

Background: Oligomerization of DHHC palmitoyltransferases has been suggested.
Results: Self-association of DHHC2 and DHHC3 was detected in intact cells and in vitro. Purified enzymes were found predominately as monomers and dimers, with the monomer displaying higher activity than the dimer.
Conclusion: DHHC proteins may exist in a monomer-oligomer equilibrium, with the monomers having higher enzyme activity.
Significance: Oligomerization may represent a mechanism to regulate DHHC enzyme activity.
Keywords: Bioluminescence Resonance Energy Transfer (BRET), Enzyme Structure, Fatty Acid, Membrane Enzymes, Posttranslational Modification, Palmitoylation
Abstract
The formation of dimers or higher-order oligomers is a property of numerous integral membrane proteins, including ion channels, transporters, and receptors. In this study, we examined whether members of the DHHC-S-acyltransferase family oligomerize in intact cells and in vitro. DHHC-S-acyltransferases are integral membrane proteins that catalyze the addition of palmitate to cysteine residues on proteins at the cytoplasmic face of cell membranes. Bioluminescence resonance energy transfer (BRET) experiments revealed that DHHC2 or DHHC3 (Golgi-specific DHHC zinc finger protein (GODZ)) self-associate when expressed in HEK-293 cells. Homomultimer formation was confirmed by coimmunoprecipitation. Purified DHHC3 resolved predominately as a monomer and dimer on blue native polyacrylamide gels. In intact cells and in vitro, catalytically inactive DHHC proteins displayed a greater propensity to form dimers. BRET signals were higher for the catalytically inactive DHHC2 or DHHC3 than their wild-type counterparts. DHHC3 BRET in cell membranes was decreased by the addition of its lipid substrate palmitoyl-CoA, a treatment that results in autoacylation of the enzyme. Enzyme activity of a covalently linked DHHC3 dimer was less than that of the monomeric form, suggesting that enzyme activity may be modulated by the oligomerization status of the protein.
Introduction
Palmitoylation is the posttranslational addition of long-chain fatty acids such as palmitate to cysteine residues of proteins via a thioester linkage. In contrast to other lipid modifications, palmitoylation is reversible and, thus, is a dynamic process that regulates protein subcellular localization, trafficking, stability, and activity. Genetic and biochemical studies have established that palmitoylation of proteins on the cytoplasmic face of cell membranes is catalyzed by a family of integral membrane proteins with a conserved Asp-His-His-Cys (DHHC) motif embedded in a cysteine-rich domain (reviewed in Ref. 1–3). These so-called DHHC proteins are present in all eukaryotes as multienyzme families that range in size from five members in Schizosaccharomyces pombe to 24 in mouse and Arabidopsis (4).
Evidence is accumulating that DHHC proteins play multiple critical roles in physiology and disease (reviewed in Refs. 3, 5). Mutations in the genes encoding DHHC9 and DHHC15 cause X-linked intellectual disability in humans. DHHC17 and DHHC13 are huntingtin-interacting proteins that palmitoylate huntingtin and other neuronal substrates. Mice lacking DHHC17 or DHHC13 have behavioral and neuropathological deficits that are similar to those of a mouse model of Huntington's disease and exhibit reduced palmitoylation of key neuronal substrates, suggesting that altered palmitoylation by DHHC17 may contribute to human Huntington's disease. Links to schizophrenia (DHHC8), tumorigenesis (DHHC9, 11, and 14), and metastasis (DHHC2) have also been reported. Accordingly, elucidating the mechanism and regulation of the DHHC proteins is important for understanding how palmitoylation manifests in physiology and pathophysiology.
The DHHC cysteine-rich domain is essential for protein acyltransferase (PAT)2 activity (6, 7). The mechanism of palmitate transfer to the protein substrate is through an acylenzyme intermediate that is dependent upon the cysteine of the DHHC motif (8, 9). Mutation of the DHHC cysteine to serine or alanine blocks enzyme activity in vitro and is essential for function in vivo (10). Sequence homology of the DHHC proteins is limited outside the DHHC cysteine-rich domain. Most DHHC proteins have a predicted topology of four transmembrane domains with the N- and C termini exposed to the cytoplasm. A few DHHC proteins have an extended N-terminal domain that contains ankyrin repeats and two additional transmembrane domains, a topology confirmed experimentally for the yeast DHHC protein Akr1 (11). The 51-amino acid DHHC cysteine-rich domain begins in the cytoplasmic loop between the second and third transmembrane domains, and its hydrophobic C-terminal region is thought to extend into the membrane.
Evidence suggests that the catalytically inactive mutant in which the DHHC cysteine is mutated acts as a dominant negative when expressed in cultured cells (12–14). However, the mechanism by which the mutation interferes with the function of the wild-type protein is unknown. One hypothesis is that DHHC proteins function as oligomers and that mixed oligomers of the wild-type and mutant enzyme are inactive (13). Support for this argument comes from coimmunoprecipitation experiments that suggest that DHHC3 (also named GODZ (Golgi-specific DHHC zinc finger protein)) forms homomultimers and heteromultimers with DHHC7 (13).
In this study, we investigated whether DHHC proteins oligomerize in cells and in detergent solution. We used DHHC2 and DHHC3 proteins as examples of the larger DHHC protein family. DHHC2 and DHHC3 are localized in different membrane compartments and have distinct sets of protein substrates (3). These proteins express well in mammalian cells and are biochemically tractable, enabling analysis of purified protein. Our results suggest that DHHC proteins form oligomers in cell membranes and may exist in a monomer-dimer equilibrium in which the active enzyme favors the monomeric state.
EXPERIMENTAL PROCEDURES
Reagents
Coelenterazine h was purchased from Nanolight. Human α-thrombin was purchased from Hematologic Technologies, Inc. The following antibodies were used: anti-FLAG M2 from Sigma (1:3,000), anti-GODZ (DHHC3) (catalog no. ab31837, 1:2000) from Abcam, anti-GFP/YFP from Clontech (catalog no. 632592, 1:1000), anti-DHHC2 (catalog no. sc-98220, 1:500) from Santa Cruz Biotechnology, and goat anti-mouse or goat anti-rabbit IgG conjugated to HRP from MP Biomedicals (1:1000). [9,10-3H]palmitate (47.7 Ci/mmol, 105,000 dpm/pmol) was purchased from PerkinElmer Life Sciences. [3H]palm-CoA was synthesized and purified as described (15). A protease inhibitor mixture of leupeptin (5 μg/ml), phenylmethylsulfonylfluoride (0.2 mm), Pepstain A (2 μM), and aprotinin (Sigma, catalog no. A6279, 1:2000) was prepared from stock solutions and used at the final concentrations indicated.
Cell Culture and Transfection
HEK-293 cells were cultured in DMEM supplemented with 10% fetal bovine serum at 37 °C with 5% CO2. HEK-293 cells were transfected with Effectene transfection reagent (Qiagen) according to the instructions of the manufacturer. TriEXTM insect cells were cultured in TriEXTM insect cell medium (Novagen) at 27 °C.
