

Background: Proteases can cleave human IgG1 antibodies, resulting in loss of cell-killing functions.
Results: Mutation of the lower hinge of IgG1 confers protease resistance but disrupts Fc effector functions.
Conclusion: Compensating mutations in the CH2 domain can selectively restore Fc effector functions on a protease-resistant backbone.
Significance: Protease-resistant antibodies may be desirable for microenvironments with high protease content and/or when selected cell-killing functions are needed.
Keywords: Antibody Engineering, Immunology, Innate Immunity, Macrophages, NK Cells
Abstract
Molecularly engineered antibodies with fit-for-purpose properties will differentiate next generation antibody therapeutics from traditional IgG1 scaffolds. One requirement for engineering the most appropriate properties for a particular therapeutic area is an understanding of the intricacies of the target microenvironment in which the antibody is expected to function. Our group and others have demonstrated that proteases secreted by invasive tumors and pathological microorganisms are capable of cleaving human IgG1, the most commonly adopted isotype among monoclonal antibody therapeutics. Specific cleavage in the lower hinge of IgG1 results in a loss of Fc-mediated cell-killing functions without a concomitant loss of antigen binding capability or circulating antibody half-life. Proteolytic cleavage in the hinge region by tumor-associated or microbial proteases is postulated as a means of evading host immune responses, and antibodies engineered with potent cell-killing functions that are also resistant to hinge proteolysis are of interest. Mutation of the lower hinge region of an IgG1 resulted in protease resistance but also resulted in a profound loss of Fc-mediated cell-killing functions. In the present study, we demonstrate that specific mutations of the CH2 domain in conjunction with lower hinge mutations can restore and sometimes enhance cell-killing functions while still retaining protease resistance. By identifying mutations that can restore either complement- or Fcγ receptor-mediated functions on a protease-resistant scaffold, we were able to generate a novel protease-resistant platform with selective cell-killing functionality.
Introduction
The mechanism of action for several therapeutic monoclonal antibodies (mAbs) is thought to be due in part to Fc-mediated effector functions (1, 2). The most common human immunoglobulin isotype used for therapeutic intervention is IgG1 (3), which contains two Fab arms linked to a single Fc domain by a flexible hinge region. The Fc domain of IgG1 can bind to FcγRs2 expressed on immune effector cells and mediate target cell destruction by cellular means, including antibody-dependent cellular cytotoxicity (ADCC) and/or antibody-dependent cellular phagocytosis (ADCP) (4). Antigen-engaged mAbs can also recruit serum components of the immune system and mediate target cell destruction by initiating the complement cascade (5–7). The interactions of both immune cell FcγRs and the C1q component of complement require key amino acid recognition motifs in the lower hinge and proximal CH2 region of human IgG (2, 8–21). Our group and others have shown that the human IgG1 subclass is susceptible to limited proteolysis in the hinge region by a number of physiologically relevant proteases associated with microbial infections (e.g. GluV8 of Staphylococcus aureus and IdeS of Streptococcus pyogenes) and invasive cancers (e.g. the matrix metalloproteinases (MMPs)) (22–24). Cleavage within the lower hinge of IgG1 occurs in a two-step process where first one heavy chain is cleaved, resulting in a singly cleaved intermediate (8, 22, 23, 25). Cleavage of the second heavy chain separates the Fc from the Fab arms, resulting in an Fc fragment and an F(ab′)2 fragment. Previous studies have indicated that the singly cleaved intermediate is the dominant cleavage product generated on the cell surface (8) and that single cleavage results in abrogated binding to FcγRs (8, 26) and a loss of complement-dependent cytotoxicity (CDC) (8). Accordingly, the singly cleaved intermediate displays a profound loss of function in terms of cell killing both in vitro and in vivo (8, 26). However, the singly cleaved intermediate retains antigen binding capabilities as well as the long circulating half-life of the intact IgG1 counterpart (8). For these reasons, our group and others have hypothesized that antibody cleavage by tumor-associated and microbial proteases can potentially function as an immune evasion mechanism (for reviews, see Refs. 27 and 28).
Previously, we have shown that the human IgG2 subclass is resistant to cleavage by a number of physiologically relevant proteases, including MMP-3, MMP-7, MMP-12, and MMP-13 as well as GluV8 (29). Sequence alignments comparing the lower hinge of IgG1 and IgG2 suggested that the resistance to cleavage may be due to amino acid differences between IgG1 and IgG2 (29). However, the IgG2 subclass has very weak binding to FcγRIIIa (30) and thus low to undetectable ADCC capacity. The IgG2 subclass also has greatly reduced CDC activity compared with IgG1 (31). Several groups have exchanged the lower hinge/proximal CH2 of IgG1 and IgG2 (18, 32) and characterized the loss of function associated with this domain exchange. Armour et al. (32) demonstrated that introduction of the lower hinge/proximal CH2 of IgG2 into IgG1 resulted in a profound loss of function and proposed that these substitutions could serve as a silent Fc platform. Shields et al. (18) introduced the lower hinge/proximal CH2 of IgG2 into IgG1 and showed a greater than 20-fold loss of binding to FcγRs. Therefore, additional efforts would be required to generate protease-resistant variants containing an IgG2 lower hinge/proximal CH2 region that retain Fc-dependent cell-killing functions.
Antibody engineering has long been recognized as a means to improve mAb-based therapies (2, 33), particularly with regard to antibodies that target tumor antigens (4, 34). Efforts to engineer anti-tumor mAbs in the Fc domain are often directed toward increasing the cell killing capacity of the mAb by augmenting binding to FcγRs (18, 35–37) or the C1q component of complement (5, 6). One common method to augment cell-killing functions is to mutate amino acids in the CH2 region and screen for variants with increased binding to C1q or FcγRs (5, 6, 18, 35, 36). A number of published reports have also documented efforts to engineer the hinge region to improve effector function or increase antibody stability (38, 39). In this study, we demonstrate that mutation of the lower hinge of IgG1 confers protease resistance but also results in the loss of Fc-mediated cell-killing functions. We show that specific mutations incorporated into the CH2 region of engineered mAbs with a protease-resistant lower hinge cannot only restore functional activities to IgG1 but in some cases substantially enhance Fc effector functions. Furthermore, we show that cleaved IgGs are detected within the tumor microenvironment of human head and neck squamous cell carcinoma, highlighting the need for a protease-resistant platform for diseases characterized by the presence of IgG-cleaving proteases.
EXPERIMENTAL PROCEDURES
Antibodies
The V-region cDNA sequences of the anti-CD20 variants were the same as those used in rituximab (VL GenBankTM accession number AR015962 and VH GenBank accession number AR000013), and the heavy and light chains were engineered onto IgG subclasses and variants by molecular cloning. Transient transfection and expression in 293T and/or CHO cells were performed with standard procedures at Janssen Research and Development, LLC. mAbs were purified using protein A columns and underwent in-house quality controls for >95% purity prior to further experimental analyses. The complementarity-determining region sequences of the humanized, complementarity-determining region-grafted anti-CD142 mAb were derived from the murine anti-human CD142 mAb TF8-5G9, which originated at the Scripps Research Institute and has been described previously (40, 41).
Protease Digestions
Protease digestions were performed at pH 7.5 at 37 °C for 24 h in PBS for Ides and GluV8 or in Tris-buffered saline with 5 mm CaCl2 for the MMP reactions. Anti-CD142 antibodies were used at a concentration of 0.5 mg/ml, and protease concentrations of 10% molar ratio for MMP-3 (Janssen Research and Development, LLC) and MMP-12 (Enzo Life Sciences), 20% molar ratio for GluV8 (Biocentrum), and 1% (w/w) for IdeS (Genovis) were used. Kinetic digests were performed with a 10% molar ratio for MMP-3 and 0.1% (w/w) IdeS and were quenched at the indicated time points using a final concentration of 10 mm EDTA for the MMP-3 digest and 10 mm iodoacetamide for the IdeS digest. The percentage of intact IgG remaining was calculated as done previously (29).
Immunohistochemistry
All immunohistochemistry was performed by QualTek Molecular Laboratories (Newtown, PA). Four-micrometer sections were dewaxed through four changes of xylene (5 min each) followed by a graded alcohol series to distilled water. Steam heat-induced epitope recovery was used for 20 min in the capillary gap in the upper chamber of a Black and Decker steamer. Sections were incubated with primary rabbit polyclonal anti-hinge detection antibodies (125 ng/ml) directed against three cleavage sites in the hinge that were described previously (23). An anti-rabbit biotinylated secondary antibody was applied followed by avidin-biotin complex-HRP. Secondary antibodies were detected with 3,3′-diaminobenzidine chromogen. Positive staining was indicated by the presence of a brown chromogen reaction product. Sections were counterstained with hematoxylin for 1 min. Slides were analyzed under a microscope using a 40× objective.
ADCC
The ADCC assays were performed as described previously with several modifications (42). ADCC assays were performed with increasing anti-CD20 IgG1 antibody variant concentrations. Briefly, human PBMCs purified from leukopaks were used as effector cells, and WIL2-S cells were used as targets in a 50:1 ratio. The WIL2-S cells were labeled with 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate reagent (PerkinElmer Life Sciences) for 30 min, washed twice, and resuspended in RPMI 1640 medium supplemented with GlutaMAX, 10% heat-inactivated FBS, 0.1 mm nonessential amino acids, and 1 mm sodium pyruvate (Invitrogen). 0.5 × 106 PBMCs, 1 × 104 labeled WIL2-S, and antibody at the indicated concentration were combined in 200 μl total in U-shaped 96-well plates, centrifuged for 2 min at 200 × g, and incubated at 37 °C for 2 h. At the end of the assay, plates were centrifuged again at 200 × g for 5 min, and 20 μl of supernatant were mixed with 200 μl of DELPHIA europium-based reagent. The fluorescence signal was measured using an Envision 2101 Multilabel Reader (PerkinElmer Life Sciences). The percentage of lysis was calculated as (Experimental release − Spontaneous release)/(Maximal release − Spontaneous release) × 100. Data were log-transformed and fit to a sigmoidal dose-response curve using GraphPad Prism v5.
