

Background: DJ-1 is associated with recessive early onset Parkinson disease.
Results: DJ-1 acts as a metal-binding protein, binding copper and mercury, and DJ-1, but not mutated clinical variants, protects cells against metal-induced cytotoxicity.
Conclusion: Small genetic alterations in a central Parkinson disease protein can sensitize cells to metal-induced cell death.
Significance: Genetic alterations in DJ-1 can sensitize cells to metal-induced cell death in Parkinson disease.
Keywords: Cell Death, Dj-1, Metals, Oxidative Stress, Parkinson Disease, Cytotoxicity
Abstract
The progressive loss of motor control due to reduction of dopamine-producing neurons in the substantia nigra pars compacta and decreased striatal dopamine levels are the classically described features of Parkinson disease (PD). Neuronal damage also progresses to other regions of the brain, and additional non-motor dysfunctions are common. Accumulation of environmental toxins, such as pesticides and metals, are suggested risk factors for the development of typical late onset PD, although genetic factors seem to be substantial in early onset cases. Mutations of DJ-1 are known to cause a form of recessive early onset Parkinson disease, highlighting an important functional role for DJ-1 in early disease prevention. This study identifies human DJ-1 as a metal-binding protein able to evidently bind copper as well as toxic mercury ions in vitro. The study further characterizes the cytoprotective function of DJ-1 and PD-mutated variants of DJ-1 with respect to induced metal cytotoxicity. The results show that expression of DJ-1 enhances the cells' protective mechanisms against induced metal toxicity and that this protection is lost for DJ-1 PD mutations A104T and D149A. The study also shows that oxidation site-mutated DJ-1 C106A retains its ability to protect cells. We also show that concomitant addition of dopamine exposure sensitizes cells to metal-induced cytotoxicity. We also confirm that redox-active dopamine adducts enhance metal-catalyzed oxidation of intracellular proteins in vivo by use of live cell imaging of redox-sensitive S3roGFP. The study indicates that even a small genetic alteration can sensitize cells to metal-induced cell death, a finding that may revive the interest in exogenous factors in the etiology of PD.
Introduction
Parkinson disease (PD)3 is the second most common neurodegenerative disease, classically described as a progressive movement disorder, and with increased prevalence in the aging population. Selective degeneration of dopamine-producing neurons in the substantia nigra pars compacta (SNpc) and depletion of striatal dopamine levels are the cardinal features of PD (1). It has been estimated that 50–60% of the SNpc dopaminergic neurons are irreversibly lost, and about 80–85% of the dopamine content of the striatum is depleted by the time of clinical diagnosis (2). However, there are also widespread alterations in other brain regions observed in a predominant caudal to rostral progression that may influence the development of non-motor symptoms, such as pain, sleep disorders, cognitive decline, or depression (3, 4). Because the progression of the disease is slow, but irreversible, the underlying factors resulting in cell death need to be clarified to enable new treatment strategies for the affected patients.
PD etiology remains obscure and appears in most instances sporadic in nature. Environmental risk factors, including prolonged pesticide exposure and lead or manganese intoxication, have been observed to cause parkinsonian symptoms (5–8). Altered zinc, copper, and iron levels have been found specifically in the substantia nigra of PD patients compared with controls (9). Increased levels of mercury measured in blood or urine have also been associated with an increased risk of PD (10). However, several studies of occupational exposure to metals have been inconclusive in relation to PD development, as reviewed previously (11). A combination of genetic components that could sensitize cells to environmental risk factors and induced cell death at an older age has been discussed, but evidence is still scarce.
Several genetic lesions, directly associated with early onset PD, have given clues about molecular mechanisms affected early in the disease progression (12–17). Mutations of the PARK7 gene, which encodes the protein DJ-1, have been linked to forms of early onset recessive autosomal PD (12, 18, 19). Even though PARK7 mutation carriers are considered to be rare, their presence highlights the molecular function of DJ-1 as a crucial component for disease prevention.
DJ-1 is a predominantly cytosolic, homodimeric protein, ubiquitously expressed in both brain and peripheral tissues (20, 21). DJ-1 has been reported to be a multifunctional protein with putative roles in, for example, Ras-dependent cell transformation (22), neuroprotection (23, 24), fertility (25), control of protein-RNA interactions (26), and modulation of androgen receptor signaling (27, 28), or it acts as a protein chaperone (29) and protease (29–31). DJ-1 has also been shown to interact with other known PD-linked proteins, PINK1 and Parkin (32, 33). A clear consensus on what DJ-1 actually does within the cells has not been established. DJ-1-deficient mice display dopamine re-uptake dysfunction but lack any overt sign of neurodegeneration, suggesting that loss of DJ-1 alone is insufficient to induce cell death and that additional extrinsic stress factors are required (23, 34, 35). Multiple studies have demonstrated that DJ-1 is responsive to oxidative stress and that DJ-1 may act as an antioxidant, protecting cells against reactive oxygen species (ROS) (23, 36–39). In human cells, DJ-1 residues Cys-46, Cys-53, and Cys-106 have been shown to be oxidized upon hydrogen peroxide treatment (40). The Cys-106 is the most sensitive to oxidation and forms a cysteine-sulfinic acid upon exposure to ROS. DJ-1 has further been described as an atypical peroxiredoxin-like peroxidase that scavenges mitochondrial H2O2 through oxidation of Cys-106 (34). Oxidation of Cys-106 has also been suggested to be important for DJ-1's relocalization to mitochondria in response to oxidative stress (36, 41).
In a previous study, we showed that DJ-1 interacts with Cu,Zn-superoxide dismutase 1 and that this interaction can be enhanced by titration of copper ions, a known ligand for the superoxide dismutase 1 enzyme (42). Considering this and the fact that increased metal exposure or deficient metal regulation as well as mutations of DJ-1 are underlying factors for PD development, we sought to find out whether DJ-1 itself could be influenced by directly interacting with metals. We also characterized the effects of metal-induced cytotoxicity, in relation to DJ-1, by utilizing a cell model system deficient of DJ-1 or with wild type (WT) or PD mutated variants of human DJ-1 protein expression.
EXPERIMENTAL PROCEDURES
Special Reagents
Polyclonal goat anti-DJ-1 (AB4150, Abcam), monoclonal mouse anti-actin AC-40 (A3853), dopamine-hydrochloride (H8502), and MG132 (C2211) were from Sigma. Rabbit anti-DJ-1-N was a gift from Dr. Darren Moore at the Brain Mind Institute, EPFL, Switzerland. MEF cells from WT and DJ-1−/− mice were obtained from Dr. Huaibin Cai, NIA, National Institutes of Health, Bethesda.
Cloning
The human PARK7 gene was cloned and inserted into bacterial expression vectors pET28a as described previously (42). For mammalian cell expression, PARK7 was amplified with DJ1-F-XhoI (5′-ATCTCGAGATGGCTTCCAAAAGAGCTC-3′) and DJ1-R-KpnI (5′-ATGGTACCCTAGTCTTTAAGAACAAGTGG-3′) and inserted into pCDNA3.1(−) hygro. Restriction sites added to the primers are underlined. PARK7 point mutations were generated by site-directed mutagenesis. The sequence of redox-sensitive S3roGFP was kindly provided by Dr. C. Plieth (University if Kiel, Germany). The reporter was generated from the smGFP sequence described previously (66) using previously published technology (52), with additional cysteine mutations at positions Cys-149 and Cys-202 that decreased the dependence of the fluorescence to intracellular pH variations. S3roGFP does not carry the S65T mutation that was previously shown to be pH-sensitive. For our experiments, the sequence of S3roGFP was PCR-amplified and cloned into pcDNA 3.1/Hygro(+) vector (Invitrogen) using forward primer 5′-TTAGGTACCATGAGTAAAGGAGAAGAACTTTTCACTG-3′ and reverse primer 5′-ATAGCGGCCGCTTTATTTGTATAGTTCATCCATGCCATG-3′ carrying KpnI and NotI sites, respectively (as underlined). All constructs were verified by DNA sequencing at Genome Enterprise Ltd., Norwich, UK, or at Macrogen, the Netherlands.
