

Background: New strategies are needed to combat antibiotic-resistant bacteria.
Results: This work identified a small molecule inhibitor, regacin, that specifically disrupts the action of a master virulence regulator from an intestinal pathogen.
Conclusion: Inhibition of virulence gene expression by small molecules is a valid therapeutic strategy.
Significance: Inhibitors of this kind can be developed into drugs to prevent or treat bacterial infections.
Keywords: Antibiotics, Bacterial Pathogenesis, Gene Regulation, Infectious Diseases, Small Molecules, AraC-like Regulator, Bacterial Infection, Enteric Pathogen, Small Molecule Inhibitors, Virulence Gene Regulation
Abstract
The misuse of antibiotics during past decades has led to pervasive antibiotic resistance in bacteria. Hence, there is an urgent need for the development of new and alternative approaches to combat bacterial infections. In most bacterial pathogens the expression of virulence is tightly regulated at the transcriptional level. Therefore, targeting pathogens with drugs that interfere with virulence gene expression offers an effective alternative to conventional antimicrobial chemotherapy. Many Gram-negative intestinal pathogens produce AraC-like proteins that control the expression of genes required for infection. In this study we investigated the prototypical AraC-like virulence regulator, RegA, from the mouse attaching and effacing pathogen, Citrobacter rodentium, as a potential drug target. By screening a small molecule chemical library and chemical optimization, we identified two compounds that specifically inhibited the ability of RegA to activate its target promoters and thus reduced expression of a number of proteins required for virulence. Biophysical, biochemical, genetic, and computational analyses indicated that the more potent of these two compounds, which we named regacin, disrupts the DNA binding capacity of RegA by interacting with amino acid residues within a conserved region of the DNA binding domain. Oral administration of regacin to mice, commencing 15 min before or 12 h after oral inoculation with C. rodentium, caused highly significant attenuation of intestinal colonization by the mouse pathogen comparable to that of an isogenic regA-deletion mutant. These findings demonstrate that chemical inhibition of the DNA binding domains of transcriptional regulators is a viable strategy for the development of antimicrobial agents that target bacterial pathogens.
Introduction
Bacterial infections are a major cause of morbidity and mortality worldwide. In developing countries, in particular, bacterial infections of the gastrointestinal tract pose a significant health problem (1). Current treatment of these infections is focused on oral rehydration, which reduces mortality but has no effect on morbidity or the progression to chronic diarrhea and its associated complications (1). Although some gastrointestinal infections respond to treatment with antibiotics, such treatment is discouraged because it leads to the development and spread of antimicrobial resistance.
Conventional antimicrobial agents that kill bacteria or impair their growth exert strong selective pressure for the development and maintenance of resistance. By contrast, a drug that inhibits microbial virulence without inhibiting growth, such as one that influences virulence gene regulation, would generate minimal selective pressure for the development of resistance. Thus, virulence gene regulators offer a potential target for novel antimicrobials (2, 3).
The AraC family of regulators control transcription of genes involved in carbon metabolism, stress responses, and pathogenesis (4). Those AraC-like regulators, which control the expression of virulence determinants of Gram-negative and Gram-positive bacteria that infect humans, animals, or plants (4, 5), are frequently encoded on pathogencity islands, prophage elements, and/or plasmids that are acquired through horizontal gene transfer (6–8). In enteric bacterial pathogens, AraC-like proteins, such as PerA and RegR from enteropathogenic Escherichia coli (EPEC)4 (9, 10), Rns from enterotoxigenic E. coli (ETEC) (11), AggR from enteroaggregative E. coli (EAEC) (12), and ToxT from Vibrio cholerae (13), are required for the expression of factors essential for intestinal colonization and virulence.
To study the regulation of virulence gene expression by AraC-like proteins, we have used the prototypical AraC-like regulator, RegA, from the EPEC-like mouse pathogen, Citrobacter rodentium (14). Like its human-specific counterparts, enterohemorrhagic E. coli (EHEC) and EPEC, C. rodentium carries a pathogenicity island known as the locus for enterocyte effacement (LEE), which is required for the intimate attachment of bacteria to intestinal epithelial cells and the formation of characteristic attaching and effacing lesions (15).
