

Abstract
Functional gastrointestinal disorders are complex symptom-based disorders without agreed upon biomarkers or pathophysiology. A better understanding of the genetic architecture of these disorders would help to better identify their complex biology and explain the common comorbidity with other disorders of persistent pain, mood, and affect, as well as possibly make it possible to identify subgroups of patients who respond to customized therapies. In contrast to monogenic diseases, polygenic diseases and traits are characterized by the contribution of common variants in a large number of genes, as well as environmental factors, to the vulnerability of an individual. Family and twin studies have clearly established a genetic component in irritable bowel syndrome. Although candidate gene studies have identified a few gene polymorphisms that may be correlated with the syndrome, small sample size, lack of reproducibility in large data sets, and the unreliability of the clinical phenotype require caution when extrapolating to a major role of any of the reported polymorphisms in the pathophysiology of irritable bowel syndrome. Future progress in this area will require better characterization of intermediate phenotypes with large effect size for the clinical phenotype, as well as consideration of gene-gene, environment-gene (epigenetics), and sex-gene interactions, genome-wide association, and whole genome sequencing approaches in large data sets.
Keywords: Irritable Bowel Syndrome, Intermediate Phenotypes, Candidate Genes, Genome-wide Association Studies
Functional gastrointestinal disorders (FGIDs) are part of a spectrum of complex syndromes that are characterized by persistent pain and discomfort referred to different visceral (irritable bowel syndrome [IBS], functional dyspepsia, interstitial cystitis/painful bladder syndrome) and somatic structures (including fibromyalgia and temporomandibular joint disorder).1 In the absence of reliable biological markers, or a full understanding of the pathophysiology of any of these syndromes, they are currently defined by symptom criteria and sometimes by the exclusion of “organic” disease.2 In addition to overlap with each other, these syndromes share several clinical and epidemiologic features, including high comorbidity with symptoms of anxiety and depression, somatization, and stress sensitivity. These similarities have led to the concept that these syndromes, including IBS, share certain pathophysiologic features (eg, central pain amplification) as well as common genetic and environmental vulnerability factors, in addition to having syndrome-specific environmental and genetic factors that are responsible for the individual syndrome manifestation.1 In the case of FGIDs, such syndrome-specific factors may be related to gastrointestinal function.
There are several fundamental problems when trying to dissect the genetic basis of complex symptom-based disorders, in particular in the case of “functional” disorders that do not have any agreed upon biological marker. The biological mechanisms presenting as symptom-based phenotypes are likely to be heterogeneous. For example, it is likely that certain individuals with mucosal immune activation (eg, lymphocytic colitis; increased mast cell numbers) and those without such changes both meet IBS symptom criteria.3 In contrast to monogenic diseases, such as cystic fibrosis or Mediterranean fever, in which the variance of the phenotype is completely explained by a single gene, polygenic disorders are determined by many common (and possibly rare) gene variants, each contributing less than 5% and oftentimes much less than 1% of the variance of the clinical phenotype. The common disease/common variant hypothesis states that complex traits result from underlying genetic variants that occur with a relatively high frequency, but the effect size of each of the variants is relatively small.4,5 A clinical phenotype only emerges if a critical threshold of multiple genetic and environmental factors is reached. The clinical phenotype is not stable. For example, affected patients commonly meet symptom criteria for several of these syndromes at a given time, or at different stages of their lives, and the clinical phenotypes in an individual are not stable over time.6 Similarly, family histories suggest that relatives and different generations of patients with FGID often do not experience gastrointestinal symptoms but meet criteria of one of the other syndromes. In this review, we will present the current status of our knowledge of the genetic basis of IBS (the FGIDs in which most studies have been conducted), make a critical assessment of these findings, and discuss novel approaches and future directions.
Family and Twin Studies
Familial aggregation has been reproducibly shown in several clinic-based studies of patients with IBS (Table 1), while family studies of other FGIDs are lacking. However, clustering of IBS in families does not necessarily implicate a genetic basis, but could also result from environmental exposures shared in a household, including diet or lifestyle behaviors, exposure to adverse life events within the family, learned cognitions about disease and illness behavior, or even shared exposure to microorganisms.7,8
Table 1.
Family Studies of IBS
| Citation | Finding |
|---|---|
| Whorwell et al, Gut 198649 | 33% of patients with IBS vs 2% of controls have a family history of IBS |
| Bellentani et al, Fam Pract 199050 | IBS in a first-degree relative predicts a diagnosis of IBS in patients with abdominal pain |
| Locke et al, Mayo Clin Proc 200051 | Reporting a relative with abdominal pain or altered bowel habit predicts IBS (odds ratio, 2.5; 95% confidence interval, 1.5–4.2) |
| Levy et al, Am J Gastroenterol 200052 | Health maintenance organization database study: children of parents with IBS are more likely to present to providers with abdominal pain than children of parents without IBS |
| Kanazawa et al, Dig Dis Sci 200453 | Familial aggregation of IBS is not due to the result of psychological factors or psychiatric disorders |
| Saito et al, Neurogastroenterol Motil 20088 | Case relatives are 3 times more likely to meet Rome criteria for IBS than control relatives by direct survey |
Twin studies are the traditional method used to estimate the genetic and environmental contributions to disease. To date, 5 twin studies of FGIDs, mostly of IBS, have been published (Table 2). Based on these studies, the genetic liability of IBS and FGIDs ranges between 0 and 20% based on concordance rates, and the genetic heritability of FGIDs was estimated to be between 22% and 57%. The variation in heritability estimates likely is due to differences in study methodology. Importantly, in these studies, general shared environment was not a significant predictor of phenotypic variance. However, in the Virginia Twin Study, it was also observed that having a mother or father with IBS was an independent predictor of IBS status, which was stronger than having a twin with IBS, suggesting that social learning or stability of the family environment (which might be influenced by anxiety or depression in one of the parents, disorders often associated with IBS) may have greater influence on the development of IBS than genetics alone.9
Table 2.
