

Background: Cyclomarin A is a mycobactericidal natural product that binds to ClpC1 in vitro.
Results: ClpC1 binds to the N-terminal domain of ClpC1 and the CymA-ClpC1 co-crystal structure identifies CymA-binding residues that, when mutated, confer resistance to CymA in vivo.
Conclusion: CymA targets ClpC1 inside mycobacteria.
Significance: This is the first direct evidence that cyclomarin kills mycobacteria by binding to ClpC1.
Keywords: Crystal Structure, Drug Discovery, Enzyme Inhibitors, Enzymes, Natural Products, ClpC, Cyclomarin
Abstract
Cyclomarin A (CymA) was identified as a mycobactericidal compound targeting ClpC1. However, the target was identified based on pulldown experiments and in vitro binding data, without direct functional evidence in mycobacteria. Here we show that CymA specifically binds to the N-terminal domain of ClpC1. In addition we have determined the co-crystal structure of CymA bound to the N-terminal domain of ClpC1 to high resolution. Based on the structure of the complex several mutations were engineered into ClpC1, which showed reduced CymA binding in vitro. The ClpC1 mutants were overexpressed in mycobacteria and two showed resistance to CymA, providing the first direct evidence that ClpC1 is the target of CymA. Phe80 is important in vitro and in cells for the ClpC1-CymA interaction and this explains why other bacteria are resistant to CymA. A model for how CymA binding to the N-terminal domain of ClpC1 leads to uncontrolled proteolysis by the associated ClpP protease machinery is discussed.
Introduction
Mycobacterium tuberculosis that causes tuberculosis (TB)3 is one of the most significant killers worldwide, second only to HIV that causes AIDS (1). Inappropriate usage of anti-TB drugs for decades has resulted in the development of multi- and extensively drug-resistant TB that do not respond to the first-line anti-TB drugs such as isoniazid and rifampicin and second line treatment such as aminoglycosides and fluoroquinolones (2, 3). Suitable treatment options are limited and identification of new therapeutics against TB is of utmost importance, especially because multidrug-resistant and extensively drug-resistant TB strains are being identified from all countries surveyed (2, 3).
The cyclic antibiotic cyclomarin A (CymA) was identified as a potent antitubercular compound in a natural product whole cell screen (4). Importantly it kills both growing and dormant, non-replicating mycobacteria. CymA, a heptapeptide from Streptomyces, and an amino alcohol derivative referred to as CymA1, are potent against multidrug-resistant M. tuberculosis, suggesting that they have a novel mode of action (4). Attempts to produce spontaneous resistant mutants against CymA to identify potential target genes by genome sequencing were unsuccessful. Instead a chemical-proteomic approach identified the caseinolytic protein C1 (ClpC1) as the target of CymA (4). Protein-degradation assays suggested that CymA was increasing proteolysis mediated by the caseinolytic protease (ClpP) inside the cell. CymA binding did not affect the ATPase activity of ClpC1 and no bactericidal activity was seen in a panel of Gram-positive and Gram-negative bacteria, despite them containing ClpC (4).
Caseinolytic protein C (ClpC) is a molecular chaperone known to exist in all kingdoms of life. It belongs to the Hsp100 family of AAA+ proteins (5). clpC1 is an essential gene for mycobacterial growth (6). M. tuberculosis possesses four Clp ATPase proteins namely ClpC1, ClpX, ClpX′ (ClpC2), and ClpB and two Clp protease paralogs, ClpP1 and ClpP2 (7). In several bacteria, Clp Hsp100 proteins, together with the ClpP form 4-ring ATP-dependent proteolytic machines (8). The pathway of degradation from intact or partly folded proteins to free amino acids can be either ATP dependent or independent (9). ClpP-mediated proteolysis aids in removal of dysfunctional proteins and maintenance of protein homeostasis in bacterial cells (10, 11). Only short peptides are hydrolyzed rapidly by ClpP itself. Globular proteins need to go through energy-dependent degradation by the ClpC-ClpP machinery in mycobacteria and other Gram-positive bacteria (12). Within the complex, ClpC takes up the role of recognizing and ATP-dependent unfolding of specific proteins and translocating the unfolded polypeptides to ClpP for degradation (13). ClpC1 is well conserved across various mycobacteria and is known to be an essential protein for bacterial growth (4). M. tuberculosis ClpC1 shares 100 and 95% identity with ClpC1 from M. bovis BCG (Pasteur) and M. smegmatis, respectively.
M. tuberculosis ClpC1 is an 848-amino acid protein with inherent ATPase activity, made up of an N-terminal helical domain (N) and two distinct nucleotide-binding or ATPase domains, D1 and D2 (14). The N-terminal domain of ClpC1 is completely conserved in all known mycobacteria and has about 63% identity to that of Bacillus subtilis. Crystal structures are available for B. subtilis ClpC and its complex with the adaptor protein MecA (8). Full-length ClpC1 proteins from M. tuberculosis and B. subtilis are almost 60% identical. However, unlike B. subtilis and many other Gram-positive bacteria, M. tuberculosis does not have any known adaptor proteins for ClpC1. M. tuberculosis ClpC1 is known to stimulate protein degradation by its association with the ClpP proteins. ClpP1 and ClpP2 alone do not show protease activity. Only when the ClpP1 and ClpP2 subunits interact to form a hetero-tetradecameric complex do they exhibit protease activity. ClpP1P2 protease activity in vitro requires the presence of ATP and a dipeptide activator (15). A functional ClpP1P2 protease complex is essential for M. tuberculosis growth in vitro and during infection (16).
ClpP, under control of its associated chaperone is a tightly regulated protease and only degrades short peptides. Inhibition of ClpP or the chaperone activity will result in toxic accumulation of protein aggregates, whereas their activation leads to uncontrolled proteolysis. Both events are fatal to bacteria. The natural antibiotics, acyldepsipeptides (ADEPs), are known to target the ClpP protease and prevent its interaction with the regulatory ATPase, resulting in uncontrolled proteolysis. ADEP binding converts ClpP into a dis-regulated protease, which hydrolyzes nascent polypeptide chains that are not yet folded (17, 18). The structures of B. subtilis ClpP and its complex with ADEPs reveal a closed-to-open gate transition of the ClpP N-terminal region, and ADEPs occupy part of the ATPase subunit-binding site, thereby directly blocking binding (19). The essential bacterial cell division protein, FtsZ, is prone to degradation by the ADEP-ClpP complex. By preventing cell division, ADEPs inhibit a vital cellular process (20).