CDC
CDC assays were performed as described previously (8) with WIL2-S cells as the target. A total of 50 μl of 0.05 × 106 cells was added to the wells of 96-well U-bottom plates in RPMI 1640 medium, 10% heat-inactivated FBS, 0.1 mm nonessential amino acids, and 1 mm sodium pyruvate. An additional 50 μl of medium was added with and without mAbs, and plates were incubated at room temperature for 1 h. After incubation, a total of 50 μl of a 10% rabbit complement (Invitrogen) solution was added, and plates were incubated at 37 °C for 20 min. Plates were centrifuged again at 200 × g for 5 min, and 50 μl of supernatant were mixed with 50 μl of the lactate dehydrogenase cytotoxicity detection kit (Roche Applied Science). Plates were incubated for 15 min at room temperature and then analyzed on a SpectraMax M5 (Molecular Devices, Sunnyvale, CA) at 490 nm. Data were normalized to maximal cytotoxicity with Triton X-100 (Sigma) and minimal control containing only cells and complement in the absence of mAb. Data were log-transformed and fit to a sigmoidal dose-response model using GraphPad Prism v5.
Twenty-four-hour ADCP
Human PBMCs were isolated from leukopaks (Biologics Specialty) using Ficoll gradient centrifugation. CD14pos monocytes were purified from PBMCs by negative depletion using a CD14 isolation kit that did not deplete CD16pos monocytes (Stem Cell Technologies). Purified monocytes were plated at 0.1 × 106 cells/cm2 in X-VIVO-10 medium (Lonza) containing 10% FBS. Macrophages were differentiated from monocytes by the addition of 25 ng/ml macrophage colony-stimulating factor (R&D Systems) for 7 days. IFNγ (50 ng/ml; R&D Systems) was added for the final 24 h of differentiation. The target cells for the assay were GFP-expressing MDA-MB-231 cells (40). Isolated macrophages were incubated in a 37 °C incubator with GFP-expressing MDA-MB-231 cells at a ratio of four macrophages (0.1 × 106 cells/well) to one MDA-MB-231 cell (25,000 cells/well) for 24 h with wild-type anti-CD142 or protease-resistant variants of anti-CD142 in 96-well U-bottom plates. The final volume of medium (DMEM + 10% FBS) used for the assay was 200 μl. At the end of 24 h, plates were centrifuged at 300 × g for 5 min, and the cells were removed from the 96-well plates using Accutase (Sigma). Macrophages were identified with anti-CD11b (clone ICRF44) and anti-CD14 (clone M5E2) antibodies (both from BD Biosciences) coupled to Alexa Fluor 647 (Invitrogen), and then cells were acquired on an LSRFortessa flow cytometer (BD Biosciences). The data were analyzed using FloJo software (Tree Star). The percentage (%) of cell killing was determined by measuring the reduction in GFP fluorescence resulting from its degradation in the lysosomes after internalization using the following equation: Percentage of tumor cells killed = ((Percentage of GFPpos, CD11bneg, CD14neg cells in no mAb control) − (Percentage of GFPpos, CD11bneg, CD14neg cells in the presence of mAb))/(Percentage of GFPpos, CD11bneg, CD14neg cells in no mAb control) × 100.
Cell Binding Assays
Target cells were plated at 0.2 × 106 in 100 μl of FACS staining buffer (BD Bioscience) with the indicated antibody concentrations. Cells were incubated for 30 min at 4 °C, washed two times with PBS, and then incubated with 100 μl of a 1:10 dilution of anti-κ-phycoerythrin (Biolegend) in FACS buffer for 30 min at 4 °C. Cells were washed two times and resuspended in PBS. Acquisition was performed on a FACSCalibur (BD Biosciences), and analysis was performed using FlowJo (TreeStar). Data were fitted to a sigmoidal dose-response curve using GraphPad Prism v5.
ELISA for Antigen Binding
Nunc-Immuno MaxiSorp plates were coated overnight with 10 μg/ml streptavidin (Invitrogen) at 4 °C. At room temperature, plates were blocked for 1 h with 3% BSA, PBS and then coated with biotinylated CD142 in 3% BSA, PBS for an additional hour. Antibody variants were added at the indicated concentration for 1 h. The plate was washed three times with 0.15 m NaCl, 0.02% Tween 20 and then treated with anti-κ-HRP (Millipore) at a 1:5000 dilution for 1 h. Plates were washed three times, 3,3′,5,5′-tetramethylbenzidine reagent (Fitzgerald Industries International) was added for 5 min, and the reaction was stopped with 3 m HCl. The absorbance was read at 450 nm. Data were log-transformed and fitted to a sigmoidal dose-response curve using GraphPad Prism v5.
In Vitro Competition FcRn Binding Analysis
A competitive binding assay was used to assess relative affinities of different antibody samples to in-house recombinant human FcRn-His6 (transmembrane and cytoplasmic domains of FcRn were replaced with a polyhistidine affinity tag). Ninety-six-well copper-coated plates (Thermo Scientific) were used to capture FcRn-His6 at 5 μg/ml in PBS after which plates were washed with 0.15 m NaCl, 0.02% Tween 20, pH ∼5 and then incubated with blocking reagent (0.05 m MES, 0.025% BSA, 0.001% Tween 20, pH 6.0, 10% ChemiBLOCKER (Millipore)). Plates were washed as above, and then serial dilutions of competitor test antibody in blocking reagent were added to the plate in the presence of a fixed 1 μg/ml concentration of an indicator antibody (a biotinylated human IgG1 monoclonal antibody). Plates were incubated at room temperature for 1 h, washed three times as above, and then incubated with a 1:10,000 dilution of streptavidin-HRP (Jackson ImmunoResearch Laboratories) at room temperature for 30 min. Plates were washed five times as above, and bound streptavidin-HRP was detected by adding 3,3′,5,5′-tetramethylbenzidine peroxidase substrate with Stable Stop (Fitzgerald Industries International) and incubating for 4 min. Color development was stopped by addition of 0.5 m HCl. Optical densities were determined with a SpectraMax Plus384 plate reader (Molecular Devices) at 450-nm wavelength. Data were fitted to a sigmoidal dose-response curve using GraphPad Prism v5.
B Cell Depletion in Cynomolgus Monkeys
The B cell depletion studies were performed by Huntingdon Life Sciences (East Millstone, NJ). Four cynomolgus monkeys per group were injected with saline or rituximab variants at 1 mg/kg. At the indicated time points, blood was collected and analyzed by flow cytometry. Cell events within a lymphocyte scatter were further subdivided into T lymphocytes (CD3pos, CD20neg), natural killer lymphocytes (CD3neg, CD159apos), or B lymphocytes (CD3neg, CD19pos). Antibodies to CD3 (SP34), CD20 (2H7), CD16 (3G8), and CD40 (5C3) and corresponding isotype control antibodies were purchased from BD Biosciences. Antibodies to CD159a (Z199), CD14 (RMO52), and CD19 (J3-119) were purchased from Beckman Coulter. Individual antibodies or appropriate antibody mixtures were added to 12 × 75-mm FACS tubes. Blood samples were mixed before use (e.g. gentle inversion or rolling). Fifty microliters of blood and 50 μl of heat-inactivated FBS (Sigma) were added to each of the tubes. Sample tubes were briefly vortexed after all additions, protected from light, and incubated at ambient temperature for a minimum of 10 min. Red blood cells were then lysed, and samples were fixed using the TQ Prep WorkstationTM (Coulter). Samples were stored at 2–8 °C protected from light until analysis. All sample acquisition took place on an FC500 flow cytometer (Coulter). FlowCount beads (Coulter) were utilized to determine cell concentrations. Data analysis was performed using FCS Express (v3.0).
RESULTS
Detection of Cleaved IgGs in Human Squamous Cell Carcinoma
Based on the observation that a number of physiologically relevant proteases associated with invasive cancers can cleave human IgGs in vitro, we assessed the presence of cleaved IgGs in human tumor tissue. We had previously generated anti-hinge antibodies that bind to cleaved IgGs but do not react with the intact IgG counterpart (8, 23). The anti-hinge antibodies were used for immunohistochemical detection of cleaved IgGs in human head and neck squamous cell carcinoma (HNSCC). As shown in Fig. 1A (left panel), cleaved IgGs were detected in HNSCC with an enrichment of detection at the tumor/stromal interface. To demonstrate that the staining was specific for cleaved IgGs, we added a pool of hinge-cleaved F(ab′)2 fragments (generated with MMP-3, GluV8, and IdeS) to block the antigen-binding arm of the anti-hinge antibodies (Fig. 1A, right panel). Inclusion of pooled F(ab′)2 fragments effectively blocked detection of cleaved IgGs, confirming the specificity of the detection reagent. Positive staining for cleaved IgGs was observed in 19 of 51 (37%) individual HNSCC cases (Fig. 1B). It was not unexpected that cleavage was not seen in all of the sections due to the heterogeneity of human tumor samples (e.g. total levels of EGF receptor vary widely in human tumor samples and are not uniform among different individuals (43)). This finding indicated that human IgGs were subject to proteolytic cleavage in the lower hinge region within the tumor microenvironment, especially at the expanding edge of the tumor where protease expression is known to be heightened (44).