Twin Studies of IBS
| No. of twin pairs | Case definition | Data collection method | Genetic liability (%) | Heritability in best-fit model (%)
|
|||
|---|---|---|---|---|---|---|---|
| Additive genetic | Shared environment | Individual environment | |||||
| Australian Twin Study | 343 | Functional bowel disease | No. of symptoms reported by interview | 20 | 57 | 0 | 43 |
| Virginia Twin Study | 6060 | IBS | Check box by IBS | 8.8 | — | — | — |
| Minnesota Twin Study | 986 | IBS by Rome II | Validated bowel disease questionnaire | 16 | 22 | 0 | 78 |
| Norwegian Twin Study | 3334 | IBS | Check box by IBS | 13.3 | 48 | 0 | 52 |
| British Twin Study | 1870 | IBS by Rome II | Validated questionnaire | 1 | 0 | 26 | 74 |
Candidate Gene Studies
Candidate gene studies are studies whereby investigators postulate that a specific gene, and typically a specific functional gene polymorphism that results in alterations in protein function or quantity, may play a role in disease pathophysiology. The most commonly used approach in IBS to date has been to search for correlations of a candidate gene polymorphism with the symptom-based phenotype IBS (Table 3). More recently, a few studies have aimed to identify correlations between gene polymorphism and biological intermediate phenotypes (see the following text), such as colonic transit, enhanced perception of visceral stimuli, or altered brain responses.10,11
Table 3.
Candidate Gene Studies in IBS
| Protein (gene) | Polymorphism | Reference | Case sample size | Finding summary |
|---|---|---|---|---|
| Serotonin pathway | ||||
| Serotonin transporter (SLC6A4) | 5-HTT LPR | Pata et al, Am J Gastroenterol 200254 | 54 | No association; LS associated with IBS-D |
| Kim et al, Gut 200455 | 256 | No association | ||
| Yeo et al, Gut 200456 | 186 | Only patients with IBS-D; SS associated with IBS-D | ||
| Lee et al, Korean J Gastroenterol 200457 | 33 | No association | ||
| Park et al, Neurogastroenterol Motil 200658 | 190 | |||
| Saito et al, Neurogastroenterol Motil 200759 | 50 | No association; S associated with IBS with a mixed bowel pattern | ||
| Li et al, Dig Dis Sci 200760 | 87 | No association; LL associated with IBS-C | ||
| Sikander et al, J Clin Gastroenterol 200961 | 151 | No association; SS associated with IBS-C | ||
| Niesler et al, Eur J Gastroenterol Hepatol 200962 | 196 | N/A; no association with IBS-C or IBS-D; lower frequency of SS in male IBS-D | ||
| Kohn et al, Dig Dis Sci 200963 | 186 | No association with IBS | ||
| Saito et al, Gastroenterology 200964 | 312 | No association with IBS or IBS subtypes | ||
| STin2 VNTR | Pata et al, Am J Gastroenterol 200254 | 54 | No association with IBS or IBS subtypes | |
| Niesler et al, Eur J Gastroenterol Hepatol 200962 | 196 | No overall IBS analysis performed; no association with IBS-C or IBS-D | ||
| Kohn et al, Dig Dis Sci 200963 | 186 | No association | ||
| rs25531 | Kohn et al, Dig Dis Sci 200963 | 186 | G allele associated with IBS | |
| 5-HT2A receptor subunit (HTR2A) | 102T>C | Pata et al, J Clin Gastroenterol 200465 | 54 | No association |
| −1438G>A | Pata et al, J Clin Gastroenterol 200465 | 54 | No association | |
| 5-HT3A receptor Type A subunit (HT3A) | −42C>T | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-C; C/T genotype increased frequency in UK IBS-D, not replicated in another cohort |
| −25C>T | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-D or IBS-C | |
| *70C>T | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-D or IBS-C | |
| *503C>T | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-D or IBS-C | |
| 5-HT3B receptor Type B subunit | 386A>C | Fukudo et al, Neuroimage 200925 | 28 | AC and CC subjects had differential brain activation compared with AA subjects |
| 5-HT3E receptor Type E subunit (HT3E) | *76G>A | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; G/A genotype more common in IBS-D (odds ratio, 8.53; P <.05), replicated in separate cohort |
| 5-HT3C receptor Type C subunit (5-HT3C) | 489C>A | Kapeller et al, Gastroenterology 200967 | 197 | CC associated with female patients with IBS-D |
| *115T>G | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-D or IBS-C | |
| *138C>T | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-D or IBS-C | |
| −189G>A | Kapeller et al, Hum Mol Genet 200866 | 200 | No overall IBS analysis performed; no association with IBS-D or IBS-C | |
| Inflammatory markers | ||||
| Interleukin-10 | −1082G>A | Gonsalkorale et al, Gut 200368 | 230 | AA or GA associated |
| Van der Veek et al, Gastroenterology 200469 | 111 | No association | ||
| −819C>T | Van der Veek et al, Gastroenterology 200469 | 111 | No association | |
| Transforming growth factor β1 | +869 T>C | Gonsalkorale et al, Gut 200368 | 230 | No association |
| +915G>C | Gonsalkorale et al, Gut 200368 | 230 | No association | |
| Tumor necrosis factor α | −308G>A | Van der Veek et al, Gastroenterology 200469 | 111 | GA associated |
| Adrenergic pathway | ||||
| Norepinephrine transporter (NET) | 237G>C | Kim et al, Gut 200455 | 276 | Not detected |
| α2A-adrenergic receptor (A2AR) | 753C>G | Kim et al, Gut 200455 | 276 | Not detected |
| 1291C>G | Kim et al, Gut 200455 | 276 | Borderline association with IBS-C | |
| Sikander et al, Clin Chim Acta 200970 | 151 | Not associated with IBS overall; non-CC genotype associated with IBS-D | ||