Constructs
BRET constructs were fusions with humanized Renilla luciferase (RLuc) or YFP. A linker sequence encoding -GGGGS was introduced between the protein of interest and RLuc or YFP. For DHHC3-RLuc and DHHC2-RLuc, murine DHHC3 or human DHHC2 cDNA was subcloned into pRLuc(h)N2 or pRLuc(h)N3 (BRET2TM, BioSignal Packard). For DHHC3-YFP and DHHC2-YFP, the YFP coding sequence was first cloned into pCDNA3.1A(-)myc-his as a BamHI-HindIII fragment to generate a vector for C-terminal YFP fusion proteins (pML1676). Murine DHHC3 or human DHHC2 cDNA was subcloned into pML1676. To generate constructs for N-terminal YFP fusion proteins, the coding sequence of YFP was subcloned into pCDNA3.1A(-)myc-his, yielding pML1675. The murine DHHC3 cDNA, the C-terminal domain of human Golgin 84 (amino acids 569–731, PCR-amplified from HeLa cells), or human DHHC2 cDNA was subcloned into pML1675. The plasmids pCDNA3.1-GABABR2-YFP, pEAK10 HA-FLAG-CXCR4-hRLuc, and pIRES puro3 FLAG-CXCR4-Venus were gifts from Dr. Michel Bouvier (16). For FLAG-DHHC3 and FLAG-G84, the murine DHHC3 cDNA or C-terminal domain of human Golgin 84 (amino acids 569–731) was subcloned into the pCMV5-FLAG vector. For DHHC3-GFP, the murine DHHC3 cDNA was subcloned into pEGFP-N3 (Clontech). Plasmids for murine myrLckNT, mouse DHHC3-FLAG-His6, DHHS3-FLAG-His6, and DHHC2-FLAG-His6 were described previously (15). The plasmids encoding catalytically inactive DHHC2(C156S) and DHHC3(C157S) were generated by site-directed mutagenesis (QuikChange). To construct a tandem DHHC3 dimer, the DHHC3 coding sequence was amplified with PCR primers to generate an XhoI and BamHI fragment that encodes DHHC3 flanked by a FLAG epitope at the N terminus and a thrombin cleavage site at the C terminus. The fragment was inserted into a plasmid in-frame with a linker sequence (GGGGS) fused to DHHC3. The integrity of all plasmid constructs was confirmed by DNA sequence analysis.
Bioluminescence Resonance Energy Transfer
BRET assays were conducted following published protocols (17). HEK-293 cells seeded on 12-well dishes were transfected with 5 ng of luciferase donor plasmid (DHHC3-RLuc, DHHC2-RLuc, DHHS3-RLuc, or DHHS2-RLuc) and increasing amounts of acceptor plasmid (DHHC3-YFP, YFP-DHHC3, YFP-G84, DHHC2-YFP, YFP-DHHC2, GABABR2-YFP, DHHS3-YFP, or DHHS2-YFP). Transfections were supplemented with empty vector to normalize total DNA added. For BRET control experiments with CXCR4, 100 ng of CXCR4-RLuc and 0–400 ng of CXCR4-Venus were used. Forty-eight hours after transfection, cells were detached from the plates with PBS-EDTA (5 min at 37 °C), transferred to a microcentrifuge tube, and collected by centrifugation at 300 × g for 5 min at room temperature. Cells were suspended in PBS-CaCl2/MgCl2 and 40 μl (∼25,000 cells) was dispensed into each well of a white 96-well plate (Costar 3912). After adding 10 μl of 25 μM coelenterazine h solution, the samples in the plate were incubated for 10 min at 37 °C. Emission was detected using a Biotek Gen5 plate reader equipped with the appropriate BRET filter set (460/40 nm and 528/20 nm). The BRET signal was calculated by the ratio of emission at 528 nm to emission at 460 nm minus the BRET in the absence of the acceptor. Luciferase expression was determined after 10 min of adding coelenterazine h. For YFP expression measurements, 40 μl of the cell suspension was distributed into black 96-well plates (Costar 3915), and readings were detected by excitation at 485/20 nm and emission at 528/20 nm. Background fluorescence measured in cells not expressing the acceptor (YFP0) was subtracted from fluorescence values measured in cells expressing the increasing amounts of acceptor (YFP) to obtain YFP-YFP0 values. BRET ratios were then plotted as a function of (YFP-YFP0)/LUC (luciferase) values. The data for saturation curves are fit using a nonlinear regression equation with a single binding site (GraphPad Prism) (17). BRET curves with DHHC proteins coexpressed with Golgin84 (Fig. 2A) or GABABR2 (C) were fit to a straight line.
FIGURE 2.
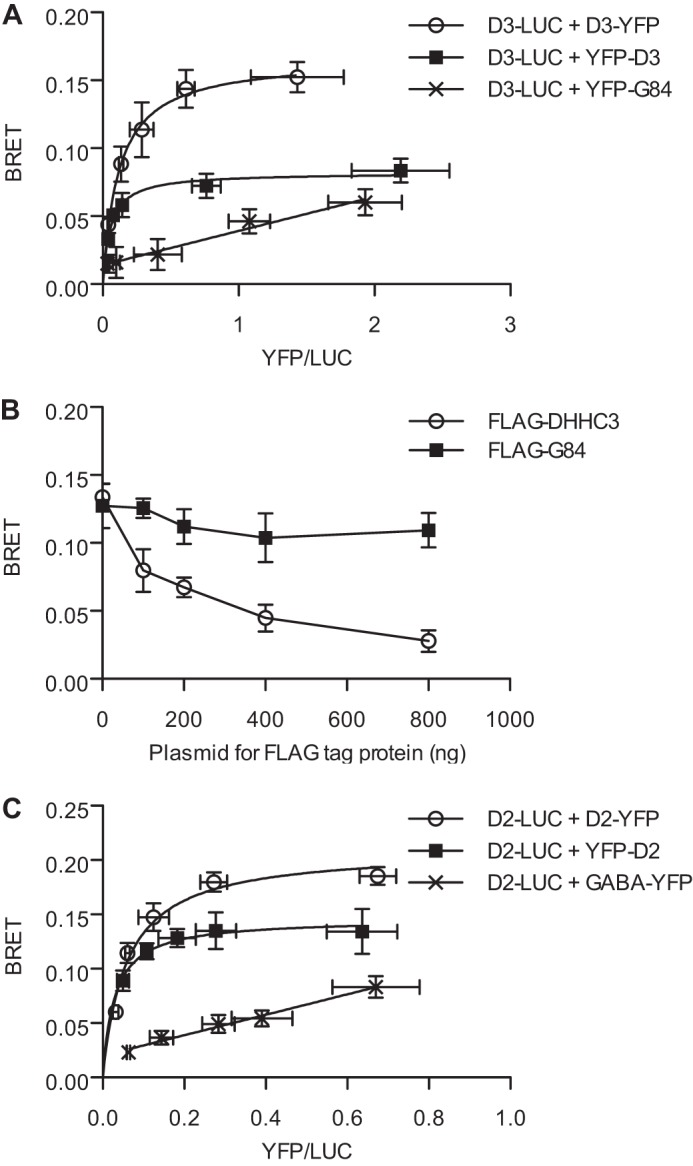
BRET assays to assess oligomerization of DHHC proteins. A, BRET saturation curve for DHHC3. BRET was measured in HEK-293 cells transfected with a fixed amount (5 ng) of DHHC3-RLuc (D3-LUC) with increasing concentrations (0–400 ng) of DHHC3-YFP (D3-YFP, ○) or YFP-DHHC3 (YFP-D3, ■). YFP-Golgin 84 (YFP-G84, ×) was coexpressed with D3-RLuc as a control to assess random interactions in Golgi membranes. Data shown are the net BRET ratio as a function of (YFP-YFP0)/LUC expression. B, competition BRET assay. HEK cells were transfected with fixed amounts of DHHC3- RLuc (5 ng) and DHHC3-YFP (200 ng), with increasing concentrations (0–800 ng) of competitor FLAG-DHHC3 (○) or FLAG-G84 (■) (0–800 ng), and monitored for BRET 48 h post-transfection. C, BRET saturation curve for DHHC2. BRET was measured in HEK-293 cells transfected with a fixed amount (5 ng) of DHHC2-RLuc plasmid DNA, and increasing concentrations (0–400 ng) of DHHC2-YFP (D2-YFP, ○), YFP-DHHC2 (YFP-D2, ■) or GABABR2-YFP (GABA-YFP, ×) were transfected into HEK cells. Data are presented as mean ± S.D. of results of three independent experiments.
