

Background: Structural determinants of ADAM17 substrate specificity are unknown.
Results: ADAM17 activity affected by noncatalytic domains and secondary structure of substrates.
Conclusion: Noncatalytic domains and substrate conformation are potentially the key structural elements that determine ADAM17 specificity.
Significance: Understanding interaction of ADAM17 with its substrates will assist in discovery ADAM isoform- and substrate-specific inhibitors.
Keywords: ADAM ADAMTS, Cell Surface Enzymes, Enzyme Inhibitors, Enzyme Kinetics, Enzyme Mechanisms, Exosite Inhibitor, Exosite Substrate, Noncatalytic Domains, Substrate Conformation
Abstract
ADAM proteases are implicated in multiple diseases, but no drugs based on ADAM inhibition exist. Most of the ADAM inhibitors developed to date feature zinc-binding moieties that target the active site zinc, which leads to a lack of selectivity and off target toxicity. Targeting secondary substrate binding sites (exosites) can potentially work as an alternative strategy for drug discovery; however, there are only a few reports of potential exosites in ADAM protease structures. In the study presented here, we utilized a series of TNFα-based substrates to probe ADAM10 and 17 interactions with its canonical substrate to identify the structural features that determine ADAM protease substrate specificity. We found that noncatalytic domains of ADAM17 did not directly bind the substrates used in the study but affected the binding nevertheless, most likely because of steric hindrance. Additionally, noncatalytic domains of ADAM17 affected the size/shape of the carbohydrate-binding pocket contained within the catalytic domain of ADAM17. This suggests that noncatalytic domains of ADAM17 play a role in substrate specificity and might help explain differences in substrate repertoires of ADAM17 and its closest homologue, ADAM10. We also addressed the question of which substrate features can affect ADAM protease specificity. We found that all ADAM proteases tested (i.e., ADAM10, 12, and 17) significantly decreased activity when the TNFα-derived sequence was induced into α-helical conformation, suggesting that conformation plays a role in determining ADAM protease substrate specificity. These findings can help in the discovery of ADAM isoform- and substrate-specific inhibitors.
Introduction
ADAM17 is a prototypical member of a disintegrin and metalloproteinase family of metzincin proteases (1), which has been implicated in several aggressive forms of cancer (2, 3), rheumatoid arthritis (4), and Alzheimer disease (5). The main contribution of ADAM17 to biological and pathophysiological processes stems from its ability to “shed” cell surface-bound molecules such as EGF receptor ligands (6, 7) or TNFα (8, 9). However, very little is known about which features of ADAM17 or its substrates can play the role in determination of substrate specificity. Most of the structural and biochemical studies of ADAM17 focused on its catalytic domain or substrate sequence as potential drivers of specificity; however, no conclusive evidence has been presented.
The role of ADAM17 noncatalytic domains in enzyme-substrate interactions has not been addressed. Crystallographic and modeling studies by Takeda et al. (10, 11), demonstrated the possibility that noncatalytic domains of membrane-bound ADAM17 can contribute to substrate recognition and binding of cell surface proteins, which necessitates studies with substrates that can interact with noncatalytic domains. Cleavage site sequence specificity has been addressed for several members of the ADAM family (12–14), but most of the substrates utilized for these studies were short (∼10 residues) and therefore were likely to interact only with the catalytic domain of ADAMs.
Similarly, ADAM17 substrate studies mostly focused on amino acid sequence as a potential specificity determinant (12), and no forays were made into other areas, such as substrate secondary structure. We previously reported that substrate glycosylation can differentially affect ADAM activity (15); therefore, it stands to reason that substrate secondary structure can be another, as yet undescribed specificity determinant.
Studies of enzyme-substrate interactions can lead to the development of selective inhibitors of ADAM17. Most of the ADAM17 inhibitors developed to date feature zinc-binding moieties targeting the zinc of the active site (4, 16, 17). There are ∼70 known human metalloproteases (ADAM, ADAMTS, and MMP) (1) that have zinc in their active site, which explains off target toxicities of zinc-binding inhibitors (18).
Targeting secondary substrate-binding sites (exosites) can potentially work as an alternative strategy for drug discovery (19–24). Selective exosite inhibitors of ADAM-related MMP and ADAMTS proteases were discovered by several groups (25–30), including ours. Exosites are defined as sites outside of the active site that participate in substrate recognition and binding (19). In the case of metalloproteases, an “exosite-binding compound” may simply imply a lack of interaction with the active site zinc (26, 28).
Although currently there are only a few reports of potential exosites in ADAM protease structures (11, 31), a recent paper by Tape et al. (32) demonstrated that it is possible to achieve selective binding to the ADAM17 ectodomain by an antibody that exploits exosites. Our group recently reported a discovery of a small molecule that inhibits ADAM17 in a non-zinc-binding fashion, which also supports exosite targeting strategies for ADAM17 drug and probe discovery (15).
TNFα, canonical substrate of ADAM17 (also called “TNFα-converting enzyme” or TACE), was used as the basis for the substrates in the studies presented herein (9). Pro-TNFα is a cell surface-bound transmembrane protein of 233 residues discovered by Carswell et al. (33). Cleavage of pro-TNFα by ADAM17 releases its mature, biologically active form of 157 residues organized as a trimer (34). Although shedding of TNFα from the cell surface by ADAM17 is an important biological event, virtually nothing is known about the structural determinants of this molecular interaction. In the study presented here, we utilized a series of TNFα-based substrates that span from juxtamembrane to mature domain of TNFα to ascertain the effect, if any, of interactions with noncatalytic domains of ADAM17. Additionally, we were interested to see whether there are differences in the way ADAM10 and ADAM17 interact with these substrates that can be exploited for selective exosite-binding inhibitor discovery.
EXPERIMENTAL PROCEDURES
Substrate Synthesis, Purification, and Characterization
Substrate synthesis was performed on a Protein Technology PS3 Peptide Synthesizer using Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid phase peptide chemistry methodology. Substrates were purified using reversed phase HPLC. Fractions were analyzed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) and by analytical reversed phase HPLC.
Circular Dichroism Spectroscopy
CD spectra were recorded over the range λ = 190–250 nm with a Jasco J-810 spectropolarimeter using a 0.2-cm path length quartz cell. Substrates solutions were prepared in deionized water and 50% TFE.2 Raw CD data (mdeg) was normalized for respective substrate concentrations to obtain molar ellipticity (θ) to allow for direct comparison of CD signatures of different substrates.