| Alphaα2C-adrenergic receptor (A2CR) | Del 332-335 | Kim et al, Gut 200455 | ||
| Other | ||||
| β3 subunit G protein (GNβ3) | 825C>T | Holtmann et al, Gastroenterology 200471 | 20 | No association |
| Andresen et al, Gastroenterology 200672 | 233 | No association | ||
| Saito et al, Neurogastroenterol Motil 200759 | 50 | No association | ||
| Saito et al, Gastroenterology 200964 | 312 | No association with IBS overall, but there was a gene-environment interaction between this SNP and prior gastrointestinal infection with IBS | ||
| Cholecystokinin type 1 receptor (CCK1-R) | 266G>C 8641G>A 984T>C |
Colucci et al, Gastroenterology 200973 | 60 | No association between these 3 SNPs with IBS or IBS symptom severity |
| Nav1.5 channel (SCN5A) | G298S | Saito et al, Am J Physiol Gastrointest Liver Physiol 200974 | 49 | Reported in 1 patient with IBS-D |
| 43 genes (inflammatory, intestinal barrier, serotonin) | 78 SNPs | Villani et al, Gastroenterology 200975 | SNPs in CDH1, TLR9, and IL6 were associated with postinfectious IBS | |
| 43 genes (inflammatory, intestinal barrier, serotonin) | 78 SNPs | Villani et al, Gastroenterology 200975 | SNPs in IL10 and HTR2A associated with IBS-D in female patients; SNPs in CDH1, TLR9, IL6 were not associated with non–postinfectious IBS | |
IBS-C, constipation-predominant irritable bowel syndrome.
Associations of Gene Polymorphisms With Clinical Phenotype
As shown in Table 3, more than a dozen candidate genes have been studied for possible associations with the clinical phenotype of IBS. The majority of genes studied encode serotonin-related proteins (eg, serotonin trans-porter, 5-HT3 receptor, 5-HT2 receptor), proteins involved in noradrenergic signaling (eg, α-adrenergic receptor), and genes encoding immune markers (eg, interleukin-10, transforming growth factor β1). Others genes include the β3 subunit of the G protein (GNβ3), cholecystokinin type 1 receptor (CCK1-R), Nav1.5 sodium channel (SCN5A), and other intestinal epithelial barrier factors.
The best-studied polymorphisms are those related to serotonin (5-hydroxytryptamine [5-HT]) signaling in the brain-gut axis, including the serotonin transporter (SERT), and several 5-HT receptors. This emphasis on genetic variations within the 5-HT signaling system is based on the excellent construct validity of this system from both preclinical12 and clinical13 studies. Parallel alterations in central and gut-related 5-HT signaling could explain the typical constellation of altered motility and secretion associated with increased stress responsiveness and increased anxiety. The 5-HTT LPR polymorphism (for serotonin transporter–linked polymorphic region) in the promoter region of the serotonin transporter gene SLC6A4 is the most intensively studied polymorphism in IBS and has also been studied in psychiatric syndromes,14 fibromyalgia, and temporomandibular joint disorder.15–17 Surprisingly, several small case-control studies performed in patients with IBS have shown variable results, and a recent meta-analysis suggests that there is no simple association between this genetic variant and the clinical phenotype of IBS.18 There has been one study showing a potential association between another functional single nucleotide polymorphism (SNP) (rs25531) on the SLC6A4 gene with IBS and another study linking the 76G>A SNP on the 5-HT3 receptor type E subunit gene (replicated in a second study) and the 489C>A SNP on the HTR3C gene with diarrhea-predominant IBS (IBS-D).
The majority of the other polymorphisms shown in Table 3 have not been found to be associated with the IBS clinical phenotype. However, the majority of these candidate gene/clinical phenotype association studies were small (<200 subjects), did not take into account the caveats addressed in the following section, and with the exception of the 5-HTT LPR and the 5-HT3E gene polymorphism studies have never been replicated in a separate cohort. Due to these limitations, very few of these reported findings allow us to make any conclusions about their validity and, in the opinion of the authors, should not be implicated in the pathophysiology of IBS at this point.
Association of Candidate Gene With Intermediate Phenotype
Intermediate phenotypes, also known as endophenotypes, are biologically related to the clinical phenotype of interest, stable over time and reproducible in different laboratories, detectable at baseline or in the presence of a provocative stimulus, and often present in unaffected family members.19,20 In contrast to the clinical phenotype, which is based on subjective and unstable patient reports (see the previous text), intermediate phenotypes are biological or physiologic variables that can be objectively measured and are believed to be more closely related to genetic factors (see Figure 1). This candidate genotype/intermediate phenotype approach has been successfully applied to many polygenic disorders, including chronic pain and schizophrenia.21 For example, polymorphisms of the COMT gene have been associated with objective measures of pain sensitivity17 and with endogenous opioid release in the brain.22 Unfortunately, no intermediate endophenotype has been described to date for FGIDs that meets all the criteria previously listed.
Figure 1.
