

Abstract
Background
Complex psychiatric traits have long been thought to be the result of a combination of genetic and environmental factors, and gene-environment interactions are thought to play a crucial role in behavioral phenotypes and the susceptibility and progression of psychiatric disorders. Candidate gene studies to investigate hypothesized gene-environment interactions are now fairly common in human genetic research, and with the shift towards genome-wide association studies, genome-wide gene-environment interaction studies are beginning to emerge.
Methods
We summarize the basic ideas behind gene-environment interaction, and provide an overview of possible study designs and traditional analysis methods in the context of genome-wide analysis. We then discuss novel approaches beyond the traditional strategy of analyzing the interaction between the environmental factor and each polymorphism individually.
Results
Two-step filtering approaches that reduce the number of polymorphisms tested for interactions can substantially increase the power of genome-wide gene-environment studies. New analytical methods including data-mining approaches, and gene-level and pathway-level analyses, also have the capacity to improve our understanding of how complex genetic and environmental factors interact to influence psychological and psychiatric traits. Such methods, however, have not yet been utilized much in behavioral and mental health research.
Conclusions
Although methods to investigate gene-environment interactions are available, there is a need for further development and extension of these methods to identify gene-environment interactions in the context of genome-wide association studies. These novel approaches need to be applied in studies of psychology and psychiatry.
Keywords: Gene-environment interaction, gene-level, pathway, gene-set, data-mining
Introduction
Genetic association studies of complex psychiatric traits, including those affecting children and adolescents, have identified specific genetic variants associated with many of these phenotypes (see e.g. (Banaschewski, Becker, Scherag, Franke, & Coghill, 2010; Hebebrand, Scherag, Schimmelmann, & Hinney, 2010; State, 2010)). However, relatively few of these findings have been consistently replicated, and confirmed associations account for a small proportion of the heritability of complex traits (see e.g. (Franke, Neale, & Faraone, 2009)). Thus, researchers are continuously working towards improving study designs and analyses to further our understanding of the etiology of behavioral traits and psychiatric illness.
It is well known that both genetic and environmental factors contribute to complex psychiatric traits, with multiple environmental and genetic influences playing a role in an individual’s risk of illness. It is also widely believed that gene-environment (G-E) interactions exist, meaning that for some genetic variants, the effect of the variant depends on certain environmental exposures, or equivalently, that the effect of an environmental factor depends on an individual’s genetic makeup. Zuk et al. (Zuk, Hechter, Sunyaev, & Lander, 2012) demonstrated that genetic interactions can inflate heritability estimates leading to what they call “phantom heritability”, and may thus contribute to the missing heritability phenomenon. As an example, they show that “limiting pathway models” can give rise to many small interaction effects (pairwise and higher-order), and that these models produce marginal effect sizes consistent with those seen in complex trait GWAS. Although their discussion focuses primarily on gene-gene (G-G) interaction effects, G-E effects can similarly contribute to the phantom heritability.
There are several key reasons for studying G-E interactions. Inclusion of G-E interactions in statistical models of factors contributing to complex traits may increase the power to detect novel genes or environmental factors that influence the trait through an interaction, which may otherwise be undetectable when the interaction is ignored. Furthermore, quantifying G-E interactions may help develop improved predictive models, either for disease onset or for response to treatment. Improved predictive models of disease risk can enable preventive strategies, particularly when the risk factors include modifiable environmental exposures. Improved predictive models of response to treatment can advance individualized medicine by assessing a patient’s chance of responding to a particular treatment regimen. However, it has been suggested that inclusion of G-G or G-E interaction effects is unlikely to substantially improve predictive models (Aschard, Chen, et al., 2012). Nevertheless, studying G-E interactions can help uncover disease-causing mechanisms, by demonstrating which factors work together to increase/decrease risk. Such knowledge can direct functional work to further resolve how an environmental factor may have a particular effect in a specific genetic context.
Despite the widespread belief that G-E interactions play an important role in human psychology and mental health, relatively few large-scale investigations of G-E have been conducted. Most G-E studies are performed in relatively small samples and investigate few candidate genetic variations and specific environmental factors. Although these small hypothesis-driven studies can contribute to our understanding of specific G-E effects, they often fail to uncover novel disease-causing mechanisms and tend to result in many un-replicated findings of limited importance (Duncan & Keller, 2011). While non-replication of genetic association findings is a big concern in all complex trait studies, it is particularly prevalent in studies of G-E (and G-G) effects. The replication problem is well exemplified in studies of the interaction between stress and the 5HTTLPR variation in the serotonin transporter gene (SLC6A4) in their effect on depression (Caspi, Hariri, Holmes, Uher, & Moffitt, 2010; Dunn et al., 2011), as well as studies of the interaction between the variation in the MAOA gene and childhood maltreatment in their effect on antisocial behavior and conduct disorder (Kim-Cohen et al., 2006). While in both of these areas of research there has been controversy, with both positive and negative studies being published for the 5HTTLPR-stress (Fisher et al., 2012; Peyrot et al., 2012) and MAOA-abuse (Fergusson, Boden, Horwood, Miller, & Kennedy, 2012; Kieling et al., 2013) interactions, recent literature on the MAOA-maltreatment interaction has been fairly consistent in support of the original findings. One factor contributing to inconsistent findings in G-E studies is the use of different definitions of the outcome (e.g. outcomes observed in childhood vs. adulthood, or antisocial behavior defined as conduct disorders, violent behavior, or arrests/convictions) and environmental exposure (e.g. childhood maltreatment defined as physical abuse, sexual abuse, or neglect) across studies. However, numerous other reasons contribute to the non-replication problem in G-E studies. Duncan and Keller provide a thorough discussion of reasons behind the lack of consistent G-E interaction findings in psychiatric studies (Duncan & Keller, 2011). In addition to inconsistent definitions of phenotypes and environmental variables, key factors contributing to this problem include low power (inadequate sample sizes), publication bias, and a high extent of multiple testing. Many of the candidate gene and genome-wide interaction studies conducted to date are underpowered. This dramatically affects the interpretations that can be drawn from much of the published literature, because significant findings from underpowered studies are likely to be false positives. The multiple testing issue goes beyond simply the number of single nucleotide polymorphisms (SNPs) analyzed, but also includes the tendency of researchers to investigate multiple outcomes/phenotypes and environmental factors and multiple characterizations of those factors, along with fitting a multitude of statistical models, and failure to properly account for such exploratory analyses. This practice is common in psychiatric G-E studies, including the MAOA literature. Investigators often define both abuse/stress exposures and behavioral outcomes in several ways, or use several alternative instruments and scales to measure the outcomes, usually without accounting for the resultant multiple testing as they consider the analyses exploratory (e.g. (Enoch, Steer, Newman, Gibson, & Goldman, 2010)). However, this level of multiple testing can contribute to false positive findings, including false replications, and thus makes the literature difficult to interpret.
In genetic association studies there has been a shift from small candidate gene studies to more agnostic genome-wide searches for polymorphisms contributing to complex traits. Furthermore, spurred by the fact that genetic association findings from genome-wide association studies (GWAS) explain a small proportion of the genetic variation contributing to complex traits, there has been increased interest in secondary analyses (Cantor, Lange, & Sinsheimer, 2010). It has become clear that the standard approach of analyzing GWAS data one SNP at a time is not adequately powered for identifying most genetic effects, at least not with the currently available sample sizes. Thus, creative approaches that make use of prior knowledge such as the gene to which a SNP belongs, or the function of the gene in relation to other genes’ functions (i.e. pathway annotation) are becoming more widely used. These approaches include gene-level tests (Neale & Sham, 2004) and pathway-based gene-set analyses (Fridley & Biernacka, 2011). It is also often suggested that incorporation of G-G and G-E interactions into analyses of GWAS data may improve the ability to detect relevant genetic variants (Cantor, et al., 2010; Cordell, 2002; Thomas, 2010a).
The shift from candidate gene to genome-wide studies seen in purely genetic studies is expanding to studies of G-E interactions. The concepts of the “GWEI” (genome-wide environmental interaction) study (Aschard, Lutz, et al., 2012) or the “GEWI” (gene-environment wide interaction) scan (Khoury & Wacholder, 2009; Thomas, 2010a) have been introduced, and genome-wide studies of G-E interactions have begun to emerge in psychiatric research literature (e.g. (Sonuga-Barke et al., 2008)). However, these approaches are still not routinely used, and most reports of G-E interactions in the psychiatric literature continue to focus on a few specific genetic variations. More importantly, the ideas of modeling G-E interactions and using advanced multi-SNP analyses to uncover novel mechanisms contributing to complex trait etiology have rarely been brought together in the context of GWAS data analysis.
In this review we begin by briefly introducing the most commonly used study designs and analysis techniques, with examples from the MAOA-maltreatment literature. We then discuss novel types of analyses that need to be further developed and applied in the context of G-E GWAS. A number of excellent reviews on G-E interactions in general (Thomas, 2010a, 2010b), or G-E effects in psychiatry (Dunn, et al., 2011; Wermter et al., 2010) have been published. In addition to genetic association studies, G-E interactions can be studied in other contexts, including family-based studies of heritability and linkage. These types of analyses have been reviewed by others (see e.g. (Dick, 2011)), and are outside the scope of this review. We limit our discussions to G-E studies of unrelated individuals with available genetic and environmental data (e.g. population-based case-control or cohort studies). Following the overview of the commonly used statistical approaches in G-E interaction studies, we focus on analysis methods that have been underutilized and may offer opportunities for greater insights into our understanding of how the genome and environment interact to result in complex observable behaviors and psychological and psychiatric traits. We emphasize the need for the development and application of such novel approaches, and suggest several possible avenues.