FIGURE 1.
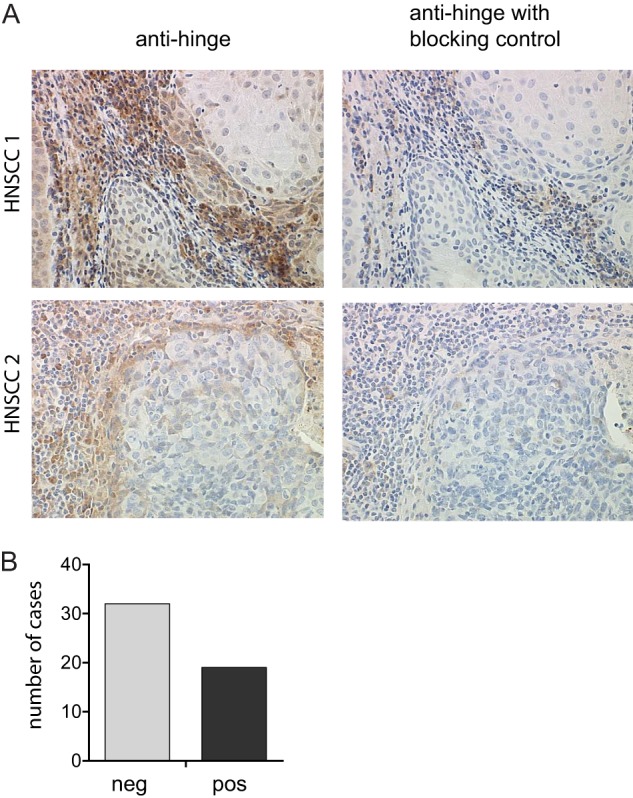
IgG hinge cleavage is detected at the tumor/stroma interface. A, representative 40× images of human head and neck squamous cell carcinoma tissue sections were assessed for the presence of cleaved IgGs using anti-hinge antibodies specific for lower hinge-cleaved IgGs but not the intact IgG counterpart (left panels). Representative adjacent sections were treated with anti-hinge antibodies in the presence of excess cleaved IgGs to determine the specificity of staining (right panels). B, bar graph depicting the number of cases with or without detection of IgG cleavage from a total of 51 sections from individual patients.
Mutation of the Lower Hinge of IgG1 Confers Protease Resistance
We next sought to determine whether we could mutate the lower hinge/proximal CH2 region of human IgG1 to confer protease resistance. Initially, three variants were generated for this purpose. The first variant contained the IgG1 to IgG2 domain exchange E233P/L234V/L235A with Gly236 deleted (EU numbering (45)); this substitution was designated 2h. To compensate for the loss of function associated with such a domain swap (18, 32), additional variants were generated with select mutations in the CH2 region. The variant 2h-DE contained the IgG2 lower hinge and the CH2 mutations S239D/I332E, which were previously shown to enhance FcγR binding to IgG1 (35) (supplemental Fig. S1). The variant 2h-AA contained the IgG2 lower hinge and the CH2 mutations K326A/E333A, which were previously shown to enhance IgG1 CDC activity (5) (supplemental Fig. S1).
To assess protease susceptibility of the newly generated variants, mAbs were incubated with proteases capable of cleaving human IgG1 (29). Similar to previous studies, IgG1 was cleaved by MMP-3, MMP-12, GluV8, and IdeS to varying degrees after a 24-h incubation (Fig. 2, A and B). IgG2, 2h, 2h-DE, and 2h-AA were all resistant to cleavage by MMP-3 and MMP-12. The variant 2h-DE was uniquely resistant to the Group A streptococcal protease IdeS but had increased susceptibility to S. aureus protease GluV8 compared with IgG2, 2h, and 2h-AA. These results indicated that lower hinge mutations augmented protease resistance and that incorporation of mutations into the CH2 influenced protease resistance as well.
FIGURE 2.
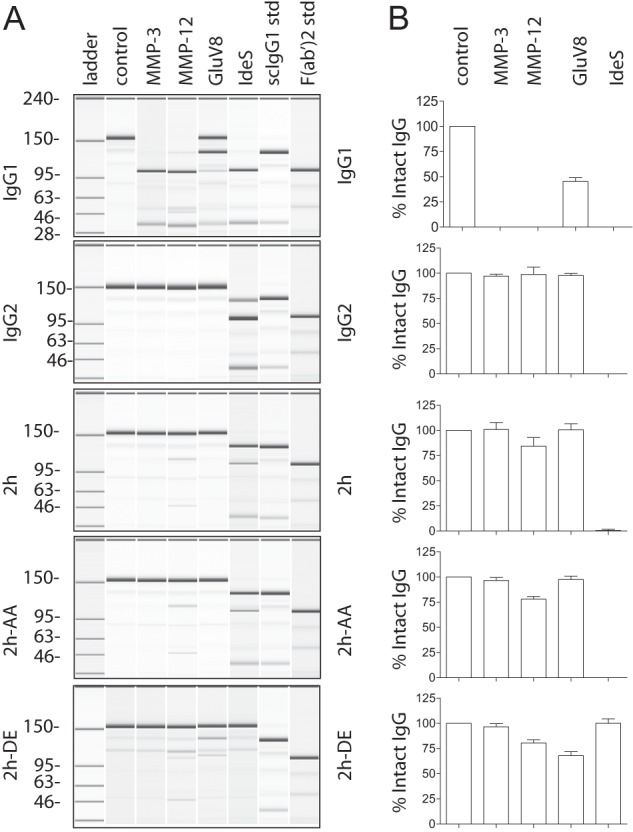
Replacement of the lower hinge/proximal CH2 amino acids of IgG1 with those of IgG2 confers protease resistance. A, purified human IgG1, IgG2, 2h, 2h-AA, and 2h-DE were incubated with different proteases and analyzed by capillary electrophoresis under denaturing, non-reducing conditions. Enzymes are listed above individual lanes, and all digestions were carried out for 24 h at 37 °C. The far right two lanes represent the purified human IgG1 standards (std) of single cleaved IgG1 and F(ab′)2 fragment of IgG1, respectively. B, bar graph representation of the percentage of intact IgG remaining after the 24 h digests (n = 3). Error bars represent S.D.
Select Protease-resistant Variants Have Restored CDC or Enhanced ADCC
We probed the ability of the protease-resistant variants to mediate Fc-dependent immune effector functions using cell-based assays. For CDC activity, we opsonized WIL2-S lymphoma cells with anti-CD20 antibody variants containing the V-region of rituximab. IgG1 mediated CDC activity, whereas the IgG2 did not, and as expected, the 2h mutation resulted in a loss of CDC activity (Fig. 3, left). However, the K326A/E333A mutations (variant 2h-AA) were able to restore CDC activity albeit at a reduced level relative to IgG1. The incorporation of the S239D/I332E mutations (variant 2h-DE) did not restore CDC activity.
FIGURE 3.
Mutation of specific amino acids can restore cell-killing functions to the 2h protease-resistant backbone. Concentration-dependent CDC (A) and ADCC (B) activities against WIL2-S target cells using anti-CD20 IgG1 (black circles), IgG2 (green squares), 2h (brown triangles), 2h-DE (red inverted triangles), and 2h-AA (orange diamonds) compared with an IgG1 isotype control (light gray crosses). Shown are representative graphs of two experiments using two different PBMC donors (for ADCC) (n = 2). Error bars represent S.D.
ADCC was assessed with anti-CD20 variants using peripheral blood mononuclear effector cells allotyped to be heterozygous for the FcγRIIIa 158V/F polymorphism and WIL2-S target cells. IgG1 mediated ADCC activity, whereas IgG2 and the variants 2h and 2h-AA did not (Fig. 3, right). The 2h-DE variant had enhanced ADCC activity with an ∼11-fold average increase in potency as compared with IgG1 (supplemental Table S1). Thus, this engineering strategy resulted in a novel methodology to select for specific cell-killing functions that are at times enhanced.
Protease-resistant Variants That Elicit Both CDC and Enhanced ADCC
To develop a protease-resistant platform that could mediate Fc effector functions more broadly (i.e. both ADCC and CDC), we combined the CDC-restoring K326A/E333A mutations with either of the ADCC-restoring mutations, S239D or I332E, resulting in variants that were designated 2h S239D/K326A/E333A (2h-DAA) or 2h K326A/I332E/E333A (2h-AEA). Twenty-four-hour proteolytic digests confirmed that both 2h-DAA and 2h-AEA maintained protease resistance to MMP-3, MMP-12, and GluV8 compared with IgG1 (Fig. 4A). The 2h-AEA variant showed greater protease resistance to IdeS compared with IgG1 and 2h-DAA, suggesting that the I332E mutation on the 2h backbone influenced protease resistance to IdeS.
FIGURE 4.

Protease resistance is maintained in variants with combinations of ADCC- and CDC-restoring mutations on the 2h protease-resistant backbone on the anti-CD142 V-region. A, IgG1 (white), 2h-DAA (gray), and 2h-AEA (black) were incubated with MMP-3, MMP-12, GluV8, and IdeS for 24 h. The percentage of intact IgG1 was calculated after capillary electrophoresis. Shown is a summary of three independent experiments. Wild-type IgG1 (black), 2h-DAA (blue), and 2h-AEA (purple) were incubated with a 10% molar ratio of MMP-3 (B) or 0.1% (w/w) IdeS (C). At the indicated time points, reactions were stopped with 10 mm EDTA (for MMP-3) or 10 mm iodoacetamide (for IdeS) and frozen prior to analysis. The percentage of intact IgG1 was calculated after capillary electrophoresis (n = 3). Error bars represent S.D.