RegA is a global regulator that controls the expression of >60 genes and facilitates colonization of the intestine by C. rodentium (16, 17). In the presence of bicarbonate, which acts as a small intestinal-specific environmental trigger, RegA coordinately activates transcription of virulence determinants and represses the expression of various housekeeping genes (16). Among the most strongly activated genes by RegA are the kfc and aap operons, which encode a putative K99-like pilus and Aap, an anti-aggregation protein, respectively (16). RegA also indirectly enhances the expression of the LEE by stimulating the GrlA-Ler regulatory cascade and thus activating the transcription of the LEE1 to LEE5 operons (18). The critical importance of RegA in C. rodentium virulence and the relevance of the C. rodentium/mouse infection model to EPEC/EHEC infections make RegA an ideal target for investigating the potential usefulness of antimicrobial agents that interfere with AraC-like virulence regulators in pathogenic bacteria. In this study we identified two small molecule compounds that specifically inhibited the activation function of RegA in vitro. One of these compounds, which we named regacin, also inhibited colonization of mouse intestine by C. rodentium in vivo.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, Primers, and Proteins
The strains and plasmids used in this study are listed in Table 1 (19–28). The primers we used are listed in Table 2. The MBP::RegA and TyrR proteins were purified as described previously (27, 29).
TABLE 1.
Strains and plasmids used in this study
NalR, nalidixic acid resistance; SmR, streptomycin resistance; CmR, chloramphenicol resistance; KnR, kanamycin resistance; TcR, tetracycline resistance; TpR, trimethoprim resistance.
| Strains/plasmids | Relevant characteristics | Source/reference |
|---|---|---|
| Strains | ||
| C. rodentium ICC169 | Spontaneous NalR derivative of wild-type C. rodentium ICC168, NalR | (19) |
| C. rodentium EMH1 | C. rodentium ICC169 regA::aphA-2, NalR, KnR | (17) |
| E. coli E22 | EPEC serotype O103:H2 | (20) |
| E. coli H10407 | ETEC serotype O78:H11, LT+, ST+ | (21) |
| E. coli 042 | EAEC serotype O44:H18, SmR, TetR, CmR | (22) |
| V. cholerae 395 | Serogroup O1, biotype Classical | (23) |
| E. coli MC4100 | F−araD139Δ (argF-lac)U169 rpsL150 relA1 flbB5301 deoC1 ptsF25 rbsR thiA | (24) |
| E. coli JP8042 | ΔlacU169 recA56 tyrR366 | (25) |
| E. coli XL1-Red | endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac mutD5 mutS mutT::Tn10, TcR | Stratagene |
| Plasmids | ||
| pCR2.1-TOPO | High copy number vector, Apr, Kanr | Invitrogen |
| pACYC184 | Medium copy number vector, p15A ori, CmR, TcR | (26) |
| pACYC184-regA (pEH6) | regA in pACYC184, CmR | (17) |
| pACYC177-tyrR (pMU1065) | tyrR in pACYC177, KnR | (25) |
| kfc-lacZ (pMU2385-kfcC) | kfc promoter region in pMU2385, TpR | (27) |
| mtr-lacZ (pMU3190) | mtr promoter region in pMU2385, TpR | (28) |
| cooB-lacZ | cooB promoter region in pMU2385, TpR | This study |
| sefA-lacZ | sefA promoter region in pMU2385, TpR | This study |
| aap-lacZ | aap promoter region in pMU2385, TpR | This study |
| tcpA-lacZ | tcpA promoter region in pMU2385, TpR | This study |
| pACYC184-rns | rns in pACYC184, CmR | This study |
| pACYC184-regR | regR in pACYC184, CmR | This study |
| pACYC184-aggR | aggR in pACYC184, CmR | This study |
| pACYC184-toxT | toxT in pACYC184, CmR | This study |
TABLE 2.