Enzyme Kinetics
ADAM enzymes were purchased from R & D Systems (Minneapolis, MN) with the exception of ADAM17 catalytic domain construct, purchased from Enzo Life Sciences (Farmingdale, NY). ADAM12 was activated as per the manufacturer's instructions. Active enzymes concentrations were determined as described elsewhere (14). Substrate stock solutions were prepared at various concentrations in R & D Systems-recommended assay buffers. The assays were conducted by incubating a range of substrate concentrations (2–100 μm) with various ADAM enzyme concentrations at 25 °C. Experiments with free l-galactose were conducted as above, but in presence of 60 μm l-galactose and an additional 1-h incubation step at room temperature with ADAM10 and ADAM17. Fluorescence was measured on a multimode microplate reader Synergy H4 (Biotek Instruments, Winooski, VT) using λexcitation = 360 nm and λemission = 460 nm. Rates of hydrolysis were obtained from plots of fluorescence versus time, using data points from only the linear portion of the hydrolysis curve. The slope from these plots was divided by the fluorescence change corresponding to complete hydrolysis and then multiplied by the substrate concentration to obtain rates of hydrolysis in units of μm/s. Kinetic parameters were calculated by nonlinear regression analysis using the GraphPad Prism 5.01 suite of programs. ADAM10 and ADAM17 substrate cleavage sites were established by MALDI-TOF MS.
Inhibition Studies
Substrate and ADAM17 working solutions were prepared in R & D Systems recommended assay buffers. 5 μl of 3× ADAM17 solution (30 nm) in assay buffer were added to solid bottom white 384 low volume plates (Nunc catalog number 264706). Next, 5 μl of test compounds (N-hydroxyacetamide (AHA) or compound 15) or pharmacological controls were added to corresponding wells. After 30 min of incubation at room temperature, the reactions were started by the addition of 5 μl of 3× solutions of respective substrates (30 μm). Fluorescence was measured every 30 min for 2 h using the multimode microplate reader Synergy H4 (Biotek Instruments, Winooski, VT) using λexcitation = 360 nm and λemission = 460 nm. Rates of hydrolysis were obtained from plots of fluorescence versus time, and inhibition was calculated using rates obtained from wells containing substrates only (100% inhibition) and substrates with enzyme (0% inhibition). Three parameters were calculated on a per plate basis: (a) the signal-to-background ratio (S/B); (b) the coefficient for variation (CV, where CV = (standard deviation/mean) × 100)) for all compound test wells; and (c) the Z′ factor (18). The IC50 value of the pharmacological control ((N-hydroxy-1-(4-methoxyphenyl)sulfonyl-4-(4-biphenylcarbonyl)piperazine-2-carboxamide;Calbiochem catalog number 444252) was also calculated using GraphPad Prism to ascertain the assay robustness.
RESULTS AND DISCUSSION
Substrate Design and Synthesis
Glycosylated substrate 1 and its nonglycosylated counterpart 4 (Fig. 1) were previously utilized to study the effects of glycosylation on ADAM protease activity and successfully used as HTS substrates to discover selective exosite-binding inhibitors of ADAM17 (15). In the present study, these substrates were used in combination with free l-galactose to probe the potential carbohydrate-binding site. Glycosylated substrates 2 and 3 were synthesized to assess the effects of size/shape and hydrophobicity of carbohydrate-binding pocket and effects of noncatalytic domains of ADAM17 on carbohydrate binding.
FIGURE 1.

Sequences of TNFα-based fluorogenic substrates and α-helical peptide 10. All of the amino acid numbers correspond to Uniprot entry P01375.
Caescu et al. (12) have previously shown that ADAM10 and 17 do not appear to recognize a specific amino acid signature within the P4-P5′ subsites, even though some preferences toward particular amino acids in certain positions do exist. It is therefore a mystery why certain cell surface molecules are cleaved exclusively by either ADAM10 or ADAM17. Is there a previously undescribed substrate recognition mechanism that differentiates ADAM10 and ADAM17?
It has been proposed in the literature (10, 11) that the noncatalytic domains of ADAM proteases might participate in the binding of their substrates and therefore might help ADAM proteases to discriminate between substrates. We hypothesized that substrate sequence beyond the P4-P5′ subsite residues would be able to interact outside of the catalytic domain and engage noncatalytic domains of ADAM10 and ADAM17. To investigate this possibility, we synthesized substrates that expanded from the original TNFα-based substrate (Fig. 1, substrate 4) (15) to cover juxtamembrane and link domains (Fig. 1, substrate 6), link and partial mature domain (Fig. 1, substrate 7), and the substrate where Pro72 was eliminated and Arg82 was introduced as compared with substrate 4 (Fig. 1, substrate 5). Substrate 5 was used to ascertain the effect of small local sequence changes on ADAM protease activities by comparing the kinetics of its hydrolysis to the ones of substrate 4.
Substrates 8 and 9 and peptide 10 (Fig. 1) were designed to assess the effect of substrate secondary structure on activity of ADAM proteases. For comparative studies, the following ADAM constructs were utilized: ADAM17 ectodomain (ECD, Arg215–Asn671), ADAM17 catalytic domain (CD, Arg215–Val477), ADAM10 ectodomain (ECD, Thr214–Gln672), and ADAM12 catalytic + disintegrin domain (Glu208–Ser519).
Circular Dichroism Spectroscopic Analysis
To determine the effect of glycosylation and juxtamembrane region sequence of TNFα on substrate conformation, CD spectra of substrates utilized in the study (Fig. 1) in water and TFE were recorded (Figs. 2 and 3). In water only substrate 9 exhibited characteristics of an α-helix with two minima at λ = 208 and 222 nm (Fig. 2D), whereas all other substrates exhibited characteristics of random coils featuring a single large negative peak at around λ = 198 nm (Figs. 2 and 3). We further characterized substrates by subjecting them to the conditions emulating intramembrane environment (i.e., 50% TFE). Interestingly, all substrates underwent significant structural rearrangement from random coil to an α-helix upon addition of 50% TFE (Fig. 2). Substrate 9 (3 α-helix-inducing heptads), although not fully α-helical under 100% aqueous conditions, nevertheless exhibited positive molar absorptivity in the 190-nm region. Absorption in this region is due to the peptide bond; therefore, the difference in molar absorptivity in this region signifies a change in dihedral angles of peptide bonds between the substrates used for this study. Peptide 10 was fully α-helical under both conditions (Fig. 2E).
FIGURE 2.

Circular dichroism spectroscopy of juxtamembrane, partial mature, and α-helical TNFα-based substrates. A, juxtamembrane substrate (#6). B, partial mature substrate (#7). C, (EIEALKA)2 substrate (#8). D, α-helical substrate (#9). E, (EIEALKA)3 peptide (#10). Substrates solutions were prepared in deionized water and 50% TFE. Raw CD data (mdeg) were normalized for respective substrate concentrations to obtain molar ellipticity (θ) to allow for direct comparison of CD signatures of different substrates. All of the substrates underwent significant structural rearrangement from random coil to an α-helix upon addition of 50% TFE. Substrate 9 (three α-helix-inducing heptads), although not fully α-helical under 100% aqueous conditions, nevertheless exhibited positive molar absorptivity in the λ = 190 nm region.