We next assessed IgG proteolysis in kinetic digest assays comparing IgG1 with the protease-resistant variants 2h-DAA and 2h-AEA. MMP-3 cleavage of IgG1 was detected within 15 min, and no intact IgG1 was detected at the 24-h time point, whereas both 2h-DAA and 2h-AEA were resistant to MMP-3 cleavage throughout the 24-h assay (Fig. 4B). Because IdeS rapidly cleaves human IgG1 (8, 24), kinetic IdeS digests were performed with shorter time points at an enzyme concentration of 0.1% (w/w). Approximately 60% intact IgG1 was detected after 1 min, and complete loss of intact IgG occurred by 30 min. In contrast, the variant 2h-AEA maintained nearly complete resistance to IdeS throughout the assay. The 2h-DAA variant displayed some loss of intact IgG after 2 h with ∼90% intact mAb remaining (Fig. 4C). These results confirmed that the combination of lower hinge mutations with I332E imparted the greatest level of resistance to IdeS.
Next, we characterized the Fc effector functions of the all the protease-resistant variants. The protease-resistant variants 2h-DAA and 2h-AEA displayed CDC activity at potencies comparable with the 2h-AA variant (Fig. 5A and supplemental Table S1). Either the S239D or the I332E mutation alone was capable of restoring ADCC activity to the 2h backbone with each variant demonstrating higher potency than IgG1 although to levels lesser than the 2h-DE variant (Fig. 5B and supplemental Table S1). The ADCC activities for both the 2h-DAA and 2h-AEA variants were ∼4-fold higher than IgG1 with heterozygous FcγRIIIa 158V/F PBMC donors. Human PBMC donors that were homozygous for the high affinity FcγRIIIa 158V/V polymorphism displayed similar relative differences in ADCC enhancements between the different Fc variants (Fig. 5C). ADCC was further enhanced by the protease-resistant variants compared with IgG1 when human PBMC donors that were homozygous for the low affinity FcγRIIIa 158F/F polymorphism were used as effector cells (Fig. 5D). The 2h-DE variant displayed an ∼30-fold average increase in potency compared with IgG1, whereas the 2h-DAA and 2h-AEA variants displayed average increases of ∼23- and ∼10-fold, respectively, among the two different donors tested. Therefore, three protease-resistant variants demonstrated increased ADCC potency compared with IgG1, and the increased potency was most apparent using low affinity FcγRIIIa 158F/F polymorphism PBMC donors.
FIGURE 5.
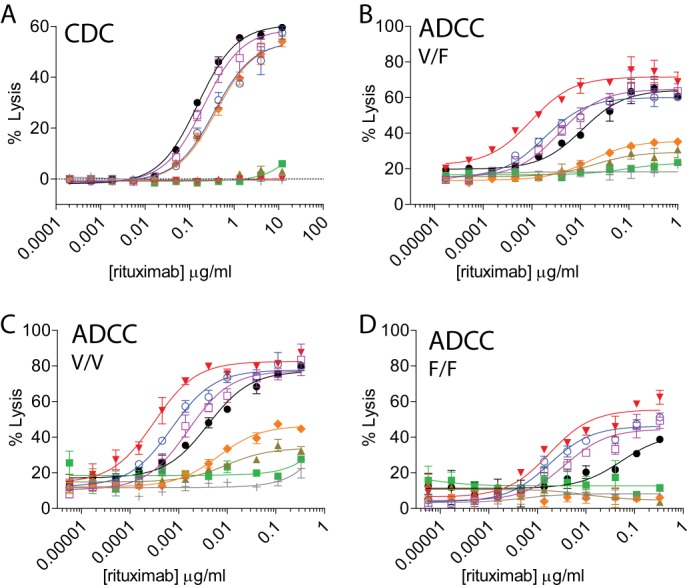
Protease-resistant variants differ in the ability to mediate Fc-dependent cell killing. A, the K326A/E333A mutations restore CDC activity to the 2h backbone. CDC activity was measured using rituximab variants against WIL2-S target cells. Shown is a representative experiment of two independent experiments, each performed in duplicate. B–D, either the S239D or I332E mutation is sufficient to restore ADCC activity to the 2h protease-resistant backbone. ADCC activity was determined using anti-CD20 variants, WIL2-S targets cells, and PBMC donor cells that were FcγRIIIa 158V/F heterozygous (B), high affinity FcγRIIIa 158V/V homozygous (C), or low affinity FcγRIIIa 158F/F homozygous (D) as effector cells. Shown are representative plots of two independent assays from two individual donors. Anti-CD20 (A–D) were as follows: IgG1 (black circles), IgG2 (green squares), and protease-resistant variants 2h (brown triangles), 2h-DE (red inverted triangles), 2h-AA (orange diamonds), 2h-DAA (blue open circles), and 2h-AEA (purple open squares) compared with an IgG1 isotype control (light gray crosses) (n = 2 for all assays). Error bars represent S.D.
Antibody-dependent Macrophage Killing of Human Tumor Cell Lines
We recently demonstrated that tumor-associated macrophages display potent antitumor activity in the presence of an anti-tumor mAb (40). Therefore, we wanted to assess the function of the protease-resistant variants against mAb-opsonized tumor cells using macrophage effector cells. Fc-dependent effector functions of macrophages against mAb-opsonized tumor cells are often assessed by ADCP. This is typically accomplished by co-incubating macrophages with mAb-opsonized tumor cells and assessing internalization of the tumor cells within macrophages over a time frame typically no longer than 4 h (40, 46–48). Our group previously developed a flow cytometry and microscopy-based ADCP assay where the target tumor cells (MDA-MB-231) expressed GFP to obviate the need to label the tumor cells with a dye (29, 46). Macrophage internalization of MDA-MB-231 cells opsonized with a tumor targeting anti-CD142 mAb was assessed by either flow cytometry or fluorescence microscopy (29, 40). We had previously noted that the GFP fluorescence within macrophages was punctate, whereas the GFP fluorescence of non-internalized tumor cells was uniform (29, 40). Because the process of ADCP results in internalization and the subsequent lysosomal destruction of the tumor cell (4) and GFP fluorescence is dependent on the structural integrity of the GFP protein, we hypothesized that the punctate GFP signal was due to breakdown of the tumor target cell within the macrophage. To test the ability of macrophages to kill anti-CD142 mAb-opsonized, GFP-expressing MDA-MB-231 tumor cells, we extended a traditional ADCP assay to 24 h. In this assay, we could observe complete destruction of the tumor cell both by flow cytometry and immunofluorescence. This was accomplished by incubating effector macrophages in the presence of anti-CD142-opsonized, GFP-expressing MDA-MB-231 target cells and assessing the loss of GFP detection after 24 h (Fig. 6, A and B). The variants 2h-DE, 2h-DAA, and 2h-AEA displayed tumor cell killing comparable with IgG1. The tumor cell killing of the IgG2 was slightly reduced, whereas the variants 2h and 2h-AA had the lowest level of tumor cell killing (Fig. 6C). These results demonstrated that the variants with ADCC-restoring mutations were capable of killing MDA-MB-231 tumor cells in the 24-h ADCP assay.
FIGURE 6.
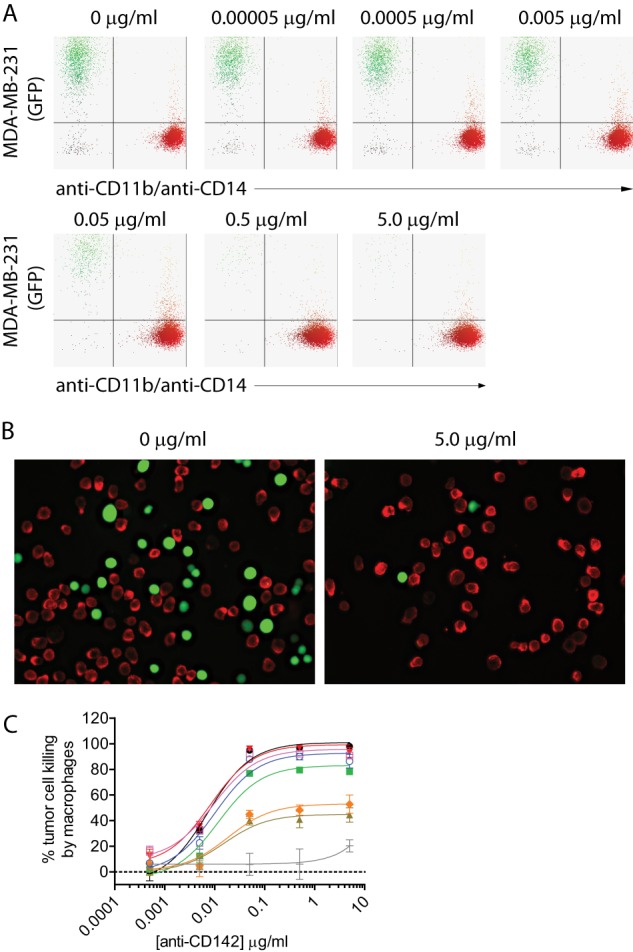
Protease-resistant variants are capable of killing tumor cells in a 24-h ADCP assay. A, representative flow cytometry dot plots depicting GFP-expressing MDA-MB-231 cells and macrophages detected with anti-CD11b and anti-CD14 antibodies coupled to Alexa Fluor 647. B, representative fluorescence microscopy images of GFP-expressing MDA-MB-231 cells (green) and macrophages (red) in the absence (left panel) or presence of an anti-CD142 IgG1 WT mAb. Macrophages were labeled with primary mouse anti-human CD11b and mouse anti-human CD14 antibodies and detected with secondary anti-mouse IgG antibody coupled to Alexa Fluor 568. C, 24-h ADCP assay using anti-CD142 variants, MDA-MB-231 GFP-expressing target cells, and effector macrophages from PBMC donors that were FcγRIIa 131H/R heterozygous and FcγRIIIa 158V/F heterozygous. Lines and symbols are as follows: IgG1 (black circles), IgG2 (green squares), and protease-resistant variants 2h (brown triangles), 2h-DE (red inverted triangles), 2h-AA (orange diamonds), 2h-DAA (blue open circles), and 2h-AEA (purple open squares) compared with an IgG1 isotype control (light gray crosses). Shown are representative plots of two independent assays (n = 2 for both assays). Error bars represent S.D.