Cell Culturing and Generation of Stable Cell Lines
MEF WT and DJ-1 knock-out cells (48) were cultured in DMEM supplemented with 7% heat-inactivated bovine calf serum (Invitrogen), 2 mm glutamine, 1% nonessential amino acids (Sigma), 50 units/ml penicillin, 50 μg/ml streptomycin, in 5% CO2 atmosphere at 37 °C. Cells were transfected using Lipofectamine 2000TM according to the manufacturer's instructions (Invitrogen). Stable DJ-1 re-expression cell lines were generated by transfecting DJ-1−/− cells with pCDNA3.1 Hygro(−) empty vector or pCDNA3.1 Hygro(−) vector containing WT or point-mutated human PARK7 genes. Stable cell lines were selected using 800 μg/ml hygromycin B and subcultured for 3 weeks. All cell culture reagents not specifically specified were from Invitrogen.
Immunofluorescent Imaging
Immunocytochemical staining was carried out as described previously (67). Briefly, cells growing on glass coverslips were washed in PBS and fixed in 4% buffered paraformaldehyde for 20 min followed by another fixation and permeabilization step in 100% methanol for 5 min at −20 °C. Samples were blocked in 5% BSA (w/v), PBS, 0.1% Tween 20 (v/v) for 1 h at room temperature prior to incubation overnight at 4 °C with primary antibody rabbit anti-DJ-1-N, raised against human DJ-1 N-terminal residues 1–13 as described previously (21, 68). 4 μg/ml anti-rabbit Alexa-488 secondary antibody was used for 1 h at room temperature prior to nuclear staining using Hoechst 33342 and mounting in Mowiol mounting media. Images were taken using an inverted Nikon A1R confocal laser scanning microscope using a ×60 oil objective and ×4 zoom. Fluorescence images of DJ-1-Alexa-488 and Hoechst 33342 nuclearly stained cells were acquired at 450/50 nm and 525/50 nm, using the same laser intensities and detection settings for all images to enable sample comparison. Maximum intensity scans are shown.
Cytotoxicity Assay
1.0 × 104 cells were seeded per well in a 96-well transparent flat bottom polystyrene noncoated tissue culture plate (3603, Corning Inc.). The cells were grown for 24 h prior to addition of stresses and grown another 18–24 h prior to assessment of cell survival. All metal salts were dissolved in water and sterile-filtered immediately prior to use. The pH of the cell media was not altered by addition of any of the chemicals used. Cell cytotoxicity was analyzed by use of the water-soluble tetrazolium salt, WST-1 (Roche Applied Science), according to the manufacturer's instructions and measured using a Thermo Scientific Multiskan Ascent at 450 nm wavelength and 630 nm as reference. Reference values, including metal treatment without cells, were subtracted from all data sets and compared with nontreated control cells, set to 100% survival. Mean values for multiple repeat ± S.E. values are shown. Cell survival data obtained using WST-1 was confirmed by microscopy. Approximate LC50 values obtained from the combined data sets are indicated in the figure legends of each experiment.
Protein Induction and Purification
Escherichia coli Rosetta(DE3)pLysS (Novagen) cells were transformed with pET28a harboring the WT human PARK7 gene. Protein expression was performed for 20 h in auto-induction ZYM-5052 media at 28 °C (69). Expressed protein was initially isolated with TALON affinity resin (Clontech) and eluted by thrombin (GE Healthcare) cleavage of the engineered N-terminal thrombin cleavage site in PBS, pH 7.5. The eluate was incubated with benzamidine-Sepharose (GE Healthcare) to remove residual thrombin, following another TALON affinity resin purification step to ensure complete removal of uncleaved protein and maximized protein purity as verified by SDS-PAGE (Fig. 1A). The purified protein was further prepared for mass spectrometric analysis (70). LC-MS/MS sequencing of the trypsinized protein was performed using an Acquity UPLC-coupled Q-TOF Micro (Waters Associates, Milford, MA) and peptide elution with acetonitrile in either a 0.1% ammonium hydroxide or a 0.1% formic acid environment. Combination of generated MS/MS data resulted in a 97.4% sequence coverage matching human DJ-1 using SwissProt database and Mascot (Matrix Sciences) search algorithm. Peptide mass tolerance was 100 ppm, and fragment tolerance was 0.5 Da for [M + 2H]2+, [M + 3H]3+, and [M + 4H]4+. Sequencing of the most N-terminal DJ-1 peptide revealed an addition of three amino acids (Gly-Ser-His) originating from the engineered N-terminal thrombin cleavage site.
FIGURE 1.

Human DJ-1 purification and metal binding analysis. A, purified recombinant human DJ-1 analyzed by SDS-PAGE and Coomassie G-250 stained for detection of total protein. High purity nontagged human DJ-1 used for XRF indicated at arrow. B, combined XRF spectra of recombinant human DJ-1 exposed to metal-containing buffer (colored solid line), metal-containing buffer only (dotted line), and backsoak buffer without added metal salt (black solid line). Prominent DJ-1 metal binding peaks for Cu(I), Cu(II), and Hg(II) were detected as well as a weaker signal for Mn(II). Asterisk marks the internal titanium peak originating from the XRF instrument.
X-ray Fluorescence
For XRF analysis, recombinant human DJ-1 WT protein was dialyzed for 24 h against a 20 mm MOPS buffer at pH 7.2 containing 1 mm metal salt, followed by another dialysis for 24 h in the same buffer without metal salt. 5 mm ascorbic acid was included in the first dialysis buffer to reduce Cu(II) to Cu(I). Both dialysis steps were done at RT at a 1:1000 (v/v) protein to buffer ratio. XRF data were collected on the ESRF protein crystallography beamlines from drops of protein or buffer frozen in liquid nitrogen, using a 30-s exposure time for the automatic procedure implemented in MxCuBE (71). Data were normalized on the Compton peak maximum and smoothed.
Atomic Absorption Spectroscopy
For AAS analysis, 0.1 mg/ml WT or point-mutated recombinant human DJ-1 was dialyzed for 24 h against AAS binding buffer (20 mm MOPS buffer, pH 7.2, 150 mm NaCl, 5 mm ascorbic acid, 10% glycerol) containing 10 μm CuCl, followed by a second dialysis for 24 h in the same buffer without metal salt. For the recombinant protein, the samples were diluted six times in distilled H2O by a Hamilton microlab 500 dispenser system prior to AAS analysis by use of an AAnalyst 300 from PerkinElmer Life Sciences. AAS data were normalized to a copper standard curve and correlated to final DJ-1 protein concentrations, as assessed by the Bradford method.
Microscale Thermophoresis
Copper (I, II) and mercury binding affinities to WT recombinant human DJ-1 or point-mutated human DJ-1 variants were measured by MST using a Monolith NT.115 instrument (NanoTemper Technologies, Germany). The proteins were fluorescently labeled by use of the Monolith NT L001 labeling assay according to the manufacturer's description (NanoTemper Technologies, Germany). Serial dilutions of metal ligands spanning a concentration of 10−2–106 nm were mixed in binding buffer (20 mm MOPS buffer, pH 7.2, 150 mm NaCl, 8% glycerol (supplemented with 5 mm ascorbic acid for reduction of Cu(I)) containing NT-647-labeled protein and collected in standard MST sample capillaries. Thermophoresis was analyzed by use of 30% LED and 50% laser intensity for all samples, and average KD values were calculated according to observed mobility shifts from multiple repeats.
