLC mass spectrometry analysis, giving a purity of >98%.
Luminescence Resonance Energy Transfer (LRET) Studies
Specific reduction of the engineered Cys residues of prothrombin mutant S101C/S210C was obtained by incubating the protein (55–80 μm) in 20 mm Tris, 500 mm NaCl, 5 mm EDTA, 0.3 mm tris(2-carboxyethyl)phosphine, 0.5 mm argatroban, pH 7.4 at 4 °C. Thiol quantification was performed using the Measure-iT thiol assay kit (Molecular Probes). When [-SH] = [prothrombin], DTPA-AMCA-maleimide2 (0.4 mm) was added, and the reaction was allowed to proceed for 15 min at room temperature. Alexa Fluor 647 maleimide dye (Molecular Probes) was then added in excess (1.2 mm), and the reaction was allowed to continue for additional 2 h. Iodoacetamide replaced Alexa Fluor 647 maleimide dye when preparing prothrombin S101C/S210C labeled with donor only. Excesses of unreacted fluorochromes, tris(2-carboxyethyl)phosphine, and argatroban were initially removed by seven cycles of MicroSpin G-25 column equilibrated with 20 mm Tris, pH 7.4, 150 mm NaCl and then extensive overnight dialysis at 4 °C. Dialyzed proteins were incubated for 15 min with slight excess of Eu3+ (100 μm, EuCl3) and finally purified by size exclusion chromatography. Incorporation of both probes was confirmed by UV–visible spectroscopy, with the spectrum showing the characteristic absorbance peaks for the protein (280 nm), donor (Eu3+)AMCA-DTPA (328 nm), and acceptor Alexa Fluor 647 (647 nm). All fluorescent derivatives were tested in the prothrombinase complex assay as reported previously (24) and returned kinetic parameters of activation practically identical to those of wild-type prothrombin. Luminescence lifetime measurements were performed with a custom-built two-channel spectrofluorometer with a pulsed nitrogen laser (NL100, Stanford Research Systems, Sunnyvale, CA) as an excitation source (25). Measurements were performed in a 300-μl cuvette in 20 mm Tris, 150 mm NaCl, 5 mm CaCl2, 0.1% PEG, pH 7.4 at 37 °C. The concentration of labeled proteins was 10–75 nm. Donor emission was recorded at 617 nm using a 620-nm, 10-nm bandwidth interference filter (Oriel, Stratford, CT), and acceptor emission was recorded at 670 nm using a 670-nm, 10-nm bandwidth interference filter (Oriel, Stratford, CT). Decays for donor-only samples were mono-exponential and were analyzed according to the expression
where α is the amplitude of the decay and τ is the luminescence lifetime. Decays of donors in the presence of acceptor and decays of sensitized acceptor emission were triple-exponential. Donor and sensitized acceptor decay curves were fitted simultaneously using global nonlinear regression with SCIENTIST (Micromath Scientific Software, Salt Lake City, UT). Such global fitting is made possible by the long (μs) lifetime of the (Eu3+)AMCA-DTPA donor and short (ns) lifetime of the Alexa Fluor 647 acceptor (26). Because the decay of sensitized acceptor in the μs time scale occurs within the lifetime of the donor engaged in energy transfer with the acceptor, the decays of the donor and sensitized acceptor obey expressions that depend on the same lifetimes but with different amplitudes. The energy transfer (E) was calculated from measurements of luminescence lifetime of the donor in the absence (τd) and presence of acceptor (τda) as
 |
The distance R between donor and acceptor was calculated according to the Förster equation
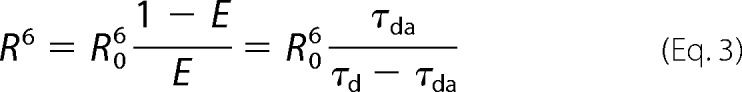 |
where R0 is the distance, where E = 0.5 (50%) and is equal to 55 Å for a completely randomized orientation of the donor-acceptor pair used (25, 27).
Proteolysis Studies
Cleavage of prothrombin mutants S525A (7.7 μm) and E311A/D318A/E323A/S525A (7.7 μm) by thrombin (1.3 μm) was studied by following consumption of the zymogen and the appearance of the bands of the activation products. Experiments were performed in 20 mm Tris, 145 mm NaCl, 2 mm Ca2+, pH 7.4 at 25 °C. Aliquots (16 μl) were quenched by the addition of loading buffer 4× (12 μl) at different time points and monitored by quantitative SDS-PAGE and densitometry. The identity of all products of activation was confirmed by N-terminal sequencing.