Primers used in this study
| Primer | Used for | Sequence 5′–3′ |
|---|---|---|
| JY-1 | EMSA (kfc) | GCGAGACAGAATATATTGTGATGG |
| JY-2 | EMSA (kfc) | CAAACAAAGACCAGAACTAGTC |
| JY-3 | N16A mutation | GTGCTTTAAGTAATCTAAGTATACCGGCTAAAGCTAGTCTTGCACATAACAATATGTTAGTAC |
| JY-4 | N16A mutation | GTACTAACATATTGTTATGTGCAAGACTAGCTTTAGCCGGTATACTTAGATTACTTAAAGCAC |
| JY-5 | W188A mutation | GAGGCCGACCTTTCCAGAAGTGCCCGGCTTGTAGACTTGGCG |
| JY-6 | W188A mutation | CGCCAAGTCTACAAGCCGGGCACTTCTGGAAAGGTCGGCCTC |
| JY-7 | R223A mutation | CAATTTCAATGCATTAGTTCTTGATATAGCGATGTATCATGCCGCAAAGT TAATC |
| JY-8 | R223A mutation | GATTAACTTTGCGGCATGATACATCGCTATATCAAGAACTAATGCATTGAAATTG |
| JY-9 | EMSA (mtr) | GTGATGATTCTACCGGTCGTCGTC |
| JY-10 | EMSA (mtr) | GCATTGCACTGTACCAGTACACGAG |
| JY-11 | cooB promoter | AGGATCCACCAAGAACTCGGGGCAG |
| JY-12 | cooB promoter | GAAGCTTCCCATCCTTGGGGAGCACTC |
| JY-13 | rns gene | AGGATCCGTCAAAGATATGTCATGCATAACC |
| JY-14 | rns gene | AGTCGACAGTTTGCATCGCAATAAATCTCTACTAG |
| JY-15 | tcpA promoter | AGGATCCACGTAGGTGGGTATAGTG |
| JY-16 | tcpA promoter | GAAGCTTGCCTAGAACGATGATCACTTCGA |
| JY-17 | toxT gene | AGGATCCGGAGATGGAAGTGGTGTGAAACT |
| JY-18 | toxT gene | AGTCGACTACCCAAAATCAGTGATACAATCG |
| JY-19 | aap promoter | AGGATCCATCAGTCAATGAGCTGTATTTAAGC |
| JY-20 | aap promoter | GAAGCTTGCGTTCCAACCGCTACCACCC |
| JY-21 | aggR gene | AGGATCCGCGTAAAAATCATATCCCACATGAC |
| JY-22 | aggR gene | AGTCGACGCAGCATCACCAACTTCAGCC |
| JY-23 | sefA promoter | GCCGAAGCTTTAGGCACTGACAATACGGCTGCAAACTT |
| JY-24 | sefA promoter | GCCGGAATTCTGGAGCCTGAACAGTAGCAACATCACCTAT |
| JY-25 | regR gene | CGGGATCCCGAGTTTACAGAGTGGATATCC |
| JY-26 | regR gene | CGGTCGACCAGTAAGAACTGCTTCTGCC |
Screening Assay for Small Molecule Inhibitors of RegA
An overnight culture of test strain MC4100(kfc-lacZ, pACYC184-regA) was diluted 1 in 100 in Luria Bertani (LB) broth (30) containing 45 mm NaHCO3 and then was dispensed in 100-μl volumes into 96-well microtiter trays (Fig. 1). Compounds (5 μl, 2 mm) from the Chembridge Microformats library (ChemBridge Corp.) were added to the wells in columns 2–11 of these test plates. The wells in columns 1 (used to determine the mean luminescence signal from untreated cells) and 12 (background) received 5 μl of 100% DMSO alone. Control plates were filled with the same volume of compounds or DMSO alone and the control strain, E. coli JP8042(mtr-lacZ, pACYC177-tyrR), diluted (1:100) in LB broth containing 1 mm tyrosine. The trays were incubated at 37 °C for 18 h, after which 8 μl of lysozyme (Sigma) solution (6 mg/ml) was added to the wells, and the mixtures were incubated at room temperature for another 20 min. The β-gal released from the bacterial cells was converted to a luminescence signal by adding 25 μl of Beta-Glo (Promega) solution into the wells of columns 1–11. The level of luminescence from each well was measured 1.43, 2.86, and 4.3 min later using the FLUOStar Omega plate reader (BMG Labtech).