Live Cell Confocal Laser Scanning Microscopy of Redox-sensitive GFP
For the in vivo measurements of cytosolic reactive oxygen species dynamics, MEF cells were grown in tissue culture-treated 8-well μ-Slide chambers with optical plastic bottom (Ibidi). After 24 h, cells were transfected with pcDNA-S3roGFP using Lipofectamine 2000 (Invitrogen). Cultures were allowed to express S3roGFP for 24 h prior to imaging. Shortly before each experiment, the culturing media of cells were replaced by Hanks' buffered saline solution (Thermo Scientific), and all further manipulations were carried out in Hanks' buffered saline solution. Imaging was done on a Nikon A1R confocal laser scanning microscopy system mounted on a Nikon Ti-E inverted epifluorescence microscope, equipped with a 408-nm diode laser and 488-nm multiline argon ion laser. Correct temperature and air/CO2 levels were maintained within the top stage electric incubation chamber with a heated glass cover mounted on the microscope stage and connected to humidity module and manual air/CO2 mixer (UNO, Okolab). Images were taken with Nikon Plan Apo VC ×60/1 40 oil objective and NIS elements AR 4.00.07 software of several different XY focal planes for 60 and 90 min at 1- and 2-min intervals, respectively. The specimen was excited in-line scanning mode with 408 and 488 nm wavelengths, and emission was collected at 525/550 nm. To minimize the fluorescence bleaching, the laser power was set at the lowest intensity and was equal for both excitation wavelengths. All other parameters that were shown to affect the resulting ratio were always kept the same for every experiment. Pixel intensities of images were controlled to vary between 500 and 3000 to avoid both saturation and false ratios due to insufficient brightness of the reporter. Metal treatments were added at minute 6 of the experiment, and imaging was continued 50 s later with almost no delays. All quantitative measurements of S3roGFP fluorescence intensity were done on NIS elements by defining the region of interest copying the shape of the cell, and the average intensities were determined for each time point. Prior to this, the mean pixel intensity of the background was subtracted from every image using the background region of interest.
Statistical Analysis
Test of sample homogeneity of variance was performed by the Levene test. Because of the nonhomogeneous sample distribution, the nonparametric Kruskal-Wallis test was performed for test of significance. In tested groups where the null hypothesis could be rejected, the differing pairs were identified by use of the Mann-Whitney U test. Statistically significant probability values (p < 0.05) are indicated with asterisk. All statistical analyses were performed using SPSS version 18.0.
RESULTS
DJ-1 Is a Metal-binding Protein That Binds Copper and Mercury
To investigate whether DJ-1 could directly interact with metals, we first cloned the human PARK7 gene into a bacterial expression vector system and purified large amounts of high purity recombinant human DJ-1 protein (Fig. 1A). The metal binding ability of DJ-1 was assessed by dialyzing the native purified protein against buffer containing metal salts (MgCl2, ZnSO4, NiSO4, AlCl3, CuCl, CuCl2, MnCl2, FeCl2, FeCl3, or HgCl2) followed by a second dialysis to wash out residual metals. XRF analysis of the metal-exposed protein revealed evident amounts of both copper as well as mercury bound to the human DJ-1 protein (Fig. 1B). Both copper(I) and copper(II) oxidation states were tested, and DJ-1 bound copper was identified by XRF as a single peak. In correlation to the internal titanium peak, a weaker signal for manganese bound to DJ-1 was also observed, although the other tested metal salts were negative. Although mercury is reactive and has high relative ion exchange selectivity for a cation exchange adsorbent, both manganese and copper have similar selectivities as iron, zinc, and magnesium. The observed metal bindings were therefore considered to be specific. XRF analysis of the metal-containing dialysis buffers against protein and back soak were done to confirm metal peak identity (Fig. 1B).
DJ-1, but Not the Clinical DJ-1 Variants A104T and D149A, Protects against Copper- and Mercury-induced Cytotoxicity
At the cellular level, metal concentrations are regulated through processes of absorption, distribution, biotransformation, and excretion (43). For metal absorption in intact tissue, copper(II) is reduced to copper(I) by metalloreductases, and copper(I) is delivered across the cells' plasma membrane by high affinity copper transporters (44). Iron, manganese, as well as nonessential heavy metals may cross the plasma membrane through binding to transferrin and divalent metal transporter 1 (45, 46). Intracellular concentrations of essential transition metals are normally maintained within a narrow range, through a complex system of metal-binding proteins. Alteration of intracellular transition metal homeostasis is cytotoxic and has been implicated in neurodegenerative diseases, such as PD (43, 47). We performed a set of cytotoxicity tests to evaluate whether DJ-1's metal binding property could serve a role in the cells' ability to maintain nontoxic metal homeostasis. To specifically assess the role of DJ-1, we made use of primary MEFs isolated from WT or DJ-1 homozygous knock-out mice (48). WT and DJ-1−/− MEF cells were exposed to increasing concentrations (0–1000 μm) of metals for 24 h before assessment of cell survival. An obvious reduction of cell survival was detected when exposing MEF DJ-1 knock-out cells to either copper or mercury, compared with WT cells (Fig. 2, A and B).
FIGURE 2.

Metal exposure and analysis of cell survival. Cell survival data compared WT and DJ-1−/− MEF cells exposed to increasing concentrations of metals for 24 h. All values were compared with nontreated control set to 100% survival and plotted against the logarithmic scale for the used metal concentrations. A, CuCl, WT LC50 = 70 μm, DJ-1−/− LC50 = 30 μm. B, HgCl2, WT LC50 = 15 μm, DJ-1−/− LC50 = 4 μm. Statistically significant change in cell survival compared with DJ-1−/− cells is indicated with *, p < 0.05.
To ascertain that the observed reduction in cell survival was due to the expression of DJ-1 protein alone, we re-introduced the human DJ-1/PARK7 gene into the DJ-1−/− MEF cells by transfection and selected monoclonal stably re-expressing DJ-1 cell lines. By doing this, we also sought to find out if the observed phenotypes could be rescued by re-expression of WT DJ-1 in the knock-out background. In addition to WT human DJ-1, we also included DJ-1 mutant A104T and D149A, previously identified in familiar forms of PD (18, 19). We also included oxidation site-mutated DJ-1 C106A, which lacks the major oxidation site of DJ-1 and has been shown to have reduced cell protective function against mitochondrially induced ROS (36, 41). Approximately equal DJ-1 protein expression was seen when comparing endogenously expressed DJ-1 in MEF WT cells to the selected human DJ-1 re-expression cell lines by Western blotting (Fig. 3A). Visualization of WT DJ-1 and the mutated DJ-1 variants in intact cells was performed by immunocytochemical staining using an antibody raised against the N-terminal part of human DJ-1. Both WT and the mutated DJ-1 variants retained the same subcellular localization, characterized as a predominantly soluble cytosolic localization (Fig. 3B and supplemental Fig. S1). A low level of DJ-1 could also be seen in the nucleus, although other types of organelle localization were not apparent. Re-introduction of human DJ-1 WT expression in the knock-out cells significantly rescued cell survival in case of both copper and mercury exposure (Fig. 3, C and D), highlighting a potential role for DJ-1 involvement in regulation of metal homeostasis and protection against metal-induced cytotoxicity. In the case of PD mutated DJ-1 A104T and D149A, both mutants showed a significant reduction in cell survival compared with DJ-1 WT upon exposure to copper or mercury, whereas the oxidation site-mutated DJ-1 C106A retained similar protective features as DJ-1 WT (Fig. 3, E and F). The results indicate that the PD mutated DJ-1 variants are not functionally active in response to these stresses. Interestingly, these results also highlight a potential dual role for DJ-1 as a mitochondrial antioxidant and a cytoplasmic metal-binding protein, because the oxidation site-mutated DJ-1 C106A variant maintained the ability to protect cells exposed to metal stresses (Fig. 3, E and F).
FIGURE 3.