To gain a better understanding of the mode of action of M. tuberculosis ClpC1 and its interaction with CymA, we undertook biophysical and crystallographic analysis. We show that CymA binds to the N-terminal domain of ClpC1 and have solved the crystal structure of the N-terminal domain of ClpC1 in complex with CymA to a resolution of 1.18 Å. The amino acids responsible for CymA binding were identified and mutants that were overexpressed in Mycobacterium bovis BCG (Pasteur) and Mycobacterium smegmatis showed resistance to CymA, directly showing that ClpC1 is the cellular target of CymA. A mechanism of action for CymA, as well as an explanation for why other bacteria are not inhibited by CymA is proposed. This work confirms ClpC1 as a promising target for anti-mycobacterial drug discovery.
EXPERIMENTAL PROCEDURES
Protein Expression in Escherichia coli and Chromatographic Purification
PCR was carried out on the ClpC1 gene optimized for overexpression in E. coli (Genscript, NJ) for the N-terminal domain (NTD; residues 1–145), D1 domain (residues 165–493), N-D1 domain (residues 1–493), D2 domain (residues 498–838), and full-length ClpC1 (residues 1–848). The genes coding for the various domains were cloned into a pET30(a) plasmid vector between NdeI and XhoI restriction sites. The proteins were overexpressed with a non-cleavable C-terminal His tag in E. coli BL21(DE3) cells (Invitrogen) by isopropyl 1-thio-β-d-galactopyranoside induction. The initial construct for ClpC1 NTD was in a pGEX-6P-1 plasmid, cloned between BamHI and EcoRI sites, and expressed as a fusion protein with Prescission protease cleavable N-terminal GST tag. F2A, F2Y, F80A, F80Y, E89A, and E89Q mutants of ClpC1 NTD protein were generated using QuikChange site-directed mutagenesis kit (Agilent Technologies, CA) on the pET30(a)-ClpC1 NTD construct. The mutations were confirmed by gene sequencing. Overexpression of the individual mutants followed the same protocol as that of wild type ClpC1 NTD.
The proteins overexpressed with a C-terminal His tag were purified using a HisTrap FF column (GE Healthcare, Uppsala, Sweden), followed by a size-exclusion purification step using a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare) for the NTD proteins and a HiLoad 26/60 Superdex 200 prep grade column (GE Healthcare) for the full-length protein and other domains. The NTD of ClpC1 overexpressed with GST tag was purified using a GSTrap FF column (GE Healthcare), followed by on-column GST tag cleavage with Prescission protease and a size exclusion purification step using a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare).
Crystallization and X-ray Structure Determination
CymA and CymA1 were prepared as described previously (4). The purified MTB ClpC1 NTD protein and the mutants were set up for crystallization at a concentration of ∼15 mg/ml. For co-crystallization, CymA was added to the protein at a 1.1:1 molar ratio and incubated in ice for about 30 min before crystallization. Crystals of ClpC1 NTD appeared in about 2–3 days at 18 °C in a condition having 0.2 m sodium acetate and 30% polyethylene glycol 3350. Crystals of ClpC1 NTD and mutants F2Y and F80Y in complex with CymA appeared in about 2–3 days in conditions having 0.2 m ammonium acetate and 20% polyethylene glycol 3350, 0.025 m magnesium formate and 20% polyethylene glycol 3350, and 0.15 m ammonium acetate and 20% polyethylene glycol 3350, respectively. The crystals were transferred to their respective crystallization reservoir solutions, supplemented with 20% glycerol, and flash frozen in liquid nitrogen.
Single crystal x-ray diffraction data were collected at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland) using the PILATUS detector on beam line X10SA. X-ray data were processed using iMOSFLM (21) and SCALA (22) from the CCP4 suite (23). The structure of the apo-form of MTB ClpC1 NTD was solved by molecular replacement using the B. subtilis ClpC1 NTD crystal structure (PDB ID 2YIQ). The structures of CymA-bound forms of the wild type, F2Y and F80Y mutants of MTB ClpC1 NTD were solved using the apo-form MTB ClpC1 NTD structure as a molecular replacement model. Model building and structure refinement were done with COOT (24) and Refmac5 (25) from the CCP4 suite. The data collection and refinement statistics can be found in Tables 1 and 2. The structure figures were prepared using PyMOL.
TABLE 1.
Summary of isothermal titration calorimetry for ClpC1 constructs binding to CymA1 at 298 K
| ClpC1 construct | n | Kd | ΔH | ΔS |
|---|---|---|---|---|
| nm | cal mol−1 | cal mol−1 degree−1 | ||
| ClpC1 (FL; 1–848) | 0.951 ± 0.004 | 17.5 ± 5.9 | −9708 ± 75 | 2.9 |
| ClpC1 (NTD; 1–145) | 1.05 ± 0.003 | 12 ± 3 | −9488 ± 57 | 4.4 |
| ClpC1 (D1; 165–493) | -a | - | - | - |
| ClpC1 (NTD-D1; 1–493) | 1.08 ± 0.003 | 19.2 ± 5.2 | −9157 ± 61 | 4.6 |
| ClpC1 (D2; 498–848) | - | − | - | - |
| ClpC1 (D1-D2; 165–848) | - | - | - | - |
a No binding detected.
TABLE 2.
Summary of data collection and refinement statistics
The numbers in parentheses refer to the highest resolution shell.