FIGURE 3.
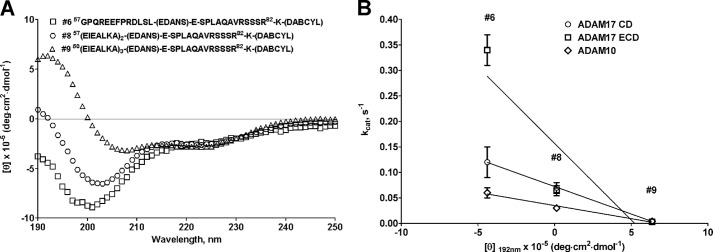
Correlation of circular dichroism spectra of juxtamembrane (substrate 6), substrate with two α-helical heptads (substrate 8), and α-helical (substrate 9) substrates. A, molar ellipticity spectra of juxtamembrane (substrate 6), substrate with two α-helical heptads (substrate 8), and α-helical (substrate 9) substrates. B, correlation plot of molar absorptivity at λ = 192 nm versus kcat of juxtamembrane (substrate 6), substrate with two α-helical heptads (substrate 8), and α-helical (substrate 9) substrates. Data points for ADAM17 ECD and CD for substrates 8 and 9 overlap in B. The linear regression analysis of a plot of molar absorptivity at λ = 192 nm versus kcat exhibited excellent R2 values, suggesting a strong correlation between the secondary structure of TNFα-based substrates and the activity of ADAM10 and 17.
Noncatalytic Domains of ADAM17 Affect Carbohydrate Binding (Pocket)
To ascertain the effect of the noncatalytic domains on activity of ADAM17 with glycosylated substrates, we compared the kinetic parameters for the hydrolysis of glycosylated substrate 1 and nonglycosylated substrate 4 by ADAM17 ECD and ADAM17 CD. Specificities of hydrolysis of both substrates by ADAM17 CD were similar, whereas ADAM17 ECD specificity was ∼6-fold greater for the glycosylated substrate (Table 1). This suggests that the presence of noncatalytic domains is required for increased specificity of ADAM17 toward the glycosylated substrate. However, the length of substrate and proximity of glycosylation to the scissile bond suggested that the actual interaction with the carbohydrate moiety most likely occurs within the catalytic domain. Interestingly, ADAM17 CD exhibited 3-fold higher affinity toward nonglycosylated substrate than ADAM17 ECD (Km = 4.0 ± 0.1 μm versus 12 ± 3 μm, for ADAM17 CD and ECD, respectively) but 2-fold lower affinity toward glycosylated substrate (Km = 6.9 ± 1.3 μm versus 3.0 ± 0.5 μm, for ADAM17 CD and ECD, respectively). This suggests that although an interaction with the carbohydrate moiety in all likelihood occurs within the catalytic domain, the noncatalytic domains affect the affinity of this interaction indirectly, perhaps by influencing the geometry of a binding pocket. This suggests cooperativity between catalytic and noncatalytic domains. To test this hypothesis, we further probed the potential carbohydrate-binding site using free 60 μm l-galactose. None of the kinetic parameters for either substrate hydrolysis by ADAM17 CD was affected by the presence of l-galactose (Table 1). In contrast, ADAM17 ECD exhibited higher affinity toward the nonglycosylated substrate (Km = 4.0 ± 0.3 μm versus 12 ± 3 μm, for l-galactose versus no l-galactose, respectively) and lower turnover of glycosylated substrate (kcat = 0.08 ± 0.01 s−1 versus 0.25 ± 0.03 s−1, for l-galactose versus no l-galactose, respectively). The lack of affinity change for binding of glycosylated substrate by ADAM17 ECD can be attributed to the inability of free l-galactose to bind to the pocket already occupied by TF antigen (Galβ1–3GalNAcα1). In the case of nonglycosylated substrate, free l-galactose potentially binds to an unoccupied pocket of ADAM17 ECD and induces affinity change to the levels of ADAM17 CD and ADAM17 CD + free l-galactose (Km = 4.0 ± 0.3, 4.0 ± 0.1, and 3.4 ± 0.9 μm for ADAM17 ECD + l-galactose, ADAM17 CD, and ADAM17 CD + l-galactose, respectively). Different effects of l-galactose on ADAM17 CD and ECD kinetic parameters thus supports the hypothesis of noncatalytic domains influencing the shape or the size of the carbohydrate-binding pocket within the catalytic domain of ADAM17. Another possibility, however small, is that free l-galactose and TF-antigen bind to different binding sites to produce the effects described above.
TABLE 1.
Kinetic parameters for glycosylated substrate 1 and nonglycosylated substrate 4 substrate hydrolysis by ADAM10 and 17 constructs in presence and absence of l-galactose
The results are reported as averages ± S.D. (n = 3).
| Enzyme |
kcat/Km |
kcat |
Km |
|||
|---|---|---|---|---|---|---|
| Glycosylated | Nonglycosylated | Glycosylated | Nonglycosylated | Glycosylated | Nonglycosylated | |
| m−1 s−1 | s−1 | μm | ||||
| ADAM17 ECD | 7.6 ± 1.7 × 104a | 1.2 ± 0.1 × 104a | 0.25 ± 0.03a | 0.14 ± 0.01a | 3.0 ± 0.5a | 12 ± 3a |
| ADAM17 CD | 3.2 ± 0.02 × 104 | 1.7 ± 0.2 × 104 | 0.22 ± 0.03 | 0.07 ± 0.01 | 6.9 ± 1.3 | 4.0 ± 0.1 |
| ADAM17 ECD + l-gal | 2.3 ± 0.14 × 104 | 1.9 ± 0.13 × 104 | 0.08 ± 0.01 | 0.1 ± 0.01 | 3.2 ± 0.3 | 4.0 ± 0.3 |
| ADAM17 CD + l-gal | 3.6 ± 0.4 × 104 | 1.8 ± 0.14 × 104 | 0.36 ± 0.03 | 0.06 ± 0.03 | 10 ± 1.3 | 3.4 ± 0.9 |
| ADAM10 ECD | 0.8 ± 0.3 × 104a | 2.5 ± 0.7 × 104a | 0.06 ± 0.01a | 0.28 ± 0.03a | 8.5 ± 1.3a | 12 ± 3a |
| ADAM10 ECD + l-gal | 2.8 ± 0.00 × 104 | 0.9 ± 0.04 × 104 | 0.34 ± 0.02 | 0.02 ± 0.00 | 12 ± 1.6 | 2.0 ± 0.5 |
a Reported in Ref. 15.