X-ray Studies
Crystallization of recombinant Gla-domainless prothrombin carrying an Ala replacement of the catalytic Ser-525 (Ser-195) to prevent autoactivation (21) was achieved at 22 °C by the vapor diffusion technique using an Art Robbins Instruments Phoenix liquid handling robot and mixing equal volumes (0.2 μl) of protein and reservoir solution. The protein solution contained 8 mg/ml Gla-domainless prothrombin S525A, 50 mm Na2PO4, 350 mm NaCl, pH 7.3, and the crystallization buffer was 100 mm Tris, pH 8.5, 25% PEG 8000. Optimization of crystal growth was obtained by the hanging drop vapor diffusion method mixing 3 μl of protein solution with equal volumes of reservoir solution (see Table 1). Crystals grew in 2 weeks in the P212121 space group and two molecules in the asymmetric unit. Diffraction quality crystals were cryoprotected in a solution similar to the mother liquor but containing 15% glycerol prior to flash-freezing. X-ray diffraction data were collected with a home source (Rigaku 1.2-kilowatt MMX007 generator with VHF optics) Rigaku Raxis IV++ detector and were indexed, integrated, and scaled with the HKL2000 software package (28). The structure was solved initially by molecular replacement using MOLREP from the CCP4 suite (29) and Protein Data Bank entry 3NXP (15) for prethrombin-1, which lacks fragment 1. The extra electron density from the 2Fo − Fc map was assigned to kringle-1 (residues 65–143) based on the amino acid composition of the recombinant construct. Kringle-1 was then refined using the high resolution Protein Data Bank entry 1NL1 (5) for bovine kringle-1 as a model and manually adjusted with COOT (30). Refinement and electron density generation were performed with REFMAC5 from the CCP4 suite, and 5% of the reflections were randomly selected as a test set for cross validation. Model building and analysis of the structures were conducted with COOT (30). The first molecule in the asymmetric unit contained no electron density for fragment 1 due to crystal packing. This was likely the result of alternative arrangements available to fragment 1 due to the extreme flexibility of the linker connecting kringle-1 and kringle-2. The description in the text refers to the second molecule in the asymmetric unit that was resolved in its entirety. Ramachandran plots were calculated using PROCHECK (31). Statistics for data collection and refinement are summarized in Table 1.
TABLE 1.
Crystallographic data for Gla-domainless prothrombin mutant S525A
NAG, n-acetylglucosamine.
| Buffer | 100 mm Tris, pH 8.5 |
| PEG | 8000 (25%) |
| PDB ID | 4HZH |
| Data collection | |
| Wavelength (Å) | 1.5418 |
| Space group | P212121 |
| Unit cell dimensions (Å) | a = 82.6, b = 103.1, c = 149.5 |
| Molecules/asymmetric unit | 2 |
| Resolution range (Å) | 40-3.3 |
| Observations | 74,483 |
| Unique observations | 19,155 |
| Completeness (%) | 96.9 (93.1) |
| Rsym (%) | 18.0 (44.9) |
| I/σ(I) | 5.4 (2.1) |
| Refinement | |
| Resolution (Å) | 40-3.3 |
| Rcryst, Rfree | 0.294, 0.329 |
| Reflections (working/test) | 18,011/980 |
| Protein atoms | 6880 |
| NAG molecules | 4 |
| r.m.s.d. bond lengthsa (Å) | 0.007 |
| r.m.s.d. anglesa (°) | 1.2 |
| r.m.s.d. ΔB (Å2) (mm/ms/ss)b | 0.74/0.06/0.28 |
| 〈B〉 protein (Å2) | 58.4 |
| 〈B〉 NAG (Å2) | 54.5 |
| Ramachandran plot | |
| Most favored(%) | 95.0 |
| Generously allowed (%) | 3.3 |
| Disallowed (%) | 1.6 |
a Root mean squared deviation (r.m.s.d.) from ideal bond lengths and angles and r.m.s.d. in B-factors of bonded atoms.
b mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain.
Molecular Dynamics Simulations
The model of prothrombin bound to prothrombinase (14) was used as the starting conformation in molecular dynamics simulations carried out using PMEMD in the AMBER12 package with the ff99SB force field. A truncated octahedron solvent box filled with TIP3P water molecules was constructed with tleap (32) and neutralized by adding the appropriate number of Na+ molecules. A 12 Å buffer was used between the protein and the edge of the solvent box. All simulations were carried out with an integration time step of 1 fs and applying periodic boundary conditions using a cutoff radius of 9 Å for the non-bonded interactions. The SHAKE algorithm (33) was used to constrain all hydrogen bond lengths. Neighbor lists were updated every 10 dynamics steps. Electrostatic interactions were calculated with the particle mesh Ewald method (34). The total number of atoms in the simulation, including water and Na+ molecules, was 299,279. Calculations were performed across 128 CPUs of the Gemini high performance cluster in the Edward A. Doisy Department of Biochemistry and Molecular Biology and took ∼330 h continuous CPU time to obtain the 24-ns trajectory.
Electrostatic Calculations
Electrostatic calculations were performed using APBS (35) for the experimental structure. Calculations were performed using a solvent dielectric of 78.14 and a protein dielectric of 2.0 at 298 K in 150 mm NaCl. Electrostatic maps were also calculated for Gla-domainless prothrombin with missing loops and side chains modeled in. The results were qualitatively similar to those obtained with the experimental structure. Final electrostatic maps were constructed by subtracting the protein self-energies from the calculated map using the dxmath utility in APBS.
RESULTS
The structure of recombinant Gla-domainless prothrombin with the active site Ser-525 (Ser-195) replaced by Ala was solved at 3.3 Å resolution with a final Rfree = 0.329 and two molecules in the asymmetric unit (Table 1). The construct (residues 45–579) can be traced in the electron density map almost in its entirety (Fig. 1), except for two disordered regions connecting the two kringles (residues 144–168) and kringle-2 to the A chain (residues 255–273). No interaction between fragments 1 and 2 is observed in the electron density map, and the only contact visible between the two domains involves the side chain of Gln-213 and the backbone of Ser-101. The overall structure spans >80 Å in length, with fragment 2 and the protease domain positioned coaxial to one another and kringle-1 sitting with its main axis at an angle of 36° relative to the rest of the molecule (Fig. 1).