FIGURE 1.

Screening system used to identify a RegA inhibitor. Test and control plates contained the same set of compounds from the Chembridge Microformats library. These were incubated with E. coli strains MC4100(kfc-lacZ, pACYC184-regA) (test plate) and JP8042(mtr-lacZ, pACYC177-tyrR) (control plate). The compound in C11 shows no inhibition of enzyme activity in either plate and represents a true negative. The compound in F8 represents a false positive, as it inhibits both RegA-mediated transcriptional activation of the kfc promoter and TyrR-mediated transcriptional activation of the mtr promoter. The compound in E11 represents a specific inhibitor of RegA (true positive) as it inhibits only RegA-mediated activation of the kfc-lacZ reporter.
Cytotoxicity Assay
The Cytotox 96 Non-radioactive Cytotoxicity Assay (Promega) was used to measure lactate dehydrogenase release, an indicator of cell death, in the culture supernatants. The assay was performed as per the manufacturer's protocol. Briefly, HeLa cells in DMEM supplemented with FBS (10%, v/v) were seeded at 0.8 × 104 cells per well. After overnight incubation, regacin suspended in DMSO was added to a final concentration of 100 μm and incubated at 37 °C with 5% CO2 for 6 h. Controls included cells incubated in the absence or presence of DMSO (1%, v/v). A substrate mix was added to each well, and plates were incubated for 30 min in the dark at room temperature before the enzymatic reaction was stopped. Absorbance (at 450 nm) from each well was measured using the FLUOStar Omega plate reader. Percentage of cell death (cytotoxicity) was determined by dividing the lactate dehydrogenase release from test samples with the maximum lactate dehydrogenase release from cells treated with Triton X-100.
β-Galactosidase Assay
Bacterial cells were grown in the presence or absence of chemical inhibitors to mid-log phase (A600 ∼ 0.6) after which β-gal activity was determined as described by Miller (31). The data shown are the results of three independent assays.
Analytical Ultracentrifugation
Sedimentation velocity experiments were conducted in a Beckman model XL-I analytical ultracentrifuge at 20 °C. 380 μl of RegA::MBP (0.15 mg/ml) and 400 μl of reference solution (20 mm Tris-Cl, 150 mm NaCl ± 30 μm regacin (pH 7.5)) were loaded into a conventional double-sector quartz cell and mounted in a Beckman 4-hole An-60 Ti rotor. Samples were centrifuged at 40,000 rpm, and the data were collected continuously at a single wavelength (280 nm) using a step size of 0.003 cm without averaging. Solvent density (1.005 g/ml at 20 °C) and viscosity (1.021 cP) as well as estimates of the partial specific volume (0.744 ml/g at 20 °C) and hydration estimate (0.403 g/g) were computed using the program SEDNTERP.31 (32). Continuous size-distribution analysis was performed using the program SEDFIT32 (33).
Electrophoretic Mobility Shift Assay (EMSA)
The 32P-labeled PCR fragments used in the EMSA were generated as follows. To generate the kfc fragment, the primer JY-1 (Table 2) was labeled with 32P at its 5′ end by using [γ-32P]ATP and T4 polynucleotide kinase. The kfc promoter fragment was generated by PCR using primer pairs (32P-JY-1 and JY-2) with plasmid kfc-lacZ as template. The mtr fragment was generated using primer pairs (32P-JY-9 and JY-10) with the chromosomal DNA of E. coli K-12 strain MC4100 as template. The end-labeled kfc and mtr fragments were incubated with 200 nm purified MBP::RegA or TyrR, respectively, in the presence of varying amounts of regacin at 30 °C for 20 min in binding buffer (10 mm Tris-HCl (pH 7.4), 50 mm KCl, 1 mm DTT, 100 μg/ml BSA, 5 ng/μl poly(dI-dC), and 45 mm NaHCO3). Glycerol was added to a final concentration of 6.5% (v/v). Samples were separated by electrophoresis on 5% native polyacrylamide gels (37.5:1) at 4 °C for ∼12 h at 10 V/cm and analyzed by using by FLA3000 phosphorimaging (Fujifilm).