We were interested to see whether l-galactose would have the same effect on the closest homologue of ADAM17, ADAM10. We utilized ADAM10 ECD construct for these studies. Similarly to ADAM17 ECD, the addition of l-galactose to the ADAM10 reaction with glycosylated substrate did not produce significant changes in affinity (Table 1; Km = 12 ± 1.6 versus 8.5 ± 1.3 μm for l-galactose versus no l-galactose, respectively). Strikingly, the effect on turnover of glycosylated substrate was opposite in the case of ADAM10 as compared with ADAM17; ∼3-fold decrease of turnover for ADAM17 and ∼6-fold increase for ADAM10. For the nonglycosylated substrate, binding of l-galactose brought on similar affinity changes for both enzymes but resulted in 14-fold drop of ADAM10 turnover while not affecting ADAM17. This suggests differences in the manner ADAM10 and 17 accommodate free l-galactose, which can be exploited for inhibitor development.
To investigate the effects of noncatalytic domains on the carbohydrate-binding pocket, we compared kinetic parameters for the hydrolysis of glycosylated substrates 2 and 3 (Fig. 1). Substrate 3 has a bulkier and more hydrophobic carbohydrate as compared with substrate 2, but the same primary structure (73LAQAVRSSSR82). The bulkier carbohydrate has no effect on kinetic parameters of the ADAM17 CD construct (Table 2), whereas ADAM17 ECD experienced ∼3-fold loss of both affinity and turnover (Table 2; Km = 7.6 ± 1.3 μm versus 22 ± 6.0 μm, for substrate 2 versus substrate 3, respectively; kcat = 0.08 ± 0.01 s−1 versus 0.06 ± 0.02 s−1, for substrate 2 versus substrate 3, respectively) resulting in ∼10-fold loss of specificity (Table 2; kcat/Km, = 6.1 ± 0.8 × 104 m−1 s−1versus 0.68 ± 0.06 × 104 m−1 s−1 for substrate 2 versus substrate 3, respectively). Because different effects on ADAM17 ECD and CD were observed, it is reasonable to propose that the shape or the size of the carbohydrate-binding pocket is different in these two constructs. It also suggests that ADAM17 CD has a larger carbohydrate-binding pocket as compared with ADAM17 ECD.
TABLE 2.
Effect of noncatalytic domains on kinetic parameters of hydrolysis for glycosylated substrates 2 and 3 by ADAM10 and 17
The results are reported as averages ± S.D. (n = 3).
| Enzyme |
kcat/Km |
kcat |
Km |
|||
|---|---|---|---|---|---|---|
| Substrate 2 (73–82) | Substrate 3 (73–82) | Substrate 2 (73–82) | Substrate 3 (73–82) | Substrate 2 (73–82) | Substrate 3 (73–82) | |
| m−1 s−1 | s−1 | μm | ||||
| ADAM17 ECD | 6.1 ± 0.8 × 104 | 0.68 ± 0.06 × 104 | 0.42 ± 0.01 | 0.15 ± 0.03 | 7.6 ± 1.3 | 22 ± 6.0 |
| ADAM17 CD | 0.11 ± 0.01 × 104 | 0.12 ± 0.01 × 104 | 0.08 ± 0.01 | 0.06 ± 0.02 | 68 ± 11 | 50 ± 9 |
The experiments with glycosylated substrates point to the possibility of noncatalytic domains affecting the size or shape of binding pockets within the ADAM17 catalytic domain. This, in turn, suggests the regulatory role of noncatalytic domains and possible allosteric effects of ligands binding to these domains.
Contribution of ADAM17 Noncatalytic Domains to Substrate Cleavage Site Specificity and Substrate Binding
We previously reported differential effects of glycosylation on cleavage sites by various ADAMs (15). Briefly, ADAM10 and 17 cleaved only at the cognate scissile bond Ala76 ↓ Val in case of both glycosylated (substrate 1) and nonglycosylated (substrate 4) substrates, whereas ADAM8 cleaved the nonglycosylated substrate only at Ala76 ↓ Val, but was able to cleave at Ala ↓ Val, Gln ↓ Ala, and Val ↓ Arg in the case of the glycosylated substrate. ADAM12 cleaved at two positions of the nonglycosylated substrate (Ala ↓ Val and Gln ↓ Ala) and three positions in the case of the glycosylated substrate (Ala ↓ Gln, Gln ↓ Ala, and Val ↓ Arg). Neither ADAM10 nor ADAM17 cleavage site specificity was affected by glycosylation. We were interested to see whether noncatalytic domains of ADAM17 played a role in its cleavage site specificity. We digested glycosylated substrates 2 and 3 and the nonglycosylated analogue substrate 5 using ADAM17 CD and ECD constructs. Additionally, we used juxtamembrane substrate 6 and substrate 7, which contains the link and a portion of mature domain of TNFα. Both ADAM17 CD and ECD constructs cleaved all of the tested substrates only at canonical scissile bond Ala76 ↓ Val, suggesting that structural features that drive ADAM17 cleavage site specificity are contained within its CD.
A small sequence shift did not appear to affect affinity of interaction for either ADAM10 or ADAM17 (Table 3; Km = 12.3 ± 3 μm versus 10.0 ± 1.2 μm for ADAM10 ECD with substrates 4 and 5 and 12.3 ± 3 μm versus 7.2 ± 0.6 μm for ADAM17 ECD with substrates 4 and 5, respectively). The effect on turnover rate was, however, different: ADAM10 exhibited ∼9-fold decrease of kcat, whereas ADAM17 remained unaffected (Table 3; kcat = 0.28 ± 0.03 s−1 versus 0.03 ± 0.004 s−1 for ADAM10 ECD with substrates 4 and 5 and 0.14 ± 0.01 s−1 versus 0.2 ± 0.02 s−1 for ADAM17 ECD with substrates 4 and 5, respectively). Because both ADAM10 and ADAM17 prefer Pro in subsite P5′ (P5′ = Pro72) (12), the effect on the ADAM10 kcat parameter can be attributed to the presence of Arg82 in the P6 position. This suggests that ADAM10 is more sensitive to local sequence variability than ADAM17.
TABLE 3.