Characterization and comparison of WT or PD-mutated DJ-1 re-expression of cells to metal stresses. A, Western blot of wild type, DJ-1−/− MEF cells, and stable DJ-1 re-expression of MEF cells expressing human DJ-1 variants as indicated. Equal protein expression was detected using protein-specific antibodies raised against human DJ-1 and for actin as loading control. B, immunocytochemical staining of DJ-1−/− MEF cells or DJ-1 re-expression of cells expressing human DJ-1 variants as indicated. Green, DJ-1; blue, nucleus. Analyzed DJ-1 variants show equal subcellular localization, predominantly soluble and cytoplasmic. Scale bars, 10 μm. −1° = minus primary antibody. C and D, cell survival data comparing empty vector transfected DJ-1−/− cells or stably re-expressing human DJ-1 WT cells exposed to increasing concentrations of metal stresses for 24 h. C, CuCl, WT LC50 = 65 μm, DJ-1−/− LC50 = 32 μm. D, HgCl2, WT LC50 = 7 μm, DJ-1−/− LC50 = 3 μm. E and F, cell survival data comparing DJ-1−/− or re-expression of cells expressing human DJ-1 WT, PD-mutated human DJ-1 A104T, D149A, or the oxidation site-mutated human DJ-1 C106A. Cells were exposed to increasing concentrations of CuCl (E) and HgCl2 (F) metal stresses for 24 h before cell survival analysis. Statistically significant increase in cell survival was compared with DJ-1−/− cells indicated with *, p < 0.05.
Clinically Important DJ-1 Mutant Variants Show Altered Metal Binding Potential
To further investigate whether the reduced cytoprotective function of DJ-1 PD mutants was a result of altered metal binding ability, recombinant human DJ-1 A104T, D149A, and C106A were produced and purified without an affinity tag (Fig. 4A). The in vitro metal binding ability of the DJ-1 proteins was again assessed by dialyzing the native purified proteins against buffer containing copper salt, followed by a second dialysis without metal salt to wash out residual metal. To obtain quantitative data for the total amount of copper bound to DJ-1 after 24 h of exposure, AAS was used instead of XRF. The AAS analysis affirmed the presence of copper bound to DJ-1 WT, and it detected ∼3 μm copper per μm DJ-1 (Fig. 4B). The analysis also confirmed that all other tested point-mutated DJ-1 proteins retained their copper binding ability (Fig. 4B). Surprisingly, the copper binding to PD mutant A104T was higher than WT, although copper binding to C106A was significantly reduced. These alterations were not large but still statistically significant due to small sample to sample variations. A His6-tagged DJ-1 WT protein was included in the analysis as a positive control for copper binding, and no residual copper was found in dialyzed metal binding buffer lacking DJ-1 protein after washing (Fig. 4B). Because the described AAS analysis only reflects the equilibrium of copper bound to DJ-1 at the end point of the dialysis, we also determined the initial binding affinities of the metal ligands. For this purpose, we made use of the MST technology that is based on direct movement of particles in a temperature gradient, also known as the Ludwig-Soret effect (49). Any change of the size, charge, and hydration shell of biomolecules due to binding to interacting molecules results in a relative change of movement along the temperature gradient and is used to determine ligand to protein binding affinities. In our study, the dissociation constant (KD) of monovalent copper(I) was found to be in the range of 4–5.7 μm for all DJ-1 variants, except for A104T that showed an ∼200-fold enhancement in binding affinity (Fig. 4C). The binding affinity of copper(II) was also enhanced in DJ-1 mutant A104T and D149A compared with WT DJ-1, whereas DJ-1 C106A was not found to bind divalent copper(II). In the case of mercury binding, WT DJ-1 and D149A showed a KD value of 0.06–0.12 μm, respectively, whereas A104T and C106A were not found to bind mercury. These data point out that DJ-1 residues Ala-104–Cys-106 may be important for divalent mercury binding and in the case of Cys-106 also divalent copper binding (Fig. 4D). Moreover, our data indicate that copper(I) binds to another region of the protein, not directly affected by the studied point mutations. Our AAS study of the total amount of copper bound to DJ-1 after 24 h of exposure reveals that copper is found in a 3:1 ratio bound to DJ-1. This analysis would suggest that the second copper-binding site would reside within the interface of the two DJ-1 proteins in the dimeric complex (Fig. 4D). A smaller amplitude in the thermophoresis shift was observed for A104T compared with WT DJ-1, which indicates that the A104T mutant may have lower thermostability, which could be linked to the altered binding affinity observed here.
FIGURE 4.
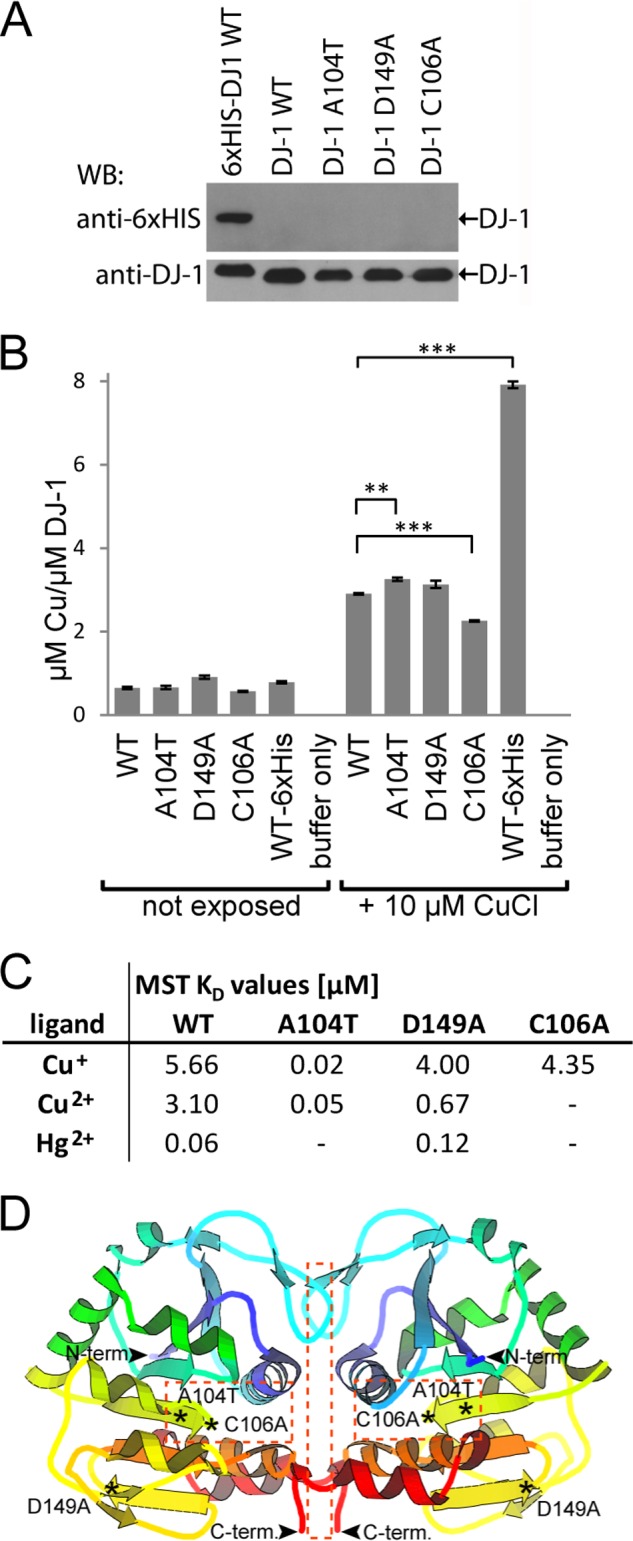
Point-mutated DJ-1 metal binding analysis. A, Western blot (WB) analysis, using anti-His6- and anti-DJ-1-specific antibodies of purified recombinant human DJ-1 WT and point-mutated DJ-1 A104T, DJ-1 D149A, DJ-1 C106A, and DJ-1 WT-His6 tag as positive control. B, AAS analysis of total amount of copper bound to recombinant human DJ-1 proteins, as shown in A, after 24 h exposure time. Statistically significant changes in total copper binding compared with WT DJ-1 are indicated with asterisks, **, p < 0.01; ***, p < 0.001. C, initial binding affinities for metal binding to DJ-1 recombinant human DJ-1 proteins, as shown in A. Calculated average MST dissociation constants (KD) for recombinant human DJ-1 WT and point-mutated variants using Cu(I), Cu(II), and Hg(II) as ligands. Lack of detected binding indicated with − sign. D, crystal structure model of dimeric human DJ-1 with the studied mutation sites indicated with *, and potential metal binding areas indicated within squares (dotted line). Protein structure model was adopted from Protein Data Bank code 2R1V.