For BRET experiments in cell membranes, cells were transfected with 5 ng of donor plasmid (DHHC3-RLuc or DHHS3-RLuc) and 200 ng of acceptor plasmid (DHHC3-YFP or DHHS3-YFP). For CXCR4 control experiment, 100 ng of CXCR4-RLuc and 200 ng of CXCR4-Venus plasmids were used. Cells expressing donor alone were used to calculate the BRET background. The cells were collected as described above, suspended in buffer A (20 mm Tris (pH7.4), 1 mm EDTA, protease inhibitors), and broken using a ball bearing cell homogenizer on ice. After centrifugation at 100,000 × g at 4 °C for 30 min, the membrane pellet was suspended in buffer A, and the protein concentration was measured by Bradford assay. Membranes (40 μg) were incubated with different concentration of palmitoyl-CoA for 10 min at room temperature. The samples were used immediately for the BRET assay as described above.
In Vitro PAT Assay
Recombinant baculoviruses expressing DHHC2 and DHHC3 were described previously. DHHC2-FLAG-His was expressed in insect Sf 9 cells and sequentially purified by nickel-nitrilotriacetic acid metal chelate and FLAG affinity chromatography as described (15). An N-terminal fragment encoding the first 226 amino acids of the N-myristoylated protein tyrosine kinase Lck, myrLckNT, was expressed in Escherichia coli with N-myristoyltransferase and purified as C-terminal His-6-tagged proteins using established protocols (15, 18).
For the PAT assay in Fig. 1, HEK-293 cells in 6-well plates were transfected with plasmid DNA (400 ng). After 48 h, the cells were collected, and membrane fractions prepared as above. The membrane pellet was suspended in buffer B (20 mm Tris (pH7.4), 1 mm EDTA, 300 mm sucrose, protease inhibitors), and the protein concentration was measured by Bradford assay. Membranes (22.5 μg in 15 μl) were incubated with 5 μl of myrLckNT (final concentration, 0.5 μM) and 30 μl of reaction mix (250 mm MES, 1.8 μM [3H]palm-CoA) at 25 °C for 10 min. The reactions were stopped by addition of 5× gel loading buffer with 10 mm TCEP and heated for 1 min at 50 °C. Reactions were divided for two SDS-PAGE gels and processed for fluorography or scintillation counting using established methods.
FIGURE 1.
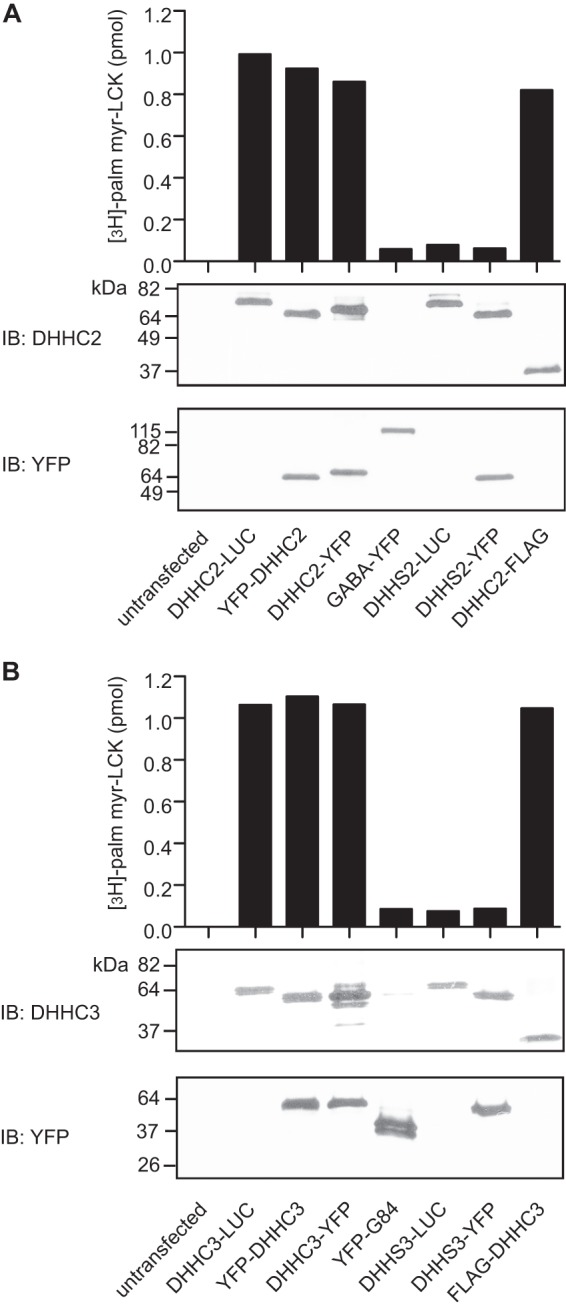
DHHC2 and DHHC3 fused to Renilla luciferase or YFP retain PAT activity. HEK-293 cells were transfected with plasmids encoding DHHC2 (A) or DHHC3 (B) fusion proteins. After 48 h, cell membranes were prepared and assayed for PAT activity using myrLckNT as a substrate (bar graph). The PAT activity is reported as picomoles of radioactive palmitate incorporated into myrLckNT from cell membranes expressing fusion proteins minus the background PAT activity for myrLckNT in membranes from untransfected cells (0.34 pmol (A) and 0.33 pmol (B)). Protein expression of DHHC fusion proteins was confirmed by immunoblot analysis (IB) (lower panels). DHHC fusion proteins were detected using DHHC2- or DHHC3-specific antibodies. YFP fusions were detected using an anti-GFP antibody. The GPCR GABABR2 (GABA) and Golgi structural protein Golgin 84 (G84) fused to YFP were used as controls in subsequent BRET experiments.
For the PAT assay in Fig. 6, the cross-linked enzyme preparations (plus or minus TCEP) were diluted in 5 μl of enzyme dilution buffer (50 mm MES (pH6.4), 100 mm NaCl, 10% glycerol, 0.1% DDM). Final assay concentrations were 25 nm DHHC2, 1 μM [3H]palm-CoA, and 0.5 μM myrLckNT in a total of 25 μl. After incubation for 10 min at 25 °C, the reactions were stopped, the samples were resolved by SDS-PAGE, and radioactive palmitate incorporated into the protein substrate was quantified by scintillation counting.
FIGURE 6.
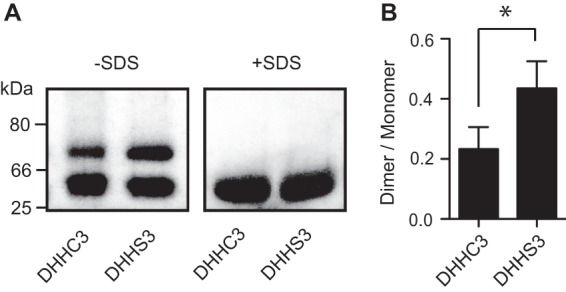
Purified DHHC3 behaves as a monomer and a dimer on blue native gel electrophoresis. A, purified DHHC3-FLAG-His-6 or DHHS3-FLAG-His-6 were resolved by blue native gel electrophoresis in the presence or absence of SDS as described under “Experimental Procedures.” Transferrin (80 kDa), BSA (66 kDa), and chymotrypsin (25 kDa) were used as molecular weight markers. B, blue native gels were scanned directly, and the ratio of dimer and monomer bands was quantified using ImageJ. Data are presented as mean ± S.D. of three experiments. *, p < 0.05.