Effect of juxtamembrane and part of mature domain of TNFα on Km and kcat parameters for hydrolysis of substrates 4, 5, 6, and 7 by ADAM10 and 17
The results are reported as averages ± S.D. (n = 3).
| Enzyme |
kcat |
Km |
||||||
|---|---|---|---|---|---|---|---|---|
| Substrate 4 (72–81) | Substrate 5 (73–82) | Substrate 6 (57–82) | Substrate 7 (72–108) | Substrate 4 (72–81) | Substrate 5 (73–82) | Substrate 6 (57–82) | Substrate 7 (72–108) | |
| s−1 | μm | |||||||
| ADAM10 ECD | 0.28 ± 0.03 | 0.03 ± 0.04 | 0.06 ± 0.01 | 0.013 ± 0.001 | 12 ± 3 | 10 ± 1.2 | 13 ± 0.5 | 4.3 ± 1.5 |
| ADAM17 ECD | 0.14 ± 0.01 | 0.2 ± 0.02 | 0.34 ± 0.03 | 0.07 ± 0.005 | 12 ± 3 | 7.2 ± 0.6 | 24 ± 6.2 | 3.3 ± 0.2 |
| ADAM17 CD | 0.07 ± 0.01 | 0.05 ± 0.01 | 0.12 ± 0.03 | 0.03 ± 0.001 | 4.0 ± 0.1 | 16 ± 5 | 20 ± 0.2 | 4.3 ± 1.5 |
In the model of interaction between cell surface-bound ADAM protease and its substrates put forward by Takeda et al. (10, 11), the hypervariable region of the enzyme binds the juxtamembrane region of cell-bound substrate when cleavage occurs in cis. If cleavage were to occur in trans, the hypervariable region would interact with portion of the substrate most distant from the membrane, which in the case of TNFα would mean within or in the vicinity of β-sheet domains. We were interested to see whether the TNFα-based substrate containing juxtamembrane sequence (Fig. 1, substrate 6) would exhibit different kinetic parameters as compared with shorter substrate 5, which likely interacts only with the catalytic domain. ADAM10 ECD did not exhibit significant differences in hydrolysis of either substrate, whereas ADAM17 ECD had less affinity toward juxtamembrane substrate 6 (Table 3; Km = 7.2 ± 0.6 μm versus 24 ± 6.2 μm for ADAM17 ECD with substrates 5 and 6, respectively). This suggests that the juxtamembrane sequence does not contribute additional productive interactions with ADAM10 and ADAM17 noncatalytic domains and might actually create a steric clash in the case of ADAM17. To investigate this further, we compared kinetic parameters of hydrolysis of substrates 5 and 6 by ADAM17 ECD to ADAM17 CD. ADAM17 CD had almost identical affinity for both substrates (Table 3), which supports the idea of juxtamembrane substrate clashing with noncatalytic domains of ADAM17 ECD. These observations are in agreement with Lee et al. (36), who suggested that C-terminal (i.e., noncatalytic) domains of ADAM17 can create a steric obstacle for interactions between N-TIMP-3 and ADAM17 ECD.
Substrate 7 (Fig. 1, Pro72–Arg108) lacks the juxtamembrane region but instead extends into mature domain of TNFα. Both ADAM10 and ADAM17 gain affinity as compared with substrate 4 (Table 3), but in the case of ADAM10 it resulted in ∼20-fold decrease of turnover, whereas ADAM17 turnover was slightly affected (Table 3; kcat = 0.28 ± 0.03 s−1 versus 0.013 ± 0.001 s−1 for ADAM10 ECD with substrates 4 and 7 and 0.14 ± 0.01 s−1 versus 0.07 ± 0.005 s−1 for ADAM17 ECD with substrates 4 and 7, respectively). Again, ADAM17 CD exhibited identical affinity toward both substrates.
The experiments described herein suggest that noncatalytic domains of ADAM10 and ADAM17 can affect the way these enzymes interact with their respective substrates. Moreover, substrates interacting with noncatalytic domains of ADAM10 and ADAM17 affected enzyme activity in different ways, suggesting that noncatalytic domains might contain the key structural elements that determine ADAM substrate repertoires.
Effect of Substrate Secondary Structure on ADAM17 Specificity
The cleavage of TNFα by ADAM17 on the cell surface occurs in its juxtamembrane region 20 amino acids away from transmembrane domain (9). The juxtamembrane region of TNFα is disordered according to the Uniprot database entry based on the primary structure composition (P01375, TNFA_HUMAN), whereas the transmembrane region is predicted to be α-helical. We have previously shown that TNFα-based substrate can form an α-helix under conditions emulating the intramembrane environment (15). We were interested to see whether activity of ADAM17 can be modulated by a substrate possessing a distinct secondary structure. The α-helical substrate 9 exhibited significant improvement of affinity toward ADAM17 ECD as compared with the random coil substrates 6 and 8 (juxtamembrane substrate and substrate with 2 α-helix-inducing heptads, respectively). Km values were 24 ± 6.2, 7.5 ± 1.3, and 2.0 ± 0.1 μm for substrates 6, 8, and 9, respectively (Table 4), which constitutes 12- and 4-fold improvement of affinity as compared with juxtamembrane substrate and the substrate with two α-helix-inducing heptads, respectively. The rate of turnover of α-helical substrate 9 by ADAM17 ECD was affected much more drastically as compared with random coil substrates. The α-helical substrate demonstrated greater than 100- and 20-fold decrease of kcat as compared with juxtamembrane substrate and substrate with 2 α-helix-inducing heptads, respectively (Table 4). Such a dramatic effect of α-helical structure on enzyme activity is likely a consequence of tight binding exhibited by this substrate. It is therefore entirely possible that the repertoire of substrates of membrane-bound ADAM17 can be limited to sequences located within disordered regions of cell surface proteins. By the same token, it is conceivable that ADAM17 activity can be regulated by α-helical structure. Additional investigations of effects of substrate conformation on ADAM17 activity are therefore warranted.
TABLE 4.
Kinetic parameters for hydrolysis of substrates containing juxtamembrane region of TNFα and α-helix-inducing heptads by ADAM10, 12, and 17
The results are reported as averages ± standard deviation (n = 3).CDD, catalytic domain + disintegrin domain.