| WT | WT-CymA | F2Y-CymA | F80Y-CymA | |
|---|---|---|---|---|
| Space group | P212121 | P212121 | P212121 | P212121 |
| Unit cell (Å) | 42.45, 57.09, 65.05 | 33.88, 58.69, 63.62 | 33.78, 58.84, 64.53 | 34.02, 58.86, 63.99 |
| Wavelength (Å) | 1.0 | 1.0 | 1.0 | 1.0 |
| Rsym (%) | 4.8 (48.0) | 6.5 (18.4) | 8.4 (25.7) | 10 (49.2) |
| Mean I/σ(I) | 15.4 (3.3) | 14.4 (7.4) | 11.7 (6.2) | 9.1 (3.5) |
| Observed reflections | 198,304 (28379) | 244,126 (35074) | 243,163 (36361) | 135,220 (17576) |
| Unique reflections | 33,696 (4834) | 42,423 (6057) | 42,684 (6110) | 23,876 (3394) |
| Multiplicity | 5.9 (5.9) | 5.8 (5.8) | 5.7 (6.0) | 5.7 (5.2) |
| Solvent content (%) | 38.7 | 10.9 | 18.5 | 19.4 |
| Resolution range for refinement (Å) | 30.0–1.37 (1.41–1.37) | 30.0–1.18 (1.21–1.18) | 30.0–1.18 (1.21–1.18) | 29.5–1.44 (1.52–1.44) |
| Completeness for range (%) | 99.3 (99.3) | 99.8 (98.7) | 99.4 (98.3) | 99.6 (99.2) |
| No of reflections | ||||
| Used for refinement | 31935 | 40221 | 40467 | 22599 |
| Used for Rfree calculation | 1709 | 2141 | 2154 | 1217 |
| R factor (%) | 14.3 | 12.2 | 12.5 | 12.7 |
| Rfree (%) | 18.2 | 15.9 | 15.4 | 17.7 |
| Average B factors (Å2) | 22.2 | 13.1 | 12.6 | 13.8 |
| Protein | 19.9 | 11.4 | 10.7 | 11.7 |
| Solvent | 39.1 | 28.3 | 27.9 | 32.4 |
| Ligands | 11.7 | 6.8 | 6.7 | 7.3 |
| No. of nonhydrogen atoms | 1391 | 1623 | 1507 | 1488 |
| Water molecules | 170 | 167 | 168 | 160 |
| R.m.s. deviations from ideality | ||||
| Bond lengths (Å) | 0.013 | 0.016 | 0.015 | 0.014 |
| Bond angles (°) | 1.556 | 1.985 | 2.057 | 1.818 |
| Ramanchandran plot | ||||
| Most flavored (%) | 96.9 | 96.8 | 96.8 | 96.8 |
| Additional allowed (%) | 3.1 | 3.2 | 3.2 | 3.2 |
| Disallowed (%) | 0 | 0 | 0 | 0 |
| PDB code | 3WDB | 3WDC | 3WDD | 3WDE |
Isothermal Titration Calorimetry
Isothermal titration calorimetry experiments were performed using an iTC200 (GE Healthcare) at 25 °C, with 15–20 μm ClpC1 (in 20 mm Tris-HCl, pH 7.5, 300 mm NaCl, 1 mm EDTA, 1 mm Tris(2-carboxyethyl)phosphine, 9% DMSO) in the sample cell. Typically, 19 injections of 2 μl of 150–200 μm CymA1 solution in the same buffer were injected into the sample cell at 2.5-min intervals. The data were fitted to a single-site binding equation with the aid of Origin software (OriginLab, MA).
Overexpression in M. bovis BCG (Pasteur) and M. smegmatis
The DNA sequence encoding ClpC1 from M. tuberculosis was cloned into the multicopy mycobacterial expression vector, pMV262, between BamHI and HindIII restriction sites. The mutants F2A, F2Y, F80A, F80Y, E89A, and E89Q were generated using the QuikChange site-directed mutagenesis kit (Agilent Technologies, CA), on the pMV262-ClpC1 construct and the mutations were confirmed by sequencing.
To check and compare CymA1 susceptibility of overexpression cells, empty pMV262 plasmid and the various constructs of pMV262-ClpC1 were electroporated into M. bovis BCG (Pasteur) and M. smegmatis MC2 155 and selected on Middlebrook 7H11 agar plates containing 0.5% glycerol and 10% (v/v) OADC supplement and with 30 μg/ml of kanamycin. Colonies were picked and grown in Middlebrook 7H9 broth containing 0.05% Tween 80, 10% (v/v) albumin, dextrose, and sodium chloride, 0.2% glycerol, and 30 μg/ml of kanamycin. The cultures of all mutants and wild type ClpC1 overexpression cells in logarithmic phase were diluted to an optical density at 600 nm (A600) of 0.02 with Middlebrook 7H9 broth and taken in 96-well test plates. CymA1 and streptomycin stocks in 90% dimethyl sulfoxide and deionized water, respectively, were introduced at varying concentrations, separately for each cell type. Compound susceptibility experiments for each cell type were done in duplicate and with two different clones of the same type. The cells with and without compounds were allowed to incubate at 37 °C for 2 days for M. smegmatis MC2 155 and 5 days for M. bovis BCG (Pasteur). Compound-treated and -untreated cells from various strains were plated on to Middlebrook 7H11 agar plates and incubated at 37 °C in an incubator for 3 weeks before counting the colony forming units. Statistical analyses were performed using a Student's t test.
RESULTS
ClpC1 N-terminal Domain Binds Cyclomarin A
To identify the domain(s) that interact with CymA, we separately cloned and expressed the individual domains of ClpC1. The full-length ClpC1 protein (residues 1–848), NTD (residues 1–145), D1 domain (residues 165–493), N-D1 domain (residues 1–493), D2 domain (residues 498–848), and D1-D2 domain (residues 165–848) were expressed with a C-terminal His tag, purified to homogeneity, and subjected to binding experiments using isothermal titration calorimetry (ITC) with CymA1 (amino-alcohol derivative of CymA (4)). CymA1 was chosen over CymA due its better solubility. The structure of CymA and CymA1, as well as results from ITC experiments can be seen in Fig. 1 and Table 1. The data clearly show that the N-terminal domain of ClpC1 specifically binds to CymA1. The D1 and D2 domains alone do not bind. The data indicate that the NTD binds to CymA1 with a similar affinity to full-length ClpC1 or the N-D1 domains, suggesting that the D1 and D2 domains do not contribute to binding.
FIGURE 1.

Structures of cyclomarin A and examples of ITC titrations. A, structures of cyclomarin A (CymA; left) and the amino-alcohol derivative (CymA1; right). B, representative titrations for CymA1 binding to ClpC1 (1–145; left) and ClpC1 (1–493; right).
Structure of M. tuberculosis ClpC1 N-terminal Domain
Because the ClpC1 NTD binds to CymA with high affinity, we attempted to crystallize the ClpC1 NTD with and without CymA. We obtained crystals for both the apo- and holo- forms by standard vapor-diffusion methods, making use of commercially available sparse-matrix screens. The crystallization conditions were further optimized to improve the diffraction quality of the crystals. The crystals used for structure determination belonged to the orthorhombic space group P212121. The data collection and refinement statistics are summarized in Table 2.
The crystal structure of M. tuberculosis ClpC1 NTD was solved by molecular replacement, using the B. subtilis NTD structure (PDB code 2Y1Q) as the search model. The high resolution (1.37 Å) structure shows that the ClpC1 N-terminal domain (residues 1–145; Fig. 2A) consists of eight α-helices. The fold of the NTD contains two repeats of a 4-helix motif that share 58% identity. A 14-amino acid loop between α4 and α5 connects the two motifs. Residues 4–68 and 79–143 superpose with a r.m.s. deviation of about 2.04 Å (Fig. 2B). The overall structure of the M. tuberculosis ClpC1 NTD is similar to the ClpC NTD from B. subtilis, with a r.m.s. deviation of 0.48 Å (Fig. 2C). All residues of the NTD, as well as the two residues of the C-terminal His tag could be seen in the electron density, allowing a complete model to be built.