The entire architecture of fragment 2 and the protease domain is very similar (r.m.s.d. = 0.7 Å) to the structure of prethrombin-1 (15). After molecular replacement of the Gla-domainless prothrombin structure using the coordinates 3NXP of prethrombin-1, extra electron density was clearly detected to assign fragment 1 with confidence from Gly-68 to Cys-143, covering kringle-1 (Cys-65 to Cys-143) almost entirely. Kringle-1 assumes its expected fold (36) and overlaps well (r.m.s.d. = 0.7 Å) with previously published structures of bovine fragment 1 (5, 6) with the distinguishing Pro-94 in the cis configuration (Fig. 2) that is unique to the entire kringle. Because of the absence of Cys-65 in the electron density map, Cys-143 is unpaired, and only two disulfide bonds (Cys-86–Cys–125, Cys-114–Cys-138) are detected. Fragment 2 is traced in the electron density map from Gln-169 to Glu-254, missing a total of 13 residues on the N terminus and 17 residues on the C terminus connecting to the A chain, including the site of cleavage at Arg-271, as reported previously for prethrombin-1 (15) and meizothrombin devoid of fragment 1 (20). Fragment 2 contains the second kringle (Cys-170 to Cys-248) with an architecture similar (r.m.s.d. = 0.8 Å) to that reported in prethrombin-1 (15) and with all three disulfide bonds (Cys-170–Cys-248, Cys-191–Cys-231, Cys-219–Cys-243) visible in the structure. The two kringles align with r.m.s.d. = 1.2 Å and differ only in a helical segment in kringle-2 replaced by an unstructured coil (residues 92–106) in kringle-1, and residues 130–134 of kringle-1 shifted relative to the homologous region in kringle-2 by ∼3 Å (Fig. 2). The linker connecting the two kringles is 26 residues long, from Gly-144 to Gln-169, and completely disordered. A high degree of disorder also extends to kringle-1, which could not be traced in the first molecule of the asymmetric unit presumably due to a different arrangement prevented by crystal packing.
FIGURE 2.

a, representative extra electron density detected for fragment 1 after molecular replacement and before refinement. The extra electron density shown as 2Fo − Fc map contoured at 1 σ enables assignment of residues of fragment 1 of Gla-domainless prothrombin with confidence. Shown as sticks are residues Ser-91–His-104 of kringle-1 with the distinguishing Pro-94 that is the single Pro in the cis conformation in the entire kringle, as first identified in the high resolution structure of bovine kringle-1 (6). These residues refer to the structure after refinement, deposited in the Protein Data Bank as entry 4HZH, and show the quality of the model built on the extra electron density detected before refinement after initial molecular replacement. b, overlay of kringle-1 (red) and kringle-2 (green) of Gla-domainless prothrombin reveals the basic similarity between the two domains. Because of the absence of Cys-65 in the electron density map, Cys-143 is unpaired, and only two disulfide bonds (1 = Cys-86–Cys-125, 2 = Cys-114–Cys-138) are detected in kringle-1. All three disulfide bonds (3 = Cys-170–Cys-248, 4 = Cys-191–Cys-231, 5 = Cys-219–Cys-243) are detected for kringle-2. The two kringles align with an r.m.s.d. = 1.2 Å, and the most notable differences (noted by arrows) are a helical segment in kringle-2 replaced by an unstructured coil (residues 92–106) in kringle-1 (see also panel a) and residues 130–134 of kringle-1 shifted relative to the homologous region in kringle-2 by ∼3 Å.
The protease domain has a well defined architecture, practically identical to that observed in prethrombin-1 (15) and prethrombin-2 (16). The A chain is traced in the electron density map in almost its entirety, from Thr-274 to the site of cleavage at Arg-320 (Arg-15), missing only Thr-272 and Ala-273. Thr-274 is three residues downstream of the Arg-271 cleavage site, and the Cα-Cα distance between Thr-274 and Arg-320 (Arg-15) is 39 Å, implying that factor Xa must dock differently on the prothrombin surface to access Arg-271 and Arg-320 (Arg-15). There are no direct interactions between fragment 2 and the A chain, and 20 residues are missing from the highly acidic linker connecting these domains. The B chain is visible in its entirety, from Ile-321 (Ile-16) to Glu-579 (Glu-247), including the autolysis loop from Glu-466 (Glu-146) to Lys-474 (Lys-149e) that is typically disordered in structures of thrombin and its precursors.