Study Design
Many of the standard study designs used to test either genetic or environmental effects separately can also be used to study G-E interactions—for example, the retrospective case-control design, the prospective cohort design, and family-based designs such as case-sibling, case-parent, trio or twin designs. Some designs, such as the case-only design, were specifically developed for testing interaction effects (Umbach & Weinberg, 1997). The MAOA-maltreatment interaction on antisocial behavior has primarily been evaluated with prospective cohort or matched cohort designs (Caspi et al., 2002; Nikulina, Widom, & Brzustowicz, 2012; Widom & Brzustowicz, 2006) including studies of twins (Foley et al., 2004). The various types of study designs for investigating G-E interactions and their advantages and disadvantages are discussed in detail by Thomas (Thomas, 2010a); in this review we focus only on the most commonly used designs.
In studies of child and adolescent psychology or psychiatry, a cohort design with longitudinal follow-up may be particularly useful to investigate research questions pertaining to effects that change during development. For instance, numerous longitudinal studies assessing various psychological aspects have been performed or are ongoing, with many using twin cohorts to evaluate genetic and environmental influences in behavioral phenotypes over time (Carmelli, Swan, Robinette, & Fabsitz, 1992; Greven, Rijsdijk, Asherson, & Plomin, 2012; van Soelen et al., 2012). Although some studies have considered a longitudinal design, few methods have been developed specifically for analyzing G-E effects using this type of data (Gu, Yang, Kraja, de Las Fuentes, & Davila-Roman, 2009; Mukherjee et al., 2012). For example, while some studies of the interaction between MAOA genotype and childhood maltreatment used longitudinal data on outcomes, most collapsed such information into a single summary outcome measure (Fergusson, et al., 2012), or analyzed more than one time point independently (Enoch, et al., 2010), rather than jointly modeling the longitudinal outcome trajectories. While these are valid approaches to data analysis, analyzing multiple time points separately can increase false positive rates if the multiple testing is not accounted for. Furthermore, fully modeling the data across time points may increase power and offer new insights into the G-E effects. Thus, development of methods to detect G-E interactions in longitudinal data is an important area of research, particularly for child psychology.
G-E interaction studies often use samples of convenience collected for prior studies. Even when testing G-E effects is included in the original study protocol, study design is often driven by considerations of cost and time efficiency. However, it is important to recognize that the study design affects the interpretation of the results, and thus the choice of study design should also take into account the research hypothesis of interest. For example, for researching how genetic and environmental effects change over time, a longitudinal cohort study might be necessary. A case-only design is a powerful approach for testing for G-E (or G-G) interactions when the environmental factor is not under genetic influence (or when the two genes of interest are independent in the general population). However, this design cannot be used to test for genetic or environmental main effects. If the disease or disorder of interest is rare, a case-control or family-based design with case ascertainment is more powerful than a cohort design. Although few studies of the MAOA-maltreatment interaction have used the case-control design, it is the most commonly used study design due to its efficiency and cost-effectiveness, and most statistical methods for detecting G-E interactions have been developed for data arising from this design. Thus, in discussing analysis methods we focus primarily on case-control data.
Regardless of the particular study design chosen, an important component of the study will be the measurement and collection of environmental data. In psychiatry, the most frequently studied environmental exposures are those related to stress, in particular childhood maltreatment and sexual abuse. However, other relevant exposures such as the prenatal environment have also been shown to play a role in a number of psychiatric traits, and are being investigated in G-E studies (Choudhry et al., 2012; Vuillermot et al., 2012). Definition of the environmental factor is critical to the success of G-E investigations, and careful thought must be given to ensure that the environmental factor is measured reliably and consistently and is appropriate for the research question. This is especially important in replication studies, where the environment needs to be measured as similarly as possible to the original study. Studies of the MAOA-maltreatment interaction have used a variety of environmental risk-factor definitions, including sexual or physical abuse (Fergusson, et al., 2012) neglect (Nikulina, et al., 2012), and family adversity (Enoch, et al., 2010). Differences in the definition of the environmental risk factor may contribute to failure to replicate G-E effects. However, demonstration of similar G-E effects with comparable, but not identical, environmental exposures can also validate findings and strengthen conclusions. For example, demonstrating similar patterns and directions of G-E interaction effects with exposure to childhood abuse and family adversity may indicate a general response to stressful early-life events. Although this type of validation (with differently-defined environmental factors) is strictly speaking not a replication, such parallel findings offer support for the existence of G-E effects, perhaps with more broadly defined environmental exposures.
Measurement error or misclassification of the environmental component can lead to bias or cause a reduction in power (Greenland, 1980; Wong, Day, Luan, & Wareham, 2004). When the underlying environmental exposure is quantitative, ideally it should be measured on a quantitative scale, as dichotomization or categorization of quantitative variables results in a loss of power. It is also critical to consider aspects such as timing of environmental exposure, and whether multiple measurements are necessary for each subject. An additional challenge is that the environmental variable is usually measured retrospectively, which can be prone to recall bias—a particular challenge in psychiatric studies. In a recent paper on the next generation of genetic epidemiology studies of complex traits, Mechanic et al identified exposure assessment as a key challenge in G-E studies, and recommend improved methods of exposure assessment that incorporate repeated measures of exposures over time in cohorts (Mechanic et al., 2012). While some studies of the interaction between MAOA and childhood maltreatment have relied on retrospective assessment of child abuse (e.g. (Beach et al., 2010; Fergusson, et al., 2012)), others had the advantage of using prospectively collected data from longitudinally-studied cohorts (e.g. (Enoch, et al., 2010; Nikulina, et al., 2012)). Some studies have also supplemented self-reported or parent-reported abuse with data from additional sources such as records from juvenile or adult criminal courts (Nikulina, et al., 2012). Mechanic et al. (Mechanic, et al., 2012) also stressed that new approaches to measuring environmental exposures need to be considered, such as the use of biomarkers to assess certain types of environmental exposures, or use of novel technological tools to aid in longitudinal data collection. When an environmental factor is difficult to assess, the trade-off between sample size and precision of data collection must be considered to maximize power, and two-stage designs involving more accurate exposure assessment in a subsample may be advantageous (Mechanic, et al., 2012).
Additionally, as in any risk-factor analysis, confounding factors should be considered as they can also bias the results. In particular, potential population stratification is a well-known concern in genetic studies (Tiwari et al., 2008). Although population stratification is less likely to bias estimates of interaction effects (Thomas, 2010a), it can bias G-E interaction analysis if a specific subpopulation or ancestral group is associated with the phenotype, genotype and the environmental exposure. Studies of the MAOA-maltreatment interaction have indicated that these effects may differ across ethnic sub-groups. In particular, a matched cohort study of mixed ethnicities examined the MAOA-abuse interaction on violent and antisocial behavior in white and non-white subjects separately, and observed evidence for an interaction in white subjects, but not in non-white subjects (Widom & Brzustowicz, 2006). A recent study utilizing data from the 1993 Pelotas Birth Cohort investigated the interaction between MAOA and maltreatment on conduct problems in a Brazilian sample, and observed no statistically significant evidence of a G-E interaction (Kieling, et al., 2013). It is possible that the ethnicity of subjects in this study contributed to the negative findings.
In G-E studies, appropriate adjustment for population stratification is not obvious (Aschard, Lutz, et al., 2012). Recently, however, principal component-based methods used for genome-wide association studies have been used to adjust for bias due to population stratification in studies of G-G interactions, and such ideas could be extended to G-E interactions (Bhattacharjee et al., 2010). While adjustment for population stratification using genomic data is a powerful technique that can account for even minor population substructure, family-based studies or studies limited to ethnically-homogenous subgroups can also guard against confounding by population stratification. The literature on MAOA-maltreatment has been variable in terms of adequately dealing with potential population stratification. For example, the primary analysis of Fergusson et al. included male subjects with mixed ancestry from the Christchurch Health and Development Study, which may have been confounded by population stratification effects; therefore secondary analyses were performed within a subset of Caucasian subjects after eliminating Maori, Pacific Island, and Asian ethnicities (Fergusson, et al., 2012). The similar results of the two analyses confirmed that association findings were unlikely to have arisen from confounding by population structure.
Traditional Analytical Methods used in Studies of G-E Interactions
Broadly speaking, a G-E interaction occurs if the genotype at a locus modifies the effect of an environmental factor on the phenotype, or equivalently, if an environmental factor modifies the effect of a genetic variant (e.g. SNP) on the phenotype. Statistically, a G-E interaction is defined as a deviation from a given model on a particular scale. For example, for a quantitative trait Y, an interaction is usually conceptualized as a departure from a model with an additive genetic and environmental effect on the phenotype, and is tested using the following model for the expected value of Y:
| Model (1) |
where E is the environmental factor (e.g. E=0,1 for a binary exposure), such as child abuse, and G is the genotype (typically coded as 0,1,2 representing the number of copies of the minor allele, although other codings are possible). The parameter βGE is the G-E interaction effect; if βGE=0, no interaction is present, while βGE≠0 indicates that the effect of E modifies the effect of G on the trait Y, and vice versa.