FIGURE 2.
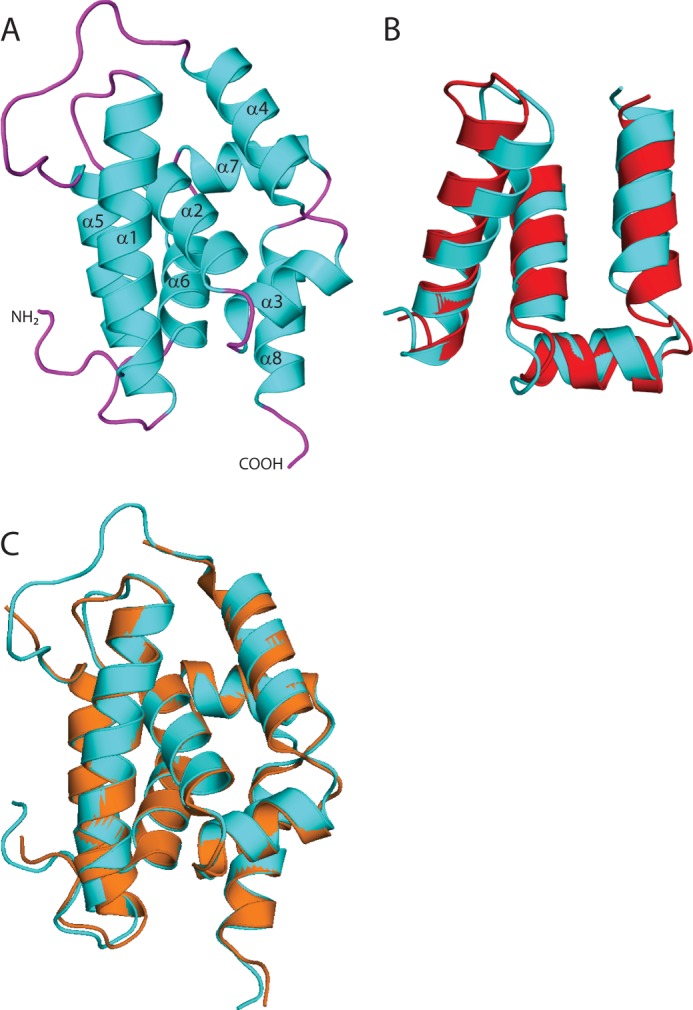
Structure of the ClpC1 N-terminal domain. A, overview of the structure of M. tuberculosis ClpC1 NTD, shown in schematic representation with the α-helices (cyan) and loops (magenta). The amino and carboxyl termini are indicated and the α-helices are labeled. B, structural alignment of ClpC1 residues 4–68 (cyan) with residues 79–143 (red; r.m.s. deviation 2.035). C, structural alignment of M. tuberculosis ClpC1 NTD (cyan) with B. subtilis ClpC NTD (orange; r.m.s. deviation 0.48).
Structural Evidence for CymA Binding
ClpC1 NTD was co-crystallized with CymA and the crystals diffracted to about 1.18-Å resolution. The structure was solved using the ClpC1 NTD as a search model for molecular replacement. The overall structure of the N-terminal domain does not change due to CymA binding. However, modest changes are seen in the region where CymA binds (Fig. 3A) suggesting CymA binding alters the conformation of the ClpC1 NTD. The construct used to solve the structure of the free ClpC1 NTD contained an N-terminal tag, some of which could be seen in the electron density, whereas the final refined structures of the complex were determined using constructs with a C-terminal His tag. No difference in affinity was seen for these different constructs, indicating that the N-terminal tag does not interfere with binding (data not shown). CymA occupies a site between the short N-terminal loop before α1 and the long loop between helices α4 and α5 (Fig. 3, B and C). Details of the interactions between CymA and ClpC1 are shown in Fig. 3D. Residues Phe2 and Phe80 of the protein are in close proximity to CymA, and the backbone amides make direct polar contacts with the carbonyls of the N-reverse prenylated tryptophan and l-alanine, respectively, of the cyclic heptapeptide. Glu89 of ClpC1 NTD α5 appears to exist in alternate side chain conformations, with only one of them forming a direct polar interaction with the compound via its N-methylhydroxyleucine residue. The side chain of Lys85 also makes two direct polar contacts with CymA. In addition to these direct contacts, several water-mediated interactions are also seen. These include the side chain of Glu3 forming a single water-mediated interaction, and the side chain of Gln17 forming two. The carbonyl of Phe80 also makes a water-mediated contact. The side chains of Phe2 and Phe80 form a hydrophobic platform, which the compound enfolds.
FIGURE 3.
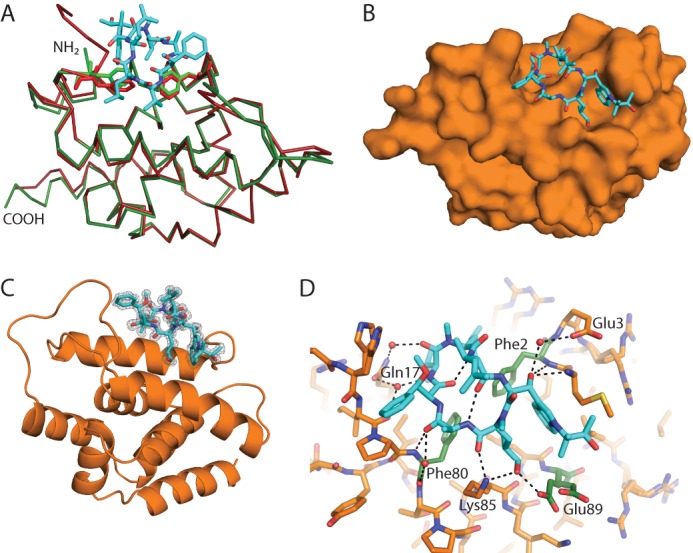
Structure of cyclomarin A bound to ClpC1 NTD. A, comparison of the structure of free ClpC1 NTD (red) and ClpC1 NTD bound to CymA (green). The protein is shown in ribbon representation, with CymA shown as cyan sticks. Phe2 and Phe80 are shown as sticks. The amino and carboxyl termini are indicated. B and C, overview of the structure of ClpC1 bound to CymA shown as surface and schematic representations, respectively, with CymA shown as cyan sticks and ClpC1 NTD orange. The difference density is shown in C at 3 σ after CymA was removed from the final round of refinement. D, details of the interactions between CymA and ClpC1, with CymA shown as cyan sticks and ClpC1 as orange sticks. Potential hydrogen bond interactions are shown as dashed lines. The side chains that contact CymA directly or via water-mediated interactions are labeled and the residues mutated in this study are highlighted as green sticks. For clarity only one orientation of Met1 is shown.