Copper- and Mercury-induced Cytotoxicity Protection Is Compromised in Response to Dopamine but Does Not Involve the Proteasome
In this study we also attempted to find an answer to the key question of PD, which is why dopaminergic neurons are the main target for degeneration. In parallel to normal aging, neurodegeneration of the dopaminergic cells of the SNpc has been linked to various types of cellular injury due to toxin exposure, oxidative stress, mitochondrial dysfunction, or dysfunction of the ubiquitin proteasome system, as reviewed recently (50). A key reason to why in particular dopamine-producing neurons are vulnerable is likely their production of dopamine itself. It is known that products of dopamine degradation are highly toxic for cellular environment. Dopamine metabolism and oxidation into dopamine quinone have been suggested to enhance ROS production, which needs to be controlled by various intrinsic and extrinsic antioxidants. For this reason, in addition to the cytotoxic effects observed in relation to metal exposure, we analyzed additional stresses such as dopamine-induced oxidative stress and proteasome inhibition in our cell system. Exposing DJ-1 WT or DJ-1−/− cells to increasing concentrations of dopamine showed a significant reduction of cell survival in the DJ-1-deficient cells (Fig. 5A), whereas cells exposed to the cell-permeable proteasome inhibitor MG132 showed no change in survival rates either with or without expressed DJ-1 (Fig. 5B). Because MEF cells lack the neuron-specific dopamine transporters used for active and rapid re-uptake of dopamine from the synaptic cleft, the observed effect seen in this cell model system relies on passive diffusion through organic cation transporters, where the driving force is supplied by the electrochemical gradient of the transported cation (51). Our analysis of dopamine-induced cytotoxicity showed the highest no observed effect concentration value of 33 μm in both WT and DJ-1−/− cells. However, a significant change in the LC50 value of 95 μm for the DJ-1−/− cells compared with 185 μm for the DJ-1 WT cells was observed after 24 h of exposure (Fig. 5A). To further analyze a possible cumulative effect of dopamine exposure and metal-induced cytotoxicity, we exposed our cell model system to nontoxic concentrations (6.6 or 33 μm) of dopamine in combination with metal stresses and analyzed cell survival (Fig. 5, C–M). In the case of combined copper and dopamine exposure (Fig. 5, C–H), a clear dopamine dose-dependent reduction of cell survival was observed in both DJ-1−/− and DJ-1 re-expression cells. The dopamine dose-dependent reduction of cell survival was less evident in DJ-1−/− cells or PD-mutated DJ-1 re-expression mutants, which already showed reduced survival rates. In the case of combined mercury and dopamine exposure, a similar inter-relation in dose-dependent cytotoxicity between DJ-1−/−, PD-mutated DJ-1 A104T and D149A, and DJ-1 WT and C106A re-expression cells was observed (Fig. 5, C and I–M). With nontoxic dopamine exposure, the LC50 values for copper dropped to the same levels for DJ-1 WT and C106A re-expression cells as for the DJ-1−/− cells (Fig. 5C). For combined dopamine and mercury exposure, both WT and C106A retained a certain degree of cytoprotection compared with DJ-1−/− and PD-mutated DJ-1 variants (Fig. 5C). The observed reduction in cell survival due to metal-induced toxicity, dopamine, and combined metal and dopamine-induced toxicity could possibly be linked to DJ-1's proposed antioxidative function. As shown here, DJ-1's ability to protect cells from copper- and mercury-induced cytotoxicity is dramatically reduced upon combined nontoxic dopamine exposure, whereas DJ-1 is not involved in the cytoprotective mechanism linked to nonoxidative insults in the form of inhibition of proteasomal degradation.
FIGURE 5.

Comparison of WT- or DJ-1-deficient cell survival in response to dopamine, MG132, or combined dopamine and metal-induced stress. A, cell survival data comparing DJ-1−/− and WT DJ-1-expressing MEF cells exposed to increasing concentrations of dopamine. The highest no observed effect concentration of dopamine observed at 33 μm in both DJ-1 knock-out and DJ-1 WT-expressing cells. WT LC50 = 185 μm, DJ-1−/− LC50 = 95 μm. Statistically significant change in cell survival indicated with *, p < 0.05. B, no change is cell survival was observed upon treatment with proteasome inhibitor MG132. C, table of LC50 values calculated from combined dopamine and metal-induced cell toxicity data shown in D–M. D–M, cell survival data comparing DJ-1−/− or cells stably re-expressing human DJ-1 WT, A104T, D149A, or C106A, exposed to 0, 6.6, or 33 μm dopamine in combination with increasing concentrations of copper (D–H) or mercury (I–M) stresses for a total of 18 h. Dopamine dose-dependent reduction of cell survival was observed in re-expressing DJ-1 WT and DJ-1 C106A cells, as well as in DJ-1−/−, DJ-1 A104T, and D149A re-expression cell lines exposed to copper or mercury.
Dopamine Enhances Metal-induced Oxidative Stress and DJ-1 Contributes to Redox Homeostasis
Most cellular compartments maintain a reducing environment under normal conditions. The intracellular redox state can, however, be influenced by internal factors, like mitochondrial hydrogen peroxide production, or external factors, like pesticide- or metal-catalyzed ROS production. To enable dynamic measurements of the cellular redox environment in real time upon our stress conditions, we made use of the reduction-oxidation-sensitive green fluorescent protein (roGFP) technology (52). When imaged by live cell CLSM, the roGFP probe exhibits significant opposing shifts in the fluorescence intensities at two excitation maxima, 408 and 488 nm. The shift in the fluorescence intensity is a result of the formation of an oxidation-dependent intramolecular disulfide bridge. The formation of the disulfide bridge is thus directly responsive to the redox state within the analyzed cellular compartment in which roGFP is expressed, which enables real time ROS production monitoring at 408/488 nm. In our hands, the used cytosolic S3roGFP probe exhibited clear changes in fluorescence ratio arising directly from changes in intracellular redox state (Fig. 6, A and B, and supplemental Movie M1). Treating DJ-1−/− or DJ-1 WT-expressing cells with up to 100 μm dopamine alone did not result in any effect on oxidative conditions in the cells during 1 h of imaging (Fig. 6C). However, exposing the cells to 100 μm CuCl resulted in an evident enhancement of the intracellular oxidative state of the cells (Fig. 6, D and E (blue line)). Interestingly, exposing the cells to a nontoxic concentration of 33 μm dopamine together with 100 μm CuCl resulted in an almost immediate and dramatic surge of intracellular oxidation (Fig. 6, D and E (red line)). The oxidative stress observed upon combined copper and dopamine exposure was both faster and larger in magnitude in the DJ-1−/− cells compared with DJ-1 WT. Similar S3roGFP experiments were performed using 5 μm HgCl2 treatments together with 33 μm dopamine. Contrary to the copper treatment experiment where oxidation can be explained by enhanced Fenton reaction, mercury exposure alone shifted the intracellular redox status slightly toward a more reduced state (Fig. 6, F and G (blue line)). Interestingly, combined mercury exposure with dopamine treatment resulted again in an obvious enhancement of intracellular oxidation in the DJ-1−/− cells that was absent in DJ-1 WT cells (Fig. 6, F and G (red line)). The live cell measurements of the intracellular redox state were done during a relative short time span of 90 min, due to the cytotoxicity of the treatments. The results clearly indicate that the presence of nontoxic concentration of dopamine enhances metal-induced oxidative stress and that DJ-1 is important for the maintenance of the cells redox buffering capacity.