Protease-resistant Variant Fab Arm Function, FcRn Binding, and Biophysical Property Assessment
To test that the protease-resistant mutations did not affect Fab arm function, we assessed the ability of the variants to bind to antigen in both plate-based and cell-based assays. The results indicated that variants engineered onto two different V-regions bound identically to their respective targets (Fig. 7, A and B). We next assessed the ability of the variants to bind to the neonatal Fc receptor, FcRn, which is thought to contribute to the long circulating half-life of IgGs. Fc binding to FcRn occurs proximal to the junction of the CH2 and CH3 regions (49), a region that is distal from the mutations present in all of the protease-resistant variants. All of the variants displayed FcRn binding comparable with IgG1 (Fig. 7C). These results demonstrate that the protease-resistant mutations do not affect the ability of the mAbs to engage either antigen or FcRn.
FIGURE 7.
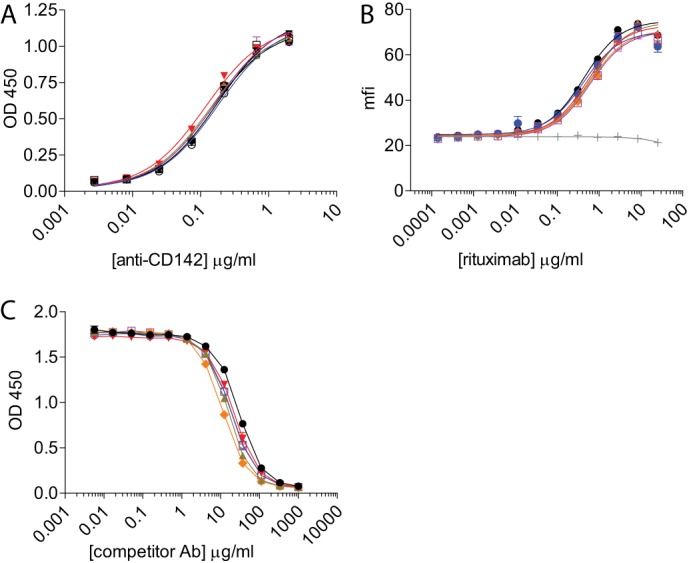
Protease-resistant variant binding to antigen and FcRn is not affected. A, the variants 2h, 2h-AA, 2h-DE, 2h-DAA, and 2h-AEA containing the anti-CD142 V-region bound similarly to IgG1 on a plate-bound ELISA assay using CD142 antigen. Shown is a representative plot of two independent experiments with increasing concentrations of anti-CD142 IgG variants. B, antigen binding was also unaffected with variants containing the anti-CD20 V-region using WIL2-S target cells. Opsonized WIL2-S cells were detected with a fluorescently conjugated anti-κ antibody, and the mean fluorescence intensity (mfi) was measured using flow cytometry. Shown is the mean fluorescence intensity of anti-κ-phycoerythrin of an experiment performed in duplicate. C, competitive binding to human FcRn at pH 6.0. Shown is a representative of two independent assays. Lines and symbols are as follows: IgG1 (black circles), IgG2 (green squares), and protease-resistant variants 2h (brown triangles), 2h-DE (red inverted triangles), 2h-AA (orange diamonds), 2h-DAA (blue open circles), and 2h-AEA (purple open squares) compared with an IgG1 isotype control (light gray crosses). Error bars represent S.D. Ab, antibody.
We next assessed several biophysical properties of select variants (IgG1, IgG2, 2h-DE, 2h-DAA, and 2h-AEA). Cross-interaction chromatography was used to assess protein-protein interactions, and the results indicated that all of the assessed variants displayed minimal protein-protein interactions (data not shown). Differential scanning calorimetry was performed to assess the thermal stability of the variants (supplemental Table S2). The results indicated varying degrees of thermal stability in the CH2 region with variants containing the I332E mutation having the lowest Tm (55.6 °C for 2h-DE and 60.7 °C for 2h-AEA). Of the variants tested, 2h-DAA decreased ∼7 °C compared with IgG1 (64.9 and 72.0 °C, respectively). Finally, the expression titers for each of the variants were comparable with the parent IgG1 mAb (data not shown).
Protease-resistant Variants Mediate B Cell Depletion in Cynomolgus Monkeys
Because of sequence differences between mouse and human FcγRs, the in vivo function of mAbs engineered for binding to human FcγRs are often assessed in non-human primates (35, 50, 51) whose FcγRs are more homologous to the human counterparts. Because many anti-human CD20 mAbs are cross-reactive with cynomolgus CD20 expressed on B cells, the cytotoxic potential of engineered anti-CD20 variants is often assessed in cynomolgus monkey B cell depletion studies (52). A previous B cell depletion study in cynomolgus monkeys using an Fc silent anti-CD20 variant did not result in depletion of B cells in vivo (53), further supporting that this model is appropriate for the assessment of Fc-dependent effector functions of anti-CD20 mAbs. An anti-CD20 mAb was not used for B cell detection because murine-derived anti-CD20 mAbs, including rituximab, a murine/human chimeric antibody, often bind to a common epitope on the large extracellular loop of CD20 and can either fully or partially mask CD20 (54). Therefore, we used an anti-CD19 antibody (clone J3-119) to detect cynomolgus monkey B cells. A single dose intravenous injection of a 1.0 mg/kg concentration of anti-CD20 IgG1 or the variants 2h-DE or 2h-DAA resulted in nearly complete loss of detection of B cells by the 6-h time point (Fig. 8 and supplemental Table S3). Depletion was sustained through day 7 of the study with B cell recovery starting at day 14 of the study. These results indicated that the protease-resistant variants 2h-DE and 2h-DAA were capable of depleting cynomolgus monkey B cells in vivo comparably with IgG1 at the 1.0 mg/kg dose.
FIGURE 8.
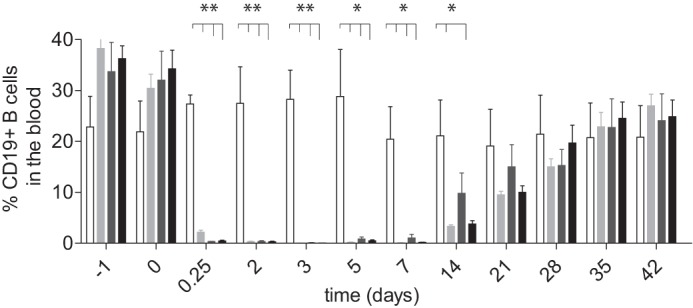
B cell depletion in cynomolgus monkeys using anti-CD20 IgG1 and anti-CD20 protease-resistant variants. Each mAb (1.0 mg/kg) was administered on day 0. Bar charts indicate the frequency of CD19pos B cells present in the blood after the indicated time points. The symbols designate the following: saline control (open bars), 2h-DE (light gray bars), 2h-DAA (dark gray bars), and IgG1 (solid black bars). Bar heights correspond to the mean ± S.D. of four animals per group. Two asterisks indicates p < 0.01, and one asterisk indicates p < 0.05 as determined by unpaired, two-tailed Student's t test. Error bars represent S.D.
DISCUSSION
Both tumors and pathologic microorganisms use multiple mechanisms to invade host tissues and evade immune responses. In the case of tumors, proteases can promote invasion, receptor shedding, and breakdown of the basement membrane (55). Because of high interstitial tumor pressure, antibodies often concentrate in the perivascular regions (56) and at the invasive front of the tumor, presumably in close proximity to proteases (44). In this study, we detected cleaved IgG in the tumor microenvironment with an enrichment of cleaved IgG at the invasive front of HNSCC tumors. Fan et al. (26) recently detected cleaved trastuzumab in human breast cancer tumor tissue, demonstrating that mAb proteolysis also occurs in vivo. Although the quantitative extent of cleavage remains to be determined, these observations coupled with the profound loss of function associated with IgG cleavage (8, 26, 27) suggest that proteolytic disablement of anti-tumor antibodies within human tumors could impair the ability of IgGs to facilitate tumor cell killing. This would be particularly problematic for the sustained efficacy of tumor-targeting mAbs (8, 26, 27). Given that many anti-tumor mAbs have very high affinities for their respective tumor antigens and that several groups have argued that high affinities restrict mAb localization within the tumor microenvironment (56), the accumulation of cleaved IgGs bound to tumor cells could both abrogate cell-killing functions and mask the tumor antigen from Fc functional mAbs. It has been well documented that HNSCC tumors can become resistant to anti-EGF receptor mAb therapy (57), and these data suggest that mAb proteolysis could also be a contributing mechanism of resistance. Therefore, we believe that the generation of a protease-resistant antibody platform could provide significant improvements to current IgG-based cytotoxic therapeutics.