Structure-based Mutagenesis and Isothermal Titration Calorimetry
M. tuberculosis ClpC1 residues Phe2, Phe80, and Glu89 are directly involved in CymA binding in the co-crystal structure and were mutated to find out how important these residues are for CymA binding. Lys85 was not selected for mutagenesis because this appeared to be less important for CymA binding (see “Discussion”). We generated alanine mutants within the ClpC1 NTD (F2A, F80A, and E89A) as well as more conservative mutations (F2Y, F80Y, and E89Q). All six mutants were expressed and purified to homogeneity. ITC experiments were carried out for the mutants and the results can be seen in Table 3. F2A resulted in ∼300 times weaker binding to CymA1 and even the very minor change, F2Y, resulted in ∼60 times weaker binding. Mutations at Phe80 had less effect: F80A reduced CymA1 binding ∼160 times, whereas F80Y reduced binding ∼10 times. E89A reduced binding affinity ∼10 times compared with wild type and E89Q caused only a ∼5 times reduction in binding affinity. These data indicate that Phe2 is more important than Phe80 for binding CymA, which in turn is more important than Glu89.
TABLE 3.
Summary of isothermal titration calorimetry experiments for ClpC1(1–145) binding to CymA1 at 298 K
| ClpC1(1–145) | n | Kd | ΔH | ΔS |
|---|---|---|---|---|
| nm | cal mol−1 | cal mol−1 degree−1 | ||
| WT | 1.05 ± 0.003 | 12 ± 3 | −9488 ± 57 | 4.4 |
| F2A | 0.894 ± 0.029 | 3900 ± 500 | −7010 ± 323 | 1.2 |
| F80A | 1.09 ± 0.013 | 2100 ± 200 | −6041 ± 108 | 5.7 |
| E89A | 1.01 ± 0.005 | 127 ± 14 | −8590 ± 63 | 2.8 |
| F2Y | 0.964 ± 0.012 | 780 ± 80 | −6257 ± 104 | 7.0 |
| F80Y | 0.983 ± 0.004 | 124 ± 12 | −7865 ± 49 | 5.2 |
| E89Q | 0.871 ± 0.004 | 65 ± 8 | −10630 ± 81 | −2.7 |
Crystal Structure of ClpC1 NTD Mutants F2Y and F80Y in Complex with CymA
To understand how the F2Y and F80Y mutants affect CymA binding, we attempted to determine high-resolution structures of these mutants bound to CymA. Crystals were obtained in the same orthorhombic space group, P212121, as the structures of the free ClpC1 NTD and ClpC1 NTD-CymA structures. The data collection and refinement statistics are shown in Table 2. As expected, the two mutations had no major effect on the overall structure compared with the wild type ClpC1 NTD-CymA complex; the r.m.s. deviation was 0.099 and 0.082 Å for the F2Y and F80Y mutants, respectively, compared with the wild type (Fig. 4A).
FIGURE 4.
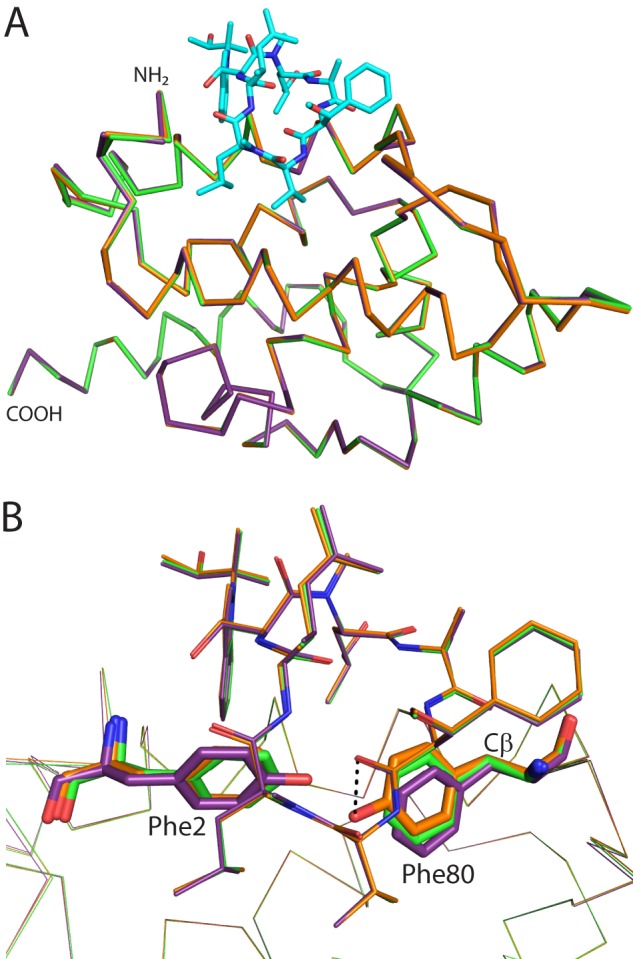
Comparison of CymA binding to WT ClpC1 versus F2Y or F80Y mutants. A, backbone alignment of the structures of WT (green), F2Y (purple), and F80Y (orange) ClpC1 NTD bound to CymA. CymA is shown as cyan sticks and ClpC1 as ribbons. The amino and carboxyl termini are indicated. B, the same orientation as in A with CymA from each structure shown as lines and ClpC1 shown as ribbons. Phe2 and Phe80 (or their Tyr mutants) are shown as sticks. CymA and ClpC1 are colored: WT, green; F2Y, purple; F80Y, orange. A potential hydrogen bond interaction between Tyr80 and a carbonyl of CymA is shown as a dashed line. The Cβ atom discussed in the text is indicated.
Both mutations affect the conformations of both aromatic residues, however, smaller changes are seen for the F80Y mutation. In both mutants there is rotation about the Cβ atom in residue 80, causing the planar ring to rotate 13° away from CymA in the F2Y mutant and 8° toward CymA in the F80Y mutant (Fig. 4B). In addition, the hydroxyl of Tyr80 makes a weak hydrogen bond with a free carbonyl of CymA. For residue 2 in the F80Y mutant, it is in almost the same position as the WT. It is co-planar and has moved 0.2 Å. For the F2Y mutant, residue 2 has moved 0.9 Å and is now out of plane compared with the WT, due to rotation away from the side chain of the modified Trp of CymA. The combination of an additional polar interaction between F80Y and CymA combined with the greater movement of residues 2 and 80 in F2Y is responsible for the 60 times lower CymA1 binding affinity for the F2Y mutant, compared with only a 10 times reduction for the F80Y mutant.