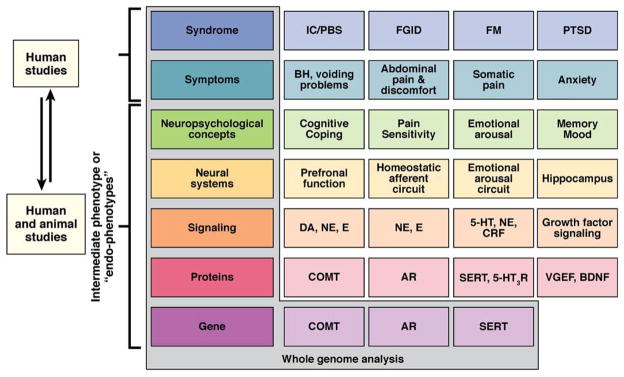
Intermediate phenotype strategy for FGIDs and frequently comorbid disorders. Shown is the progressive deconstruction of clinical, symptom-based syndromes (several examples are shown in the first row) into intermediate phenotypes (examples are shown in each row) all the way down to the genome. For clarity, only a small number of candidate genes are shown. Although the syndrome and the symptoms can only be studied in humans, the neurobiological endophenotypes can be studied both in humans and in animal models. It has been postulated that the correlation between neurobiological endophenotypes and genes is significantly higher than the correlation between clinical syndromes and genes. IC/PBS, interstitial cystitis/painful bladder syndrome; FM, fibromyalgia; PTSD, posttraumatic stress syndrome; DA, dopamine; NE, norepinephrine; E, epinephrine; CRF, corticotropin-releasing factor; COMT, catechol-O-methyltransferase; AR, adrenoreceptor; VGEF, vascular endothelial growth factor; BDNF, brain-derived neurotrophic factor. Mayer EA, Bushnell MC. Functional pain disorders: time for a paradigm shift? In: Mayer EA, Bushnell MC, eds. Functional pain syndromes: presentation and pathophysiology. Seattle, WA: IASP Press, 2009:531–565. This figure has been reproduced with permission of the International Association for the Study of Pain® (IASP®). The figure may not be reproduced for any other purpose without permission.
Camilleri et al pioneered the intermediate endophenotype approaches in gastrointestinal disorders.10,11 For example, in one study, several significant gene-sensory and gene-motor associations were observed in IBS and healthy subjects involving several adrenergic and serotonergic gene polymorphisms.23 Fukudo et al studied the relationship between 5-HTT LPR genotype and regional cerebral blood flow using positron emission tomography during and without colorectal balloon inflation, with the idea that altered brain activation by rectal stimulation might serve as an IBS endophenotype. The 5-HTT LPR polymorphism s/s genotype appeared to be associated with altered brain activation24,25 and with connectivity within an emotional arousal network with disinhibition of the amygdala,26 suggesting that this area of the brain may mediate central pain amplification seen in patients with IBS.
Limitations and Interpretation of Current Candidate Gene Studies
False-positive findings
The great majority of findings from small samples (such as those used in most FGID studies) represent false positives that cannot be replicated in a second sample. Because of the large number of genes (20,000) and the even larger number of testable genetic variants (millions), investigators will observe positive associations between hypothesized polymorphisms and complex FGID by chance. Many advocate the use of a higher statistical threshold for significance if multiple genetic variants are tested and/or comparisons (eg, cases vs controls, case-subtype A vs controls, case-subtype B vs controls, and so on) are performed. For example, if 100 SNPs are tested, then the P value to be considered statistically significant after a Bonferroni correction would be .0005. Such high thresholds require either a large genetic effect whereby the variant is very common in cases but relatively uncommon in controls (highly unlikely for FGIDs) and/or very large sample sizes (thousands of patients). Less conservative corrections for multiple comparisons such as the Benjamini–Hochberg approach to controlling the false discovery rate26 are also available and appropriate but do not entirely alleviate the multiple-testing burden. In addition to sample size, replication of positive studies (lacking for most FGID studies) is critically important, if not an absolute requirement to rule out type I and II errors. Although candidate gene association studies are attractive to test specific hypotheses related to the pathophysiology of FGIDs, the results of one well-designed study still require additional field-based and laboratory-based corroboration before firmly concluding that a genetic variant truly lies in the causal pathway of disease or disorder development.
Disease phenotype
Most studies to date have used conventional symptom-based criteria for defining the presence and type of FGID. The problem with such an approach is that it is dichotomous (eg, affected or unaffected), with no accommodation for intermediate states that are common in polygenic disorders. Similar to many syndromes in psychiatry, such as depression and attention-deficit disorders, such categorical disease definitions are probably artificial because they do not take into account (1) the continuum of symptoms from healthy individuals to health care–seeking patients; (2) the spectrum and overlap of FGIDs with each other as well as other disorders (eg, depression, anxiety, somatization, fibromyalgia); (3) the lack of stability of symptoms over time if disease definition is based on symptom reporting for a set period (eg, “last year,” “past 3 months”); and (4) the underlying biological mechanisms, grouping people with similar symptoms but different mechanisms for symptom experience or reporting.
Small sample size
Most studies (including all the studies listed in Table 3) do not have sufficiently large sample sizes to address whether a genetic variant may be associated with a specific disorder subtype. It is biologically plausible that a genetic variant may not be causally relevant for the overall disorder but may be responsible for a specific disorder subtype or symptom, particularly if there is clinical and presumed underlying biological heterogeneity. For example, the 5-HTT LPR s allele is not associated with IBS as a group but may be associated with IBS-D or with patients with IBS with high comorbid anxiety. Furthermore, the magnitude of effect of genetic contributors for IBS and other FGIDs is likely quite modest. If at best a few genes contribute about 5% of the variance and the majority less than 1%, and the clinical phenotypes are unreliable (see the introduction), a very large number of subjects (several thousand) are required. In addition, identification of susceptibility loci for FGIDs will likely require a genome-wide approach, testing thousands if not millions of SNPs, whereby multiple-testing issues apply. Finally, analyses studying multiple interactions (eg, gene 1 × gene 2 × environment × sex) will also need to be enlarged to take into account additional variables.