| Enzyme |
kcat/Km |
kcat |
Km |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Substrate 6 | Substrate 8 | Substrate 9 | Substrate 6 | Substrate 8 | Substrate 9 | Substrate 6 | Substrate 8 | Substrate 9 | |
| m−1 s−1 | s−1 | μm | |||||||
| ADAM17 ECD | 1.5 ± 0.25 × 104 | 0.9 ± 0.04 × 104 | 0.16 ± 0.08 × 104 | 0.34 ± 0.03 | 0.064 ± 0.01 | 0.003 ± 0.001 | 24 ± 6.2 | 7.5 ± 1.3 | 2.0 ± 0.1 |
| ADAM17 CD | 0.62 ± 0.08 × 104 | 0.9 ± 0.04 × 104 | 0.16 ± 0.00 × 104 | 0.12 ± 0.03 | 0.07 ± 0.01 | 0.004 ± 0.001 | 20 ± 0.2 | 7.9 ± 0.4 | 2.3 ± 0.6 |
| ADAM10 ECD | 0.5 ± 0.04 × 104 | 0.39 ± 0.006 × 104 | 0.06 ± 0.00 × 104 | 0.06 ± 0.01 | 0.03 ± 0.004 | 0.004 ± 0.001 | 13 ± 0.5 | 6.2 ± 0.7 | 5.5 ± 1.5 |
| ADAM12 CDD | 0.03 ± 0.001 × 104 | 0.05 ± 0.004 × 104 | ND | 0.006 ± 0.01 | 0.003 ± 0.002 | ND | 18 ± 5 | 6.2 ± 1.6 | ND |
As discussed above under “Contribution of ADAM17 Noncatalytic Domains to Substrate Cleavage Site Specificity and Substrate Binding,” the noncatalytic domains did not contribute to the binding of juxtamembrane substrate 6. We were curious whether the same is true for the substrate containing the α-helical juxtamembrane sequence. Indeed, ADAM17 CD exhibited almost identical kinetic parameters for all three substrates (Table 4) as compared with ADAM17 ECD. This advocates the idea that noncatalytic domains of ADAM17 do not contribute to interactions with juxtamembrane region of TNFα, at least in an in vitro setting. The circular dichroism spectra of substrate 9 (Fig. 2D) indicated α-helices at both 100% aqueous and 50% TFE, and thus the portion of the TNFα-based substrate that interacts with catalytic domain is likely α-helical. This in turn suggests that the catalytic domain of ADAM17 potentially contains secondary binding sites responsible for ∼10-fold improved affinity of ADAM17 toward α-helical substrate (substrate 9) as compared with a random coil one (substrate 6) (Table 4).
Effects of substrate α-helicity on activity of ADAM10 were less pronounced as compared with ADAM17. Observed were 15- and 7.5-fold decreases of kcat and 2.5- and 1.1-fold decreases of Km as compared with juxtamembrane and substrate with two α-helix-inducing heptads, respectively (Table 4). For comparison, ADAM12 did not cleave the α-helical substrate and its activity toward juxtamembrane and substrate with two α-helix-inducing heptads was weak (kcat = 0.006 ± 0.01 s−1 and 0.003 ± 0.002 s−1, for juxtamembrane and substrate with two α-helix-inducing heptads, respectively).
To confirm that the effect of substrate 9 on ADAM10 and ADAM17 kinetics was due to the induction of α-helical character upon TNFα native sequence (71SPLAQAVRSSSR82) rather than interactions of α-helical heptads (EIEALKA)3 with the enzymes, we synthesized just the (EIEALKA)3 portion of the molecule and tested it in ADAM10 and ADAM17 inhibition assays. (EIEALKA)3 formed an α-helix in water (Fig. 2E). If interaction of substrate 9 with ADAMs was due to just an α-helical secondary structure, we could expect competition between α-helical substrate 9 and peptide 10, resulting in inhibition of hydrolysis of 9. The addition of (EIEALKA)3 did not induce significant inhibition of hydrolysis of α-helical substrate 9 by ADAM10 ECD, ADAM17 CD, or ADAM17 ECD constructs (data not shown), suggesting that α-helicity alone is not enough for binding to either ADAM. We further tested (EIEALKA)3 for inhibition of hydrolysis of substrate 8 (data not shown) and commercial ADAM substrate Mca-KPLGL-Dpa-AR (data not shown). In both cases (EIEALKA)3 (up to a concentration of 100 μm) did not exhibit significant inhibition of substrate hydrolysis.
The data presented in this section demonstrate that the secondary substrate structure can play a role in ADAM protease activity and specificity. Significant differences in kinetic parameters for hydrolysis of juxtamembrane substrate (substrate 6) and its analogues (substrates 8 and 9) (Table 4) can be attributed to a difference in the peptide bond angles rather than additional interactions with ADAM noncatalytic domains. As witnessed by the CD spectra of three abovementioned substrates (Fig. 3A), the main difference lies in the 190–200-nm region where peptide bonds absorb. Substrate 6 exhibited negative molar absorptivity, substrate 8 was close to 0, whereas substrate 9 showed a positive signal consistent with α-helical secondary structure. This suggests that a scissile bond (Ala76–Val) in all three substrates is potentially presented to ADAM proteases in a somewhat different environment, which resulted in drastic loss of turnover and increase of affinity by ADAM17. The linear regression analysis of a plot of molar absorptivity at λ = 192 nm versus kcat exhibited excellent R2 values for ADAM17 CD and ADAM10 ECD (Fig. 3B; R2 = 0.999 and 0.982, for ADAM17 CD and ADAM10 ECD, respectively) and a very good R2 value of 0.815 for ADAM17 ECD. This suggests a strong correlation between the secondary structure of TNFα-based substrates and activity of ADAM10 and 17.
Inhibition Studies
Both α-helical (substrate 9) and glycosylated (substrate 1) substrates exhibited higher affinity toward ADAM17 as compared with their nonglycosylated, random coil counterpart (substrate 4) potentially because of the additional interactions with ADAM17 exosites. Based on the existing model of ADAM17 interactions with its substrates (10, 11), it is highly likely that the carbohydrate moiety of glycosylated substrate and the α-helical juxtamembrane sequence interact with exosites of ADAM17, which in turn suggests the existence of multiple exosites. We investigated this hypothesis by using exosite and active site inhibitors of ADAM17 in the presence of α-helical (substrate 9), glycosylated (substrate 1), and nonglycosylated (substrate 4) substrates. AHA, a known zinc binder and competitive millimolar range inhibitor of many metalloproteases (37), inhibited hydrolysis of all three substrates equipotently (Fig. 4A) (IC50 = 1.6 ± 0.1, 1.2 ± 0.02, and 0.8 ± 0.02 mm for glycosylated, nonglycosylated, and α-helical substrates, respectively). Compound 15, a noncompetitive ADAM17 inhibitor recently reported by our group (15), appreciably inhibited hydrolysis only of the glycosylated substrate (Fig. 4B). AHA most likely only binds within the active site of ADAM17 because of its size and therefore is unable to interact with ADAM17 exosites that glycosylated and potentially α-helical substrates bind to. The equipotent inhibition of all three substrates, therefore, is unsurprising. The noncompetitive exosite inhibitor (compound 15) preferentially abrogated hydrolysis of glycosylated substrate and spared hydrolysis of nonglycosylated and α-helical substrate (Fig. 4B). This suggests that α-helical and glycosylated substrates bind to ADAM17 in a different fashion and that the α-helical substrate binding does not interact with a pocket where compound 15 can be localized. These data, in combination with loss of turnover and increase of affinity of the α-helical substrate toward ADAM17, as compared with non-α-helical substrates 6 and 7 (Table 4), led us to believe that the α-helical substrate interacts with unique exosites compared with a glycosylated substrate. Thus, an α-helical substrate can potentially lead to discovery of inhibitors that interact with exosite(s) different from the one that compound 15 interacts with. Because ADAM17 has a wide repertoire of substrates (e.g. Notch, EGF receptor ligands, TNFα), a small molecule inhibitor capable of selectively inhibiting hydrolysis of a particular substrate while sparing hydrolysis of other substrates can be of value for both therapy and research.