A key challenge faced in the generation of a protease-resistant platform was associated with the observation that cleavage by physiologically relevant proteases mapped to the lower hinge/proximal CH2 region spanning amino acids Pro232–Gly237 (27), a region otherwise considered critical for human IgG interactions with both complement and FcγRs (8–21). We had previously demonstrated that the lower hinge of IgG2 was resistant to many proteases (29); however, numerous studies have documented that exchanging the lower hinge/proximal CH2 of IgG1 with corresponding amino acids in IgG2 resulted in a profound decrease in binding to FcγRs (16, 18, 32). In this study, we demonstrate that discrete mutations in the CH2 region can compensate for the loss of function associated with mutating the lower hinge of IgG1, challenging the long-standing observation that the lower hinge sequence is critical for initiating the complement cascade and facilitating potent ADCC relative to an IgG1. Furthermore, by identifying which mutations were capable of restoring either ADCC activity or CDC activity, we were capable of generating combinatorial variants with both ADCC and CDC capacity while still maintaining protease resistance. Others have demonstrated that mutations in either the CH2 or CH3 regions can restore effector functions to Fc modifications typically associated with a loss of function, particularly with regard to removal of the Fc glycan at Asn297, a traditional Fc silencing approach (58). Wittrup and co-workers (59) demonstrated that aglycosylated IgGs generated in yeast could selectively bind to FcγRs by introducing compensating mutations proximal to the Asn-X-Ser/Thr N-linked glycosylation motif. Georgiou and co-workers (60) demonstrated that point mutations in the CH3 region could restore FcγRI binding to aglycosylated IgGs expressed in Escherichia coli, whereas binding to other FcγRs was limited. Together, these studies not only demonstrate that point mutations in CH2 and/or CH3 regions can compensate for traditional silencing mutations, but restoring function to traditionally silent Fc platforms introduces an extraordinary amount of flexibility in terms of fine-tuning Fc-dependent effector functions.
The locations of the specific CH2 mutations used to restore Fc effector function are shown in supplemental Fig. S1. The Lys326 and Glu333 residues are solvent-exposed, and Idusogie et al. (5) have previously demonstrated that mutation of these residues can modulate CDC function in both the IgG1 and IgG2 subclasses. The authors hypothesized that that these two positions may play a structural role in Fc-C1q interactions. Both of the Ser239 and Ile332 residues are solvent-exposed, and mutations at these positions can profoundly affect ADCC and ADCP (35). A crystal structure of an ADCC-enhanced human Fc fragment was previously solved that contained the mutations S239D/A330L/I332E (61). By modeling a complex of the mutated Fc with human CD16, the authors suggested that the enhanced ADCC of the mutated Fc may be attributed to the formation of additional hydrogen bonds, hydrophobic contacts, and/or electrostatic interactions (61). Taken together, these observations may provide some rationale for the restoration of function seen with these mutations; however, additional studies would be needed to confirm these hypotheses.
The ability of IgG-cleaving proteases to subvert IgG effector functions in vivo has been most clearly established with the S. pyogenes protease IdeS. Björck and co-workers (24, 62, 63) have demonstrated that IdeS is a highly potent protease with unique specificity for all human IgG subclasses (24) that is also capable of cleaving distinct IgG subclasses within other species (e.g. IdeS cleaves rabbit IgG (62) and murine IgG2a and IgG3 but not murine IgG1 (63)). Intravenous administration of IdeS into rabbits resulted in the complete loss of detection of intact endogenous rabbit IgG within 6 h, demonstrating the extraordinary catalytic activity of this protease (62). In the same study, IdeS treatment of rabbits was capable of abrogating the ability of polyclonal anti-platelet antibodies to clear platelets (62). In a separate study, Nandakumar et al. (63) reported that IdeS treatment of mice was capable of alleviating the joint inflammation induced by collagen antibody-induced arthritis. Because of the potency of IdeS in vivo, the authors of the aforementioned studies have proposed that IdeS could be used therapeutically to ameliorate autoantibody-mediated pathology in autoimmune disorders (64), highlighting the perhaps underappreciated ability of IgG-cleaving proteases to subvert host humoral immunity. In this study, we describe variants that are resistant to IdeS but were still capable of Fc-mediated effector functions. The 2h-DE and 2h-AEA variants in particular were highly resistant to cleavage by IdeS. Unlike MMPs, which can cleave short peptides with recognition motifs as small as 6 amino acids (65), IdeS does not cleave peptide analogues of the IgG hinge (25). The smallest known substrate of IdeS is a papain-generated Fc fragment. This led to speculation that IdeS recognizes an exosite in the Fc domain (25). The observation that the I332E mutation in combination with the protease-resistant IgG2 lower hinge renders a human IgG resistant to IdeS cleavage suggests that the IdeS exosite may reside in or near the Leu328-Pro329-Ala330-Pro331-Ile332-Glu333 region deemed critical for C1q and FcγR binding (2). Furthermore, this study demonstrates that, at least for proteases that utilize exosite binding sites, mutations distal from the cleavage site can profoundly influence proteolytic susceptibility.
Numerous investigations have now shown that human IgG2 is resistant to cleavage by several proteases compared with IgG1 (24, 27, 66–68). It is intriguing to note that during the course of many microbial infections antibodies directed against bacterial polysaccharide antigens are often of the IgG2 subclass (31). Additionally, in some cases, patients bearing the higher affinity FcγRIIa polymorphism, His131, have improved clinical responses to bacterial infections (69) and improved IgG2-mediated phagocytosis of opsonized bacteria (70). These correlations between higher affinity FcγR-mAb interactions and efficacy are also in line with the observation that in some cases patients bearing higher affinity Fc receptor polymorphisms have a more favorable clinical response to IgG1 mAb-mediated cancer therapies (71–73). For these reasons, one common goal for Fc engineering efforts is to generate Fc variants that demonstrate improved effector functions in patients bearing lower affinity FcγR polymorphisms (35, 74). In this study, we have shown that several of the protease-resistant variants have substantially improved potency in ADCC assays when assessed with FcγRIIIa 158F/F PBMC donors compared with both IgG2 and IgG1 (Fig. 4D). Therefore, not only have we generated variants possessing the protease-resistant properties of human IgG2 but several of these same variants display potent ADCC and CDC effector functions, two qualities that are limited or absent with IgG2 antibodies or the 2h variant.
These variants also provide a unique tool kit to determine the contribution of specific mechanisms of action for any given target. For instance, the influence of CDC activity on the efficacy of rituximab is debatable. Some murine tumor xenograft studies have demonstrated that depletion of complement by cobra venom factor or the use of complement-deficient mice decreases the effectiveness of B cell depletion by rituximab (75–77). Meanwhile, other studies have shown that complement may not be required for B cell depletion in vivo (78–80). In the present study, a variant of rituximab with ADCC activity that lacked detectable CDC activity (2h-DE) was able to deplete B cells in the peripheral blood of cynomolgus monkeys similarly to IgG1 WT rituximab. Therefore, at a 1 mg/kg dose, CDC activity is not required for peripheral blood B cell depletion in cynomolgus monkeys.
In summary, we have engineered variants that can selectively restore either ADCC and/or CDC on a protease-resistant backbone, resulting in a versatile platform amenable to the fine-tuning of specific Fc-mediated effector functions. Furthermore, this study demonstrates that FcγR- and C1q-binding motifs localized in the lower hinge of IgG deemed critical for effector function are in fact dispensable but only in the context of compensating mutations engineered into the CH2 region. Because many cancers and invasive microorganisms are associated with proteases and FcγR-dependent functions are often considered a critical mechanism of action for mAbs against invasive diseases, our protease-resistant, Fc-functional antibodies present a novel fit-for-purpose platform for particular disease microenvironments. Additionally, the improved ADCC activity may be useful for indications where IgG1 WT is insufficient in eliciting potent ADCC independent of protease expression. The utility of protease-resistant mAbs as a therapeutic platform will be further assessed for stability, pharmacodynamic properties when targeting antigens in invasive diseases, immunogenicity, and biodistribution. The results of this study suggest novel directions for enhanced functionality for monoclonal antibodies for cancer and infectious diseases.
Acknowledgments
We thank Zhiqiang An, Ningyan Zhang, Russell Lingham, and Barry Springer for thoughtful discussions and Michael Naso, Jill Carton, and Tadas Panavas in the Molecular Protein and Biosciences group at Janssen Research and Development, LLC for cloning, expression, purification, and quality control of IgG variants. We also thank Diedra Bethea and Salman Muzammil for performing cross-interaction chromatography and differential scanning calorimetry, respectively.
M. Kinder, A. R. Greenplate, K. D. Grugan, K. L. Soring, K. A. Heeringa, S. G. McCarthy, R. E. Jordan, W. R. Strohl, and R. J. Brezski are employees of Janssen Research and Development, LLC. G. Bannish and M. Perpetua are employees of Huntingdon Life Sciences. F. Lynch is an employee of QualTek Molecular Laboratories.

This article contains supplemental Fig. S1 and Tables S1–S3.
- FcγR
- Fcγ receptor
- ADCC
- antibody-dependent cellular cytotoxicity
- ADCP
- antibody-dependent cellular phagocytosis
- MMP
- matrix metalloproteinase
- IdeS
- immunoglobulin G-degrading enzyme of S. pyogenes
- PBMC
- peripheral blood mononuclear cell
- CDC
- complement-dependent cytotoxicity
- pos
- positive
- neg
- negative
- HNSCC
- head and neck squamous cell carcinoma.