Control groups
Some genetic studies use “convenience” samples of DNA collected from other studies without information about disease/disorder status. Contamination by misclassification may be particularly important for common symptom-based disorders such as FGIDs. Ideally, control subjects should not have a present or past history of an FGID or other comorbid somatic or psychiatric disorders, or a family history of such disorders, and should be recruited prospectively in a similar manner to case recruitment. Also, because functional symptoms present in adulthood and are female predominant, and because genetic variation may differ by race, cases and controls should ideally be matched by age, sex, and race. For example, confounding due to population stratification arises when differences in both allele and disease frequencies exist in a population of mixed racial/ethnic subpopulations. The presence of population stratification could potentially severely bias genetic effect estimates, leading to both false-negative and false-positive results. Most commonly used approaches for adjusting for population stratification include principal components analysis27 and multidimensional scaling.28
Lack of gene-gene, gene-environment, and gene-sex interactions
FGIDs are complex polygenic disorders with significant sex and environmental effects. Prior epidemiologic studies and the previously cited twin studies9 support this concept, and an extensive literature supports the importance of epigenetic factors in determining the clinical phenotype. For example, it has been shown that unless the environmental factor of adverse life events is taken into account, there is no relationship between the 5-HTT LPR polymorphism and the clinical trait of depression.29 The effect of adverse early life events influences the adult phenotype through gene modification, such as methylation,30 and the phenotype of SERT knockout mice varies with the sex of the animal.31
Future Directions
Genome-wide Association Studies and Whole Genome Sequencing
More than 100 genome-wide association studies have been conducted for a large number of human diseases, identifying hundreds of polymorphisms with influence on disease vulnerability.32 However, in most of these studies, one or two genes that had already been known were confirmed, while a large number of common gene variants found in these studies are responsible only for a small fraction of the genetic variation that we know from family and twin studies. It is likely that a large number of these genes are connected by pathways forming extensive networks with each other and with the various phenotypes. In FGIDs, this approach is further complicated by the fact that in contrast to many of the disease studies with genome-wide association studies (including inflammatory bowel disease), there are few biological markers (such as gastrointestinal transit, gene expression profiles) or validated intermediate phenotypes for FGIDs. Nevertheless, with the anticipated continuing decrease in the costs of performing such genome-wide studies (genome-wide association studies and whole genome sequencing), combined with a consortium approach to study a large number of well-phenotyped patients (as is under way in other complex diseases such as Alzheimer’s disease or major psychiatric diseases), such approaches are likely to be applied to FGIDs and related persistent pain disorders in the near future. Systems biological approaches will be required to characterize the complex interactions between hundreds of genes and multiple intermediate phenotypes.33
Better Phenotyping
With the rapid advances in large-scale genotyping approaches, the bottleneck in the better understanding of the genetic underpinnings of complex diseases such as FGIDs is the identification and validation of robust intermediate phenotypes (see the previous text). This will require a shift of emphasis from categorical syndrome definition of individual FGIDs (“agnostic approach”) and of individual persistent pain disorders to a view of these syndromes as a “mosaic” made up of a large number of interrelated phenotypes.34 Such correlations between intermediate phenotypes have been reported in a variety of diseases, including disorders of metabolism35 and schizo-phrenia,36 and the approach has been adopted in FGID studies by some investigators (see Association of Candidate Genes With Intermediate Phenotype). Among many highly correlated phenotypes, the one with the highest reliability and effect size for the clinical syndrome should be optimal to use for detecting associations with particular genotypes.34
Novel Analytic Approaches
As genotyping technology rapidly advances and improves, novel analytic methods need to be developed and appropriately applied. Single SNP analyses using χ2 tests, Armitage trend tests, and multivariable logistic regression are often sufficient to gain a first impression of genetic associations; however, haplotype-based37,38 and gene-based and pathway-based methods39 – 44 often allow a more comprehensive and integrated view of the network of associations and interactions that may exist. Importantly, focus has recently been shifting to the discovery of rare variants via sequencing, and attention should be given to optimal strategies for analyzing next-generation sequencing data.45– 47
It is also suggested that some thought be given to whether a purely frequentist approach for assessing evidence for a genetic association should be adopted or whether a Bayesian approach would be more informative. In the frequentist approach, a P value is computed for the null hypothesis of no association. However, a P value alone does not fully quantify how confident we should be that the variant is truly associated with the phenotype. Bayesian methodology provides an alternative approach that overcomes the limitations of the P value (but with the added cost of additional modeling assumptions). The Bayesian approach provides a measure of evidence (the Bayes factor) that allows for a practical and quantitative way of incorporating biological information in the association estimate, allows the direct comparison of SNPs across studies, and provides a comprehensive approach to combining results over several studies in a meta-analysis and across genes in pathway-based analysis.48 New and innovative statistical methods are rapidly being developed in the hope of facilitating more robust conclusions and interpretable results in future genetic association studies. Investigators are advised to stay abreast of these statistical advances and to adopt them into their studies.
Summary and Conclusions
Gene discovery for complex polygenic disorders such as FGIDs remains a priority but represents a vast challenge. The field, in particular the individual candidate gene hunting approach, has been hampered by use of discrete and narrow symptom-based definitions—focusing on one disorder such as IBS—instead of objective biological phenotypes. Future studies seeking to identify causal genetic loci for FGIDs will likely use a combination of approaches, with candidate gene as well as genome-wide approaches. Whole genome sequencing in combination with expression profiling and novel analysis techniques will not only enable researchers to identify variations in individual genes, but also the effect of epigenetic processes such as gene methylation on gene expression, reflecting environmental influences (Figure 2).
Figure 2.