These ideas can be extended to other types of outcome variables. For example, for a binary trait/disease D (such as the presence/absence of depression), the logarithm of the odds of the diseased state D can be modeled, and the interaction can be tested using logistic regression (see Figure 1). Under this model, a G-E interaction is present if the effects of G and E on the log-odds of disease are not additive, which is evaluated by testing whether βGE=0 in the following model:
| Model (2) |
Figure 1.
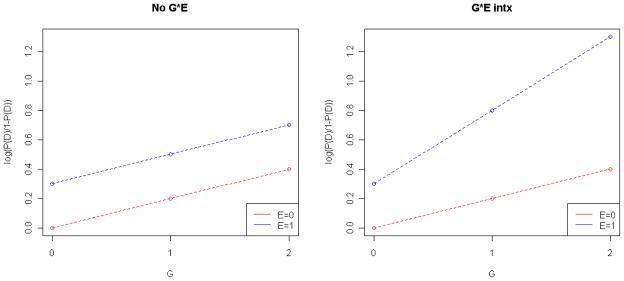
Logistic regression model log(P(D)/1-P(D)) = 0 + 0.2*G + 0.2*E + βGE*G*E for a gene-environment interaction on a binary trait with (A) βGE=0 and (B) βGE≠0.
Typically, studies of G-E interactions fit Model (1) using linear regression (for a quantitative trait) or Model (2) using logistic regression (for a binary trait), estimate the parameters including βGE (the G-E interaction effect), and test the null hypothesis that βGE=0. It should be noted that the parameters in this model are scale dependent, and that a value of βGE=0 in Model (2) may in fact correspond to an interaction when transformed from the logit to the penetrance or probit scale (Cordell, 2002). Conversely, an interaction on the additive scale in a linear or logistic regression model may be removable by applying an appropriate transformation. For certain types of interactions, use of such transformations may lead to more parsimonious models giving better fit and greater power to detect association with the contributing factors (Satagopan & Elston, 2013).
Although single-locus G-E analyses of quantitative and binary traits are usually based on models (1) & (2), alternative biological/functional model formulations have been proposed, such as those based on dominant or recessive, rather than additive, allelic effects. Further statistical parameterizations are also possible, which may improve power to detect genetic effects. For instance, Ma et al (Ma et al., 2012) developed models for testing G-E interaction effects with binary environmental variables in the “Natural and Orthogonal Interaction” (NOIA) framework. While the interpretation of the model parameters differs in this framework, these models were shown to be generally more powerful for detecting additive allelic effects in the presence of interactions.
Suppose a logistic regression model including a G-E interaction effect on a disease D (Model (2)) is fit to the data, with G=0,1,2 being the genotype at a particular locus (e.g. AA, AT, TT) and E=0,1 being a binary environmental exposure (such as smoking no/yes). If no G-E interaction is present (i.e. βGE = 0), exp(βG) is the odds ratio for the genetic effect, which describes the odds of disease for an individual with genotype G=1 compared to an individual with genotype G=0, regardless of environmental exposure status E. Similarly, exp(βE) is the odds ratio for the environmental effect, which gives the ratio of the odds of disease for an exposed individual (E=1) compared to an unexposed individual (E=0), regardless of the value of genotype G. However, if a G-E interaction exists then βGE ≠ 0 and the effect of G modifies the effect of E (and vice versa). In this case the OR of G=1 vs. G=0 is given by OR=exp(βG*1+βGE*E), which is dependent on the exposure status E. If E=0, then the OR for the genetic effect is OR=exp(βG); whereas if E=1, then the OR for the genetic effect is OR= exp(βG+βGE). A numerical example is shown in Table 1 and Figure 1, where the odds ratios of any G*E combination is given relative to the baseline G=0, E=0. For the particular models in this example, the genetic effect becomes higher in the presence of the environmental exposure under the model with a G-E interaction (Figure 1b). When no interaction is present, the odds ratio of disease attributed to the genetic effect is 1.22 regardless of exposure status; for an individual with G=1 as compared to G=0, the OR for an unexposed individual (E=0) is 1.22/1=1.22--the same as for an exposed individual with E=1 (1.65/1.35=1.22). However, under the model with G-E interaction, the odds ratio of disease attributed to the genetic effect is higher for an exposed individuals; when E=0, the OR for an individual with G=1 compared to G=0 is again 1.22/1=1.22, but when E=1, the respective OR is 2.23/1.35=1.65 (Table 1).
Table 1.
ORs for the model log(P(D)/1-P(D)) = 0 + 0.2*G + 0.3*E + βGE*G*E
| No G-E interaction (β GE=0) | G-E interaction (βGE= 0.3) | |||
|---|---|---|---|---|
| E=0 | E=1 | E=0 | E=1 | |
| G=0 | e0=1 | e0.3=e0.2*0+0.3*1=1.35 | e0=1 | e0.3=e0.2*0+0.3*1+0.3*0*1= 1.35 |
| G=1 | e0.2=e0.2*1+0.3*0=1.22 | e0.5=e0.2*1+0.3*1=1.65 | e0.2=e0.2*1+0.3*0+0.3*1*0=1.22 | e0.8=e0.2*1+0.3*1+0.3*1*1= 2.23 |
| G=2 | e0.4=e0.2*2+0.3*0=1.49 | e0.7=e0.2*2+0.3*1=2.01 | e0.4=e0.2*2+0.3*0+0.3*2*0=1.49 | E1.3=e0.2*2+0.3*1+0.3*2*1= 3.67 |
In a case-control analysis, when either Model (1) or Model (2) is used to evaluate the G-E interaction by testing the null hypothesis H0: βGE=0, a one degree of freedom (1df) test is performed, since a single parameter is tested. However, a researcher may be interested in a different hypothesis. In particular, the goal may be to identify relevant genetic variants while allowing for the possibility of a G-E interaction, which can be investigated by jointly testing H0: βG=βGE=0. This two degree of freedom (2df) joint test of two parameters has been shown to be a statistically powerful analytical method (Kraft, Yen, Stram, Morrison, & Gauderman, 2007). Which of these two tests is employed depends on the research hypothesis and the distinction between an explicit interest in testing for G-E interaction or simply detecting a genetic effect while allowing for a possible interaction.
Although we have primarily focused on the case-control design, the case-only design is also a popular strategy. This design requires different analytical methods. By restricting the analysis to cases, only hypotheses of interactions can be tested, and this design cannot be used to evaluate marginal genetic or environmental effects. Case-only methods examine potential correlations or associations between the genetic factor and the environmental component; assuming that the genetic and environmental factors are independent in the general population, an association between G and E in cases implies a G-E interaction effect on the binary trait. Young et al used a case-only design to examine the MAOA genotype-maltreatment interaction on conduct severity (Young et al., 2006). However, they did not apply the usual case-only analysis to examine the G-E interaction effect on the presence of conduct disorder by investigating the association between genotype (MAOA) and environment (maltreatment); instead, they modeled the G-E interaction effect on conduct disorder severity within cases as the outcome. The analysis approach of Young et al. (Young, et al., 2006) may have lead to a loss in power, potentially contributing to the observed negative result. More importantly, the results should not be considered as a failure to replicate prior findings of MAOA-maltreatment interaction effect, as the outcome in the study of Young et al. was severity of conduct disorder within a group of adolescents with persistent conduct and substance use problems, rather than presence of conduct disorder.
While a case-only analysis has been shown to be more powerful than the case-control approach, it is prone to bias if the assumption of G-E independence fails (Albert, Ratnasinghe, Tangrea, & Wacholder, 2001). This assumption is likely to be invalid in certain psychiatric studies, when the exposures of interest are behavioral factors that are themselves under genetic influence. To avoid this potential bias while retaining good statistical power, a number of hybrid methods have been developed to combine the estimates derived from the case-control and case-only designs (Dai et al., 2012). For example, an empirical Bayes approach that calculates a weighted average of both estimates was proposed by Mukherjee and Chatterjee (Mukherjee & Chatterjee, 2008).
Extensions to Improve Power for Genome-Wide Gene-Environment Interaction Studies
Unlike candidate G-E interaction studies such as those focused on the MAOA-maltreatment effect on antisocial behavior, genome-wide G-E studies seek to identify interaction effects in the absence of pre-defined hypotheses. Such G-E interaction studies are often conducted by performing a test of interaction with each genotyped SNP. However, this one-SNP-at-a-time 16 analysis approach inevitably requires stringent significance thresholds to account for the multiple testing burden; therefore sample size requirements to achieve 80% power for G-E interactions can be very large due to the sheer number of interactions considered, requiring thousands of samples to detect even moderate effect sizes (Murcray, Lewinger, Conti, Thomas, & Gauderman, 2011). Thus, a number of screening approaches have been developed to reduce the number of G-E effects tested (and consequently the multiple testing correction) and therefore improve power of G-E studies. These two-stage analytical approaches first use a screening procedure to select a smaller group of SNPs to be tested for G-E effects at the second stage. In the approach of Kooperberg and Leblanc, only genetic variants that exhibit some marginal evidence of a genetic effect are considered for an interaction (Kooperberg & Leblanc, 2008). Screening based on marginal effects can miss G-E interactions with SNPs that do not exhibit main effects on the trait, and thus other variations of this two-stage approach utilize two different analyses of interactions in the available data at the two stages to complement marginal effects screening (Murcray, Lewinger, & Gauderman, 2009). Further extensions of such methods have been proposed, including a “hybrid” method (Murcray, et al., 2011) and a “cocktail” method (Hsu et al., 2012) that combine multiple estimators (case-control, case-only, Empirical Bayes, etc.) in the two-stage screening procedure. As compared to a standard approach of testing for interaction of the environmental variable with each SNP throughout the genome, which requires a very stringent multiple testing correction, such two-step approaches can substantially increase power of genome-wide G-E interaction studies.