REFERENCES
- 1. Presta L. G. (2008) Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol. 20, 460–470 [DOI] [PubMed] [Google Scholar]
- 2. Strohl W. R. (2009) Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 20, 685–691 [DOI] [PubMed] [Google Scholar]
- 3. Reichert J. M. (2010) Metrics for antibody therapeutics development. MAbs 2, 695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weiner L. M., Surana R., Wang S. (2010) Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 10, 317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Idusogie E. E., Wong P. Y., Presta L. G., Gazzano-Santoro H., Totpal K., Ultsch M., Mulkerrin M. G. (2001) Engineered antibodies with increased activity to recruit complement. J. Immunol. 166, 2571–2575 [DOI] [PubMed] [Google Scholar]
- 6. Moore G. L., Chen H., Karki S., Lazar G. A. (2010) Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs 2, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Natsume A., Shimizu-Yokoyama Y., Satoh M., Shitara K., Niwa R. (2009) Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. 100, 2411–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brezski R. J., Vafa O., Petrone D., Tam S. H., Powers G., Ryan M. H., Luongo J. L., Oberholtzer A., Knight D. M., Jordan R. E. (2009) Tumor-associated and microbial proteases compromise host IgG effector functions by a single cleavage proximal to the hinge. Proc. Natl. Acad. Sci. U.S.A. 106, 17864–17869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burton D. R., Jefferis R., Partridge L. J., Woof J. M. (1988) Molecular recognition of antibody (IgG) by cellular Fc receptor (FcRI). Mol. Immunol. 25, 1175–1181 [DOI] [PubMed] [Google Scholar]
- 10. Canfield S. M., Morrison S. L. (1991) The binding affinity of human IgG for its high affinity Fc receptor is determined by multiple amino acids in the CH2 domain and is modulated by the hinge region. J. Exp. Med. 173, 1483–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duncan A. R., Winter G. (1988) The binding site for C1q on IgG. Nature 332, 738–740 [DOI] [PubMed] [Google Scholar]
- 12. Duncan A. R., Woof J. M., Partridge L. J., Burton D. R., Winter G. (1988) Localization of the binding site for the human high-affinity Fc receptor on IgG. Nature 332, 563–564 [DOI] [PubMed] [Google Scholar]
- 13. Hezareh M., Hessell A. J., Jensen R. C., van de Winkel J. G., Parren P. W. (2001) Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 75, 12161–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hulett M. D., Witort E., Brinkworth R. I., McKenzie I. F., Hogarth P. M. (1994) Identification of the IgG binding site of the human low affinity receptor for IgG FcγRII. Enhancement and ablation of binding by site-directed mutagenesis. J. Biol. Chem. 269, 15287–15293 [PubMed] [Google Scholar]
- 15. Jefferis R., Lund J., Pound J. (1990) Molecular definition of interaction sites on human IgG for Fc receptors (huFcγR). Mol. Immunol. 27, 1237–1240 [DOI] [PubMed] [Google Scholar]
- 16. Lund J., Winter G., Jones P. T., Pound J. D., Tanaka T., Walker M. R., Artymiuk P. J., Arata Y., Burton D. R., Jefferis R. (1991) Human FcγRI and FcγRII interact with distinct but overlapping sites on human IgG. J. Immunol. 147, 2657–2662 [PubMed] [Google Scholar]
- 17. Partridge L. J., Woof J. M., Jefferis R., Burton D. R. (1986) The use of anti-IgG monoclonal antibodies in mapping the monocyte receptor site on IgG. Mol. Immunol. 23, 1365–1372 [DOI] [PubMed] [Google Scholar]
- 18. Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., Presta L. G. (2001) High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 [DOI] [PubMed] [Google Scholar]
- 19. Tamm A., Kister A., Nolte K. U., Gessner J. E., Schmidt R. E. (1996) The IgG binding site of human FcγRIIIB receptor involves CC′ and FG loops of the membrane-proximal domain. J. Biol. Chem. 271, 3659–3666 [DOI] [PubMed] [Google Scholar]
- 20. Wines B. D., Powell M. S., Parren P. W., Barnes N., Hogarth P. M. (2000) The IgG Fc contains distinct Fc receptor (FcR) binding sites: the leukocyte receptors FcγRI and FcγRIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J. Immunol. 164, 5313–5318 [DOI] [PubMed] [Google Scholar]
- 21. Woof J. M., Partridge L. J., Jefferis R., Burton D. R. (1986) Localisation of the monocyte-binding region on human immunoglobulin G. Mol. Immunol. 23, 319–330 [DOI] [PubMed] [Google Scholar]
- 22. Gearing A. J., Thorpe S. J., Miller K., Mangan M., Varley P. G., Dudgeon T., Ward G., Turner C., Thorpe R. (2002) Selective cleavage of human IgG by the matrix metalloproteinases, matrilysin and stromelysin. Immunol. Lett. 81, 41–48 [DOI] [PubMed] [Google Scholar]
- 23. Ryan M. H., Petrone D., Nemeth J. F., Barnathan E., Björck L., Jordan R. E. (2008) Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol. Immunol. 45, 1837–1846 [DOI] [PubMed] [Google Scholar]
- 24. von Pawel-Rammingen U., Johansson B. P., Björck L. (2002) IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J. 21, 1607–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vincents B., von Pawel-Rammingen U., Björck L., Abrahamson M. (2004) Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry 43, 15540–15549 [DOI] [PubMed] [Google Scholar]
- 26. Fan X., Brezski R. J., Fa M., Deng H., Oberholtzer A., Gonzalez A., Dubinsky W. P., Strohl W. R., Jordan R. E., Zhang N., An Z. (2012) A single proteolytic cleavage within the lower hinge of trastuzumab reduces immune effector function and in vivo efficacy. Breast Cancer Res. 14, R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brezski R. J., Jordan R. E. (2010) Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? MAbs 2, 212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. von Pawel-Rammingen U. (2012) Streptococcal IdeS and its impact on immune response and inflammation. J. Innate Immun. 4, 132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brezski R. J., Oberholtzer A., Strake B., Jordan R. E. (2011) The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. MAbs 3, 558–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bruhns P., Iannascoli B., England P., Mancardi D. A., Fernandez N., Jorieux S., Daëron M. (2009) Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725 [DOI] [PubMed] [Google Scholar]
- 31. Jefferis R. (2007) Antibody therapeutics: isotype and glycoform selection. Expert Opin. Biol. Ther. 7, 1401–1413 [DOI] [PubMed] [Google Scholar]
- 32. Armour K. L., Clark M. R., Hadley A. G., Williamson L. M. (1999) Recombinant human IgG molecules lacking Fcγ receptor I binding and monocyte triggering activities. Eur. J. Immunol. 29, 2613–2624 [DOI] [PubMed] [Google Scholar]
- 33. Strohl W. R., Strohl L. M. (2012) Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharmaceutical Industry, Woodhead Publishing, Cambridge, UK [Google Scholar]
- 34. Natsume A., Niwa R., Satoh M. (2009) Improving effector functions of antibodies for cancer treatment: enhancing ADCC and CDC. Drug Des. Devel. Ther. 3, 7–16 [PMC free article] [PubMed] [Google Scholar]
- 35. Lazar G. A., Dang W., Karki S., Vafa O., Peng J. S., Hyun L., Chan C., Chung H. S., Eivazi A., Yoder S. C., Vielmetter J., Carmichael D. F., Hayes R. J., Dahiyat B. I. (2006) Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 103, 4005–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stavenhagen J. B., Gorlatov S., Tuaillon N., Rankin C. T., Li H., Burke S., Huang L., Vijh S., Johnson S., Bonvini E., Koenig S. (2007) Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. 67, 8882–8890 [DOI] [PubMed] [Google Scholar]
- 37. Umaña P., Jean-Mairet J., Moudry R., Amstutz H., Bailey J. E. (1999) Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 17, 176–180 [DOI] [PubMed] [Google Scholar]
- 38. Dall'Acqua W. F., Cook K. E., Damschroder M. M., Woods R. M., Wu H. (2006) Modulation of the effector functions of a human IgG1 through engineering of its hinge region. J. Immunol. 177, 1129–1138 [DOI] [PubMed] [Google Scholar]
- 39. Yan B., Boyd D., Kaschak T., Tsukuda J., Shen A., Lin Y., Chung S., Gupta P., Kamath A., Wong A., Vernes J. M., Meng G. Y., Totpal K., Schaefer G., Jiang G., Nogal B., Emery C., Vanderlaan M., Carter P., Harris R., Amanullah A. (2012) Engineering upper hinge improves stability and effector function of a human IgG1. J. Biol. Chem. 287, 5891–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grugan K. D., McCabe F. L., Kinder M., Greenplate A. R., Harman B. C., Ekert J. E., van Rooijen N., Anderson G. M., Nemeth J. A., Strohl W. R., Jordan R. E., Brezski R. J. (2012) Tumor-associated macrophages promote invasion while retaining Fc-dependent anti-tumor function. J. Immunol. 189, 5457–5466 [DOI] [PubMed] [Google Scholar]
- 41. Ngo C. V., Picha K., McCabe F., Millar H., Tawadros R., Tam S. H., Nakada M. T., Anderson G. M. (2007) CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int. J. Cancer 120, 1261–1267 [DOI] [PubMed] [Google Scholar]