The conformation of Gla-domainless prothrombin in the free state with the domains not vertically stacked spans a total length of about 80 Å and was not anticipated by previous studies. A long axis of 113 Å was inferred from measurements of the distance of fluorescein probes anchored to the plane of a membrane and the C-terminal of the zymogen on the assumption that all domains are vertically stacked (37). A recent model of prothrombin bound to the prothrombinase complex predicts a long axis of about 110 Å, again with all domains coaxial to one another (14). The crystal structure of Gla-domainless prothrombin offers an essential starting point to probe the conformation of the zymogen in solution. To this end, we carried out LRET measurements of a full-length prothrombin mutant carrying the double substitution S101C/S210C and conjugated donor and acceptor to Cys-101 and Cys-210. The Cα-Cα distance between Ser-101 in kringle-1 and Ser-210 in kringle-2 is 12 Å in the structure of Gla-domainless prothrombin, but considerably longer (60 Å) in the model of prothrombin bound to prothrombinase (14) where the domains are fully extended and vertically stacked. LRET is particularly suited to measure distances in water up to 60 Å, at which energy is transferred between a luminescent Eu3+ chelate used as donor and the organic dye CY-5 used as acceptor (25–27, 38). Lifetime data for the LRET donor-acceptor pair are reported in Fig. 3. The donor quenching and acceptor sensitization both fit to the same triple-exponential decay comprising a short lifetime of 33.8 ± 0.5 μs with almost complete (94%) energy transfer and a population of 40% and a long lifetime of 272 ± 4 μs with 55% energy transfer and a population of 60%. The third, slowest lifetime (606 ± 9 μs) is identical to the decay of the donor-only control and free Eu3+ chelate. The donor-acceptor distance associated with the slow 272 ± 4 μs decay can be calculated accurately from the Förster equation (Equation 3) as 54 ± 2 Å and is comparable with the 60 Å distance of the fully extended model of prothrombin bound to the prothrombinase complex (14). The donor-acceptor distance associated with the fast 33.8 ± 0.5 μs decay can only be assigned as an upper bound of 34 Å due to the extremely large (94%) energy transfer that produces a weak dependence on distance in the Förster sigmoidal equation (26, 38). Within experimental error, the distance of 12 Å detected in the structure of Gla-domainless prothrombin is entirely consistent with the energy transfer of 94% associated with the fast 33.8 ± 0.5 μs decay. Analysis of the amplitudes of the decay reveals that about 60% of prothrombin molecules in solution have the two kringles separated by a distance compatible with a fully extended conformation, and 40% of the molecules assume a more compact conformation where the two kringles are at most 34 Å apart, or at least 20 Å closer than in the fully extended conformation. The significant variation in the distance between residues 101 in kringle-1 and residue 210 in kringle-2 detected by LRET measurements is caused by the intrinsic flexibility of the connecting linker between the two kringles that appears as disordered in the crystal structure. Because of the constraints from crystal packing, the x-ray crystal structure only reveals a snapshot of the ensemble of distinct arrangements accessible to kringle-1 that likely moves like a dumbbell relative to kringle-2 and the rest of the prothrombin molecule. Additional support to these conclusions comes from molecular dynamics simulations of free prothrombin. Starting from the fully extended conformation in the model bound to the prothrombinase complex (14), prothrombin relaxes into a partially collapsed conformation where the Cα-Cα distance between Ser-101 in kringle-1 and Ser-210 in kringle-2 decreases from 60 Å to an average of 36 Å within 7 ns (Fig. 3).
FIGURE 3.

a and b, semilog plot of lifetime data for the LRET donor-acceptor pair conjugated to residues 101 in kringle-1 and 210 in kringle-2 of the full-length prothrombin mutant S101C/S210C. The donor quenching (a, green circles) and acceptor sensitization (b, green circles) both obey the same triple-exponential decay comprising a short lifetime of 33.8 ± 0.5 μs with almost complete (94%) energy transfer and a population of 40% and a long lifetime of 272 ± 4 μs with 55% energy transfer and a population of 60%. The third, slowest lifetime (606 ± 9 μs) is identical to the decay of the donor-only control and free Eu3+ chelate (gray circles). The donor-acceptor distances associated with the slow (272 ± 4 μs) and fast (33.8 ± 0.5 μs) decays derived from the Förster equation (see “Experimental Procedures”) are 54 ± 2 and ≤34 Å, respectively. c, molecular dynamics simulations of the prothrombin conformation shown as the Cα-Cα distance between Ser-101 in kringle-1 and Ser-210 in kringle-2 as a function of time, over a 24-ns trajectory. Horizontal red lines depict the values of 54 and 34 Å determined by LRET measurements using probes attached to residues 101 and 210 mutated to Cys. d, overlay of the starting (with domains colored as in Fig. 1) and ending (cyan) prothrombin structures from the molecular dynamics simulation (see panel c). The starting structure is the model of prothrombin bound to prothrombinase (14), and the ending structure is the average of the last 5 ns of the simulation. Residues Ser-101 and Ser-210 are rendered as sticks with Cα-Cα distances indicated in Å.