Random and Site-directed Mutagenesis
Random mutations in the regA gene were generated by transforming plasmid pACYC184-regA into competent cells of XL1-Red (Stratagene) according to the manufacturer's instructions. Individual chloramphenicol-resistant colonies were cultured overnight in LB broth, subcultured, and reincubated overnight. These cultures were used to prepare a regA-mutant plasmid library. After screening, regA-mutant plasmids were extracted from isolates that displayed the desired phenotype, retransformed into E. coli strain MC4100(kfc-lacZ), and plated on X-gal indicator plates in the presence of 100 μm regacin to confirm the phenotype.
Site-directed mutagenesis of the regA gene was performed by using the QuikChange mutagenesis kit (Stratagene). Plasmid pACYC184-regA was used as the template. Experiments were done according to the manufacturer's instructions using the primers listed in Table 2. All mutations were confirmed by sequencing analysis.
Construction of Various pACYC184 Derivatives
DNA fragments containing the rns, regR, aggR, and toxT genes were amplified from genomic DNA of H10407 (ETEC), E22 (EPEC), 042 (EAEC), and 395 (V. cholerae), respectively, by PCR using primers listed in Table 2. These fragments were then each cloned into pCR2.1-TOPO and sequenced. The fragments were each excised by BamHI/SalI digestion and ligated into the same sites of pACYC184 to generate the plasmids pACYC184-rns, pACYC184-regR, pACYC184-aggR, and pACYC184-toxT.
Construction of Promoter-lacZ Fusion Plasmids
The transcriptional-fusion plasmids were constructed by PCR amplification of the regulatory region of cooB, sefA, aap, and tcpA from genomic DNA of H10407 (ETEC), E22 (EPEC), 042 (EAEC), and 395 (V. cholerae), respectively, using primers listed in Table 2. tk;2The resulting PCR fragments were each cloned into pCR2.1-TOPO, and these fragments were sequenced. The fragments were then excised from the pCR2.1-TOPO derivatives by BamHI/HindIII digestion and cloned into the same sites of the single-copy transcriptional-fusion vector pMU2385 to create the cooB-lacZ, sefA-lacZ, aap-lacZ, and tcpA-lacZ transcriptional fusions.
Computational Modeling and Docking
A Hidden Markov Model was created from the amino acid sequence of the RegA protein of C. rodentium and was then compared with all of the Hidden Markov Models in the Protein Data Bank (accessed October 16, 2012). HHpred, a homology detection program (34), detected the ToxT crystal structure (PDB code 3GBG (35)) as the closest homolog. A PIR alignment was generated from the 3GBG.pdb HHpred model and submitted to MODELLER v 9.10 (36). The resulting RegA model was then minimized sequentially (hydrogens, then side chains, and then main chains) under the Merck Molecular Force Field for 10,000 iterations and assessed using Procheck (37), which found that >99% of the residues resided in the allowed regions of the Ramachandran plot. Chi1-Chi2 plots, main-chain parameters, side-chain parameters, G-factors, bond angles, and bond lengths were also all within the allowed parameters.
The inhibitor compound was sketched into SYBYL-X 1.2 (Tripos) and minimized under the Tripos forcefield for 1000 iterations. To find potential docking receptors, SiteID was performed in SYBYL-X 1.2, which highlighted nine potential binding pockets under default conditions. The Surflex-Dock program (38) within SYBYL-X 1.2 was used for docking. A protomol (docking receptor) was generated around each of these pockets with the threshold reduced to 0.3 and the bloat increased to 3 Å for each protomol. Regacin was docked into each of these pockets with the protein hydrogen and heavy atom movement allowed, along with compound ring flexibility. Five additional starting conformers per molecule were flagged, and the density of search was increased to 9.00. All other parameters were kept at default values. The top 10 poses were optimized and analyzed visually.