Simplified model for interaction between genes, peripheral and central endophenotypes, and environmental factors resulting in IBS symptoms. Individual genes influence the function of neural networks in the brain and in the enteric nervous system (including closely related gut cells). Alterations in these neural systems result in alterations in central and peripheral systems related to pain perception, autonomic arousal, peristalsis, and secretion. Significant interactions of genetic with epigenetic factors (including microflora environmental stressors) and with the sex of the individual are likely. PFC, prefrontal cortex; OFC, orbitofrontal cortex; BA 25, Brodman area 25; AM, amygdala. TPH2, tryptophan hydroxylase 2. Mayer EA, Bushnell MC. Functional pain disorders: time for a paradigm shift? In: Mayer EA, Bushnell MC, eds. Functional pain syndromes: presentation and pathophysiology. Seattle, WA: IASP Press, 2009:531–565. This figure has been reproduced with permission of the International Association for the Study of Pain® (IASP®). The figure may not be reproduced for any other purpose without permission.
Acknowledgments
Funding
Supported by a grant from the National Institutes of Health (DK76797 to Y.A.S.) and (DK48351, DK64539, and AT002681 to E.A.M.).
Abbreviations used in this paper
- FGID
functional gastrointestinal disorder
- 5-HT
5-hydroxytryptamine
- IBS
irritable bowel syndrome
- IBS-D
diarrhea-predominant irritable bowel syndrome
- SNP
single nucleotide polymorphism
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Mayer EA, Bushnell MC. Functional pain disorders: time for a paradigm shift? In: Mayer EA, Bushnell MC, editors. Functional pain syndromes: presentation and pathophysiology. Seattle, WA: IASP Press; 2009. pp. 531–565. [Google Scholar]
- 2.Longstreth GF, Thompson WG, Chey WD, et al. Functional bowel disorders. Gastroenterology. 2006;130:1480–1491. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 3.Chadwick VS, Chen W, Shu D, et al. Activation of the mucosal immune system in irritable bowel syndrome. Gastroenterology. 2002;122:1778–1783. doi: 10.1053/gast.2002.33579. [DOI] [PubMed] [Google Scholar]
- 4.Diatchenko L, Nackley AG, Slade GD, et al. Idiopathic pain disorders—pathways of vulnerability. Pain. 2006;123:226–230. doi: 10.1016/j.pain.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 5.Diatchenko L, Slade GD, Nackley AG, et al. Genetic basis for individual variations in pain perception and the development of chronic pain condition. Hum Mol Genet. 2005;14:135–143. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 6.Halder SL, Locke GR, III, Schleck CD, et al. Natural history of functional gastrointestinal disorders: a 12-year longitudinal population-based study. Gastroenterology. 2007;133:799–807. doi: 10.1053/j.gastro.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Kalantar JS, Locke GR, III, Zinsmeister AR, et al. Familial aggregation of irritable bowel syndrome: a prospective study. Gut. 2003;52:1703–1707. doi: 10.1136/gut.52.12.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saito YA, Zimmerman JM, Harmsen WS, et al. Irritable bowel syndrome aggregates strongly in families: a family-based case-control study. Neurogastroenterol Motil. 2008;20:790–797. doi: 10.1111/j.1365-2982.2007.1077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy RL, Jones KR, Whitehead WE, et al. Irritable bowel syndrome in twins: heredity and social learning both contribute to etiology. Gastroenterology. 2001;121:799–804. doi: 10.1053/gast.2001.27995. [DOI] [PubMed] [Google Scholar]
- 10.Camilleri M, Carlson P, Zinsmeister AR, et al. Neuropeptide s receptor induces neuropeptide expression and associates with intermediate phenotypes of functional gastrointestinal disorders. Gastroenterology. 2010;138:98–107. doi: 10.1053/j.gastro.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papathanasopoulos A, Camilleri M, Carlson PJ, et al. A preliminary candidate genotype-intermediate phenotype study of satiation and gastric motor function in obesity. Obesity (Silver Spring) 2009 Oct 29; doi: 10.1038/oby.2009.360. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci. 2008;9:85–96. doi: 10.1038/nrn2284. [DOI] [PubMed] [Google Scholar]
- 13.Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397–414. doi: 10.1053/j.gastro.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Hariri AR, Holmes A. Genetics of emotional regulation: the role of the serotonin transporter in neural function. Trends Cogn Sci. 2006;10:182–191. doi: 10.1016/j.tics.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Cohen H, Buskila D, Neumann L, et al. Confirmation of an association between fibromyalgia and serotonin transporter promoter region (5-HTTLPR) polymorphism, and relationship to anxiety-related personality traits. Arthritis Rheum. 2002;46:845– 847. doi: 10.1002/art.10103. [DOI] [PubMed] [Google Scholar]
- 16.Offenbaecher M, Bondy B, de Jonge S, et al. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999;42:2482–2488. doi: 10.1002/1529-0131(199911)42:11<2482::AID-ANR27>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 17.Diatchenko L, Nackley AG, Slade GD, et al. Catechol-O-methyl-transferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain. 2006;125:216–224. doi: 10.1016/j.pain.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 18.Van Kerkhoven LA, Laheij RJ, Jansen JB. Meta-analysis: a functional polymorphism in the gene encoding for activity of the serotonin transporter protein is not associated with the irritable bowel syndrome. Aliment Pharmacol Ther. 2007;26:979–986. doi: 10.1111/j.1365-2036.2007.03453.x. [DOI] [PubMed] [Google Scholar]