Recent Developments and New Directions
Just as GWAS of marginal genetic effects have traditionally studied the association between the phenotype and each SNP individually, G-E analyses of GWAS data tend to evaluate the interaction of the environmental factor with each SNP, one at a time. Because this approach tends to be severely under-powered for analysis of G-E effects on the genome-wide scale, two-step filtering strategies have been proposed to alleviate some of the multiple-testing burden in genome-wide G-E studies. In the genetic-only realm, it has also been recognized that analyzing one SNP at a time has low power to detect small genetic effects, and methods such as gene-level tests (Neale & Sham, 2004) and pathway-based gene-set analyses (Biernacka, Jenkins, Wang, Moyer, & Fridley, 2012; Fridley & Biernacka, 2011) have thus been suggested as a way to incorporate prior biological or functional knowledge and enhance power to detect genetic association. Data-mining techniques have also emerged as a promising avenue for high dimensional genetic data analysis that allows for simultaneous modeling of effects of multiple genetic or environmental risk factors. Yet most of the gene-level and pathway-level approaches have not been extended to include G-E interactions, and G-E GWAS that have been performed have rarely looked beyond interactions of the environmental variable with individual SNPs. One notable exception was the recent work of Wei et al (Wei et al., 2012) on lung cancer, which used GWAS data to investigate interactions between asbestos exposure and genetic variation at the SNP, gene, and pathway levels. G-E interaction searches of psychiatric traits may benefit from similar approaches that incorporate information on the relationships between SNPs (e.g. belonging to the same gene, or gene-set). Similarly, data-mining approaches have rarely been utilized for G-E interaction studies. While these methods have been advocated for investigation of G-G effects, the current versions are less appropriate for G-E investigation. In this section we discuss these emerging approaches and propose areas where additional development of methods is needed.
Data Mining Methods
Because many of the traditional analytical approaches are underpowered for testing for interaction effects, a number of novel analysis methods have recently been proposed for detection of G-G and G-E interactions, including data-mining methods. These approaches search for patterns in high dimensional data and are most appropriate for exploratory analysis, and therefore can be considered as hypothesis-generating, rather than hypothesis testing, and should be validated in independent data. Additionally, these methods typically do not involve a framework to assess interactions explicitly, but rather consider joint models that allow for an interaction. Thus, subsequent statistical modeling should be used to gain further understanding of the effect of the identified factors on the disease risk or phenotype.
Many data-mining methods were designed to address issues of multiple testing and computational efficiency related to the analysis of high-dimensional data, and therefore can be used for both studies of candidate genes and GWAS. This ability to handle high throughput data is a major advantage of such approaches; however, most data-mining methods for genetic data analysis were developed for predictive modeling or identification of interacting genetic risk factors, and were not specifically designed to detect G-E interactions. These methods have sometimes been applied to search for interactions between a gene and the environment, where the environmental variable is treated no differently than a SNP. Hence, methods development and extensions for data-mining approaches specific to G-E interactions is an area that warrants further research.
Nevertheless, such data-mining methods for G-G interactions remain promising alternatives to traditional analytical techniques to discover potentially interacting genetic and environmental factors. Two popular non-parametric data-mining approaches are Random Forests (RF) and Multifactor Dimensionality Reduction (MDR). MDR was originally designed to detect SNP-SNP interactions by reducing combinations of genotypes at multiple loci into a single class variable representing high/low risk categories (Ritchie et al., 2001). MDR examines the association between a binary phenotype and this new collapsed variable defined by multilocus genotypes. Therefore the MDR method is actually evaluating combinations of genetic factors jointly, allowing for the possibility of an interaction; it does not test for an interaction explicitly, which should be examined post-hoc. MDR has often been used to rank SNPs and combinations of SNPs based on their predictive ability, thus functioning as a screening tool. Specific interactions of identified SNPs can then be further examined in a second stage or replication study. A number of freely available software packages exist to apply MDR and examine post-hoc relationships (Bush, Dudek, & Ritchie, 2006; Winham & Motsinger-Reif, 2011).
Most applications of MDR thus far involved the identification of G-G interactions, although MDR has been utilized for G-E analysis. Most applications of MDR to environmental data consider the environmental factor in the same manner as another categorical variable/SNP, where it is one of many factors that are considered in the analysis; because the environmental factor is not included in every combination evaluated, this is not a direct examination of G-E interaction. Such an approach has been applied in the psychiatric literature to candidate gene studies of BDNF, GSK3B, and the serotonin transporter to investigate the effect of negative life events in major depressive disorder (Yang et al., 2010; K. R. Zhang et al., 2009). This strategy requires that the environmental component be represented categorically rather than as a continuous trait. The traditional non-parametric MDR approach has been extended to model-based MDR (MBMDR), which incorporates a regression framework to allow for adjustment for covariates and quantitative traits (Calle, Urrea, Malats, & Van Steen, 2010). However, these extensions were not designed to address a search for G-E interaction, and therefore further methodological extension is needed for the application of MDR in this context.
Another popular data-mining approach that has been proposed for the study of G-G interactions is Random Forests (RF), which was originally designed with the goal of prediction using high-dimensional data (Breiman, 2001). RF is an ensemble or ‘forest’ of many classification or regression trees, each constructed on a bootstrap sample of subjects from the original data set, with a random subset of predictors (e.g. SNPs) considered at each node. The hierarchical splits of the classification or regression trees allow for non-linear interactions to be incorporated into the prediction model. Hypothesis testing is not performed as part of an RF analysis. Rather, if an interaction is present, then it may be captured by the model. Furthermore, RF does not perform variable selection, but instead computes variable importance measures that assess the predictive ability of each variable. These measures can be used to rank SNPs, and have been suggested as a way of detecting interacting genetic variants. However, in high dimensional data they have been shown to only rank interacting factors highly if they have strong marginal effects (Winham et al., 2012). Additionally, most variable importance measures do not reveal which factors are interacting (i.e. which SNPs are interacting with E); a variable importance measure to assess the joint contribution of all possible pairs of SNPs has been proposed (Bureau et al., 2005), although the intensive computation required to evaluate such a measure is prohibitive in analysis of high-dimensional data resulting from GWAS.
Extension of RF to consider G-E interactions is further complicated by the fact that it is not clear how best to incorporate environmental covariates into a RF analysis of genetic data. A covariate can be regressed out of the phenotype, such that a regression model is fit between the phenotype and covariate, and then the residual is used as the new trait value, but this strategy precludes the investigation of G-E interaction. Instead, the environmental factor can be incorporated into the prediction model, allowing for the possibility of G-E interaction. However, it has been shown that using predictors on vastly different scales (such as SNPs and quantitative environmental variables) can bias results when considered together in the same forest; solutions have been proposed to avoid such biases through an alternative implementation of RF that uses sub-sampling with replacement rather than bootstrap sampling and an alternative permutation scheme to assess variable importance (Strobl, Boulesteix, Kneib, Augustin, & Zeileis, 2008; Strobl, Boulesteix, Zeileis, & Hothorn, 2007). Nevertheless, how environmental variables and their interactions with genetic factors should be modeled in the context of RF is a critical, but open research question.
Because of the difficulty in interpretation of interactions, as well as the computational restrictions and multiple testing burden associated with high-dimensional genetic data, RF is often utilized as a filter or screening tool that allows for interactions; SNPs and environmental factors ranked highly with RF in the first stage can be carried forward to the second stage where their interactions are explicitly modeled with traditional statistical methods. However, such two stage designs have a tendency towards over-fitting because of the selection of the top SNPs in the same dataset in which they are subsequently further analyzed; to avoid over-fitting, either cross-validation or permutation techniques should be used to evaluate significance, or each stage should be applied to an independent data set. Few applications of RF have focused on G-E interactions, but examples exist where RF was used as a screening tool with subsequent tests of interactions. For example, Maenner et al used RF variable importance measures to screen for relevant genetic and environmental factors in a study of age of onset of coronary heart disease; interactions between genetic factors and smoking were then modeled with generalized estimating equations (Maenner et al., 2009). Similarly, Zhai et al used RF to screen for SNP predictors of esophageal cancer, and then modeled G-E interactions using the top SNPs with a case-only analysis in a second stage (Zhai et al., 2012). While current implementations of RF can be utilized to investigate both genetic and environmental effects, these implementations were not designed with the detection and interpretation of G-E interactions in mind, thus further research is necessary to optimize these and related methods in this setting.
Gene-Level G-E Analysis
It has long been recognized that gene-level tests may offer power gains over analyses of single variants (Neale & Sham, 2004). In particular, if there are several causal variants in a gene, combining these effects into a single test can increase power to detect associations with relevant genes. Although these methods do not identify specific SNP-trait associations, they are designed to identify relevant genes to improve understanding of disease mechanisms and enable further investigation of polymorphisms within these genes. Such gene-level tests are routinely used in psychiatric data analysis (see e.g. (Karpyak et al., 2012)); however, the gene-level approach has rarely been implemented for the study of G-E interactions.