FIGURE 6.

Real time analysis of oxidative stress levels by use of CLSM live cell imaging of redox-sensitive S3roGFP. A, CLSM live cell imaging experiment showing redox-sensitive S3roGFP 408/488 nm ratios in transfected MEF DJ-1 WT cells. The cells were exposed to 0.5 mm H2O2 to mimic enhanced oxidative stress conditions 5 min after initiating image acquisition. After 35 min of imaging acquisition, the cells were reduced by treatment with 2 mm DTT. A clear change in the 408/488 nm ratio was observed upon oxidation and reduction of the imaged cells. B, CLSM images of S3roGFP-expressing cells treated as shown in A. The observed redox-dependent change in 408/488 nm ratio is visualized by pseudo-coloring according to included ratio scale bar 0–1.2 (blue, reduced; oxidized, red). Image size scale bar, 20 μm. C, CLSM live cell imaging experiment showing S3roGFP 408/488 nm ratios in DJ-1−/− and DJ-1 WT-expressing cells exposed to 100 μm dopamine, arrow. Mean value, ± S.E. for five cells per cell type are shown. D, S3roGFP 408/488 nm ratios in DJ-1 WT-expressing cells exposed to 100 μm CuCl, with or without addition of 33 μm dopamine. Control indicates nonexposed cells. Mean value, ± S.E. for 10 cells per treatment are shown. E, S3roGFP 408/488 nm ratios in DJ-1−/− cells treated as in D. Mean value, ± S.E. for 10 cells per treatment are shown. F, S3roGFP 408/488 nm ratios in DJ-1 WT-expressing cells exposed to 5 μm HgCl2, with or without addition of 33 μm dopamine. Mean value, ± S.E. for 12–17 cells per treatment are shown. G, S3roGFP 408/488 nm ratios in DJ-1−/− cells treated as in F. Mean value, ± S.E. for 13–19 cells per treatment are shown. Cells that died during the analysis were excluded from the shown quantified data.
DISCUSSION
Our study identifies human DJ-1 as a metal-binding protein able to evidently bind copper and mercury in vitro. We also show that expression of DJ-1 protects the cells against mercury- and copper-induced cytotoxicity and that PD-mutated DJ-1 A104T and D149A have lost this function. To our knowledge this is the first report identifying DJ-1 as a metal-binding protein involved in metal homeostatic processes and able to protect cells from induced metal cytotoxicity.
Previous cross-sectional as well as longitudinal population-based studies of heritability of PD in twins have reported no or a very low genetic component in typical late onset PD (53–55). It has therefore been suggested that environmental factors are most important in typical late onset PD, whereas genetic factors are substantial in early onset PD (53). The primary environmental effectors believed to be involved are toxic pollutants such as pesticides and toxic metals, causing brain inflammation and oxidative damage to neurons (56, 57). Mercury, in particularly the lipophilic methylmercury, bioaccumulates in the food chain and is stored in biological tissue, especially in the brain (58). Dose-response association between PD and blood mercury levels has been reported (10). Alterations in brain iron homeostasis has also been linked to PD, where up to a 2-fold elevation of iron levels specifically in the SNpc and lateral globus pallidus in comparison with age-matched controls has been reported previously (47, 59). Chronic occupational exposure to manganese or copper, individually or combinations of lead, iron, and copper, has been associated with PD (7). Because both copper and iron are capable of catalyzing free radical formation via the Fenton reaction, excess accumulation or deficient regulation of these metals can induce production of ROS and trigger cell death. Our study indicates that even a small genetic alteration can sensitize cells to metal-induced cell death. This finding supports the assumption that a combination of genetic predisposition may sensitize cells to environmental toxin-induced cell death and subsequent susceptibility for disease development.
As outlined previously, mutation of the human DJ-1/PARK7 gene has been associated with familial early onset PD. In some patients, the DJ-1/PARK7 gene is deleted (12). In the case of the DJ-1 point mutations M26I and L166P, the mutations alter the proteins' stability leading to enhanced degradation of DJ-1 and can therefore be considered clear loss of function mutations (60). The reasons for PD development in other DJ-1 point mutation carriers are less clear. The ability to dimerize and expression levels of the studied PD mutants A104T and D149A are comparable with nonmutated DJ-1 and do not per se support a loss of function hypothesis (60, 61). However, circular dichroism analyses have revealed reduced thermostability of mutant A104T and D149A, indicating secondary structural changes (62). DJ-1 Ala-104 and Asp-149 are completely conserved residues between a broad spectrum of species, highlighting a potentially important structure-function relationship. In our cell model system, both the A104T and the D149A mutant forms are normally expressed as compared with WT DJ-1. However, our data show that both PD mutants have lost their ability to protect cells against copper- and mercury-induced stresses, resembling DJ-1-deficient cells. These results indicate that the mutants are not functionally active in terms of cytoprotection. Mutating DJ-1 cysteine 106 to alanine has been shown to abolish DJ-1's ability to protect cells from MPP+- and rotenone-induced oxidative stress as a result of mitochondrial damage (36, 41). Our data show that C106A-mutated DJ-1 retains its cytoprotective function against induced metal exposure, indicating that DJ-1s' ability to function as an antioxidant through Cys-106 oxidation is not needed for metal-induced stress protection. Exposing cells to nontoxic concentrations of dopamine enhances the cytotoxic effect of metal exposure. Oxidized dopamine itself has been shown to bind directly to cysteine residues in proteins (63). It has also been suggested that dopamine bound to proteins may be involved in redox alteration under oxidative stress, generating a redox-active cysteinyl-dopamine adduct that enhances metal-catalyzed oxidation of proteins (64). Our live cell analysis of redox-sensitive S3roGFP confirms these suggestions and provides direct in vivo evidence of this enhanced intracellular oxidation.
The observation of enhanced cell toxicity upon combined metal and dopamine treatment may also be interesting from a pharmacological perspective. The dopamine concentrations used in this study were not higher than the blood plasma dopamine concentrations measured in PD patients on conventional levodopa therapy, where mean plasma levodopa concentrations ranging between 2.3 and 35.9 μm (0.45–7.07 μg/ml) and peak concentrations up to 69.7 μm (13.75 μg/ml) have been reported (65). Our data show that nontoxic concentrations (6.6–33 μm) of dopamine in combination with metals dramatically enhanced ROS production and reduced cell survival in comparison with metals alone. Although this in vitro study hints that individual bodily accumulation of toxic metals could accelerate cell death upon dopamine replacement therapy, the relevance of this observation to the in vivo situation in PD patients cannot be assessed without further studies in clinical material. We hope that our findings may revive the interest in exogenous factors in the etiology of PD and encourage new research aiming for a better understanding of metal homeostatic processes and their role in the etiology of idiopathic PD.
Acknowledgments
We thank Dr. Darren Moore at the Brain Mind Institute, EPFL, Switzerland, for providing the rabbit anti-DJ-1-N antibody. We thank Dr. Huaibin Cai, National Institute on Aging, National Institutes of Health, Bethesda, MD, for MEF cells from WT and DJ-1−/− mice. We also thank Dr. C. Plieth, University of Kiel, Germany, for the sequence of redox-sensitive S3roGFP.
This work was supported by Western Norway Regional Health Authority Helse Vest Project Grant 911575.

This article contains supplemental Fig. S1 and Movie S1.
- PD
- Parkinson disease
- SNpc
- substantia nigra pars compacta
- ROS
- reactive oxygen species
- MEF
- mouse embryonic fibroblasts
- AAS
- atomic absorption spectroscopy
- XRF
- x-ray fluorescence
- MST
- microscale thermophoresis
- CLSM
- confocal laser scanning microscopy
- roGFP
- reduction-oxidation-sensitive GFP.