Overexpression in M. bovis BCG (Pasteur) and M. smegmatis
The ClpC1 protein of M. tuberculosis is 100% identical to that of M. bovis BCG (Pasteur) and about 96% identical to that of M. smegmatis. The pMV262 plasmid vector expresses a cloned gene under control of a constitutively expressed mycobacterial heat shock promoter, hsp60. Because pMV262 is a multicopy, constitutively expressed plasmid, the clpC1 gene and its mutants were cloned into pMV262 so that the genes are overexpressed relative to the WT copy of ClpC1. Mycobacterial transformants with the pMV262, pMV262-ClpC1, and the various ClpC1 mutants were selected on Middlebrook 7H11 agar plates with kanamycin. Mycobactericidal experiments were carried out using CymA1 rather than CymA, due to its improved solubility. We determined the 50% minimum inhibitory concentration (MIC50) for CymA1 against M. smegmatis and M. bovis BCG to be 1.2 and 0.28 μm, respectively (data not shown). The M. smegmatis and M. bovis strains were treated with 1.0 and 0.16 μm CymA1, respectively. This is below the MIC50 value so that the compound would only have a modest effect on growth inhibition. Overexpression of ClpC1 mutants F80Y and E89A conferred statistically significant resistance to CymA1, whereas other mutations produced no significant effect in M. smegmatis MC2 155 (Fig. 5A). In M. bovis BCG, apart from F80Y, E89A and another mutant, F2Y, showed statistically significant resistance to CymA1 (Fig. 5B). The other mutants did not show significant resistance. This shows that modulating the binding of CymA to ClpC1 in cells changes their sensitivity to CymA. This is the first direct evidence that ClpC1 is the cellular target of CymA leading to a bactericidal effect. All the strains were equally sensitive to streptomycin, a standard aminoglycoside, which is used as a first line anti-TB drug (data not shown).
FIGURE 5.
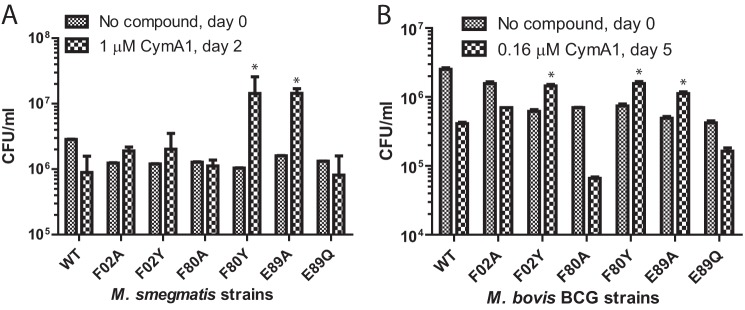
Overexpression of WT ClpC1 or ClpC1 mutants in (A) M. smegmatis or (B) M. bovis BCG (Pasteur). Colony-forming units (cfu) were counted without CymA1 at day 0, then with sub-MIC50 concentrations of CymA1 at the day indicated. M. smegmatis strains expressing F80Y and F89Y mutants and M. bovis BCG strains expressing F02Y, F80Y, and E89A mutants showed statistically significant resistance (p < 0.05) compared with WT, indicated by an asterisk.
DISCUSSION
The structure of the M. tuberculosis ClpC1 N-terminal domain is very similar to that of the B. subtilis ClpC N-terminal domain (8) and it also has a similar fold to the ClpA and ClpB N domain from other bacteria (26–28). At a resolution of 1.37 Å, all the residues could be placed into the electron density without any ambiguity. Several residues such as Ser, Arg, Lys, Gln, and Glu were found to have alternate conformations for their side chains.
The overall structure of ClpC1 NTD and its CymA-bound form are very similar (r.m.s. deviation ∼0.6 Å). However, CymA binding stabilizes the structure of the NTD, especially within the flexible loop regions, and it is likely that this has contributed to the improved diffraction quality of crystals of the NTD-CymA complex. Subtle differences were seen due to CymA binding, primarily in the loops and α-helix 5 that directly contact the compound. The key residues for CymA binding are apparent from the structure. This was confirmed by mutagenesis and binding experiments. Phe2, Phe80, and Glu89 were selected for mutagenesis and of these Phe2 contributed most to CymA1 binding, followed by Phe80, then Glu89. The overall sequence identity of ClpC1 from various Mycobacterium species is close to 95%, but the N-terminal domain of mycobacterial ClpC1 is 100% conserved. This explains why all tested mycobacteria were found to be sensitive to CymA (4).
The residues for mutagenesis were selected based on an initial co-crystal structure of the ClpC1 NTD bound to CymA, using a construct that left additional N-terminal amino acids on the protein (data not shown). Subsequent experiments used a C-terminal tag to avoid possible artifacts caused by the additional N-terminal residues. In this initial structure the side chain of Lys85 was not involved in specific interactions with CymA so this residue was not selected for mutagenesis (data not shown).
A comparison of the crystal structures of CymA bound to WT ClpC1 NTD and to the F2Y and F80Y mutants showed that small movements in the positions of these residues are responsible for the reduced CymA affinity. This is compensated in part for F80Y by Tyr forming an additional polar interaction with a carbonyl of CymA. This is in agreement with the ITC data, which indicate that both the F2Y and F80Y mutants have a reduced binding enthalpy. Polar interactions typically contribute to favorable binding enthalpy (29), and the enthalpy is more favorable for F80Y than F2Y. The enthalpic penalty compared with WT may be from breaking hydrogen bonds between waters bound to the OH groups of the Tyr residues in the mutants when CymA binds. The entropy term (ΔS) is slightly more favorable for the mutants, especially F2Y, which could result from conformational entropy of the ClpC1 NTD, including residues 2 and 8, when CymA is bound. Alternatively desolvation of waters bound to the hydroxyl of the Tyr residue may lead to an increase in entropy.
ClpC1 is an essential gene in mycobacteria and CymA treatment did not produce any spontaneous resistance mutations in M. tuberculosis (4). As a result, this study was unable to directly show that ClpC1 is the cellular target of CymA. Our structural approach has identified important residues involved in CymA binding. In both M. smegmatis and M. bovis BCG, overexpression of ClpC1 F80Y and E89A conferred resistance to CymA1. Both mutations reduced the affinity of ClpC1 for CymA to a similar extent. The ClpC1 proteins were constitutively expressed using the same pMV262 vector under control of an hsp60 promoter, so it is reasonable to assume that all the constructs will have a similar level of expression. However, it remains a possibility that the expression levels of the different proteins could vary, which could influence the degree of resistance.