A number of methods for gene-level analysis have been proposed, including the use of the minimum SNP-specific p-value, use of a summary measure of the individual SNP p-values, or joint modeling of the effects of all SNPs in the gene (Ballard, Cho, & Zhao, 2010). Furthermore, rare variants are typically analyzed using gene-level tests (Asimit & Zeggini, 2010; Bansal, Libiger, Torkamani, & Schork, 2010), and development of new gene-level rare variant analysis methods is an active area of research. In addition to gene-level analyses that do not require phased genotype data, haplotype analyses have been widely used as a multi-marker alternative to single SNP tests (see e.g. (N. Liu, Zhang, & Zhao, 2008)). Of the many multi-marker approaches that have been developed for genetic data analysis, few have been extended, or applied, to the investigation of G-E interactions. However, several methods have recently been introduced for G-G interaction assessment at the gene level, and some of these can also be applied to G-E interaction studies.
Chatterjee et al. (Chatterjee, Kalaylioglu, Moslehi, Peters, & Wacholder, 2006) proposed a one-degree of freedom test (based on Tukey’s model) that makes use of multiple SNPs in a region to perform a single test of (or accounting for) G-G or G-E interaction. This approach can improve the power to detect genetic associations in the presence of G-G or G-E interaction by using a single interaction parameter, but it assumes that a SNP’s interaction effect is proportional to its marginal effect on the trait. Because this assumption is restrictive, Wang et al. suggested an extension of the Tukey approach to model G-G (or G-E) interactions (Wang, Ho, Ye, Strickler, & Elston, 2009). This approach exploits the joint association between two genes or between a gene and an environmental factor, in addition to marginal SNP associations with the trait, using the partial least squares algorithm applied to data from multiple SNPs in a gene. These approaches are similar in nature to gene-level tests of marginal genetic effects that jointly model the effects of multiple SNPs in a gene in a regression framework; however, here the goal is to test the G-E effects rather than just the G effects.
Dimension reduction techniques such as principal components analysis (PCA), which has been shown to be a powerful technique for gene-level analysis (Gauderman, Murcray, Gilliland, & Conti, 2007), can also be extended to study interactions. For example, in a study of rheumatoid arthritis Li et al. derived principal components (PCs) representing each of the genes of interest, and then searched for G-G interactions by testing the PC-PC interaction effects in a logistic regression model (Li, Tang, Biernacka, & de Andrade, 2009). Along these lines, He et al. used a gene-level approach that applies “optimal” LD-based weights to markers prior to performing PCA, and extended it to G-G interaction analysis by testing for interaction between pairs of the resulting PCs (He et al., 2011). Other dimension reduction methods that have been used in the context of G-G interaction analysis include a canonical correlation based method proposed by Peng et al. (Peng, Zhao, & Xue, 2010). Similar approaches can be considered in the context of G-E interaction studies.
Extensions to G-E analysis are also emerging for gene-level approaches involving both common and rare variants. For instance, Tzeng et al. proposed an approach based on gene-trait similarity regression for analysis of quantitative traits, which evaluates the similarity between pairs of individuals to assess gene-level gene-environment interactions (Tzeng et al., 2011). This gene-trait similarity method aggregates multiple markers in the gene, which can be weighted based on allele frequency, and is equivalent to a haplotype random-effects model. The proposed test statistics, derived from the haplotype random-effects model, allow for both joint tests of genetic effects as well as explicit tests of G-E interaction. Haplotypes can also be analyzed directly for interactions with the environment, using extensions of traditional approaches for single variants, although care must be taken to correctly account for phase uncertainty. Recently, such methods have been extended to more efficiently detect haplotype-haplotype and haplotype-environment interactions using Bayesian hierarchical modeling (Li, Zhang, & Yi, 2011).
In the framework of data-mining approaches, a gene-based MDR method was recently introduced (Oh et al., 2012), which uses a two-step procedure consisting of within-gene and between-gene MDR analysis. The first step uses MDR to jointly model effects of multiple SNPs within a gene, while the second step performs MDR analysis on these summarized gene-level effects. This method has the advantages of reducing computation time and providing an interpretation at the gene-level, but because it may be biased due to the use of the two-stage approach, the statistical properties of the methods need to be evaluated.
Wei et al. (Wei, et al., 2012) performed G-E analyses at the gene level in a GWAS of lung cancer using a modification of the VErsatile Gene-based Association Study (VEGAS) approach of Liu et al. (J. Z. Liu et al., 2010). The VEGAS method combines the p-values from multiple SNPs within a gene and corrects for LD without the need for permutations; Wei et al, on the other hand, used it to combine SNP-environment interaction p-values within each gene. Although the performance of this modified use of the VEGAS test needs to be evaluated in the context of G-E interaction analysis, this study provides a good example of the type of innovative analyses that should be considered in genome-wide G-E studies of psychiatric traits.
Various other gene-level tests that have been proposed for analysis of SNP data could potentially be extended to test for G-G or G-E interactions. Simple gene-level tests can be performed by analyzing each SNP individually, and then taking the minimum p-value or a combination of SNP-level p-values as the test statistic for the gene (Zaykin, Zhivotovsky, Westfall, & Weir, 2002). These methods could be extended to G-E interaction studies by using SNP-environment interaction p-values instead of p-values of SNP-specific marginal effects. With such approaches, care must be taken to correct for the number of SNPs and linkage disequilibrium (LD) pattern in the gene, which is usually done by using permutations. However, appropriate permutations are more challenging--or in fact not possible--when testing a hypothesis regarding interactions (Anderson, 2001; Buzkova, Lumley, & Rice, 2011). Buzkova et al. (Buzkova, et al., 2011) show that it is not typically possible to construct exact permutation tests of G-G and G-E interaction effects. Instead, they propose a parametric bootstrap method that can be applied to tests of interactions, including those between an environmental factor and multiple correlated SNPs.
Further research is required to improve the detection of G-E interactions at the gene level, as such approaches may be considerably more powerful for identifying genes that have effects on complex traits that are dependent on the environmental context. While many of the methods proposed for gene-level tests of marginal genetic effects or gene-level G-G interaction analysis can be extended to evaluate G-E effects, it is important to assess whether these novel extensions have correct type 1 errors, and determine which methods are most powerful under various G-E interaction models.
Pathway-Level Analyses Incorporating G-E Effects
Beyond testing for effects at the gene-level, pathway-based approaches were introduced as a way to assess the overall evidence of association of a trait with genetic variation in a group of functionally related genes (i.e. a gene set), thus incorporating prior biological knowledge (Fridley & Biernacka, 2011). Pathway-based approaches could also be used in the context of G-E interaction analysis to determine which environmental factors interact with which sets of genes to influence a complex trait. Pathway-level analyses can be used to perform an agnostic search for gene sets that interact with a specific environmental exposure in their effect on the outcome of interest by evaluating G-E effects in the context of all gene sets annotated in existing pathway databases. Alternatively, a candidate pathway approach can be taken by performing a global test of G-E effects with a specific pre-defined set of genes. In such studies, the candidate gene set should be carefully selected, taking into account both the environmental exposure and the outcome under consideration. For example, if one aims to test whether a particular set of genes interacts with exposure to stress in its effect on psychological or behavioral outcomes, a gene set representing a stress-response pathway, such as genes involved in the hypothalamic-pituitary-adrenocortical axis (Ising & Holsboer, 2006) may be a reasonable candidate.
In addition to the definition of candidate pathways or gene sets, deciding which specific genetic variants should be included is an important challenge for pathway-level G-E analysis. Traditionally, after selection of genes for a specific pathway, all genotyped SNPs within those genes, and within a pre-specified distance from those genes, are included in the pathway analysis (Fridley & Biernacka, 2011). However, other approaches for SNP selection could be used, such as inclusion of SNPs that regulate expression of the genes in the pathway (eQTLs), or limiting SNPs in the analysis to those with a putative functional role, or weighing SNPs in the analysis according to their functional role. External bioinformatics resources, such as those emerging from the ENCODE project, may be helpful in guiding the SNP selection.
Although pathway-level analyses may not identify specific SNPs (or even genes) that interact with the environmental factor, they can identify pathways through which the environmental factor exerts its effect on the phenotype. More focused studies can then be undertaken to determine which specific genetic variants have functional effects, and to characterize the variant-environment interactions. Development of new Gene Set Analysis (GSA) methods is an active area of research (Biernacka, et al., 2012; Fridley & Biernacka, 2011; K. Zhang, Cui, Chang, Zhang, & Wang, 2010). However, the methods developed to date have focused on genetic associations, and have not incorporated G-E interactions. Wei et al. (Wei, et al., 2012) performed pathway-level analyses of G-E interaction effects using data from a GWAS of lung cancer. They used an implementation of an extension of the popular Gene Set Enrichment Analysis (GSEA) approach (K. Zhang, et al., 2010). However, given the complexities of permutation in the analysis of interaction effects discussed above (Buzkova, et al., 2011), the assessment of significance of the G-E effect at the pathway level in such analyses needs to be further evaluated. Thomas (Thomas, 2010a, 2010b) reviewed the ideas behind studying G-E interactions in the context of biological pathways, noting that GSEA and hierarchical models may be particularly promising for empirical evaluation of effects in the context of pre-defined pathways, but had not yet been applied to studies of G-E effects. He also discussed Bayesian network analysis as a promising approach for discovering novel pathways in the context of G-E studies.