FIGURE 4.

Results of inhibition study of ADAM17 with AHA (A) and compound 15 (B) in the presence of various substrates. Inhibition of ADAM17-mediated hydrolysis of glycosylated (substrate 1), nonglycosylated (substrate 4), and α-helical (substrate 9) substrates was measured using 10-point 1:3-fold dilutions of AHA and compound 15. The data were analyzed using GraphPad Prism. The noncompetitive exosite inhibitor (compound 15) preferentially abrogated hydrolysis of the glycosylated substrate and spared hydrolysis of nonglycosylated and α-helical substrates, suggesting that α-helical and glycosylated substrates bind to ADAM17 in a different fashion and that the α-helical substrate binding does not interact with a pocket where compound 15 can be localized.
Significance
To develop molecules capable of selective inhibition of ADAM17-mediated hydrolysis, one needs to understand how different members of the ADAM protease family interact with their substrates and whether there are unique features in the way these interactions occur. Although utilization of full-length proteins to study enzyme-protein interactions is believed to be more physiologically relevant and preferred over synthetic substrates, this approach has proved highly problematic in the field of ADAMs research. For example, it was observed that ADAM17 failed to cleave several full-length proteins in biochemical assays but was able to cleave peptides based on their cleavage sites (reviewed by Roy Black in Ref. 38). Among the full-length proteins that exhibited this behavior were pro-TGFα (35), l-selectin, p55 TNFR, and p75 TNFR (38), all validated physiological substrates of ADAM17. Moreover, the lack of suitable methodologies (long and expensive production process, sensitivity issues for HPLC-based methods, stability issues for fluorescamine labeling approach, etc.) for kinetic studies using recombinant full-length proteins justifies utilization of synthetic fluorogenic substrates for specificity studies.
In the present study we used TNFα-based fluorogenic substrates to determine that one of the key structural elements within ADAM17 structure responsible for substrate specificity are its noncatalytic domains, most likely via presenting a steric hindrance. Moreover, noncatalytic domains of ADAM17 likely affected the size/shape of the carbohydrate-binding pocket contained within the catalytic domain of ADAM17. This suggests that noncatalytic domains of ADAM17 play a role in substrate specificity and might help explain differences in substrate repertoires of ADAM17 and its closest homologue, ADAM10.
We also determined that the substrate secondary structure modulates ADAM activity. ADAMs can be differentiated based on this property, and this information can be used in the rational design of ADAM isoform-selective inhibitors.
This work was supported, in whole or in part, by National Institutes of Health Grants DA033985 (to D. M.), CA098799 (to G. B. F.), and DA031370 (to R. A. H.). This work was also supported by James and Esther King Biomedical Research Program Grant 2KN05 (to D. M.), the Multiple Sclerosis National Research Institute (to G. B. F.), and the State of Florida, Executive Office of the Governor's Office of Tourism, Trade, and Economic Development.
- TFE
- trifluoroethanol
- AHA
- N-hydroxyacetamide
- CD
- catalytic domain
- ECD
- extracellular domain.
REFERENCES
- 1. Edwards D. R., Handsley M. M., Pennington C. J. (2008) The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moss M. L., Stoeck A., Yan W., Dempsey P. J. (2008) ADAM10 as a target for anti-cancer therapy. Curr. Pharm. Biotechnol. 9, 2–8 [DOI] [PubMed] [Google Scholar]
- 3. Kataoka H. (2009) EGFR ligands and their signaling scissors, ADAMs, as new molecular targets for anticancer treatments. J. Dermatol. Sci. 56, 148–153 [DOI] [PubMed] [Google Scholar]
- 4. Moss M. L., Sklair-Tavron L., Nudelman R. (2008) Drug insight. Tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 4, 300–309 [DOI] [PubMed] [Google Scholar]
- 5. Asai M., Hattori C., Szabó B., Sasagawa N., Maruyama K., Tanuma S., Ishiura S. (2003) Putative function of ADAM9, ADAM10, and ADAM17 as APP α-secretase. Biochem. Biophys. Res. Commun. 301, 231–235 [DOI] [PubMed] [Google Scholar]
- 6. Blobel C. P. (2005) ADAMs. Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43 [DOI] [PubMed] [Google Scholar]
- 7. Murphy G. (2008) The ADAMs. Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 8, 929–941 [DOI] [PubMed] [Google Scholar]
- 8. Moss M. L., Jin S. L., Milla M. E., Bickett D. M., Burkhart W., Carter H. L., Chen W. J., Clay W. C., Didsbury J. R., Hassler D., Hoffman C. R., Kost T. A., Lambert M. H., Leesnitzer M. A., McCauley P., McGeehan G., Mitchell J., Moyer M., Pahel G., Rocque W., Overton L. K., Schoenen F., Seaton T., Su J. L., Becherer J. D. (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 385, 733–736 [DOI] [PubMed] [Google Scholar]
- 9. Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F., Castner B. J., Stocking K. L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K. A., Gerhart M., Davis R., Fitzner J. N., Johnson R. S., Paxton R. J., March C. J., Cerretti D. P. (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729–733 [DOI] [PubMed] [Google Scholar]
- 10. Takeda S. (2009) Three-dimensional domain architecture of the ADAM family proteinases. Semin. Cell Dev. Biol. 20, 146–152 [DOI] [PubMed] [Google Scholar]
- 11. Takeda S., Igarashi T., Mori H. (2007) Crystal structure of RVV-X. An example of evolutionary gain of specificity by ADAM proteinases. FEBS Lett. 581, 5859–5864 [DOI] [PubMed] [Google Scholar]
- 12. Caescu C. I., Jeschke G. R., Turk B. E. (2009) Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 424, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moss M. L., Rasmussen F. H. (2007) Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal. Biochem. 366, 144–148 [DOI] [PubMed] [Google Scholar]
- 14. Moss M. L., Rasmussen F. H., Nudelman R., Dempsey P. J., Williams J. (2010) Fluorescent substrates useful as high-throughput screening tools for ADAM9. Comb. Chem. High Throughput Screen 13, 358–365 [DOI] [PubMed] [Google Scholar]