To generate resistance to CymA inside cells it is likely that a functional ClpC1 enzyme is required, in addition to a reduced affinity for CymA. The crystal structure of ClpC1 NTD F80Y in complex with CymA is very similar to that of the wild type ClpC1 NTD in complex with CymA, suggesting that the NTD is still functional. In M. bovis BCG F2Y also showed significant resistance to CymA1, this showed an intermediate reduction in binding affinity between the modest change for F80Y and E89A and the more dramatic reduction seen for F2A and F80A. This indicates that F2Y is also tolerated in M. bovis BCG. The mutant F80A also showed a slight increased sensitivity to CymA1 in M. bovis BCG compared with WT. WT produced a 9.4% decrease in cfu with CymA1, whereas F80A gave a 16.3% decrease. The reason for this increased sensitivity is not clear, but it may be because overexpression of this mutant creates some toxicity making it more susceptible to the effects of the compound.
ClpC1 E89Q binds to CymA1 with high affinity in vitro and overexpression of ClpC1 E89Q did not provide resistance. This is likely because this minor change has not sufficiently changed the affinity of the protein for CymA. In the high-resolution crystal structures of ClpC1-NTD and its two mutants in complex with CymA, Glu89 exists in two alternate conformations with only one interacting with CymA, suggesting that this residue is less important for CymA binding. The glutamine side chain of the mutant will still be able to hydrogen bond with N-methylhydroxyleucine residue in CymA, explaining why the compound still binds with high affinity and is active against this mutant in mycobacteria. Gram-positive bacteria such as Bacillus, which are resistant to CymA, have a conserved tyrosine in ClpC, equivalent to Phe80 in M. tuberculosis ClpC1. This suggests that F80Y produces a functional ClpC1 in mycobacteria. The ClpC1 mutants F2A and F80A also did not provide resistance to CymA when overexpressed in M. smegmatis or M. bovis BCG (Pasteur). The reason for this is that these proteins are very likely non-functional. Therefore, despite showing reduced CymA binding in vitro, they do not confer resistance in cells. Phe2 is conserved in ClpC proteins from various bacteria suggesting any changes are not well tolerated. The crystal structure of the ClpC1 NTD F2Y in complex with CymA shows Tyr2 affects the position of Phe80, which may affect interactions with other proteins. The more drastic alanine mutations probably perturb the local fold of the ClpC1 NTD so again do not produce a functional ClpC1 protein. Future experiments are needed to address the effect of these mutations on the function of the protein.
The N-terminal domain of the diverse AAA+ proteins are known to be involved in substrate recognition, either directly or indirectly by serving as a binding platform for adaptor proteins that help in substrate recognition (30). Unlike other Gram-positive bacteria, mycobacterial ClpC1 does not have an adaptor protein and hence the recognition of substrates for energy-dependent proteolysis by ClpP may be carried out by the ClpC1 NTD itself. Another possibility that has been proposed previously is that the NTD might only act as a substrate discriminator by controlling access to binding sites located in the interior of the ATPase domain (30). In either case, the NTD regulates access to the tunnel made by ClpC ATPase domains and the associated Clp protease machinery. The ATPase domains D1 and D2 are involved in the ATP-dependent unfolding of the proteins for proteolysis by ClpP. We propose that CymA binding reduces the flexibility of the linker between the two N-terminal repeats, making them immobile and leaving the ClpC1 tunnel open. This could allow free access for larger proteins into the unfolding and proteolytic core of the Clp machinery. This can lead to cell death if functional or partially folded nascent proteins are degraded. This is similar to what happens to ClpP proteins upon ADEP binding. ADEP binding to the N terminus of Gram-positive bacterial ClpP widens the ClpP opening, preventing the binding to the regulatory ATPase so that essential nascent peptides and proteins are degraded. Further work is required to understand the proteins that get degraded upon treatment with CymA. It may be similar to the B. subtilis FtsZ protein and the cell-division pathway that gets affected by the ClpP-ADEP complex (20).
B. subtilis ClpC protein has a similar domain organization and N-terminal domain structure as that of M. tuberculosis ClpC1 so would be expected to bind CymA. The F80Y mutant of M. tuberculosis ClpC1 binds CymA1 with only a 10-fold lower affinity. B. subtilis ClpC protein has a tyrosine instead of Phe80 of mycobacterial ClpC1, so it should also bind CymA in vitro. Gram-positive bacteria including B. subtilis are resistant to CymA (4). All of them have an adaptor protein that binds to ClpC NTD in vivo. In B. subtilis the adaptor protein is MecA, which is required for ClpC-mediated proteolysis (31, 32). The crystal structure of the ClpC NTD-MecA complex from B. subtilis is available (8). A comparison of the ClpC1 NTD-CymA structure with B. subtilis ClpC NTD-MecA shows that the CymA-binding site is close to the MecA interacting regions of B. subtilis ClpC NTD. The association of MecA with ClpC may prevent CymA binding to ClpC, thereby making B. subtilis resistant to CymA. Alternatively CymA may have no effect because of the role played by the MecA adapter protein. MecA is known to stimulate the ATPase activity of ClpC and transfer substrates to the ATPase tunnel (32, 33). It is possible that CymA could be binding the NTD of B. subtilis ClpC, but not causing any effect because the substrate recognition role is performed by MecA and not the ClpC NTD. In either case, the presence of an adaptor protein for ClpC in other bacteria along with the F80Y mutation combine to confer resistance to CymA.
We present the crystal structure and biophysical characterization of M. tuberculosis ClpC1 NTD and have identified three residues important for binding CymA. Full-length ClpC1 proteins containing mutations at these residues were overexpressed in M. bovis BCG (Pasteur) and M. smegmatis showing that E89A and F80Y confer resistance to CymA and providing direct evidence that ClpC1 is the cellular target of CymA. The compound is a very efficient inhibitor of growth for M. tuberculosis. CymA is a natural product and lacks good pharmacokinetic properties, making it challenging for optimization into an orally bioavailable drug. However, this work chemically validates ClpC1 as a drug target in M. tuberculosis. Compounds with a similar mode of action would be very attractive drugs due to their sterilization activity on persistent non-replicating bacteria. The structural data presented here can be used to design potent compounds against this target.
Acknowledgments
We thank the beamline scientists at PXII of the Swiss Light Source for assistance with data collection.