In addition to the approaches discussed by Thomas, other types of GSA methods could be extended to investigate G-E effects. As in analyses of SNP-environment interactions, as new pathway-based methods are developed and applied to the study of complex traits, two types of approaches may be considered: gene-set analyses that incorporate G-E interaction effects (i.e. allow for interaction), versus gene-set analyses that specifically test for the combined role of GE interaction effects in a given gene set (i.e. explicitly test for interaction). Both types of approaches could lead to increased understanding of the complex etiology of psychiatric traits. Within the first class of methods (those that incorporate G-E interaction effects rather than explicitly testing for them), data mining approaches such as RF may be applicable. For example, Chang et al applied RF for pathway analysis in a study of the genetic factors underlying glioma (Chang et al., 2008); in addition, Chung and Chen recently proposed a two-stage approach based on RF for (genetic) pathway analysis (Chung & Chen, 2012). By incorporating environmental effects into the RF, the overall joint effect of environmental factors and SNPs in a pathway could be assessed. However, as discussed above, this approach would be limited by the fact that RFs are not well suited to analysis of data with different scales (for example, 3-level genotype data with quantitative environmental data). Moreover, RFs have been shown to have poor power to detect interacting factors, despite being widely considered as a method that does detect interactions (Winham, et al., 2012). To overcome these limitations, novel extensions of data-mining approaches more suitable for G-E pathway-based analyses need to be developed.
Among methods that specifically test for the combined role of G-E interaction effects in a given gene set, extensions of p-value combination methods could be considered. For example, Biernacka et al. compared a number of GSA methods for analysis of SNP data, including an approach that first applies gene-level tests, and then combines the gene-level p-values at the gene-set (pathway) level (Biernacka, et al., 2012). To extend this method to pathway-level G-E interaction analysis, the interaction of each gene with the environment could be investigated using one of the aforementioned gene-level G-E approaches; for example, one could test for interactions of gene-level PCs with the environment in a regression framework, and then combine the G-E interaction p-values for all genes belonging to a pathway using an approach such as those proposed by Biernacka et al. (Biernacka, et al., 2012). However, in extending such methods to G-E interaction analysis, it is important to investigate the statistical properties of the extended approach, especially in light of the potential difficulties with empirically assessing the significance of the tests due to the challenges with permutation testing of interaction effects (Buzkova, et al., 2011).
Discussion/Conclusions
Complex psychiatric traits are the result of multiple genetic and environmental factors that may interact in complicated ways to influence disease susceptibility. Environmental exposures throughout development likely impact psychological traits, but a person’s genetic makeup is believed to modulate the environment’s influence on these complex phenotypes. Studies of G-E interactions are therefore critical in child and adolescent psychiatry—however, the investigation of complex interacting effects is challenging.
While genome-wide studies of G-E effects are important for discovery of novel interactions between genetic and environmental risk factors, these studies tend to be under-powered and have identified few significant findings. Creative approaches for analyzing the data emerging from such studies are needed to increase the statistical power. Many prior reviews of G-E interactions have correctly concluded that larger G-E studies with more detailed characterization of environmental factors and clinical phenotypes are needed; we strongly agree with these conclusions. In discussing the implications of the MAOA-abuse interaction effects on antisocial behavior, Fergusson et al. (Fergusson, et al., 2012) noted that it is time to move beyond studies of single genes towards studies that can delineate the combined effects of multiple genetic and environmental risk factors. In line with this goal, we also emphasize the need for more advanced analyses that make better use of the available data and prior biological knowledge (e.g. pathways). In a recent paper providing recommendations for the next generation of large-scale genetic epidemiology studies, some of the challenges and issues associated with identifying G-E and G-G interactions were summarized (Mechanic, et al., 2012). The recommendations included the development of new methods and study designs for improved identification of G-E (and G-G) interactions. Here we summarized a number of promising approaches for incorporating and detecting G-E interaction effects in the context of genome-wide association studies.
In a recent exploration of why GWAS findings do not explain much of the heritability of complex traits (i.e. the missing heritability problem), Zuk et al (Zuk, et al., 2012) showed that certain types of disease risk models can lead to many small interaction effects, which would be largely undetectable in typical studies. Two-step/filtering methods that reduce the number of interactions tested at the second stage can substantially increase power of genome-wide G-E studies. In this review, we also outlined several other types of analytical methods that have the potential to accelerate the identification of genome-wide G-E interaction effects in complex psychiatric traits. These methods include extensions of data-mining methods that are more suitable for G-E interaction assessment, and approaches for evaluating G-E effects at the gene and pathway levels. Further development and extension of such novel approaches, and implementation of the methods in freely-available user-friendly software, will be critical to the identification of genome-wide G-E interaction effects in complex psychiatric traits.
Putative interactions identified using genome-wide G-E studies need to be confirmed and validated using functional studies making use of animal models (Schwartzer, Koenig, & Berman, 2012) and cellular human models (Ober & Vercelli, 2011) in order to provide further insights into the underlying biological mechanisms. Randomized experiments, such as the study of the MAOA-adversity interaction on aggression performed by Gallardo-Pujol et al. (Gallardo-Pujol, Andres-Pueyo, & Maydeu-Olivares, 2013), can provide further support for G-E interaction findings. Nevertheless, investigation of G-E effects in genome-wide studies of human populations using advanced statistical techniques has the potential to make important contributions to understanding of the complex causal pathways underlying behavioral phenotypes and psychiatric illness.
Key Points.
Numerous study designs and analysis methods can be used to investigate gene-environment interactions; most existing methods assess the interaction effects between an environmental factor and individual genetic variations
Most studies of gene-environment interactions in psychology and psychiatry have focused on few candidate genes, and few results have been consistently replicated
Genome-wide association studies of gene-environment interactions are beginning to appear in psychiatric literature. These studies primarily use the approach of investigating interactions between individual SNPs and the environment.
Although investigation of genome-wide gene-environment interaction effects has the potential for important new discoveries, analyzing each SNP individually followed by Bonferroni correction tends to be severely under-powered. Two-stage strategies that filter SNPs to reduce the number of interactions tested in the second stage have been proposed to increase power. Another possibility is to use novel multi-marker approaches to reduce the number of tests performed.
New statistical methods are being developed to investigate gene-environment interactions, including data-mining methods and gene and pathway-level analyses. Application of such methods in psychiatric research is likely to lead to new discoveries of genes and pathways that contribute to complex psychiatric phenotypes differentially depending on the environmental context.
Acknowledgments
This work was supported by the National Institute on Drug Abuse (NIDA; R21 DA019570, PI Biernacka). This article was invited by the journal, for which the principal author has been offered a small honorarium payment towards personal expenses.
Abbreviations
- G-E
Gene-environment
- G-G
gene-gene
Footnotes
Conflict of interest statement: No conflict of interest.
References
- Albert PS, Ratnasinghe D, Tangrea J, Wacholder S. Limitations of the case-only design for identifying gene-environment interactions. American journal of epidemiology. 2001;154(8):687–693. doi: 10.1093/aje/154.8.687. [DOI] [PubMed] [Google Scholar]
- Anderson MJ. Permutation tests for univariate or multivariate analysis of variance and regression. Canadian Journal of Fisheries and Aquatic Sciences. 2001;58(3):626–639. [Google Scholar]
- Aschard H, Chen J, Cornelis MC, Chibnik LB, Karlson EW, Kraft P. Inclusion of gene-gene and gene-environment interactions unlikely to dramatically improve risk prediction for complex diseases. Am J Hum Genet. 2012;90(6):962–972. doi: 10.1016/j.ajhg.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschard H, Lutz S, Maus B, Duell EJ, Fingerlin TE, Chatterjee N, et al. Challenges and opportunities in genome-wide environmental interaction (GWEI) studies. Human genetics. 2012;131(10):1591–1613. doi: 10.1007/s00439-012-1192-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimit J, Zeggini E. Rare variant association analysis methods for complex traits. Annual review of genetics. 2010;44:293–308. doi: 10.1146/annurev-genet-102209-163421. [DOI] [PubMed] [Google Scholar]