Coimmunoprecipitation
HEK-293 cells in 6-well plates were transfected with plasmids encoding fusion proteins (400 ng each) for 48 h. The cells were collected and solubilized by adding 500 μl of lysis buffer (50 mm Tris (pH7.4), 150 mm NaCl, 10% glycerol, 1% DDM, protease inhibitors), followed by an incubation on a rotator at 4 °C for 2 h. After ultracentrifugation at 100,000 × g for 30 min at 4 °C, 400 μl of the supernatant was incubated with 20 μl of anti-FLAG resin (equilibrated in lysis buffer containing 0.1% DDM) for 2 h at 4 °C. The resin was washed four times with 1 ml of lysis buffer containing 0.1% DDM. Protein was eluted from the resin with 80 μl of 1.5× gel loading buffer with 30 mm DTT, incubating at 37 °C for 30 min. After heating to 50 °C for 1 min, the samples were resolved by SDS-PAGE and processed for immunoblotting.
Cross-linking
DHHC2-FLAG-His-6 protein was purified by sequential nickel-nitrilotriacetic acid metal chelate and FLAG affinity chromatography. Protein was eluted from the anti-FLAG M2 affinity gel in buffer containing 25 mm sodium phosphate (pH7.4), 150 mm NaCl, 0.1% DDM, and 0.25 μg/μL FLAG peptide. The purified proteins were incubated with DTSSP in 80 μl at 25 °C for 30 min. The final DHHC2 concentration was 1 μm. Reactions were stopped by adding 1 m Tris buffer (pH 7.4) to 50 mm final concentration and incubated at 25 °C for 30 min. For the samples plus TCEP, a TCEP stock (100 mm TCEP dissolved in 1 m Tris (pH7.4), 0.1% DDM) was added to a final concentration of 5 mm. For the samples minus TCEP, the same volume of buffer (1 m Tris (pH7.4), 0.1% DDM) was added. All samples were incubated at 25 °C for 30 min prior to PAT assay or immunoblot analyses.
Blue Native PAGE
Blue native PAGE was performed as described, with minor modifications (19). Detergent-solubilized DHHC3-FLAG-His-6 or DHHS3-FLAG-His-6 purified by nickel-nitrilotriacetic acid metal chelate chromatography (20 μl) was mixed with 6 μl of 50% glycerol and 1 μl of 5% (w/v) Brilliant Blue G (Sigma). Half of the samples were supplemented with SDS (final concentration of 0.2%) to denature native protein complexes. The samples were applied to a 3–13% native gel and electrophoresed on ice, first in cathode buffer B (50 mm Tricine, 7.5 mm imidazole, 0.02% Brilliant blue G) for 30 min at 100 V, and then in cathode buffer B/10 (50 mm Tricine, 7.5 mm imidazole, 0.002% Brilliant blue G) for 40 min at 200 V. The gels were scanned, and the ratio of dimer and monomer bands were quantified using ImageJ.
Thrombin Treatment of Tandem DHHC3 Protein
HEK-293 cells were transfected with plasmids expressing FLAG-DHHC3 or FLAG-DHHC3-thrombin cleavage site-DHHC3. After 48 h, the cells were collected and solubilized in lysis buffer (50 mm Tris (pH7.4), 150 mm NaCl, 10% glycerol, 1% DDM, protease inhibitors), followed by an incubation on a rotator at 4 °C for 2 h. After ultracentrifugation at 100,000 × g for 30 min at 4 °C, 600 μl of the supernatant was incubated with 30 μl of anti-FLAG resin (equilibrated in lysis buffer containing 0.1% DDM) for 2 h at 4 °C. The resin was washed four times with 1 ml of lysis buffer containing 0.1% DDM. Protein was eluted from the resin by 500 ng/μl FLAG peptide. The purified proteins were incubated with thrombin at a 1:25 ratio at 25 °C. At different time points, the treatment was stopped by gel loading buffer for SDS-PAGE or used immediately for an in vitro PAT assay.
Statistics
Data are expressed as means ± S.D. For comparisons between two groups, Student's t test (two-tailed) was used. p < 0.05 was considered statistically significant.
RESULTS
DHHC Proteins Oligomerize in Mammalian Cells
To determine whether DHHC proteins exist as dimers or higher-order oligomers in living cells, BRET was used to detect homomeric interactions of DHHC palmitoyltransferases in living cells. The bioluminescent enzyme Renilla luciferase was fused to the C terminus of DHHC2 or DHHC3 (DHHC2-RLuc, DHHC3-RLuc). YFP fusions at the N terminus and at the C terminus were generated. Control fusion proteins included GABABR2-YFP, a G protein-coupled receptor (GPCR) for GABA, and YFP-Golgin 84, a tail-anchored Golgi structural protein (20). The constructs were designed so that luciferase and YFP were exposed to the cytoplasm. Following transfection in HEK-293 cells, all of the fusion proteins were expressed at the appropriate molecular weights (Fig. 1, A and B, bottom two panels). PAT activity of the DHHC fusion proteins was confirmed by in vitro assays using an N-terminal fragment of N-myristoylated Lck as a substrate (Fig. 1, A and B, top panels). Catalytically inactive DHHC protein fusions were generated by mutation of the DHHC cysteine to serine and are designated as DHHS2 and DHHS3 (Fig. 1).
BRET saturation curves were generated to determine whether DHHC proteins oligomerize. A constant amount of DHHC3-RLuc plasmid was transfected with increasing amounts of DHHC3-YFP, YFP-DHHC3, or YFP-Golgin 84 plasmid. DHHC3-RLuc displayed saturable BRET signals irrespective of whether the YFP moiety was appended at the N- or C-terminus (Fig. 2A). Because BRET is a measure of proximity, proteins that are colocalized on the same membrane may display a BRET signal because of random collisions, but the signal is typically smaller than that driven by protein-protein interaction and does not saturate with increasing expression. DHHC3 is localized at the Golgi apparatus. As a control for random interactions of DHHC3 with other Golgi-localized proteins, we tested DHHC3-RLuc with the integral membrane protein YFP-Golgin 84. Consistent with their proximity in Golgi membranes, DHHC3-RLuc and YFP-Golgin 84 coexpression resulted in a BRET signal. However, the signal was lower than that seen with DHHC3 pairs and was linear with respect to increasing ratios of YFP:RLuc expression (Fig. 2A).
A competition assay was used to confirm the specificity of the protein interaction suggested by the BRET saturation curves. In this experiment, increasing amounts of DHHC3 tagged with the FLAG epitope were coexpressed with DHHC3-RLuc and DHHC3-YFP. The BRET signal was decreased with increasing expression of the FLAG-tagged DHHC3 construct, whereas expression of FLAG-Golgin 84 had a minimal effect (Fig. 2B).
Similar experiments were performed with DHHC2 to determine whether the propensity to oligomerize was found in other DHHC proteins. Although DHHC2 and DHHC3 share similar membrane topologies, their sequence homology is only 21%, and they are localized in different membrane compartments. Although DHHC3 is localized in the Golgi, DHHC2 is found in recycling endosomes and at the plasma membrane. DHHC2-RLuc displayed saturable BRET interactions with YFP-DHHC2 and DHHC2-YFP (Fig. 2C). For a random collision control, the GPCR GABABR2-YFP was assayed with DHHC2-RLuc. The positive BRET signal for DHHC2 and the GABA receptor indicated proximity in the same membrane compartment, but this BRET pair did not display saturation kinetics (Fig. 2C). Interestingly, the maximal BRET values for both DHHC2 and DHHC3 were higher with the YFP appended to the C terminus versus the N terminus, demonstrating that the orientation of the proteins has an effect on BRET signals (Fig. 2, A and C).