The atomic coordinates and structure factors (codes 3WDB, 3WDC, 3WDD, and 3WDE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TB
- tuberculosis
- CymA
- cyclomarin A
- ClpP
- caseinolytic protease
- ClpC
- caseinolytic protein C
- ADEP
- acyldepsipeptide
- NTD
- N-terminal domain
- ITC
- isothermal titration calorimetry
- r.m.s.
- root mean square.
REFERENCES
- 1. Dye C., Bassili A., Bierrenbach A. L., Broekmans J. F., Chadha V. K., Glaziou P., Gopi P. G., Hosseini M., Kim S. J., Manissero D., Onozaki I., Rieder H. L., Scheele S., van Leth F., van der Werf M., Williams B. G. (2008) Measuring tuberculosis burden, trends, and the impact of control programmes. Lancet Infect. Dis. 8, 233–243 [DOI] [PubMed] [Google Scholar]
- 2. Ma Z., Lienhardt C., McIlleron H., Nunn A. J., Wang X. (2010) Global tuberculosis drug development pipeline. The need and the reality. Lancet 375, 2100–2109 [DOI] [PubMed] [Google Scholar]
- 3. Villemagne B., Crauste C., Flipo M., Baulard A. R., Déprez B., Willand N. (2012) Tuberculosis. The drug development pipeline at a glance. Eur. J. Med. Chem. 51, 1–16 [DOI] [PubMed] [Google Scholar]
- 4. Schmitt E. K., Riwanto M., Sambandamurthy V., Roggo S., Miault C., Zwingelstein C., Krastel P., Noble C., Beer D., Rao S. P., Au M., Niyomrattanakit P., Lim V., Zheng J., Jeffery D., Pethe K., Camacho L. R. (2011) The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed Engl. 50, 5889–5891 [DOI] [PubMed] [Google Scholar]
- 5. Schirmer E. C., Glover J. R., Singer M. A., Lindquist S. (1996) HSP100/Clp proteins. A common mechanism explains diverse functions. Trends Biochem. Sci. 21, 289–296 [PubMed] [Google Scholar]
- 6. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 7. Ribeiro-Guimarães M. L., Pessolani M. C. (2007) Comparative genomics of mycobacterial proteases. Microb. Pathog. 43, 173–178 [DOI] [PubMed] [Google Scholar]
- 8. Wang F., Mei Z., Qi Y., Yan C., Hu Q., Wang J., Shi Y. (2011) Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature 471, 331–335 [DOI] [PubMed] [Google Scholar]
- 9. Chandu D., Nandi D. (2004) Comparative genomics and functional roles of the ATP-dependent proteases Lon and Clp during cytosolic protein degradation. Res. Microbiol. 155, 710–719 [DOI] [PubMed] [Google Scholar]
- 10. Sauer R. T., Bolon D. N., Burton B. M., Burton R. E., Flynn J. M., Grant R. A., Hersch G. L., Joshi S. A., Kenniston J. A., Levchenko I., Neher S. B., Oakes E. S., Siddiqui S. M., Wah D. A., Baker T. A. (2004) Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 12. Kress W., Maglica Z., Weber-Ban E. (2009) Clp chaperone-proteases. Structure and function. Res. Microbiol. 160, 618–628 [DOI] [PubMed] [Google Scholar]
- 13. Sauer R. T., Baker T. A. (2011) AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 80, 587–612 [DOI] [PubMed] [Google Scholar]
- 14. Kar N. P., Sikriwal D., Rath P., Choudhary R. K., Batra J. K. (2008) Mycobacterium tuberculosis ClpC1. Characterization and role of the N-terminal domain in its function. FEBS J. 275, 6149–6158 [DOI] [PubMed] [Google Scholar]
- 15. Akopian T., Kandror O., Raju R. M., Unnikrishnan M., Rubin E. J., Goldberg A. L. (2012) The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 31, 1529–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raju R. M., Unnikrishnan M., Rubin D. H., Krishnamoorthy V., Kandror O., Akopian T. N., Goldberg A. L., Rubin E. J. (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brötz-Oesterhelt H., Beyer D., Kroll H. P., Endermann R., Ladel C., Schroeder W., Hinzen B., Raddatz S., Paulsen H., Henninger K., Bandow J. E., Sahl H. G., Labischinski H. (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 18. Kirstein J., Hoffmann A., Lilie H., Schmidt R., Rübsamen-Waigmann H., Brötz-Oesterhelt H., Mogk A., Turgay K. (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 1, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee B. G., Park E. Y., Lee K. E., Jeon H., Sung K. H., Paulsen H., Rübsamen-Schaeff H., Brötz-Oesterhelt H., Song H. K. (2010) Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol. 17, 471–478 [DOI] [PubMed] [Google Scholar]
- 20. Sass P., Josten M., Famulla K., Schiffer G., Sahl H. G., Hamoen L., Brötz-Oesterhelt H. (2011) Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc. Natl. Acad. Sci. U.S.A. 108, 17474–17479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM. A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo F., Maurizi M. R., Esser L., Xia D. (2002) Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J. Biol. Chem. 277, 46743–46752 [DOI] [PubMed] [Google Scholar]
- 27. Lee S., Sowa M. E., Watanabe Y. H., Sigler P. B., Chiu W., Yoshida M., Tsai F. T. (2003) The structure of ClpB. A molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240 [DOI] [PubMed] [Google Scholar]
- 28. Li J., Sha B. (2003) Crystal structure of the E. coli Hsp100 ClpB N-terminal domain. Structure. 11, 323–328 [DOI] [PubMed] [Google Scholar]
- 29. Ferenczy G. G., Keserũ G. M. (2010) Thermodynamics guided lead discovery and optimization. Drug Discov. Today 15, 919–932 [DOI] [PubMed] [Google Scholar]
- 30. Mogk A., Dougan D., Weibezahn J., Schlieker C., Turgay K., Bukau B. (2004) Broad yet high substrate specificity. The challenge of AAA+ proteins. J. Struct. Biol. 146, 90–98 [DOI] [PubMed] [Google Scholar]
- 31. Kirstein J., Schlothauer T., Dougan D. A., Lilie H., Tischendorf G., Mogk A., Bukau B., Turgay K. (2006) Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 25, 1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schlothauer T., Mogk A., Dougan D. A., Bukau B., Turgay K. (2003) MecA, an adaptor protein necessary for ClpC chaperone activity. Proc. Natl. Acad. Sci. U.S.A. 100, 2306–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turgay K., Hamoen L. W., Venema G., Dubnau D. (1997) Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 11, 119–128 [DOI] [PubMed] [Google Scholar]