- Ballard DH, Cho J, Zhao H. Comparisons of multi-marker association methods to detect association between a candidate region and disease. Genetic epidemiology. 2010;34(3):201–212. doi: 10.1002/gepi.20448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaschewski T, Becker K, Scherag S, Franke B, Coghill D. Molecular genetics of attention-deficit/hyperactivity disorder: an overview. European child & adolescent psychiatry. 2010;19(3):237–257. doi: 10.1007/s00787-010-0090-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nature Reviews Genetics. 2010;11(11):773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach SR, Brody GH, Gunter TD, Packer H, Wernett P, Philibert RA. Child maltreatment moderates the association of MAOA with symptoms of depression and antisocial personality disorder. J Fam Psychol. 2010;24(1):12–20. doi: 10.1037/a0018074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S, Wang Z, Ciampa J, Kraft P, Chanock S, Yu K, et al. Using principal components of genetic variation for robust and powerful detection of gene-gene interactions in case-control and case-only studies. Am J Hum Genet. 2010;86(3):331–342. doi: 10.1016/j.ajhg.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka JM, Jenkins GD, Wang L, Moyer AM, Fridley BL. Use of the gamma method for self-contained gene-set analysis of SNP data. European journal of human genetics: EJHG. 2012;20(5):565–571. doi: 10.1038/ejhg.2011.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Machine Learning. 2001;45(1):5–32. [Google Scholar]
- Bureau A, Dupuis J, Falls K, Lunetta KL, Hayward B, Keith TP, et al. Identifying SNPs predictive of phenotype using random forests. Genetic epidemiology. 2005;28(2):171–182. doi: 10.1002/gepi.20041. [DOI] [PubMed] [Google Scholar]
- Bush WS, Dudek SM, Ritchie MD. Parallel multifactor dimensionality reduction: a tool for the large-scale analysis of gene-gene interactions. Bioinformatics. 2006;22(17):2173–2174. doi: 10.1093/bioinformatics/btl347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzkova P, Lumley T, Rice K. Permutation and parametric bootstrap tests for gene-gene and gene-environment interactions. Annals of human genetics. 2011;75(1):36–45. doi: 10.1111/j.1469-1809.2010.00572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle ML, Urrea V, Malats N, Van Steen K. mbmdr: an R package for exploring gene-gene interactions associated with binary or quantitative traits. Bioinformatics. 2010;26(17):2198–2199. doi: 10.1093/bioinformatics/btq352. [DOI] [PubMed] [Google Scholar]
- Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86(1):6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmelli D, Swan GE, Robinette D, Fabsitz R. Genetic influence on smoling- A study of male twins. New England Journal of Medicine. 1992;327(12):829–833. doi: 10.1056/NEJM199209173271201. [DOI] [PubMed] [Google Scholar]
- Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. The American journal of psychiatry. 2010;167(5):509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297(5582):851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Chang JS, Yeh RF, Wiencke JK, Wiemels JL, Smirnov I, Pico AR, et al. Pathway analysis of single-nucleotide polymorphisms potentially associated with glioblastoma multiforme susceptibility using random forests. Cancer Epidemiology Biomarkers & Prevention. 2008;17(6):1368–1373. doi: 10.1158/1055-9965.EPI-07-2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Kalaylioglu Z, Moslehi R, Peters U, Wacholder S. Powerful multilocus tests of genetic association in the presence of gene-gene and gene-environment interactions. Am J Hum Genet. 2006;79(6):1002–1016. doi: 10.1086/509704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry Z, Sengupta SM, Grizenko N, Fortier ME, Thakur GA, Bellingham J, et al. LPHN3 and attention-deficit/hyperactivity disorder: interaction with maternal stress during pregnancy. Journal of Child Psychology and Psychiatry. 2012;53(8):892–902. doi: 10.1111/j.1469-7610.2012.02551.x. [DOI] [PubMed] [Google Scholar]
- Chung RH, Chen YE. A Two-Stage Random Forest-Based Pathway Analysis Method. PloS one. 2012;7(5) doi: 10.1371/journal.pone.0036662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11(20):2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- Dai JY, Logsdon BA, Huang Y, Hsu L, Reiner AP, Prentice RL, et al. Simultaneously testing for marginal genetic association and gene-environment interaction. American journal of epidemiology. 2012;176(2):164–173. doi: 10.1093/aje/kwr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM. Gene-environment interaction in psychological traits and disorders. Annual review of clinical psychology. 2011;7:383–409. doi: 10.1146/annurev-clinpsy-032210-104518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. The American journal of psychiatry. 2011;168(10):1041–1049. doi: 10.1176/appi.ajp.2011.11020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn EC, Uddin M, Subramanian SV, Smoller JW, Galea S, Koenen KC. Research review: gene-environment interaction research in youth depression - a systematic review with recommendations for future research. Journal of child psychology and psychiatry, and allied disciplines. 2011;52(12):1223–1238. doi: 10.1111/j.1469-7610.2011.02466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch MA, Steer CD, Newman TK, Gibson N, Goldman D. Early life stress, MAOA, and gene-environment interactions predict behavioral disinhibition in children. Genes, brain, and behavior. 2010;9(1):65–74. doi: 10.1111/j.1601-183X.2009.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergusson DM, Boden JM, Horwood LJ, Miller A, Kennedy MA. Moderating role of the MAOA genotype in antisocial behaviour. The British journal of psychiatry: the journal of mental science. 2012;200(2):116–123. doi: 10.1192/bjp.bp.111.093328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher HL, Cohen-Woods S, Hosang GM, Korszun A, Owen M, Craddock N, et al. Interaction between specific forms of childhood maltreatment and the serotonin transporter gene (5-HTT) in recurrent depressive disorder. Journal of affective disorders. 2012 doi: 10.1016/j.jad.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley DL, Eaves LJ, Wormley B, Silberg JL, Maes HH, Kuhn J, et al. Childhoodadversity, monoamine oxidase a genotype, and risk for conduct disorder. Arch Gen Psychiatry. 2004;61(7):738–744. doi: 10.1001/archpsyc.61.7.738. [DOI] [PubMed] [Google Scholar]
- Franke B, Neale BM, Faraone SV. Genome-wide association studies in ADHD. Human genetics. 2009;126(1):13–50. doi: 10.1007/s00439-009-0663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridley BL, Biernacka JM. Gene set analysis of SNP data: benefits, challenges, and future directions. European journal of human genetics: EJHG. 2011;19(8):837–843. doi: 10.1038/ejhg.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo-Pujol D, Andres-Pueyo A, Maydeu-Olivares A. MAOA genotype, social exclusion and aggression: an experimental test of a gene-environment interaction. Genes, brain, and behavior. 2013;12(1):140–145. doi: 10.1111/j.1601-183X.2012.00868.x. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ, Murcray C, Gilliland F, Conti DV. Testing association between disease and multiple SNPs in a candidate gene. Genetic epidemiology. 2007;31(5):383–395. doi: 10.1002/gepi.20219. [DOI] [PubMed] [Google Scholar]
- Greenland S. The effect of misclassification in the presence of covariates. American journal of epidemiology. 1980;112(4):564–569. doi: 10.1093/oxfordjournals.aje.a113025. [DOI] [PubMed] [Google Scholar]
- Greven CU, Rijsdijk FV, Asherson P, Plomin R. A longitudinal twin study on the association between ADHD symptoms and reading. Journal of Child Psychology and Psychiatry. 2012;53(3):234–242. doi: 10.1111/j.1469-7610.2011.02445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu CC, Yang WW, Kraja AT, de Las Fuentes L, Davila-Roman VG. Genetic association analysis of coronary heart disease by profiling gene-environment interaction based on latent components in longitudinal endophenotypes. BMC Proc. 2009;3(Suppl 7):S86. doi: 10.1186/1753-6561-3-s7-s86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Wang K, Edmondson AC, Rader DJ, Li C, Li M. Gene-based interaction analysis by incorporating external linkage disequilibrium information. European journal of human genetics: EJHG. 2011;19(2):164–172. doi: 10.1038/ejhg.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebebrand J, Scherag A, Schimmelmann BG, Hinney A. Child and adolescent psychiatricgenetics. European child & adolescent psychiatry. 2010;19(3):259–279. doi: 10.1007/s00787-010-0091-y. [DOI] [PubMed] [Google Scholar]
- Hsu L, Jiao S, Dai JY, Hutter C, Peters U, Kooperberg C. Powerful cocktail methods for detecting genome-wide gene-environment interaction. Genetic epidemiology. 2012;36(3):183–194. doi: 10.1002/gepi.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising M, Holsboer F. Genetics of stress response and stress-related disorders. Dialogues Clin Neurosci. 2006;8(4):433–444. doi: 10.31887/DCNS.2006.8.4/mising. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpyak VM, Winham SJ, Biernacka JM, Cunningham JM, Lewis KA, Geske JR, et al. Association of GATA4 sequence variation with alcohol dependence. Addiction biology. 2012 doi: 10.1111/j.1369-1600.2012.00482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, Wacholder S. Invited commentary: from genome-wide association studies to gene-environment-wide interaction studies--challenges and opportunities. American journal of epidemiology. 2009;169(2):227–230. doi: 10.1093/aje/kwn351. discussion 234–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieling C, Hutz MH, Genro JP, Polanczyk GV, Anselmi L, Camey S, et al. Gene- environment interaction in externalizing problems among adolescents: evidence from the Pelotas 1993 Birth Cohort Study. Journal of child psychology and psychiatry, and allied disciplines. 2013;54(3):298–304. doi: 10.1111/jcpp.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, et al. MAOA, maltreatment, and gene-environment interaction predicting children’s mental health: new evidence and a meta-analysis. Molecular psychiatry. 2006;11(10):903–913. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- Kooperberg C, Leblanc M. Increasing the power of identifying gene x gene interactions in genome-wide association studies. Genetic epidemiology. 2008;32(3):255–263. doi: 10.1002/gepi.20300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Human heredity. 2007;63(2):111–119. doi: 10.1159/000099183. [DOI] [PubMed] [Google Scholar]
- Li J, Tang R, Biernacka JM, de Andrade M. Identification of gene-gene interaction using principal components. BMC proceedings. 2009;3(Suppl 7):S78. doi: 10.1186/1753-6561-3-s7-s78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang K, Yi N. A Bayesian hierarchical model for detecting haplotype-haplotype and haplotype-environment interactions in genetic association studies. Human heredity. 2011;71(3):148–160. doi: 10.1159/000324841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. A Versatile Gene-Based Test for Genome-wide Association Studies. Am J Hum Genet. 2010;87(1):139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Zhang K, Zhao H. Haplotype-association analysis. Advances in genetics. 2008;60:335–405. doi: 10.1016/S0065-2660(07)00414-2. [DOI] [PubMed] [Google Scholar]