Proteomic Analysis of the Secretome and Whole-cell Fractions of C. rodentium
C. rodentium strains EMH1(pACYC184-regA) and EMH1(pACYC184) were grown in DMEM in the absence or presence of 100 μm regacin. After reaching A600 0.8, the bacterial cells were separated from supernatants by centrifugation. The clarified supernatants were filtered through a 0.22-μm pore-size filter (Sartorius), and the proteins in the supernatants were precipitated with 10% (v/v) trichloroacetic acid and washed with 25% (v/v) acetone. Cell pellets were sonicated to obtain whole-cell lysates. Proteins in the supernatants and whole-cell lysates were separated by SDS-PAGE using 4–12% Criterion XT gels (Bio-Rad) and stained with Coomassie Brilliant Blue G250. The bands of interest were excised, trypsin-digested, and analyzed by tandem mass spectrometry at the Proteomics Laboratory, Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia.
Formulation of Regacin
An oral, soluble liquid formulation of regacin was prepared by dissolving 15 mg of compound into 100 μl of DMSO. This was then introduced dropwise, with mixing to 900 μl of a solution comprising a 1:1 ratio of polysorbate 80 and propylene glycol. One milliliter of this solution was then slowly diluted further with 3 ml of 5% (v/v) glycerol in distilled water. A final concentration of 2 mg/ml regacin in ∼1.3% DMSO, ∼4% (v/v) glycerol, 12.5% (v/v) polysorbate 80, and 12.5% (v/v) propylene glycol was achieved by the subsequent dropwise addition of 3.5 ml of 5% (v/v) glycerol in 12.5% (v/v) polysorbate 80 and 12.5% (v/v) propylene glycol.
Infection of Mice
Four-week-old specific-pathogen-free C57BL/6 mice were inoculated in groups of five with ∼2 × 109 cfu of either the C. rodentium wild-type strain ICC169 or its isogenic regA mutant (EMH1) by oral gavage as described previously (17). Mice were weighed daily, and regacin (50 mg/kg) or the solvent formulation (control) was orally administered to the mice 15 min before or 12 h after inoculation and twice daily thereafter for the duration of the experiment. Feces were collected each day for enumeration of C. rodentium colonies by plating on selective media as described previously (17).
Statistical Methods
Quantitative data were compared by using Student's t test, two-tailed (GraphPad Prism 5). p values of <0.05 were taken to indicate statistical significance.
RESULTS
Design of a Phenotypic Screen and Identification of a RegA Inhibitor
To screen for RegA inhibitors we developed an approach that involved the use of promoter reporters. This utilized a single-copy plasmid in which the C. rodentium kfc promoter region was transcriptionally fused to the lacZ structural gene, which encodes β-gal (27). In the E. coli K-12 strain, MC4100, the kfc-lacZ plasmid, expresses low levels of β-gal activity. Introducing a RegA-expressing plasmid (pACYC184-regA) increases β-gal activity ∼100-fold (27). The strong RegA-mediated activation of transcription from the kfc promoter offered a convenient screen for chemical inhibitors of RegA. In our screening assay, false positive compounds that affected E. coli growth or the activity of β-gal were eliminated by using a control E. coli K-12 strain, JP8042, which contained a pair of plasmids, pACYC177-tyrR and mtr-lacZ. The housekeeping gene mtr encodes a tryptophan transporter, and transcription from the mtr promoter is strongly activated by the TyrR regulatory protein in the presence of tyrosine (39). A compound was scored positive if by 1.43 or 2.86 min it had reduced the luminescence signal to <20% of the mean luminescence value of the untreated test strain but had not reduced the signal of the control strain by >20% of the mean of the untreated control strain (Fig. 1).