The structure of prothrombin tests the validity of epitopes assigned from previous studies involving peptide fragments and site-directed mutagenesis (Fig. 4). The structure also enables an analysis of the electrostatic properties of the protein revealed only partially by the smaller versions of prothrombin, i.e. prethrombin-2 and prethrombin-1. Kringle-1 is electropositive in almost its entirety, except for a region with electronegative potential positioned on the back of the fragment relative to the active site of prothrombin (Fig. 4). Residues 205–220 on fragment 2 (10) and the fragment Tyr-557–Gln-571 (Tyr-225–Gln-239) toward the C terminus of the B chain (11) are involved in direct factor Xa binding in the prothrombinase complex (10). The portion of this epitope residing on fragment 2 is fully exposed to solvent and negatively charged, but most of the portion on the B chain is buried under fragment 2 and is unlikely to contribute to factor Xa recognition (Fig. 4). This region of the molecule deserves attention. In thrombin and prethrombin-2, the C-terminal region of the B chain is embedded in the positively charged exosite II (16, 39). The presence of fragment 2 in prothrombin covers most of this region with a strong electronegative potential generated by the sequence 249EEAVEE254 that is part of the linker between kringle-2 and the A chain containing a total of 11 acidic residues (Fig. 4). Cleavage at Arg-271 generates prethrombin-2 and removes this linker region, thereby causing a sharp reversal, from negative to positive, of the electrostatic potential of the putative epitope for factor Xa binding on the B chain (Fig. 4). On the other hand, cleavage at Arg-320 (Arg-15) to generate meizothrombin would leave fragment 2 in place and the electrostatic potential of the epitope for factor Xa unchanged (Fig. 4). Electrostatics may play a key role in determining the order of cleavage at Arg-271 or Arg-320 (Arg-15) chosen by prothrombinase in the activation of prothrombin to thrombin. Other studies have concluded that the fragment Gly-473–Val-488 (Gly-149d–Val-163) in the autolysis loop is involved in the binding of cofactor Va in the prothrombinase complex (8). However, most of this segment is buried inside the protein core, and the boundary of the epitope is too close (<15 Å away) to the site of cleavage at Arg-320 (Arg-15) where factor Xa docks (Fig. 4). A strong positive electrostatic potential is generated by charged residues in exosite I (Fig. 4) that have been implicated in the binding of cofactor Va (9). The putative epitope is thought to assume distinct conformations in prothrombin relative to the mature enzyme because fluorescein-labeled fragments of hirudin targeting exosite I bind to thrombin with an affinity >100-fold higher than prothrombin (40, 41). However, earlier NMR studies on the binding of fibrinogen fragments document smaller differences between prothrombin and thrombin (42), in keeping with the similarity of structural architecture and electrostatic properties of exosite I in the two proteins.
FIGURE 4.

a and b, x-ray crystal structure of Gla-domainless prothrombin in surface/ribbon representation oriented with active site region (white circle) in the front (a) or rotated 180° (b) showing the bent conformation of the zymogen. Epitopes for factor Xa (yellow) and cofactor Va (red) identified by functional studies are mapped on the surface. The site of cleavage at Arg-271 is in a disordered region, but Arg-320 (Arg-15) is indicated. c and d, electrostatic potential surface of Gla-domainless prothrombin revealing the properties of the epitopes for factor Xa (yellow oval) and cofactor Va (green oval). The surface appears to be predominantly negative, especially in the region connecting kringle-2 to the protease domain housing the epitope for factor Xa binding. The epitope for cofactor Va recognition involves exosite I in the protease domain and is positively charged. The epitope for factor Xa on the C-terminal of the B chain is covered by the acidic residues connecting kringle-2 to the A chain and the junction between the two kringles. The electrostatic properties of this epitope change drastically upon cleavage at Arg-271 that generates prethrombin-2. e and f, cleavage at Arg-271 generates prethrombin-2 by removing the acidic linker between kringle-2 and the A chain and exposes the positively charged surface of exosite II on the protease domain. On the other hand, cleavage at Arg-271 has no effect on the epitope for cofactor Va recognition.
Important features in the protease domain are relevant to other zymogens of the trypsin-like family. Absence of a new N terminus at residue Ile-321 (Ile-16) impedes formation of the H-bond with Asp-524 (Asp-194) in the active site, causing a flip in the peptide bond between residues Glu-522 (Glu-192) and Gly-523 (Gly-193) and disruption of the oxyanion hole defined by the tight β-turn between the backbone N atoms of the catalytic Ser-525 (Ser-195) and Gly-523 (Gly-193) (Fig. 5). As these changes compromise stabilization of the transition state, a flip of the side chain of Asp-519 (Asp-189) by almost 7 Å from the position occupied in thrombin results in a primary specificity pocket unsuitable for high affinity binding of an Arg side chain at the P1 position of substrate (Fig. 5). In addition, access to the primary specificity pocket is hindered by collapse of the 547–549 (215–217) segment, a signature of the inactive E* form of the protease and zymogen (43, 44) that brings Trp-547 (Trp-215) in hydrophobic interaction with Trp-370 (Trp-60d) in the 60-loop and Trp-468 (Trp-148) in the autolysis loop (Fig. 5). The collapse of Trp-547 (Trp-215) obliterates 26% of accessibility to the active site and is similar to that reported in prethrombin-1 (15) and prethrombin-2 (16).
FIGURE 5.

a, stereo image of the active site of Gla-domainless prothrombin. The absence of a new N terminus at residue Ile-321 (Ile-16) causes a shift in the position of Asp-524 (Asp-194) resulting in disruption of the oxyanion hole (arrow) defined by the tight β-turn between the backbone N atoms of the catalytic Ser-525 (Ser-195) and Gly-523 (Gly-193). The side chain of Asp-519 (Asp-189) in the primary specificity pocket flips almost 7 Å from the position occupied in thrombin. The 547–549 (215–217) segment collapses in the active site, a signature of the inactive E* form. Trp-468 (Trp-148) in the autolysis loop was removed for clarity. b, stereo image of the activation domain of Gla-domainless prothrombin. Arg-320 (Arg-15) is clearly visible in the electron density map with an intact Arg-320–Ile-321 (Arg-15–Ile-16) peptide bond. Ionic interaction with Glu-323 (Glu-18) reduces solvent exposure for proteolytic attack of Arg-320 (Arg-15) by 32%. Electron density 2Fo − Fc maps are contoured at 1 σ.