Oligomerization of DHHC Proteins Is Regulated by the Activation State of the Enzyme
We sought to determine whether enzyme activity influenced the oligomerization status of DHHC2 and DHHC3. BRET assays were performed to compare DHHC3 or DHHC2 with their respective catalytically inactive DHHS mutant proteins. The BRET signal for the DHHS mutants of DHHC3 and DHHC2 was significantly higher than that of the wild-type proteins (Fig. 3, A and B). When DHHC3-RLuc was expressed with catalytically inactive DHHS3-YFP, a BRET signal intermediate between the active and inactive forms of the enzyme was measured (Fig. 3C). These results suggest that the active and inactive forms of the enzyme exist as oligomers, with the inactive state of the enzyme favoring the oligomeric state.
FIGURE 3.

Regulation of DHHC protein oligomerization. A, BRET saturation curves were performed in cells coexpressing wild-type DHHC3-RLuc and DHHC3-YFP (D3, ○) or in cells coexpressing the catalytically inactive DHHS3-RLuc and DHHS3-YFP (S3, ■). B, BRET saturation curves were performed in cells coexpressing wild-type DHHC2-RLuc and DHHC2-YFP (D2, ○) or coexpressing the catalytically inactive DHHS2-RLuc and DHHS2-YFP (S2, ■). C, BRET saturation curves were performed as in A, with the inclusion of a curve for wild-type DHHC3-RLuc coexpressed with catalytically inactive DHHS3-YFP to detect DHHC3/DHHS3 hetero-oligomers (D3 + S3, ×). The effect of 2-BP was assessed on BRET pairs, wild-type DHHC3 (D), wild-type DHHC2 (E), the GPCR CXCR4 (F), catalytically inactive DHHS3 (G), and catalytically inactive DHHS2 (H). HEK-293 cells were transfected and, 24 h later, treated with or without 100 μm 2-BP. BRET saturation curves were generated ∼16 h after treatment with 2-BP (control, ○; 2-BP treatment, ■). Data are presented as mean ± S.D. of results of three independent experiments.
As an independent means of assessing the effect of enzyme activity on oligomerization, we used an inhibitor of palmitoylation, 2-bromopalmitate (2-BP). 2-BP inhibits palmitoylation of proteins in cells and DHHC PAT activity in vitro. BRET assays were performed on cells expressing DHHC protein fusions following an overnight treatment with 2-BP. The BRET signal produced by the DHHC3 constructs was modestly but significantly increased by 2-BP treatment compared with the vehicle control (Fig. 3D). A similar effect of 2-BP on DHHC2 oligomerization was observed (Fig. 3E). 2-BP is pleiotropic in its effects. To assess whether the increased BRET signal observed with 2-BP treatment was due to a general perturbation of membranes or protein aggregation, we used as a control, Chemokine (C-X-C motif) receptor 4 (CXCR4), a GPCR that was characterized previously as an oligomer using BRET (16). In contrast to the DHHC proteins, treatment with 2-BP modestly inhibited the CXCR4 BRET signal (Fig. 3F). Thus, the enhancement of the BRET signal observed with 2-BP treatment of DHHC2 and DHHC3 is not common to all BRET pairs. Importantly, 2-BP had no effect on the BRET signal of the catalytically inactive mutants (Fig. 3, G and H). Taken together, these results suggest a strong correlation between oligomerization and the inactive enzyme.
To begin to probe the mechanism that underlies oligomerization of DHHC proteins, we assessed whether multimeric forms of the enzyme could be detected in vitro. Membrane preparations from HEK-293 cells expressing DHHC3-RLuc and DHHC3-YFP were tested in a BRET assay, and those expressing a DHHC3 BRET pair displayed significantly higher signals than membranes from cells expressing DHHC3 with a Golgi-localized protein (Fig. 4A). Consistent with the observations in live cells, the BRET signal for the catalytically inactive mutant was higher than that of the active enzyme. In their reaction cycle, DHHC proteins form a palmitoyl-enzyme intermediate in the first step of the reaction, followed by transfer of the same palmitoyl moiety to the protein substrate. The reaction cycle is dependent upon the cysteine of the DHHC motif. We predicted that addition of palmitoyl-CoA to membranes would promote activation of the enzyme and diminish the BRET signal induced by oligomerization of DHHC3. BRET between DHHC3-Rluc and DHHC3-YFP decreased in a dose-dependent fashion when membranes were incubated with increasing concentrations of palmitoyl-CoA. The BRET signal for catalytically inactive DHHS3 was only diminished slightly (Fig. 4, B and C). The highest concentration of palmitoyl-CoA used in these experiments had no effect on CXCR4 BRET (Fig. 4D), excluding a nonspecific effect of palmitoyl-CoA addition to membranes.
FIGURE 4.
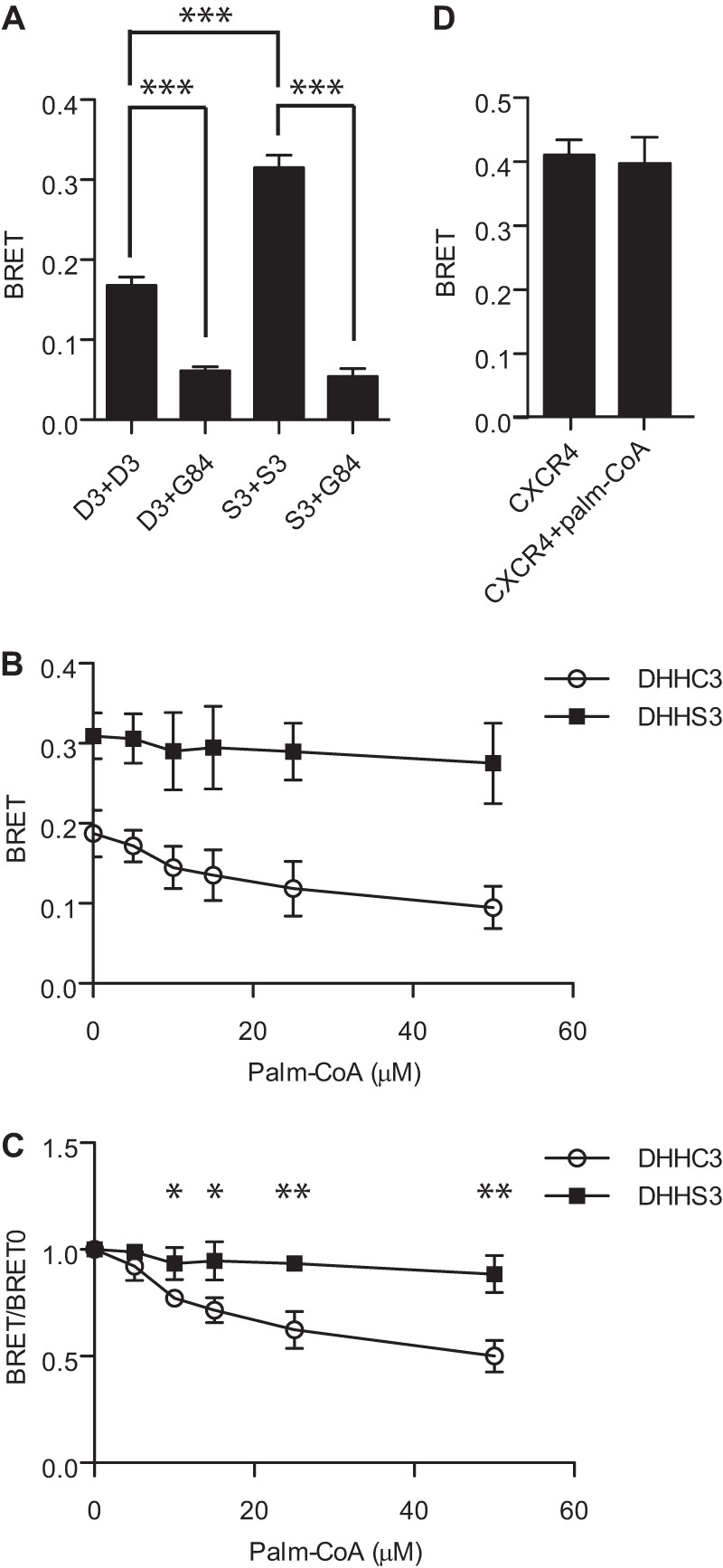
Palmitoyl-CoA inhibits BRET of wild-type but not catalytically inactive DHHC3 in cell membranes. A, detection of DHHC3 oligomerization in vitro. BRET assays were performed on membranes prepared from cells transfected with the BRET pairs DHHC3-RLuc and DHHC3-YFP, DHHC3-RLuc and YFP-Golgin 84, DHHS3-RLuc and DHHS3-YFP, and DHHS3-RLuc and YFP-Golgin 84. Transfections were performed with 5 ng of plasmid DNA for luciferase fusions and 200 ng of plasmid DNA for YFP fusions. B, BRET assays were performed on membranes from cells expressing the wild-type DHHC3 BRET pair (D3, ○) or the catalytically inactive DHHS3 BRET pair (S3, ■) following incubation with increasing concentrations of palmitoyl-CoA at 25 °C for 10 min. C, replot of data from B, in which the net BRET signal for wild-type or catalytically inactive DHHC3 are normalized to the value of the samples without palmitoyl-CoA. D, as a control for nonspecific effects of palmitoyl-CoA, membranes from cells transfected with the BRET pair CXCR4-RLuc (100 ng) and CXCR4-Venus (200 ng) were assayed immediately following treatment with or without 50 μm palmitoyl-CoA at 25 °C for 10 min. Data are presented as mean ± S.D. of results of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We also analyzed oligomerization by coimmunoprecipitation of detergent-solubilized proteins. Wild-type or catalytically inactive DHHC3 tagged with GFP was expressed with FLAG-tagged versions of DHHC3. DHHC3-GFP was found in FLAG immunoprecipitates of FLAG-DHHC3 or FLAG-DHHS3 but not with FLAG-tagged Golgin 84 (Fig. 5). Catalytically inactive DHHS3 was more abundant in the immunoprecipitates than the wild-type enzyme, which is consistent with the BRET studies and supporting the contention that oligomerization favors the inactive state of the enzyme.