- Ma J, Xiao F, Xiong M, Andrew AS, Brenner H, Duell EJ, et al. Natural and orthogonal interaction framework for modeling gene-environment interactions with application to lung cancer. Human heredity. 2012;73(4):185–194. doi: 10.1159/000339906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenner MJ, Denlinger LC, Langton A, Meyers KJ, Engelman CD, Skinner HG. Detecting gene-by-smoking interactions in a genome-wide association study of early-onset coronary heart disease using random forests. BMC Proc. 2009;3(Suppl 7):S88. doi: 10.1186/1753-6561-3-s7-s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechanic LE, Chen HS, Amos CI, Chatterjee N, Cox NJ, Divi RL, et al. Next generation analytic tools for large scale genetic epidemiology studies of complex diseases. Genetic epidemiology. 2012;36(1):22–35. doi: 10.1002/gepi.20652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee B, Chatterjee N. Exploiting gene-environment independence for analysis of case-control studies: an empirical Bayes-type shrinkage estimator to trade-off between bias and efficiency. Biometrics. 2008;64(3):685–694. doi: 10.1111/j.1541-0420.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- Mukherjee B, Ko YA, Vanderweele T, Roy A, Park SK, Chen J. Principal interactions analysis for repeated measures data: application to gene-gene and gene-environment interactions. Stat Med. 2012;31(22):2531–2551. doi: 10.1002/sim.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcray CE, Lewinger JP, Conti DV, Thomas DC, Gauderman WJ. Sample size requirements to detect gene-environment interactions in genome-wide association studies. Genetic epidemiology. 2011;35(3):201–210. doi: 10.1002/gepi.20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcray CE, Lewinger JP, Gauderman WJ. Gene-environment interaction in genome- wide association studies. American journal of epidemiology. 2009;169(2):219–226. doi: 10.1093/aje/kwn353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75(3):353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina V, Widom CS, Brzustowicz LM. Child abuse and neglect, MAOA, and mental health outcomes: a prospective examination. Biological psychiatry. 2012;71(4):350–357. doi: 10.1016/j.biopsych.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Vercelli D. Gene-environment interactions in human disease: nuisance or opportunity? Trends in genetics: TIG. 2011;27(3):107–115. doi: 10.1016/j.tig.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Lee J, Kwon MS, Weir B, Ha K, Park T. A novel method to identify high order gene-gene interactions in genome-wide association studies: gene-based MDR. BMC bioinformatics. 2012;13(Suppl 9):S5. doi: 10.1186/1471-2105-13-S9-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Zhao J, Xue F. A gene-based method for detecting gene-gene co-association in a case-control association study. European journal of human genetics: EJHG. 2010;18(5):582–587. doi: 10.1038/ejhg.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrot WJ, Middeldorp CM, Jansen R, Smit JH, de Geus EJ, Hottenga JJ, et al. Strong effects of environmental factors on prevalence and course of major depressive disorder are not moderated by 5-HTTLPR polymorphisms in a large Dutch sample. Journal of affective disorders. 2012 doi: 10.1016/j.jad.2012.08.044. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69(1):138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan JM, Elston RC. Evaluation of removable statistical interaction for binary traits. Statistics in medicine. 2013;32(7):1164–1190. doi: 10.1002/sim.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzer JJ, Koenig CM, Berman RF. Using mouse models of autism spectrum disorders to study the neurotoxicology of gene-environment interactions. Neurotoxicology and teratology. 2012 doi: 10.1016/j.ntt.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonuga-Barke EJ, Lasky-Su J, Neale BM, Oades R, Chen W, Franke B, et al. Does parental expressed emotion moderate genetic effects in ADHD? An exploration using a genome wide association scan. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2008;147B(8):1359–1368. doi: 10.1002/ajmg.b.30860. [DOI] [PubMed] [Google Scholar]
- State MW. The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron. 2010;68(2):254–269. doi: 10.1016/j.neuron.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl C, Boulesteix AL, Kneib T, Augustin T, Zeileis A. Conditional variable importance for random forests. BMC bioinformatics. 2008;9:307. doi: 10.1186/1471-2105-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl C, Boulesteix AL, Zeileis A, Hothorn T. Bias in random forest variable importance measures: Illustrations, sources and a solution. BMC bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Gene--environment-wide association studies: emerging approaches. Nature reviews Genetics. 2010a;11(4):259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Methods for investigating gene-environment interactions in candidate pathway and genome-wide association studies. Annual review of public health. 2010b;31:21–36. doi: 10.1146/annurev.publhealth.012809.103619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari HK, Barnholtz-Sloan J, Wineinger N, Padilla MA, Vaughan LK, Allison DB. Review and evaluation of methods correcting for population stratification with a focus on underlying statistical principles. Human heredity. 2008;66(2):67–86. doi: 10.1159/000119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng JY, Zhang DW, Pongpanich M, Smith C, McCarthy MI, Sale MM, et al. Studying Gene and Gene-Environment Effects of Uncommon and Common Variants on Continuous Traits: A Marker-Set Approach Using Gene-Trait Similarity Regression. Am J Hum Genet. 2011;89(2):277–288. doi: 10.1016/j.ajhg.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach DM, Weinberg CR. Designing and analysing case-control studies to exploit independence of genotype and exposure. Stat Med. 1997;16(15):1731–1743. doi: 10.1002/(sici)1097-0258(19970815)16:15<1731::aid-sim595>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- van Soelen ILC, Brouwer RM, Peper JS, van Leeuwen M, Koenis MMG, van Beijsterveldt T, et al. Brain SCALE: Brain Structure and Cognition: an Adolescent Longitudinal Twin Study into the Genetic Etiology of Individual Differences. Twin Research and Human Genetics. 2012;15(3):453–467. doi: 10.1017/thg.2012.4. [DOI] [PubMed] [Google Scholar]
- Vuillermot S, Joodmardi E, Perlmann T, Ogren SO, Feldon J, Meyer U. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32(2):436–451. doi: 10.1523/JNEUROSCI.4831-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Ho G, Ye K, Strickler H, Elston RC. A partial least-square approach for modeling gene-gene and gene-environment interactions when multiple markers are genotyped. Genetic epidemiology. 2009;33(1):6–15. doi: 10.1002/gepi.20351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Wang LE, McHugh MK, Han Y, Xiong M, Amos CI, et al. Genome-wide geneenvironment interaction analysis for asbestos exposure in lung cancer susceptibility. Carcinogenesis. 2012;33(8):1531–1537. doi: 10.1093/carcin/bgs188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermter AK, Laucht M, Schimmelmann BG, Banaschweski T, Sonuga-Barke EJ, Rietschel M, et al. From nature versus nurture, via nature and nurture, to gene x environment interaction in mental disorders. European child & adolescent psychiatry. 2010;19(3):199–210. doi: 10.1007/s00787-009-0082-z. [DOI] [PubMed] [Google Scholar]
- Widom CS, Brzustowicz LM. MAOA and the “cycle of violence:” childhood abuse and neglect, MAOA genotype, and risk for violent and antisocial behavior. Biological psychiatry. 2006;60(7):684–689. doi: 10.1016/j.biopsych.2006.03.039. [DOI] [PubMed] [Google Scholar]
- Winham SJ, Colby CL, Freimuth RR, Wang X, de Andrade M, Huebner M, et al. SNP interaction detection with Random Forests in high-dimensional genetic data. BMC bioinformatics. 2012;13:164. doi: 10.1186/1471-2105-13-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winham SJ, Motsinger-Reif AA. An R package implementation of multifactor dimensionality reduction. BioData Min. 2011;4(1):24. doi: 10.1186/1756-0381-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Day NE, Luan JA, Wareham NJ. Estimation of magnitude in gene-environment interactions in the presence of measurement error. Stat Med. 2004;23(6):987–998. doi: 10.1002/sim.1662. [DOI] [PubMed] [Google Scholar]
- Yang CX, Xu Y, Sun N, Ren Y, Liu ZF, Cao XH, et al. The combined effects of the BDNF and GSK3B genes modulate the relationship between negative life events and major depressive disorder. Brain Research. 2010;1355:1–6. doi: 10.1016/j.brainres.2010.07.079. [DOI] [PubMed] [Google Scholar]
- Young SE, Smolen A, Hewitt JK, Haberstick BC, Stallings MC, Corley RP, et al. Interaction between MAO-A genotype and maltreatment in the risk for conduct disorder: failure to confirm in adolescent patients. The American journal of psychiatry. 2006;163(6):1019–1025. doi: 10.1176/ajp.2006.163.6.1019. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Zhivotovsky LA, Westfall PH, Weir BS. Truncated product method for combining P-values. Genetic epidemiology. 2002;22(2):170–185. doi: 10.1002/gepi.0042. [DOI] [PubMed] [Google Scholar]
- Zhai RH, Zhao Y, Liu G, Ter-Minassian M, Wu IC, Wang ZX, et al. Interactions between environmental factors and polymorphisms in angiogenesis pathway genes in esophageal adenocarcinoma risk: A case-only study. Cancer. 2012;118(3):804–811. doi: 10.1002/cncr.26325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Cui S, Chang S, Zhang L, Wang J. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic acids research. 2010;38(Web Server issue):W90–95. doi: 10.1093/nar/gkq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KR, Xu Q, Xu Y, Yang H, Luo JX, Sun Y, et al. The combined effects of the 5-HTTLPR and 5-HTR1A genes modulates the relationship between negative life events and major depressive disorder in a Chinese population. Journal of affective disorders. 2009;114(1–3):224–231. doi: 10.1016/j.jad.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(4):1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]