The structure of Gla-domainless prothrombin addresses a central issue in the molecular mechanism of prothrombin activation, i.e. the solvent exposure of the two sites of cleavage at Arg-271 and Arg-320 (Arg-15). The former, responsible for the conversion of prothrombin to prethrombin-2, is positioned in a disordered region of fragment 2 readily exposed to solvent for proteolytic attack. The activation domain around the site of cleavage at Arg-320 (Arg-15), responsible for the conversion of prothrombin to meizothrombin, has a well defined architecture and shows an intact Arg-320–Ile-321 (Arg-15–Ile-16) peptide bond with Arg-320 (Arg-15) in ionic interaction with the carboxylate of Glu-323 (Glu-18) (Fig. 5). The activation domain of Gla-domainless prothrombin is more similar to that of prethrombin-1 (15), where Arg-15 is loosely engaged by Glu-18, than that of prethrombin-2 (16), where Arg-15 is caged by interactions with Glu-14e, Asp-14l, and Glu-18. We have recently shown that Ala replacement of Glu-14e, Asp-14l, and Glu-18 produces autoactivation of pretrombin-2 (16) by forcing Arg-15 out of its anionic Glu-14e/Asp-14l/Glu-18 cage and into the solvent (21). Importantly, the same E14eA/D14lA/E18A substitution causes prethrombin-1 and prothrombin to autoactivate (21), suggesting that Arg-320 (Arg-15) may not be accessible to solvent in any of the inactive thrombin precursors. In accord with previous studies (45), the S525A (S195A) mutant of Gla-domainless prothrombin is cleaved by thrombin at Arg-155 and Arg-284 with similar specificity but shows no cleavage at Arg-320 (Arg-15) (Fig. 6). When the anionic cage Glu-311/Asp-318/Glu-323 (Glu-14e/Asp-14l/Glu-18) is neutralized by Ala substitutions and the additional Ala mutation of the active site residue Ser-525 (Ser-195) is introduced to prevent autoactivation (21), the resulting quadruple mutant E311A/D318A/E323A/S525A is cleaved by thrombin at Arg-320 (Arg-15) (Fig. 6).
FIGURE 6.

a and b, cleavage of Gla-domainless prothrombin (GD-ProT) mutants S525A and E311A/D318A/E323A/S525A (EDES) by thrombin, under experimental conditions of 20 mm Tris, 200 mm NaCl, 2 mm CaCl2, pH 7.4 at 25 °C. The chemical identity of each band in the gels, labeled with the same color as the kinetic traces (c and d), was assigned by N-terminal sequencing. c and d, both mutants are cleaved with similar kinetic rates at Arg-155 to generate prethrombin-1 (Pre1) and Arg-284 to generate prethrombin-2 (Pre2), proving that these sites have similar solvent exposure. On the other hand, Arg-320 (Arg-15) is cleaved only in the mutant E311A/D318A/E323A/S525A (d), suggesting that this Arg residue is trapped by residues of the Glu-311/Asp-318/Glu-323 (Glu-14e/Asp-14l/Glu-18) anionic cage as seen in prethrombin-2 (16). Thr, thrombin.
DISCUSSION
Although prothrombin shares the Gla domain with clotting factors VII, IX, and X and protein C, it is the only vitamin K-dependent factor carrying kringles and not EGF modules as auxiliary domains. The findings reported in this study reveal some functional consequences of the presence of kringles in prothrombin that have eluded previous investigators due to the lack of structural information. The structure of Gla-domainless prothrombin features a slightly bent conformation about 80 Å long with disorder in the linker regions connecting the two kringles and kringle-2 to the A chain in the protease domain. Kringle-2 of prothrombin is anchored by interactions with the B chain in the protease domain, but kringle-1 appears free to move like a dumbbell relative to the rest of the zymogen. Disorder in kringle domains and connecting linkers has been documented in plasminogen, where four linkers connect the five kringles that fold together in a “resting” conformation that shields the activation domain from unwanted proteolysis. Two of these linkers are very short and clearly visible in the structure, but the linkers connecting kringles 3 and 4 and kringles 4 and 5 are more than 20 residues long and cannot be traced in the electron density map (46). Kringle-3 is completely missing in a low resolution structure of type I plasminogen (46), but also in the high resolution structure of angiostatin corresponding to the first three kringles of plasminogen (47). The bent conformation of Gla-domainless prothrombin with the domains not vertically stacked is a snapshot of the highly mobile kringle-1 frozen by crystal packing while sampling the ensemble of alternative arrangements relative to kringle-2 and the rest of the prothrombin molecule allowed by the flexible linker. Conformational flexibility of prothrombin in solution is supported by direct LRET measurements of the distance between residues 101 in kringle-1 and 210 in kringle-2. Prothrombin exists in two conformations where these residues are separated by either 54 Å (60% of the population) or up to 34 Å (40% of the population). The former distance is consistent with the model of fully extended prothrombin bound to the prothrombinase complex (14). The shorter distance, also detected by molecular dynamics simulations, is associated with a partially collapsed conformation where the two kringles come closer to each other (Fig. 3). The conformation of Gla-domainless prothrombin documented by our x-ray crystal structure likely captures this fraction of prothrombin in solution that becomes stabilized by crystal packing.