- 19.Gottesman, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 20.Mayer EA. The challenge of studying the biology of complex, symptom-based GI disorders. Gastroenterology. 2008;134:1826–1827. doi: 10.1053/j.gastro.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7:818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- 22.Zubieta JK, Heitzeg MM, Smith YR, et al. COMT val 158 met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- 23.Camilleri M, Busciglio I, Carlson P, et al. Candidate adrenergic, GNB3, and serotonergic polymorphisms, endophenotype, pharmacogenetics of clonidine in irritable bowel syndrome and health. Gastroenterology. 2008 [Google Scholar]
- 24.Labus JS, Mayer EA, Hamaguchi T, et al. 5-Httlpr gene polymorphism modulates activity and connectivity within an emotional arousal network of healthy control subjects during visceral pain (abstr) Gastroenterology. 2008;134:A121. doi: 10.1371/journal.pone.0123183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukudo S, Kanazawa M, Mizuno T, et al. Impact of serotonin transporter gene polymorphism on brain activation by colorectal distention. Neuroimage. 2009;47:946–951. doi: 10.1016/j.neuroimage.2009.04.083. [DOI] [PubMed] [Google Scholar]
- 26.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodological) 1995;57:12. [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Yu K. Improved correction for population stratification in genome-wide association studies by identifying hidden population structures. Genet Epidemiol. 2008;32:215–226. doi: 10.1002/gepi.20296. [DOI] [PubMed] [Google Scholar]
- 29.Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 30.Fish EW, Shahrokh D, Bagot R, et al. Epigenetic programming of stress responses through variations in maternal care. Ann N Y Acad Sci. 2004;1036:167–180. doi: 10.1196/annals.1330.011. [DOI] [PubMed] [Google Scholar]
- 31.Cornelissen LL, Brooks DP, Wibberley A. Female, but not male, serotonin reuptake transporter (5-HTT) knockout mice exhibit bladder instability. Auton Neurosci. 2005;122:107–110. doi: 10.1016/j.autneu.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 33.Papp J, Sobel E. The genetics of pain. In: Mayer EA, Bushnell MC, editors. Functional pain syndromes: presentation and pathophysiology. Seattle, WA: IASP Press; 2009. pp. 405–422. [Google Scholar]
- 34.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10:318–328. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin LJ, North KE, Dyer T, et al. Phenotypic, genetic, and genome-wide structure in the metabolic syndrome. BMC Genet. 2003;4(Suppl 1):S95. doi: 10.1186/1471-2156-4-S1-S95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aukes MF, Alizadeh BZ, Sitskoorn MM, et al. Genetic overlap among intelligence and other candidate endophenotypes for schizophrenia. Biol Psychiatry. 2009;65:527–534. doi: 10.1016/j.biopsych.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 37.Schaid DJ, Rowland CM, Tines DE, et al. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lake SL, Lyon H, Tantisira K, et al. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum Hered. 2003;55:56–65. doi: 10.1159/000071811. [DOI] [PubMed] [Google Scholar]
- 39.Conti DV, Cortessis V, Molitor J, et al. Bayesian modeling of complex metabolic pathways. Hum Hered. 2003;56:83–93. doi: 10.1159/000073736. [DOI] [PubMed] [Google Scholar]
- 40.Kooperberg C, Ruczinski I. Identifying interacting SNPs using Monte Carlo logic regression. Genet Epidemiol. 2005;28:157–170. doi: 10.1002/gepi.20042. [DOI] [PubMed] [Google Scholar]
- 41.Satagopan JM, Yandell BS, Newton MA, et al. A bayesian approach to detect quantitative trait loci using Markov chain Monte Carlo. Genetics. 1996;144:805–816. doi: 10.1093/genetics/144.2.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kesh S, Mensah NY, Peterlongo P, et al. TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann N Y Acad Sci. 2005;1062:95–103. doi: 10.1196/annals.1358.012. [DOI] [PubMed] [Google Scholar]
- 43.Ritchie MD, Hahn LW, Roodi N, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee N, Kalaylioglu Z, Moslehi R, et al. Powerful multilocus tests of genetic association in the presence of gene-gene and gene-environment interactions. Am J Hum Genet. 2006;79:1002–1016. doi: 10.1086/509704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rusk N. Focus on next-generation sequencing data analysis. Foreword Nat Methods. 2009;6:S1. doi: 10.1038/nmeth.f.271. [DOI] [PubMed] [Google Scholar]
- 46.Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6:S13–S20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- 47.Li B, Leal SM. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephens M, Balding DJ. Bayesian statistical methods for genetic association studies. Nat Rev Genet. 2009;10:681–690. doi: 10.1038/nrg2615. [DOI] [PubMed] [Google Scholar]
- 49.Whorwell PJ, McCallum M, Creed FH, et al. Non-colonic features of irritable bowel syndrome. Gut. 1986;27:37–40. doi: 10.1136/gut.27.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bellentani S, Baldoni P, Petrella S, et al. A simple score for the identification of patients at high risk of organic diseases of the colon in the family doctor consulting room. The Local IBS Study Group. Fam Pract. 1990;7:307–312. doi: 10.1093/fampra/7.4.307. [DOI] [PubMed] [Google Scholar]
- 51.Locke GR, III, Zinsmeister AR, Talley NJ, et al. Familial association in adults with functional gastrointestinal disorders. Mayo Clin Proc. 2000;75:907–912. doi: 10.4065/75.9.907. [DOI] [PubMed] [Google Scholar]
- 52.Levy RL, Whitehead WE, Von Korff MR, et al. Intergenerational transmission of gastrointestinal illness behavior. Am J Gastroenterol. 2000;95:451–456. doi: 10.1111/j.1572-0241.2000.01766.x. [DOI] [PubMed] [Google Scholar]