After screening 11,840 compounds from the ChemBridge Microformats library, we identified 13 positive hits. After a second round of β-gal assay, we focused on one compound, 5-[(2,5-dichlorobenzyl)thio]-1,3,4-thiadiazol-2-amine (DCTTA), as it was the only compound that completely inhibited the β-gal activity of the test strain at 100 μm (Fig. 2, A and C). We then queried ChemBridge's 900,000 compound collection for analogs of DCTTA by using the website Hit2Lead.com and selected six two- and three-dimensional analogs. Analysis of these chemicals in our screening assay revealed one compound, 5-[(4-chlorobenzyl)thio]-1,3,4-thiadiazol-2-amine (that we named regacin) that was a stronger inhibitor of RegA than DCTTA (Fig. 2, B and C). The concentrations required for an IC50 of RegA were ∼6.2 and 1.7 μm for DCTTA and regacin, respectively. To confirm that regacin did not inhibit bacterial growth, E. coli K-12 and C. rodentium were grown in the absence or presence of 100 μm regacin, and no differences in growth or viability were observed (Fig. 3A). We also evaluated the toxicity of regacin toward mammalian cells in vitro. No cytotoxicity was observed when HeLa cells were incubated with 100 μm regacin (Fig. 3B). Regacin was then subjected to further analyses.
FIGURE 2.
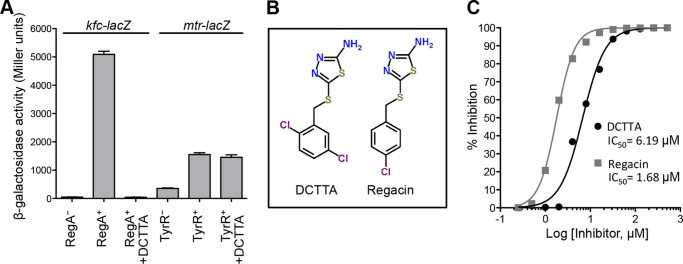
Quantitative analysis of the inhibitory effects of DCTTA and regacin on RegA activity. A, β-galactosidase assays. In the presence of 100 μm DCTTA, the kfc promoter activity in a RegA+ background (MC4100(kfc-lacZ, pACYC184-regA)) was reduced to the level seen in the RegA− background (MC4100(kfc-lacZ, pACYC184)). In contrast, in the presence of 100 μm DCTTA, the mtr promoter activity in the TyrR+ background (JP8042(mtr-lacZ, pACYC177-tyrR)) was unchanged. Data are the mean ± S.D. of three independent assays. B, chemical structures of the RegA inhibitors, DCTTA and regacin. C, dose-response curves. Data were obtained by measuring the β-gal activities of the E. coli strain MC4100(kfc-lacZ, pACYC184-regA) grown in the presence of varying concentrations of DCTTA and regacin. The results of three independent assays were used to calculate the IC50 (sigmoidal dose-response equation, GraphPad Prism 5).
FIGURE 3.
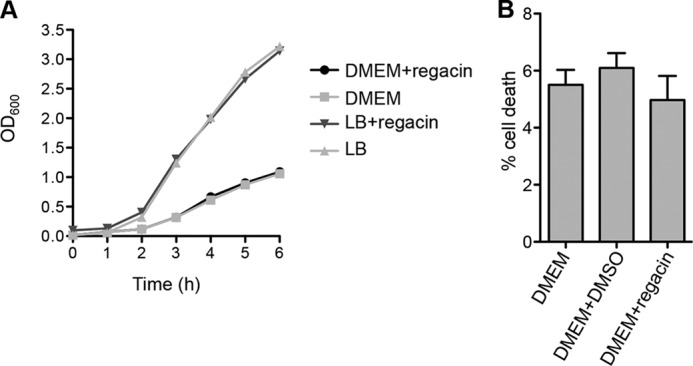
Regacin does not inhibit the growth of C. rodentium and is not cytotoxic to mammalian cells. A, the wild-type C. rodentium strain, ICC169, was grown at 37 °C either in LB broth with shaking (200 rpm) or in DMEM statically (5% CO2) in the absence or presence of 100 μm regacin. Bacterial growth (A600) was followed over a period of 6 h. B, after 6 h of incubation, HeLa cell supernatants were submitted to lactate dehydrogenase lactate dehydrogenase release quantification. The percentage of lactate dehydrogenase release for regacin (100 μm) or DMSO (1%) was compared with the lactate dehydrogenase release of HeLa cells in DMEM alone. Data are the mean ± S.D. of three independent assays, p > 0.2.