The different solvent exposure of the two sites of cleavage at Arg-271 and Arg-320 (Arg-15) and the peculiar electrostatic properties of prothrombin revealed by the x-ray crystal structure presented here suggest a plausible mechanism of activation. Cleavage at Arg-271 occurs first, causing the release of fragments 1 and 2 and the reversal of the electrostatic potential of the epitope recognizing factor Xa. This acts as a signal for prothrombinase to move on rapidly to the second site of cleavage at Arg-320 (Arg-15) and explains why activation of prothrombin under physiological conditions on the surface of platelets proceeds via the prethrombin-2 intermediate (12) and in a concerted manner (13). The alternative scenario where prothrombinase cleaves first at Arg-320 (Arg-15) to generate meizothrombin must envision a conformational change in the activation domain that exposes Arg-320 (Arg-15) for proteolytic attack and makes it preferable to cleavage at the constitutively exposed Arg-271. Such conformational change may be driven by exosite-dependent interactions between prothrombin and prothrombinase (48), but its validity should await confirmation from future functional and structural studies.
Acknowledgment
We are grateful to Tracey Baird for help with the illustrations.
This work was supported, in whole or in part, by National Institutes of Health Grants HL49413, HL73813, and HL112303 (to E. D. C.).
The atomic coordinates and structure factors (code 4HZH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DTPA
- diethylenetriaminepentaacetic acid
- AMCA
- 7-amino-4-methylcoumarin-3-acetic acid
- LRET
- luminescence resonance energy transfer
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Schmidt A. (1872) Neue Untersuchungen ueber die Fasserstoffesgerinnung. Pflüger Archiv. 6, 413–538 [Google Scholar]
- 2. Butenas S., van't Veer C., Mann K. G. (1999) “Normal” thrombin generation. Blood 94, 2169–2178 [PubMed] [Google Scholar]
- 3. Rosing J., Tans G., Govers-Riemslag J. W., Zwaal R. F., Hemker H. C. (1980) The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 255, 274–283 [PubMed] [Google Scholar]
- 4. Kotkow K. J., Furie B., Furie B. C. (1993) The interaction of prothrombin with phospholipid membranes is independent of either kringle domain. J. Biol. Chem. 268, 15633–15639 [PubMed] [Google Scholar]
- 5. Huang M., Rigby A. C., Morelli X., Grant M. A., Huang G., Furie B., Seaton B., Furie B. C. (2003) Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 10, 751–756 [DOI] [PubMed] [Google Scholar]
- 6. Soriano-Garcia M., Padmanabhan K., de Vos A. M., Tulinsky A. (1992) The Ca2+ ion and membrane binding structure of the Gla domain of Ca-prothrombin fragment 1. Biochemistry 31, 2554–2566 [DOI] [PubMed] [Google Scholar]
- 7. Deguchi H., Takeya H., Gabazza E. C., Nishioka J., Suzuki K. (1997) Prothrombin kringle 1 domain interacts with factor Va during the assembly of prothrombinase complex. Biochem. J. 321, 729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yegneswaran S., Mesters R. M., Fernández J. A., Griffin J. H. (2004) Prothrombin residues 473–487 contribute to factor Va binding in the prothrombinase complex. J. Biol. Chem. 279, 49019–49025 [DOI] [PubMed] [Google Scholar]
- 9. Chen L., Yang L., Rezaie A. R. (2003) Proexosite-1 on prothrombin is a factor Va-dependent recognition site for the prothrombinase complex. J. Biol. Chem. 278, 27564–27569 [DOI] [PubMed] [Google Scholar]
- 10. Taneda H., Andoh K., Nishioka J., Takeya H., Suzuki K. (1994) Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. J. Biochem. 116, 589–597 [DOI] [PubMed] [Google Scholar]
- 11. Yegneswaran S., Mesters R. M., Griffin J. H. (2003) Identification of distinct sequences in human blood coagulation factor Xa and prothrombin essential for substrate and cofactor recognition in the prothrombinase complex. J. Biol. Chem. 278, 33312–33318 [DOI] [PubMed] [Google Scholar]
- 12. Wood J. P., Silveira J. R., Maille N. M., Haynes L. M., Tracy P. B. (2011) Prothrombin activation on the activated platelet surface optimizes expression of procoagulant activity. Blood 117, 1710–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haynes L. M., Bouchard B. A., Tracy P. B., Mann K. G. (2012) Prothrombin activation by platelet-associated prothrombinase proceeds through the prethrombin-2 pathway via a concerted mechanism. J. Biol. Chem. 287, 38647–38655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee C. J., Wu S., Pedersen L. G. (2011) A proposed ternary complex model of prothrombinase with prothrombin: protein-protein docking and molecular dynamics simulations. J. Thromb. Haemost. 9, 2123–2126 [DOI] [PubMed] [Google Scholar]
- 15. Chen Z., Pelc L. A., Di Cera E. (2010) Crystal structure of prethrombin-1. Proc. Natl. Acad. Sci. U.S.A. 107, 19278–19283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pozzi N., Chen Z., Zapata F., Pelc L. A., Barranco-Medina S., Di Cera E. (2011) Crystal structures of prethrombin-2 reveal alternative conformations under identical solution conditions and the mechanism of zymogen activation. Biochemistry 50, 10195–10202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vijayalakshmi J., Padmanabhan K. P., Mann K. G., Tulinsky A. (1994) The isomorphous structures of prethrombin2, hirugen-, and PPACK-thrombin: changes accompanying activation and exosite binding to thrombin. Protein Sci. 3, 2254–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedrich R., Panizzi P., Fuentes-Prior P., Richter K., Verhamme I., Anderson P. J., Kawabata S., Huber R., Bode W., Bock P. E. (2003) Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature 425, 535–539 [DOI] [PubMed] [Google Scholar]