REFERENCES
- 1. Hirsch E., Graybiel A. M., Agid Y. A. (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature 334, 345–348 [DOI] [PubMed] [Google Scholar]
- 2. Marsden C. D. (1996) in Oxford Textbook of Medicine (Weatherall D. J., Ledingham J. G., Warrell D. A., eds) 3rd Ed., pp. 3998–4022, Oxford University Press, Oxford [Google Scholar]
- 3. Braak H., Del Tredici K., Rüb U., de Vos R. A., Jansen Steur E. N., Braak E. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211 [DOI] [PubMed] [Google Scholar]
- 4. Poewe W. (2007) in Parkinson's Disease and Movement Disorders (Jankovic J., Tolosa E., eds) 5th Ed., pp. 67–76, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 5. Wang J. D., Huang C. C., Hwang Y. H., Chiang J. R., Lin J. M., Chen J. S. (1989) Manganese-induced parkinsonism: an outbreak due to an unrepaired ventilation control system in a ferromanganese smelter. Br. J. Ind. Med. 46, 856–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sikk K., Haldre S., Aquilonius S. M., Taba P. (2011) Manganese-induced Parkinsonism due to Ephedrone abuse. Parkinsons Dis. 2011, 865319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gorell J. M., Johnson C. C., Rybicki B. A., Peterson E. L., Kortsha G. X., Brown G. G., Richardson R. J. (1999) Occupational exposure to manganese, copper, lead, iron, mercury, and zinc and the risk of Parkinson's disease. Neurotoxicology 20, 239–247 [PubMed] [Google Scholar]
- 8. Semchuk K. M., Love E. J., Lee R. G. (1992) Parkinson's disease and exposure to agricultural work and pesticide chemicals. Neurology 42, 1328–1335 [DOI] [PubMed] [Google Scholar]
- 9. Dexter D. T., Carayon A., Javoy-Agid F., Agid Y., Wells F. R., Daniel S. E., Lees A. J., Jenner P., Marsden C. D. (1991) Alterations in the levels of iron, ferritin, and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114, 1953–1975 [DOI] [PubMed] [Google Scholar]
- 10. Ngim C. H., Devathasan G. (1989) Epidemiologic study on the association between body burden mercury level and idiopathic Parkinson's disease. Neuroepidemiology 8, 128–141 [DOI] [PubMed] [Google Scholar]
- 11. Wirdefeldt K., Adami H. O., Cole P., Trichopoulos D., Mandel J. (2011) Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur. J. Epidemiol. 26, S1–S58 [DOI] [PubMed] [Google Scholar]
- 12. Bonifati V., Rizzu P., van Baren M. J., Schaap O., Breedveld G. J., Krieger E., Dekker M. C., Squitieri F., Ibanez P., Joosse M., van Dongen J. W., Vanacore N., van Swieten J. C., Brice A., Meco G., van Duijn C. M., Oostra B. A., Heutink P. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 [DOI] [PubMed] [Google Scholar]
- 13. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 14. Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., de Silva R., Lees A., Martí-Massó J. F., Pérez-Tur J., Wood N. W., Singleton A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 [DOI] [PubMed] [Google Scholar]
- 15. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 16. Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 17. Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 [DOI] [PubMed] [Google Scholar]
- 18. Hague S., Rogaeva E., Hernandez D., Gulick C., Singleton A., Hanson M., Johnson J., Weiser R., Gallardo M., Ravina B., Gwinn-Hardy K., Crawley A., St George-Hyslop P. H., Lang A. E., Heutink P., Bonifati V., Hardy J., Singleton A. (2003) Early-onset Parkinson's disease caused by a compound heterozygous DJ-1 mutation. Ann. Neurol. 54, 271–274 [DOI] [PubMed] [Google Scholar]
- 19. Abou-Sleiman P. M., Healy D. G., Quinn N., Lees A. J., Wood N. W. (2003) The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann. Neurol. 54, 283–286 [DOI] [PubMed] [Google Scholar]
- 20. Wilson M. A., Collins J. L., Hod Y., Ringe D., Petsko G. A. (2003) The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 9256–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang L., Shimoji M., Thomas B., Moore D. J., Yu S. W., Marupudi N. I., Torp R., Torgner I. A., Ottersen O. P., Dawson T. M., Dawson V. L. (2005) Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 14, 2063–2073 [DOI] [PubMed] [Google Scholar]
- 22. Nagakubo D., Taira T., Kitaura H., Ikeda M., Tamai K., Iguchi-Ariga S. M., Ariga H. (1997) DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 231, 509–513 [DOI] [PubMed] [Google Scholar]
- 23. Kim R. H., Smith P. D., Aleyasin H., Hayley S., Mount M. P., Pownall S., Wakeham A., You-Ten A. J., Kalia S. K., Horne P., Westaway D., Lozano A. M., Anisman H., Park D. S., Mak T. W. (2005) Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park J., Kim S. Y., Cha G. H., Lee S. B., Kim S., Chung J. (2005) Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 361, 133–139 [DOI] [PubMed] [Google Scholar]
- 25. Okada M., Matsumoto K., Niki T., Taira T., Iguchi-Ariga S. M., Ariga H. (2002) DJ-1, a target protein for an endocrine disrupter, participates in the fertilization in mice. Biol. Pharm. Bull. 25, 853–856 [DOI] [PubMed] [Google Scholar]
- 26. van der Brug M. P., Blackinton J., Chandran J., Hao L. Y., Lal A., Mazan-Mamczarz K., Martindale J., Xie C., Ahmad R., Thomas K. J., Beilina A., Gibbs J. R., Ding J., Myers A. J., Zhan M., Cai H., Bonini N. M., Gorospe M., Cookson M. R. (2008) RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 10244–10249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takahashi K., Taira T., Niki T., Seino C., Iguchi-Ariga S. M., Ariga H. (2001) DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx α to the receptor. J. Biol. Chem. 276, 37556–37563 [DOI] [PubMed] [Google Scholar]
- 28. Tillman J. E., Yuan J., Gu G., Fazli L., Ghosh R., Flynt A. S., Gleave M., Rennie P. S., Kasper S. (2007) DJ-1 binds androgen receptor directly and mediates its activity in hormonally treated prostate cancer cells. Cancer Res. 67, 4630–4637 [DOI] [PubMed] [Google Scholar]
- 29. Shendelman S., Jonason A., Martinat C., Leete T., Abeliovich A. (2004) DJ-1 is a redox-dependent molecular chaperone that inhibits α-synuclein aggregate formation. PLoS Biol. 2, e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olzmann J. A., Brown K., Wilkinson K. D., Rees H. D., Huai Q., Ke H., Levey A. I., Li L., Chin L. S. (2004) Familial Parkinson's disease-associated L166P mutation disrupts DJ-1 protein folding and function. J. Biol. Chem. 279, 8506–8515 [DOI] [PubMed] [Google Scholar]
- 31. Chen J., Li L., Chin L. S. (2010) Parkinson disease protein DJ-1 converts from a zymogen to a protease by carboxyl-terminal cleavage. Hum. Mol. Genet. 19, 2395–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang B., Xiong H., Sun P., Zhang Y., Wang D., Hu Z., Zhu Z., Ma H., Pan Q., Xia J. H., Xia K., Zhang Z. (2006) Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson's disease. Hum. Mol. Genet. 15, 1816–1825 [DOI] [PubMed] [Google Scholar]
- 33. Moore D. J., Zhang L., Troncoso J., Lee M. K., Hattori N., Mizuno Y., Dawson T. M., Dawson V. L. (2005) Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 14, 71–84 [DOI] [PubMed] [Google Scholar]
- 34. Andres-Mateos E., Perier C., Zhang L., Blanchard-Fillion B., Greco T. M., Thomas B., Ko H. S., Sasaki M., Ischiropoulos H., Przedborski S., Dawson T. M., Dawson V. L. (2007) DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 14807–14812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goldberg M. S., Pisani A., Haburcak M., Vortherms T. A., Kitada T., Costa C., Tong Y., Martella G., Tscherter A., Martins A., Bernardi G., Roth B. L., Pothos E. N., Calabresi P., Shen J. (2005) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45, 489–496 [DOI] [PubMed] [Google Scholar]