- 15. Minond D., Cudic M., Bionda N., Giulianotti M., Maida L., Houghten R. A., Fields G. B. (2012) Discovery of novel inhibitors of a disintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates. J. Biol. Chem. 287, 36473–36487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kenny P. A., Bissell M. J. (2007) Targeting TACE-dependent EGFR ligand shedding in breast cancer. J. Clin. Invest. 117, 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Georgiadis D., Yiotakis A. (2008) Specific targeting of metzincin family members with small-molecule inhibitors. Progress toward a multifarious challenge. Bioorg. Med. Chem. 16, 8781–8794 [DOI] [PubMed] [Google Scholar]
- 18. Fingleton B. (2007) Matrix metalloproteinases as valid clinical targets. Curr. Pharm. Des. 13, 333–346 [DOI] [PubMed] [Google Scholar]
- 19. Dennis M. S., Eigenbrot C., Skelton N. J., Ultsch M. H., Santell L., Dwyer M. A., O'Connell M. P., Lazarus R. A. (2000) Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature 404, 465–470 [DOI] [PubMed] [Google Scholar]
- 20. Roberge M., Peek M., Kirchhofer D., Dennis M. S., Lazarus R. A. (2002) Fusion of two distinct peptide exosite inhibitors of Factor VIIa. Biochem. J. 363, 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberge M., Santell L., Dennis M. S., Eigenbrot C., Dwyer M. A., Lazarus R. A. (2001) A novel exosite on coagulation factor VIIa and its molecular interactions with a new class of peptide inhibitors. Biochemistry 40, 9522–9531 [DOI] [PubMed] [Google Scholar]
- 22. Izaguirre G., Rezaie A. R., Olson S. T. (2009) Engineering functional antithrombin exosites in α1-proteinase inhibitor that specifically promote the inhibition of factor Xa and factor IXa. J. Biol. Chem. 284, 1550–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheer J. M., Romanowski M. J., Wells J. A. (2006) A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. U.S.A. 103, 7595–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hosseini M., Jiang L., Sørensen H. P., Jensen J. K., Christensen A., Fogh S., Yuan C., Andersen L. M., Huang M., Andreasen P. A., Jensen K. J. (2011) Elucidation of the contribution of active site and exosite interactions to affinity and specificity of peptidylic serine protease inhibitors, using non-natural arginine analogs. Mol. Pharmacol. 80, 585–597 [DOI] [PubMed] [Google Scholar]
- 25. Johnson A. R., Pavlovsky A. G., Ortwine D. F., Prior F., Man C. F., Bornemeier D. A., Banotai C. A., Mueller W. T., McConnell P., Yan C., Baragi V., Lesch C., Roark W. H., Wilson M., Datta K., Guzman R., Han H. K., Dyer R. D. (2007) Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 282, 27781–27791 [DOI] [PubMed] [Google Scholar]
- 26. Engel C. K., Pirard B., Schimanski S., Kirsch R., Habermann J., Klingler O., Schlotte V., Weithmann K. U., Wendt K. U. (2005) Structural basis for the highly selective inhibition of MMP-13. Chem. Biol. 12, 181–189 [DOI] [PubMed] [Google Scholar]
- 27. Baragi V. M., Becher G., Bendele A. M., Biesinger R., Bluhm H., Boer J., Deng H., Dodd R., Essers M., Feuerstein T., Gallagher B. M., Jr., Gege C., Hochgürtel M., Hofmann M., Jaworski A., Jin L., Kiely A., Korniski B., Kroth H., Nix D., Nolte B., Piecha D., Powers T. S., Richter F., Schneider M., Steeneck C., Sucholeiki I., Taveras A., Timmermann A., Van Veldhuizen J., Weik J., Wu X., Xia B. (2009) A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis. Evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheum. 60, 2008–2018 [DOI] [PubMed] [Google Scholar]
- 28. Lauer-Fields J. L., Minond D., Chase P. S., Baillargeon P. E., Saldanha S. A., Stawikowska R., Hodder P., Fields G. B. (2009) High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg. Med. Chem. 17, 990–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roth J., Minond D., Darout E., Liu Q., Lauer J., Hodder P., Fields G. B., Roush W. R. (2011) Identification of novel, exosite-binding matrix metalloproteinase-13 inhibitor scaffolds. Bioorg. Med. Chem. Lett. 21, 7180–7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wittwer A. J., Hills R. L., Keith R. H., Munie G. E., Arner E. C., Anglin C. P., Malfait A. M., Tortorella M. D. (2007) Substrate-dependent inhibition kinetics of an active site-directed inhibitor of ADAMTS-4 (Aggrecanase 1). Biochemistry 46, 6393–6401 [DOI] [PubMed] [Google Scholar]
- 31. Hall T., Pegg L. E., Pauley A. M., Fischer H. D., Tomasselli A. G., Zack M. D. (2009) ADAM8 substrate specificity. Influence of pH on pre-processing and proteoglycan degradation. Arch. Biochem. Biophys. 491, 106–111 [DOI] [PubMed] [Google Scholar]
- 32. Tape C. J., Willems S. H., Dombernowsky S. L., Stanley P. L., Fogarasi M., Ouwehand W., McCafferty J., Murphy G. (2011) Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. U.S.A. 108, 5578–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carswell E. A., Old L. J., Kassel R. L., Green S., Fiore N., Williamson B. (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U.S.A. 72, 3666–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wingfield P., Pain R. H., Craig S. (1987) Tumour necrosis factor is a compact trimer. FEBS Lett. 211, 179–184 [DOI] [PubMed] [Google Scholar]
- 35. Sunnarborg S. W., Hinkle C. L., Stevenson M., Russell W. E., Raska C. S., Peschon J. J., Castner B. J., Gerhart M. J., Paxton R. J., Black R. A., Lee D. C. (2002) Tumor necrosis factor-α converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 277, 12838–12845 [DOI] [PubMed] [Google Scholar]
- 36. Lee M. H., Verma V., Maskos K., Becherer J. D., Knäuper V., Dodds P., Amour A., Murphy G. (2002) The C-terminal domains of TACE weaken the inhibitory action of N-TIMP-3. FEBS Lett. 520, 102–106 [DOI] [PubMed] [Google Scholar]
- 37. Gooljarsingh L. T., Lakdawala A., Coppo F., Luo L., Fields G. B., Tummino P. J., Gontarek R. R. (2008) Characterization of an exosite binding inhibitor of matrix metalloproteinase 13. Protein Sci. 17, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Black R. A., Doedens J. R., Mahimkar R., Johnson R., Guo L., Wallace A., Virca D., Eisenman J., Slack J., Castner B., Sunnarborg S. W., Lee D. C., Cowling R., Jin G., Charrier K., Peschon J. J., Paxton R. (2003) Substrate specificity and inducibility of TACE (tumour necrosis factor α-converting enzyme) revisited. The Ala-Val preference, and induced intrinsic activity. Biochem. Soc. Symp. 2003, 39–52 [DOI] [PubMed] [Google Scholar]