- 19. Martin P. D., Malkowski M. G., Box J., Esmon C. T., Edwards B. F. (1997) New insights into the regulation of the blood clotting cascade derived from the x-ray crystal structure of bovine meizothrombin des F1 in complex with PPACK. Structure 5, 1681–1693 [DOI] [PubMed] [Google Scholar]
- 20. Papaconstantinou M. E., Gandhi P. S., Chen Z., Bah A., Di Cera E. (2008) Na+ binding to meizothrombin desF1. Cell. Mol. Life Sci. 65, 3688–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pozzi N., Chen Z., Zapata F., Niu W., Barranco-Medina S., Pelc L. A., Di Cera E. (2013) Autoactivation of thrombin precursors. J. Biol. Chem. 288, 11601–11610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1–8 Ä resolution. II. Crystallographic refinement, refined crystal structure, and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 23. Guinto E. R., Vindigni A., Ayala Y. M., Dang Q. D., Di Cera E. (1995) Identification of residues linked to the slow–>fast transition of thrombin. Proc. Natl. Acad. Sci. U.S.A. 92, 11185–11189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manithody C., Yang L., Rezaie A. R. (2002) Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry 41, 6780–6788 [DOI] [PubMed] [Google Scholar]
- 25. Heyduk E., Heyduk T. (1997) Thiol-reactive, luminescent Europium chelates: luminescence probes for resonance energy transfer distance measurements in biomolecules. Anal. Biochem. 248, 216–227 [DOI] [PubMed] [Google Scholar]
- 26. Selvin P. R., Rana T. M., Hearst J. E. (1994) Luminescence resonance energy transfer. J. Am. Chem. Soc. 116, 6029–6030 [Google Scholar]
- 27. Selvin P. R., Hearst J. E. (1994) Luminescence energy transfer using a terbium chelate: improvements on fluorescence energy transfer. Proc. Natl. Acad. Sci. U.S.A. 91, 10024–10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected by oscillation methods. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Morris A. L., MacArthur M. W., Hutchinson E. G., Thornton J. M. (1992) Stereochemical quality of protein structure coordinates. Proteins 12, 345–364 [DOI] [PubMed] [Google Scholar]
- 32. Case D. A., Darden T. A., Cheatham T. E., Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2012) AMBER 12, University of California, San Francisco [Google Scholar]
- 33. Ryckaert J. P., Ciccotti G., Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 34. Darden T., York D., Pedersen L. G. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 35. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ozhogina O. A., Bominaar E. L. (2009) Characterization of the kringle fold and identification of a ubiquitous new class of disulfide rotamers. J. Struct. Biol. 168, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Q., Lentz B. R. (1997) Fluorescence resonance energy transfer study of shape changes in membrane-bound bovine prothrombin and meizothrombin. Biochemistry 36, 4701–4711 [DOI] [PubMed] [Google Scholar]
- 38. Heyduk T. (2002) Measuring protein conformational changes by FRET/LRET. Curr. Opin. Biotechnol. 13, 292–296 [DOI] [PubMed] [Google Scholar]
- 39. Bode W., Turk D., Karshikov A. (1992) The refined 1.9-Ä x-ray crystal structure of d-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1, 426–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anderson P. J., Nesset A., Dharmawardana K. R., Bock P. E. (2000) Role of proexosite I in factor Va-dependent substrate interactions of prothrombin activation. J. Biol. Chem. 275, 16435–16442 [DOI] [PubMed] [Google Scholar]
- 41. Anderson P. J., Nesset A., Dharmawardana K. R., Bock P. E. (2000) Characterization of proexosite I on prothrombin. J. Biol. Chem. 275, 16428–16434 [DOI] [PubMed] [Google Scholar]
- 42. Ni F., Ning Q., Jackson C. M., Fenton J. W., 2nd. (1993) Thrombin exosite for fibrinogen recognition is partially accessible in prothrombin. J. Biol. Chem. 268, 16899–16902 [PubMed] [Google Scholar]
- 43. Gohara D. W., Di Cera E. (2011) Allostery in trypsin-like proteases suggests new therapeutic strategies. Trends Biotechnol. 29, 577–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pozzi N., Vogt A. D., Gohara D. W., Di Cera E. (2012) Conformational selection in trypsin-like proteases. Curr. Opin. Struct. Biol. 22, 421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petrovan R. J., Govers-Riemslag J. W., Nowak G., Hemker H. C., Tans G., Rosing J. (1998) Autocatalytic peptide bond cleavages in prothrombin and meizothrombin. Biochemistry 37, 1185–1191 [DOI] [PubMed] [Google Scholar]
- 46. Law R. H., Caradoc-Davies T., Cowieson N., Horvath A. J., Quek A. J., Encarnacao J. A., Steer D., Cowan A., Zhang Q., Lu B. G., Pike R. N., Smith A. I., Coughlin P. B., Whisstock J. C. (2012) The x-ray crystal structure of full-length human plasminogen. Cell Rep. 1, 185–190 [DOI] [PubMed] [Google Scholar]
- 47. Cnudde S. E., Prorok M., Castellino F. J., Geiger J. H. (2006) X-ray crystallographic structure of the angiogenesis inhibitor, angiostatin, bound to a peptide from the group A streptococcal surface protein PAM. Biochemistry 45, 11052–11060 [DOI] [PubMed] [Google Scholar]
- 48. Bock P. E., Panizzi P., Verhamme I. M. (2007) Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 5, Suppl. 1, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]