FIGURE 5.
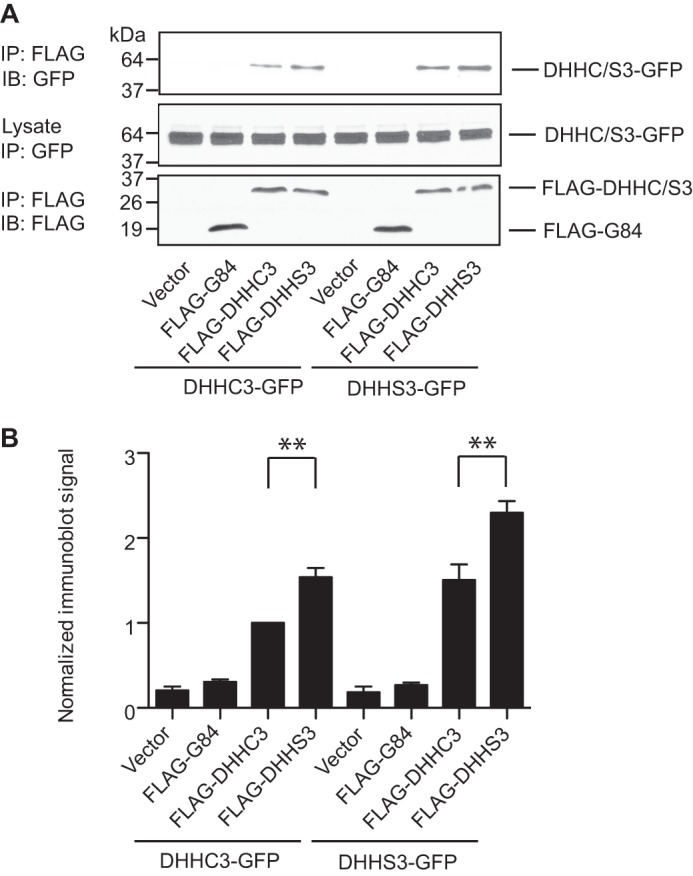
Oligomerization of DHHC3 detected by coimmunoprecipitation. A, DHHC3-GFP or DHHS3-GFP were transfected into HEK cells with empty vector, FLAG-Golgin 84, FLAG-DHHC3, or FLAG-DHHS3. After 48 h, the cells were collected, and detergent extracts were prepared by solubilization with 1% DDM and immunoprecipitated (IP) with FLAG antibodies. Proteins in the immunoprecipitates were detected by GFP and FLAG immunoblot analyses (IB). Wild-type and catalytically inactive DHHC3 tagged with GFP was coimmunoprecipitated with their FLAG-tagged counterparts but not with FLAG-tagged Golgin 84. The amount of catalytically inactive DHHS3-GFP pulled down in the immunoprecipitates was consistently higher than the wild-type protein. Data are representative of one of three independent experiments. B, statistical analysis of the coimmunoprecipitation experiments. The results from anti-GFP immunoblots for coimmunoprecipitation were quantified using ImageJ. The data were normalized to the sample of FLAG-DHHC3 + GFP-DHHC3. Error bars represent the mean ± S.D. of three independent experiments. **, p < 0.01.
We used blue native gel electrophoresis to assess the oligomeric status of DHHC3 purified from insect cells. In the absence of SDS, both wild-type and catalytically inactive DHHC3 migrated as two bands. When incubated in the presence of SDS, the two forms collapsed into a single band that migrates at a position consistent with the monomeric molecular weight of 34 kDa. The upper band migrates between the 66- and 80-kDa markers, suggesting that it represents a dimer. The dimeric form of the enzyme is more abundant in the catalytically inactive DHHS3 preparation (Fig. 6B).
Monomeric DHHC Proteins Display Higher Activity Than Their Dimeric Counterparts
The results from the blue native gel electrophoresis suggest that DHHC proteins exist in a monomer-dimer equilibrium, with catalytically inactive DHHS3 favoring formation of the dimer. To assess PAT activity of the monomeric and dimeric forms of the enzyme, we used the reversible cross-linker DTSSP to trap the oligomeric state of the enzyme. Purified DHHC2 incubated with DTSSP and resolved by SDS-PAGE displayed a concentration-dependent increase in the dimeric form of the enzyme that could be reversed by addition of the reducing agent TCEP (Fig. 7A). Parallel samples were assayed for PAT activity (Fig. 7B). PAT activity was reduced in the presence of DTSSP, but enzyme activity could be recovered partially when TCEP was added to disrupt dimerization.
FIGURE 7.