- 53.Kanazawa M, Endo Y, Whitehead WE, et al. Patients and nonconsulters with irritable bowel syndrome reporting a parental history of bowel problems have more impaired psychological distress. Dig Dis Sci. 2004;49:1046–1053. doi: 10.1023/b:ddas.0000034570.52305.10. [DOI] [PubMed] [Google Scholar]
- 54.Pata C, Erdal ME, Derici E, et al. Serotonin transporter gene polymorphism in irritable bowel syndrome. Am J Gastroenterol. 2002;97:1780–1784. doi: 10.1111/j.1572-0241.2002.05841.x. [DOI] [PubMed] [Google Scholar]
- 55.Kim HJ, Camilleri M, Carlson PJ, et al. Association of distinct alpha(2) adrenoceptor and serotonin transporter polymorphisms with constipation and somatic symptoms in functional gastrointestinal disorders. Gut. 2004;53:829–837. doi: 10.1136/gut.2003.030882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeo A, Boyd P, Lumsden S, et al. Association between a functional polymorphism in the serotonin transporter gene and diarrhoea predominant irritable bowel syndrome in women. Gut. 2004;53:1452–1458. doi: 10.1136/gut.2003.035451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee DY, Park H, Kim WH, et al. Serotonin transporter gene polymorphism in healthy adults and patients with irritable bowel syndrome [in Korean] Korean J Gastroenterol. 2004;43:18–22. [PubMed] [Google Scholar]
- 58.Park JM, Choi MG, Park JA, et al. Serotonin transporter gene polymorphism and irritable bowel syndrome. Neurogastroenterol Motil. 2006;18:995–1000. doi: 10.1111/j.1365-2982.2006.00829.x. [DOI] [PubMed] [Google Scholar]
- 59.Saito YA, Locke GR, III, Zimmerman JM, et al. A genetic association study of 5-HTT LPR and GNbeta3 C825T polymorphisms with irritable bowel syndrome. Neurogastroenterol Motil. 2007;19:465–470. doi: 10.1111/j.1365-2982.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Nie Y, Xie J, et al. The association of serotonin transporter genetic polymorphisms and irritable bowel syndrome and its influence on tegaserod treatment in Chinese patients. Dig Dis Sci. 2007;52:2942–2949. doi: 10.1007/s10620-006-9679-y. [DOI] [PubMed] [Google Scholar]
- 61.Sikander A, Rana S, Sinha S, et al. Serotonin transporter promoter variant: Analysis in Indian IBS patients and control population. J Clin Gastroenterol. 2009;43:902–904. doi: 10.1097/MCG.0b013e3181b37e8c. [DOI] [PubMed] [Google Scholar]
- 62.Niesler B, Kapeller J, Fell C, et al. 5-HTTLPR and STin2 polymorphisms in the serotonin transporter gene and irritable bowel syndrome: effect of bowel habit and sex. Eur J Gastroenterol Hepatol. 2009 Jun 25; doi: 10.1097/MEG.0b013e32832e9d6b. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 63.Kohen R, Jarrett ME, Cain KC, et al. The serotonin transporter polymorphism rs25531 is associated with irritable bowel syndrome. Dig Dis Sci. 2009 Jan 1; doi: 10.1007/s10620-008-0666-3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saito Y, Larson J, Atkinson E, et al. The role of 5-HTT LPR and GNbeta3 825C>T polymorphisms and gene environment interactions in irritable bowel syndrome. Gastroenterology. 2009;136:289. doi: 10.1007/s10620-012-2319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pata C, Erdal E, Yazc K, et al. Association of the -1438 G/A and 102 T/C polymorphism of the 5-Ht2A receptor gene with irritable bowel syndrome 5-Ht2A gene polymorphism in irritable bowel syndrome. J Clin Gastroenterol. 2004;38:561–566. doi: 10.1097/00004836-200408000-00005. [DOI] [PubMed] [Google Scholar]
- 66.Kapeller J, Houghton L, Monnikes H, et al. First evidence for an association of functional variant in the microRNA-510 target site of the serotonin receptor-type 3E gene with diarrhea predominant irritable bowel syndrome. Hum Mol Genet. 2008;17:2967–2977. doi: 10.1093/hmg/ddn195. [DOI] [PubMed] [Google Scholar]
- 67.Kapeller J, Houghton L, Walstab J, et al. A coding variant in the serotonin receptor 3C subunit is associated with diarrhea-predominant irritable bowel syndrome (abstr) Gastroenterology. 2009;136:A155–A156. [Google Scholar]
- 68.Gonsalkorale WM, Perrey C, Pravica V, et al. Interleukin 10 genotypes in irritable bowel syndrome: evidence for an inflammatory component? Gut. 2003;52:91–93. doi: 10.1136/gut.52.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van der Veek P, de Kroon Y, Verspaget H, et al. Tumor necrosis factor alpha and interleukin 10 gene polymorphisms in irritable bowel syndrome (abstr) Gastroenterology. 2004;126:A53. doi: 10.1111/j.1572-0241.2005.00257.x. [DOI] [PubMed] [Google Scholar]
- 70.Sikander A, Rana SV, Prasad KK. Role of serotonin in gastrointestinal motility and irritable bowel syndrome. Clin Chim Acta. 2009;403:47–55. doi: 10.1016/j.cca.2009.01.028. [DOI] [PubMed] [Google Scholar]
- 71.Holtmann G, Siffert W, Haag S, et al. G-protein beta 3 subunit 825 CC genotype is associated with unexplained (functional) dyspepsia. Gastroenterology. 2004;126:971–979. doi: 10.1053/j.gastro.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 72.Andresen V, Camilleri M, Kim HJ, et al. Is there an association between GNbeta3-C825T genotype and lower functional gastrointestinal disorders? Gastroenterology. 2006;130:1985–1994. doi: 10.1053/j.gastro.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 73.Colucci R, Ghisu N, Bellini M, et al. Analysis of cholecystokinin type 1 receptor gene polymorphisms in patients with irritable bowel syndrome (abstr) Gastroenterology. 2009;136:A225. [Google Scholar]
- 74.Saito YA, Strege PR, Tester DJ, et al. Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am J Physiol Gastrointest Liver Physiol. 2009;296:G211–G218. doi: 10.1152/ajpgi.90571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Villani A, Saito Y, Lemire M, et al. Validation of genetic risk factors for post-infectious irritable bowel syndrome (IBS) in patients with sporadic IBS. Gastroenterology. 2009;136:289. [Google Scholar]