- 36. Canet-Avilés R. M., Wilson M. A., Miller D. W., Ahmad R., McLendon C., Bandyopadhyay S., Baptista M. J., Ringe D., Petsko G. A., Cookson M. R. (2004) The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martinat C., Shendelman S., Jonason A., Leete T., Beal M. F., Yang L., Floss T., Abeliovich A. (2004) Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES-derived cell model of primary Parkinsonism. PLoS Biol. 2, e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meulener M. C., Xu K., Thomson L., Thompson L., Ischiropoulos H., Bonini N. M. (2006) Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. U.S.A. 103, 12517–12522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taira T., Saito Y., Niki T., Iguchi-Ariga S. M., Takahashi K., Ariga H. (2004) DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kinumi T., Kimata J., Taira T., Ariga H., Niki E. (2004) Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 317, 722–728 [DOI] [PubMed] [Google Scholar]
- 41. Blackinton J., Lakshminarasimhan M., Thomas K. J., Ahmad R., Greggio E., Raza A. S., Cookson M. R., Wilson M. A. (2009) Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J. Biol. Chem. 284, 6476–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu X. M., Lin H., Maple J., Björkblom B., Alves G., Larsen J. P., Møller S. G. (2010) The Arabidopsis DJ-1a protein confers stress protection through cytosolic SOD activation. J. Cell Sci. 123, 1644–1651 [DOI] [PubMed] [Google Scholar]
- 43. Bleackley M. R., Macgillivray R. T. (2011) Transition metal homeostasis: from yeast to human disease. Biometals 24, 785–809 [DOI] [PubMed] [Google Scholar]
- 44. Lee J., Peña M. M., Nose Y., Thiele D. J. (2002) Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 277, 4380–4387 [DOI] [PubMed] [Google Scholar]
- 45. Bridges C. C., Zalups R. K. (2005) Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 204, 274–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ballatori N. (2002) Transport of toxic metals by molecular mimicry. Environ. Health Perspect. 110, Suppl. 5, 689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sian-Hülsmann J., Mandel S., Youdim M. B., Riederer P. (2011) The relevance of iron in the pathogenesis of Parkinson's disease. J. Neurochem. 118, 939–957 [DOI] [PubMed] [Google Scholar]
- 48. Chandran J. S., Lin X., Zapata A., Höke A., Shimoji M., Moore S. O., Galloway M. P., Laird F. M., Wong P. C., Price D. L., Bailey K. R., Crawley J. N., Shippenberg T., Cai H. (2008) Progressive behavioral deficits in DJ-1-deficient mice are associated with normal nigrostriatal function. Neurobiol. Dis. 29, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duhr S., Braun D. (2006) Why molecules move along a temperature gradient. Proc. Natl. Acad. Sci. U.S.A. 103, 19678–19682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schapira A. H., Jenner P. (2011) Etiology and pathogenesis of Parkinson's disease. Mov. Disord. 26, 1049–1055 [DOI] [PubMed] [Google Scholar]
- 51. Ciarimboli G. (2008) Organic cation transporters. Xenobiotica 38, 936–971 [DOI] [PubMed] [Google Scholar]
- 52. Hanson G. T., Aggeler R., Oglesbee D., Cannon M., Capaldi R. A., Tsien R. Y., Remington S. J. (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279, 13044–13053 [DOI] [PubMed] [Google Scholar]
- 53. Tanner C. M., Ottman R., Goldman S. M., Ellenberg J., Chan P., Mayeux R., Langston J. W. (1999) Parkinson disease in twins: an etiologic study. JAMA 281, 341–346 [DOI] [PubMed] [Google Scholar]
- 54. Wirdefeldt K., Gatz M., Reynolds C. A., Prescott C. A., Pedersen N. L. (2011) Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol. Aging 32, 1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marttila R. J., Kaprio J., Koskenvuo M., Rinne U. K. (1988) Parkinson's disease in a nationwide twin cohort. Neurology 38, 1217–1219 [DOI] [PubMed] [Google Scholar]
- 56. Jomova K., Vondrakova D., Lawson M., Valko M. (2010) Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 345, 91–104 [DOI] [PubMed] [Google Scholar]
- 57. Liu B., Gao H. M., Hong J. S. (2003) Parkinson's disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: role of neuroinflammation. Environ. Health Perspect. 111, 1065–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Díez S. (2009) Human health effects of methylmercury exposure. Rev. Environ. Contam. Toxicol. 198, 111–132 [DOI] [PubMed] [Google Scholar]
- 59. Götz M. E., Double K., Gerlach M., Youdim M. B., Riederer P. (2004) The relevance of iron in the pathogenesis of Parkinson's disease. Ann. N.Y. Acad. Sci. 1012, 193–208 [DOI] [PubMed] [Google Scholar]
- 60. Blackinton J., Ahmad R., Miller D. W., van der Brug M. P., Canet-Avilés R. M., Hague S. M., Kaleem M., Cookson M. R. (2005) Effects of DJ-1 mutations and polymorphisms on protein stability and subcellular localization. Brain Res. Mol. Brain. Res. 134, 76–83 [DOI] [PubMed] [Google Scholar]
- 61. Görner K., Holtorf E., Odoy S., Nuscher B., Yamamoto A., Regula J. T., Beyer K., Haass C., Kahle P. J. (2004) Differential effects of Parkinson's disease-associated mutations on stability and folding of DJ-1. J. Biol. Chem. 279, 6943–6951 [DOI] [PubMed] [Google Scholar]
- 62. Malgieri G., Eliezer D. (2008) Structural effects of Parkinson's disease linked DJ-1 mutations. Protein Sci. 17, 855–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ito S., Kato T., Fujita K. (1988) Covalent binding of catechols to proteins through the sulfhydryl-group. Biochem. Pharmacol. 37, 1707–1710 [DOI] [PubMed] [Google Scholar]
- 64. Akagawa M., Ishii Y., Ishii T., Shibata T., Yotsu-Yamashita M., Suyama K., Uchida K. (2006) Metal-catalyzed oxidation of protein-bound dopamine. Biochemistry 45, 15120–15128 [DOI] [PubMed] [Google Scholar]
- 65. Nyholm D., Lennernäs H., Gomes-Trolin C., Aquilonius S. M. (2002) Levodopa pharmacokinetics and motor performance during activities of daily living in patients with Parkinson's disease on individual drug combinations. Clin. Neuropharmacol. 25, 89–96 [DOI] [PubMed] [Google Scholar]
- 66. Davis S. J., Vierstra R. D. (1998) Soluble, highly fluorescent variants of green fluorescent protein (GFP) for use in higher plants. Plant Mol. Biol. 36, 521–528 [DOI] [PubMed] [Google Scholar]
- 67. Björkblom B., Vainio J. C., Hongisto V., Herdegen T., Courtney M. J., Coffey E. T. (2008) All JNKs can kill, but nuclear localization is critical for neuronal death. J. Biol. Chem. 283, 19704–19713 [DOI] [PubMed] [Google Scholar]
- 68. Moore D. J., Zhang L., Dawson T. M., Dawson V. L. (2003) A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 87, 1558–1567 [DOI] [PubMed] [Google Scholar]
- 69. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 70. Björkblom B., Ostman N., Hongisto V., Komarovski V., Filén J. J., Nyman T. A., Kallunki T., Courtney M. J., Coffey E. T. (2005) Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J. Neurosci. 25, 6350–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Leonard G. A., Sole V. A., Beteva A., Gabadinho J., Guijarro M., McCarthy J., Marrocchelli D., Nurizzo D., McSweeney S., Mueller-Dieckmann C.. (2009) Online collection and analysis of x-ray fluorescence spectra on the macromolecular crystallography beamlines of the ESRF. J. Appl. Crystallogr. 42, 333–335 [Google Scholar]