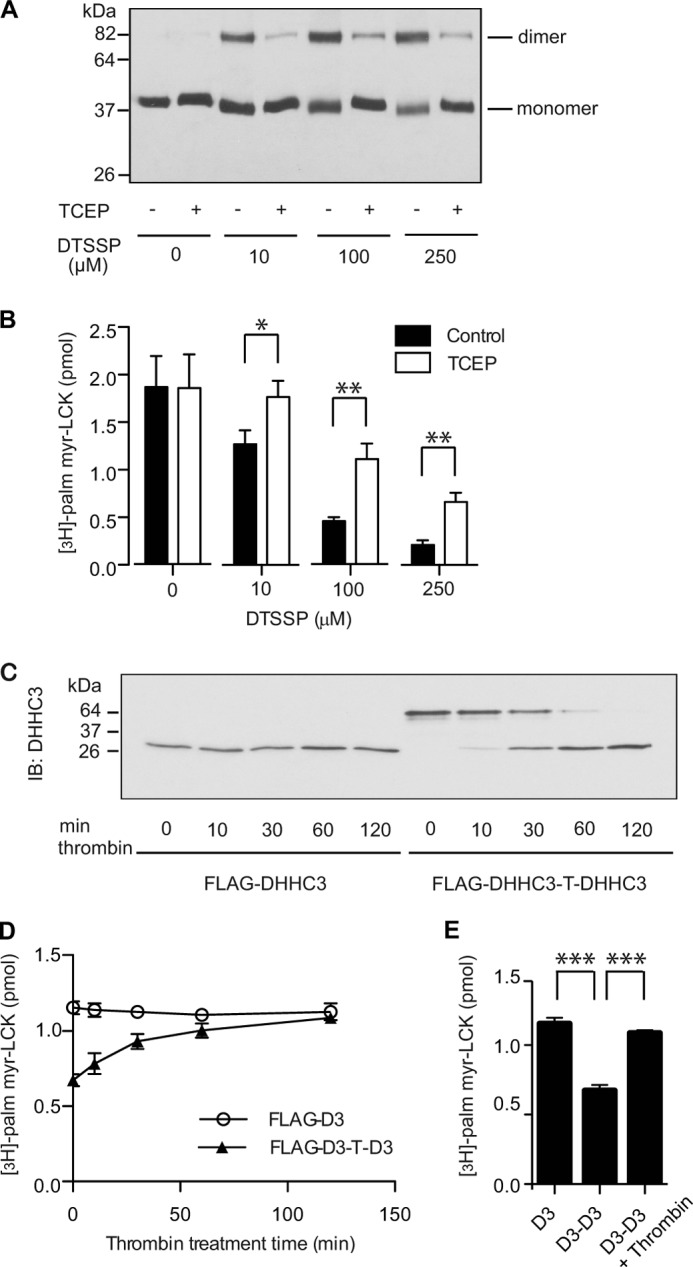
Dimeric DHHC3 is less active than monomeric DHHC3. A, cross-linking of DHHC2 with DTSSP stabilizes the dimeric form of the enzyme. Purified DHHC2-FLAG-His-6 was incubated with increasing concentration of DTSSP at 25 °C for 30 min. The reaction was stopped by Tris buffer. Half of the samples were treated with TCEP to reverse cross-linking. The samples were resolved by SDS-PAGE and detected in immunoblots with anti-FLAG antibody. B, reversal of cross-linking in DHHC2 increases PAT activity. Samples prepared as in A were assayed for PAT activity using myrLCKNT as a substrate. [3H]palmitate incorporation was quantified by scintillation spectrometry. Data are representative of one of three independent experiments. Error bars represent the mean ± S.D. of triplicate samples from a single experiment. C, D, and E, monomeric DHHC3 has higher PAT activity than dimeric DHHC3. FLAG-DHHC3 or FLAG-DHHC3-T-DHHC3 was purified from HEK-293 cells as described under “Experimental Procedures” and incubated with thrombin. The preparations were sampled at various time points and assayed by immunoblot (IB) to monitor the extent of thrombin cleavage (C) and assayed for PAT activity (D). D, data are representative of one of three independent experiments. Error bars represent the mean ± S.D. of triplicate samples from a single experiment. E, statistical analysis of the PAT activity in D of FLAG-DHHC3 (at time = 0), FLAG-DHHC3-T-DHHC3 (at time = 0), and the thrombin-treated FLAG-DHHC3-T-DHHC3 (at time = 120). Error bars represent the mean ± S.D. of triplicate samples. ***, p < 0.001.
The reduced activity of the cross-linked enzyme could be due to inactivation of the enzyme through means other than restricting dissociation of the monomer. As a second approach to assess PAT activity of the monomeric and dimeric forms of the enzyme, a stable dimer was generated by linking two DHHC3 molecules with a thrombin cleavage site. After purification from HEK-293 cells, the proteins were treated with thrombin to release monomeric DHHC3 (Fig. 7C). PAT activity was monitored over the time course of thrombin cleavage. In the absence of thrombin treatment, dimeric DHHC3 was less active than the monomer. PAT activity increased with time as the monomer was released by thrombin cleavage (Fig. 7, D and E). These results suggest that the PAT activity of DHHC3 is modulated by its oligomeric status.
DISCUSSION
In this study, we examined whether DHHC proteins self-associate in cells and in vitro. Using BRET, we detected homomultimerization of DHHC2 and DHHC3 in intact cells and in membrane preparations. Evidence that the BRET assays were reporting a protein-protein interaction as opposed to crowding of the proteins in cell membranes is as follows. First, the DHHC2 and DHHC3 BRET pairs displayed saturation kinetics when donor protein expression was held constant in the presence of increasing acceptor protein. Second, DHHC3 BRET was specifically and dose-dependently competed by expression of untagged DHHC3. Third, saturable BRET signals were not observed when DHHC2-Rluc or DHHC3-RLuc was expressed with YFP fusions to integral membrane proteins localized in the same membrane compartment as the DHHC protein. As an independent measure of oligomerization, we used coimmunoprecipitation of differentially epitope-tagged constructs to pull down homomeric complexes of DHHC3. Our results confirm a prior report of DHHC3 homomultimerization (13).
The DHHC proteins that we analyzed do not appear to be stable multimeric complexes. The BRET data suggest that DHHC2 and DHHC3 oligomers are dynamic in cell membranes. Although the number of subunits in the different oligomeric states in native membranes is unknown, our data with purified enzyme in detergent solution suggest that monomers and dimers predominate. However, Fang et al. (13) immunoprecipitated cross-linked dimers and trimers of DHHC3 from transfected mammalian cells (13). We occasionally observed molecular weight species larger than dimers in our experiments, but their appearance was inconsistent. The amount of dimer detected in detergent solution was modest relative to the monomeric protein (Fig. 6). It is unclear at present whether the proportion of monomer and dimer in detergent solution is representative of their status in cell membranes or whether detergent solubilization promotes dissociation. Future experiments will assess oligomerization of purified DHHC proteins reconstituted into artificial phospholipid bilayers.
Interestingly, a single amino acid change that renders the enzyme catalytically inactive is sufficient to increase oligomerization. We cannot exclude the possibility that a conformational change induced by the change in amino acid is sufficient to promote self-association. However, it seems more likely that posttranslational modification of the enzyme with palmitate plays a role. DHHC proteins are palmitoylated at steady state when expressed ectopically in mammalian cells and yeast (21). Palmitoylation of the catalytically inactive mutant of DHHC3 was essentially absent when expressed in yeast (21) or reduced substantially when expressed in mammalian cells3. The number and identity of the palmitoylation sites in DHHC2 and DHHC3 is unknown at present, although palmitoylation is at least partially dependent upon the DHHC cysteine. Palmitoylation of integral membrane proteins increases their affinity for lipid rafts (22), and it is possible that the lipid environment has an impact on the propensity of the enzyme to oligomerize.
It will be important to determine whether our findings of DHHC2 and DHHC3 oligomerization extend to other members of the DHHC protein family. It has been reported that DHHC3 forms homomultimers and heteromultimers with DHHC7 (13). DHHC7 is closely related by sequence to DHHC3, and they display similar subcellular localization and overlapping substrate specificity (3, 13). It was suggested that formation of heteromultimers might account for the dominant negative effects of expressing catalytically inactive DHHC proteins in cells (13). If, as our findings suggest, that the oligomer represents a less active state of the enzyme, then sequestration of a DHHC protein in oligomers with a catalytically inactive protein represents a possible mechanism to explain its dominant inhibitory behavior.
We observed that oligomerization of DHHC2 and DHHC3 was increased when enzyme activity was inhibited chemically or by mutation, whereas addition of the substrate palmitoyl-CoA to cell membranes promoted dissociation of DHHC oligomers. These results suggest that the oligomeric form of the enzyme might be inactive. However, our experiments with a stable DHHC3 dimer demonstrated that a covalently linked dimer is active as an enzyme but less active than the monomeric species. It is possible that the covalently linked dimer may have sufficient conformational flexibility to functionally dissociate, leading to a switch from an inactive to active state, but at present we can only conclude that enzyme activity is modulated by the oligomeric state of the DHHC protein.