- 42. Brezski R. J., Luongo J. L., Petrone D., Ryan M. H., Zhong D., Tam S. H., Schmidt A. P., Kruszynski M., Whitaker B. P., Knight D. M., Jordan R. E. (2008) Human anti-IgG1 hinge autoantibodies reconstitute the effector functions of proteolytically inactivated IgGs. J. Immunol. 181, 3183–3192 [DOI] [PubMed] [Google Scholar]
- 43. Ang K. K., Berkey B. A., Tu X., Zhang H. Z., Katz R., Hammond E. H., Fu K. K., Milas L. (2002) Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 62, 7350–7356 [PubMed] [Google Scholar]
- 44. Johansson N., Airola K., Grénman R., Kariniemi A. L., Saarialho-Kere U., Kähäri V. M. (1997) Expression of collagenase-3 (matrix metalloproteinase-13) in squamous cell carcinomas of the head and neck. Am. J. Pathol. 151, 499–508 [PMC free article] [PubMed] [Google Scholar]
- 45. Edelman G. M., Cunningham B. A., Gall W. E., Gottlieb P. D., Rutishauser U., Waxdal M. J. (1969) The covalent structure of an entire γG immunoglobulin molecule. Proc. Natl. Acad. Sci. U.S.A. 63, 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nesspor T. C., Raju T. S., Chin C. N., Vafa O., Brezski R. J. (2012) Avidity confers FcγR binding and immune effector function to aglycosylated immunoglobulin G1. J. Mol. Recognit. 25, 147–154 [DOI] [PubMed] [Google Scholar]
- 47. Oflazoglu E., Stone I. J., Brown L., Gordon K. A., van Rooijen N., Jonas M., Law C. L., Grewal I. S., Gerber H. P. (2009) Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br J. Cancer 100, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richards J. O., Karki S., Lazar G. A., Chen H., Dang W., Desjarlais J. R. (2008) Optimization of antibody binding to FcγRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 7, 2517–2527 [DOI] [PubMed] [Google Scholar]
- 49. Roopenian D. C., Akilesh S. (2007) FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 7, 715–725 [DOI] [PubMed] [Google Scholar]
- 50. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Natsume A., In M., Takamura H., Nakagawa T., Shimizu Y., Kitajima K., Wakitani M., Ohta S., Satoh M., Shitara K., Niwa R. (2008) Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 68, 3863–3872 [DOI] [PubMed] [Google Scholar]
- 52. Reff M. E., Carner K., Chambers K. S., Chinn P. C., Leonard J. E., Raab R., Newman R. A., Hanna N., Anderson D. R. (1994) Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 83, 435–445 [PubMed] [Google Scholar]
- 53. Anderson D. R., Grillo-López A., Varns C., Chambers K. S., Hanna N. (1997) Targeted anti-cancer therapy using rituximab, a chimaeric anti-CD20 antibody (IDEC-C2B8) in the treatment of non-Hodgkin's B-cell lymphoma. Biochem. Soc. Trans. 25, 705–708 [DOI] [PubMed] [Google Scholar]
- 54. Teeling J. L., Mackus W. J., Wiegman L. J., van den Brakel J. H., Beers S. A., French R. R., van Meerten T., Ebeling S., Vink T., Slootstra J. W., Parren P. W., Glennie M. J., van de Winkel J. G. (2006) The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J. Immunol. 177, 362–371 [DOI] [PubMed] [Google Scholar]
- 55. Kessenbrock K., Plaks V., Werb Z. (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams G. P., Schier R., McCall A. M., Simmons H. H., Horak E. M., Alpaugh R. K., Marks J. D., Weiner L. M. (2001) High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 61, 4750–4755 [PubMed] [Google Scholar]
- 57. Cooper J. B., Cohen E. E. (2009) Mechanisms of resistance to EGFR inhibitors in head and neck cancer. Head Neck 31, 1086–1094 [DOI] [PubMed] [Google Scholar]
- 58. Tao M.-H., Morrison S. L. (1989) Studies of aglycosylated chimeric mouse-human IgG Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143, 2595–2601 [PubMed] [Google Scholar]
- 59. Sazinsky S. L., Ott R. G., Silver N. W., Tidor B., Ravetch J. V., Wittrup K. D. (2008) Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 20167–20172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jung S. T., Reddy S. T., Kang T. H., Borrok M. J., Sandlie I., Tucker P. W., Georgiou G. (2010) Aglycosylated IgG variants expressed in bacteria that selectively bind FcγRI potentiate tumor cell killing by monocyte-dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 107, 604–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oganesyan V., Damschroder M. M., Leach W., Wu H., Dall'Acqua W. F. (2008) Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol. Immunol. 45, 1872–1882 [DOI] [PubMed] [Google Scholar]
- 62. Johansson B. P., Shannon O., Björck L. (2008) IdeS: a bacterial proteolytic enzyme with therapeutic potential. PLoS One 3, e1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nandakumar K. S., Johansson B. P., Björck L., Holmdahl R. (2007) Blocking of experimental arthritis by cleavage of IgG antibodies in vivo. Arthritis Rheum 56, 3253–3260 [DOI] [PubMed] [Google Scholar]
- 64. Nandakumar K. S., Holmdahl R. (2008) Therapeutic cleavage of IgG: new avenues for treating inflammation. Trends Immunol. 29, 173–178 [DOI] [PubMed] [Google Scholar]
- 65. Jiang T., Olson E. S., Nguyen Q. T., Roy M., Jennings P. A., Tsien R. Y. (2004) Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. U.S.A. 101, 17867–17872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guentsch A., Hirsch C., Pfister W., Vincents B., Abrahamson M., Sroka A., Potempa J., Eick S. (2013) Cleavage of IgG1 in gingival crevicular fluid is associated with the presence of Porphyromonas gingivalis. J. Periodontal Res. 48, 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jefferis R., Weston P. D., Stanworth D. R., Clamp J. R. (1968) Relationship between the papain sensitivity of human γG immunoglobulins and their heavy chain subclass. Nature 219, 646–649 [DOI] [PubMed] [Google Scholar]
- 68. Turner M. W., Bennich H. H., Natvig J. B. (1970) Pepsin digestion of human G-myeloma proteins of different subclasses. I. The characteristic features of pepsin cleavage as a function of time. Clin. Exp. Immunol. 7, 603–625 [PMC free article] [PubMed] [Google Scholar]
- 69. Yee A. M., Phan H. M., Zuniga R., Salmon J. E., Musher D. M. (2000) Association between FcγRIIa-R131 allotype and bacteremic pneumococcal pneumonia. Clin. Infect. Dis. 30, 25–28 [DOI] [PubMed] [Google Scholar]
- 70. Sanders L. A., Feldman R. G., Voorhorst-Ogink M. M., de Haas M., Rijkers G. T., Capel P. J., Zegers B. J., van de Winkel J. G. (1995) Human immunoglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2-mediated bacterial phagocytosis by neutrophils. Infect. Immun. 63, 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bibeau F., Lopez-Crapez E., Di Fiore F., Thezenas S., Ychou M., Blanchard F., Lamy A., Penault-Llorca F., Frébourg T., Michel P., Sabourin J. C., Boissière-Michot F. (2009) Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 27, 1122–1129 [DOI] [PubMed] [Google Scholar]
- 72. Cartron G., Dacheux L., Salles G., Solal-Celigny P., Bardos P., Colombat P., Watier H. (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood 99, 754–758 [DOI] [PubMed] [Google Scholar]
- 73. Musolino A., Naldi N., Bortesi B., Pezzuolo D., Capelletti M., Missale G., Laccabue D., Zerbini A., Camisa R., Bisagni G., Neri T. M., Ardizzoni A. (2008) Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 26, 1789–1796 [DOI] [PubMed] [Google Scholar]
- 74. Niwa R., Hatanaka S., Shoji-Hosaka E., Sakurada M., Kobayashi Y., Uehara A., Yokoi H., Nakamura K., Shitara K. (2004) Enhancement of the antibody-dependent cellular cytotoxicity of low-fucose IgG1 Is independent of FcγRIIIa functional polymorphism. Clin. Cancer Res. 10, 6248–6255 [DOI] [PubMed] [Google Scholar]
- 75. Cragg M. S., Morgan S. M., Chan H. T., Morgan B. P., Filatov A. V., Johnson P. W., French R. R., Glennie M. J. (2003) Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101, 1045–1052 [DOI] [PubMed] [Google Scholar]
- 76. Di Gaetano N., Cittera E., Nota R., Vecchi A., Grieco V., Scanziani E., Botto M., Introna M., Golay J. (2003) Complement activation determines the therapeutic activity of rituximab in vivo. J. Immunol. 171, 1581–1587 [DOI] [PubMed] [Google Scholar]
- 77. Golay J., Cittera E., Di Gaetano N., Manganini M., Mosca M., Nebuloni M., van Rooijen N., Vago L., Introna M. (2006) The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica 91, 176–183 [PubMed] [Google Scholar]
- 78. Beers S. A., Chan C. H., James S., French R. R., Attfield K. E., Brennan C. M., Ahuja A., Shlomchik M. J., Cragg M. S., Glennie M. J. (2008) Type II (tositumomab) anti-CD20 monoclonal antibody out performs type I (rituximab-like) reagents in B-cell depletion regardless of complement activation. Blood 112, 4170–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beers S. A., French R. R., Chan H. T., Lim S. H., Jarrett T. C., Vidal R. M., Wijayaweera S. S., Dixon S. V., Kim H., Cox K. L., Kerr J. P., Johnston D. A., Johnson P. W., Verbeek J. S., Glennie M. J., Cragg M. S. (2010) Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191–5201 [DOI] [PubMed] [Google Scholar]
- 80. Gong Q., Ou Q., Ye S., Lee W. P., Cornelius J., Diehl L., Lin W. Y., Hu Z., Lu Y., Chen Y., Wu Y., Meng Y. G., Gribling P., Lin Z., Nguyen K., Tran T., Zhang Y., Rosen H., Martin F., Chan A. C. (2005) Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 174, 817–826 [DOI] [PubMed] [Google Scholar]