Our working model of a potential monomer-dimer equilibrium for DHHC proteins has precedent in the behavior of other integral membrane proteins. There is emerging evidence that oligomerization is a dynamic process for some GPCRs (23). Dimerization of GPCRs has been studied extensively, but its functional consequences remain incompletely understood. There is consensus that class C GPCRs form obligate and stable dimers (24). Studies suggest that at least some members of the much larger class A family of GPCRs exist in a monomer-dimer equilibrium (23). A recent single-molecule imaging study demonstrated that the M1-muscarinic receptor dimers form and dissociate on a time scale of seconds, with about 30% of the receptors existing as dimers at a given time (25). A second example is the EGF receptor, which undergoes ligand-induced dimerization and activation. Dimerization of the EGF receptor has also been observed in the absence of ligand where the receptor is in a monomer-dimer equilibrium (26). The functional consequence of ligand-independent dimerization may be an inhibition of kinase activity (27, 28), but there is also evidence to suggest that dimerization is a prerequisite for EGF binding (29). The striking and intriguing correlation that we observe between the oligomeric state of DHHC proteins and their activity suggests that DHHC PATs may represent another example of protein activity regulated by dynamic self-association.
Acknowledgments
We thank Dr. Michel Bouvier for plasmids and advice on BRET, Wendy Greentree for technical support, and members of the laboratory for helpful discussions. We also thank Dr. Benjamin Jennings for training J. L. in the enzymology of DHHC proteins and for providing purified myr-LckNT.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1GM51466 (to M. E. L.).
C. Gottlieb and M. E. Linder, unpublished data.
- PAT
- protein acyltransferase
- RLuc
- Renilla luciferase
- LUC
- luciferase
- BRET
- bioluminescence resonance energy transfer
- TCEP
- tris(2-carboxyethyl)phosphine
- DTSSP
- 3,3′-dithiobis(sulfosuccinimidylpropionate)
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- GPCR
- G protein-coupled receptor
- 2-BP
- 2-bromopalmitate
- [3H]palm-CoA
- [9,10-3H]palmitoyl-CoA.
REFERENCES
- 1. Linder M. E., Deschenes R. J. (2007) Palmitoylation. Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 8, 74–84 [DOI] [PubMed] [Google Scholar]
- 2. Fukata Y., Fukata M. (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175 [DOI] [PubMed] [Google Scholar]
- 3. Greaves J., Chamberlain L. H. (2011) DHHC palmitoyl transferases. Substrate interactions and (patho)physiology. Trends Biochem. Sci. 36, 245–253 [DOI] [PubMed] [Google Scholar]
- 4. Batistic O. (2012) Genomics and localization of the Arabidopsis DHHC-cysteine-rich domain S-acyltransferase protein family. Plant Physiol. 160, 1597–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Young F. B., Butland S. L., Sanders S. S., Sutton L. M., Hayden M. R. (2012) Putting proteins in their place. Palmitoylation in Huntington disease and other neuropsychiatric diseases. Prog. Neurobiol. 97, 220–238 [DOI] [PubMed] [Google Scholar]
- 6. Lobo S., Greentree W., Linder M., Deschenes R. (2002) Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J. Biol. Chem. 277, 41268–41273 [DOI] [PubMed] [Google Scholar]
- 7. Roth A. F., Feng Y., Chen L., Davis N. G. (2002) The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J. Cell Biol. 159, 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mitchell D. A., Mitchell G., Ling Y., Budde C., Deschenes R. J. (2010) Mutational analysis of Saccharomyces cerevisiae Erf2 reveals a two-step reaction mechanism for protein palmitoylation by DHHC enzymes. J. Biol. Chem. 285, 38104–38114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jennings B. C., Linder M. E. (2012) DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J. Biol. Chem. 287, 7236–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitchell D. A., Vasudevan A., Linder M. E., Deschenes R. J. (2006) Protein palmitoylation by a family of DHHC protein S-acyltransferases. J. Lipid Res. 47, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 11. Politis E. G., Roth A. F., Davis N. G. (2005) Transmembrane topology of the protein palmitoyl transferase Akr1. J. Biol. Chem. 280, 10156–10163 [DOI] [PubMed] [Google Scholar]
- 12. Fukata M., Fukata Y., Adesnik H., Nicoll R. A., Bredt D. S. (2004) Identification of PSD-95 palmitoylating enzymes. Neuron 44, 987–996 [DOI] [PubMed] [Google Scholar]
- 13. Fang C., Deng L., Keller C. A., Fukata M., Fukata Y., Chen G., Lüscher B. (2006) GODZ-mediated palmitoylation of GABA(A) receptors is required for normal assembly and function of GABAergic inhibitory synapses. J. Neurosci. 26, 12758–12768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noritake J., Fukata Y., Iwanaga T., Hosomi N., Tsutsumi R., Matsuda N., Tani H., Iwanari H., Mochizuki Y., Kodama T., Matsuura Y., Bredt D. S., Hamakubo T., Fukata M. (2009) Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J. Cell Biol. 186, 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jennings B. C., Nadolski M. J., Ling Y., Baker M. B., Harrison M. L., Deschenes R. J., Linder M. E. (2009) 2-Bromopalmitate and 2-(2-hydroxy-5-nitro-benzylidene)-benzo[b]thiophen-3-one inhibit DHHC-mediated palmitoylation in vitro. J. Lipid Res. 50, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamdan F. F., Percherancier Y., Breton B., Bouvier M. (2006) Monitoring protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET). Curr. Protoc. Neurosci., Chapter 5, Unit 5.23 [DOI] [PubMed] [Google Scholar]
- 17. Achour L., Kamal M., Jockers R., Marullo S. (2011) Using quantitative BRET to assess G protein-coupled receptor homo- and heterodimerization. Methods Mol. Biol. 756, 183–200 [DOI] [PubMed] [Google Scholar]
- 18. Mumby S. M., Linder M. E. (1994) Myristoylation of G-protein a subunits. Methods Enzymol. 237, 254–268 [DOI] [PubMed] [Google Scholar]
- 19. Wittig I., Braun H. P., Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 20. Misumi Y., Sohda M., Tashiro A., Sato H., Ikehara Y. (2001) An essential cytoplasmic domain for the Golgi localization of coiled-coil proteins with a COOH-terminal membrane anchor. J. Biol. Chem. 276, 6867–6873 [DOI] [PubMed] [Google Scholar]
- 21. Ohno Y., Kashio A., Ogata R., Ishitomi A., Yamazaki Y., Kihara A. (2012) Analysis of substrate specificity of human DHHC protein acyltransferases using a yeast expression system. Mol. Biol. Cell 23, 4543–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levental I., Lingwood D., Grzybek M., Coskun U., Simons K. (2010) Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. U.S.A. 107, 22050–22054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lambert N. A. (2010) GPCR dimers fall apart. Sci. Signal. 3, pe12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gurevich V. V., Gurevich E. V. (2008) How and why do GPCRs dimerize? Trends Pharmacol. Sci. 29, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hern J. A., Baig A. H., Mashanov G. I., Birdsall B., Corrie J. E., Lazareno S., Molloy J. E., Birdsall N. J. (2010) Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. U.S.A. 107, 2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Endres N. F., Engel K., Das R., Kovacs E., Kuriyan J. (2011) Regulation of the catalytic activity of the EGF receptor. Curr. Opin. Struct. Biol. 21, 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu X., Sharma K. D., Takahashi T., Iwamoto R., Mekada E. (2002) Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 13, 2547–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., Mellman I. (2010) Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–787 [DOI] [PubMed] [Google Scholar]