Inhibition of the Synthesis and Secretion of Virulence Effectors by Regacin in Vitro
RegA activates the transcription of several genes encoding known or putative protein secretion systems and their cognate secreted proteins (16, 18). To determine if regacin blocks the synthesis and secretion of these proteins, we carried out proteomic analyses of C. rodentium. To facilitate visualization of the proteins of interest, we used the regA-mutant C. rodentium strain, EMH1, containing plasmid pACYC184 (control) and the regA-complemented strain EMH1(pACYC184-regA) in this assay. For the regA-complemented strain (RegA+), the levels of several secreted proteins from the regacin-treated sample were considerably lower than those from the control sample (Fig. 4, lanes 1 and 2). Regacin also inhibited the synthesis of a protein in the whole-cell lysate (Fig. 4, lanes 4 and 5). No differences were seen in the protein profiles of the regacin-treated sample and the RegA− control strain (Fig. 4, lanes 1 and 3 and lanes 4 and 6).
FIGURE 4.
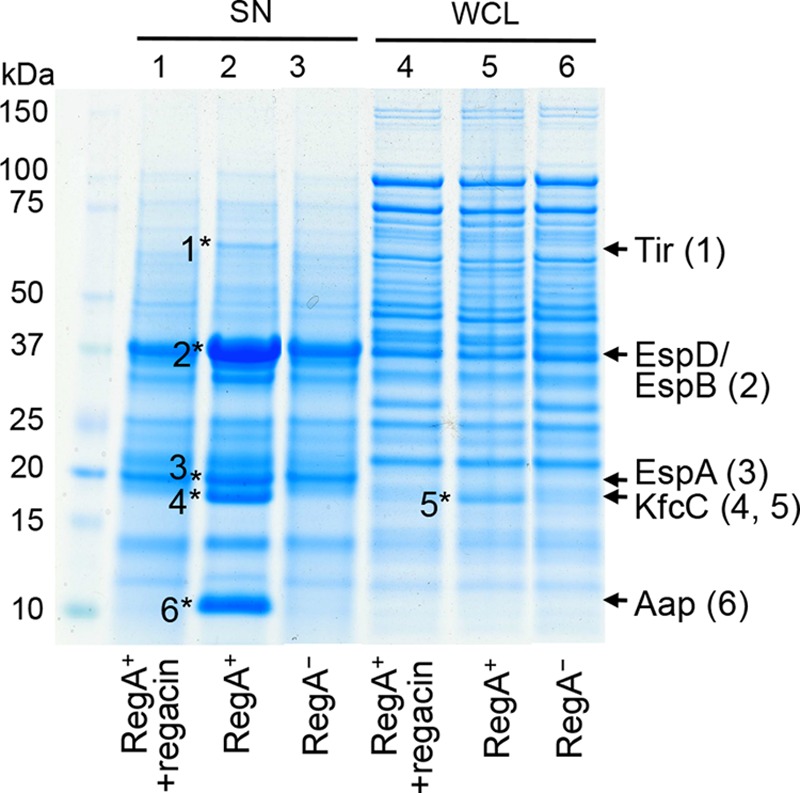
Effect of regacin on the production of virulence proteins in C. rodentium. A RegA+ (EMH1(pACYC184-regA)) strain of C. rodentium was grown in DMEM in the absence or presence of 100 μm regacin and the RegA− strain of C. rodentium (EMH1(pACYC184)) (control) was grown in DMEM alone. Proteins in the supernatants (SN) and whole-cell lysates (WCL) were separated by SDS-PAGE and stained with Coomassie Brilliant Blue G250. Six protein bands which were relatively less abundant in the presence of regacin (indicated by asterisks, in lanes 2 and 5) were excised and analyzed by tandem mass spectrometry. The identities of these proteins are shown at the right of the gel with the numbers in parentheses corresponding to those of the protein band.
To determine the identities of the regacin-affected proteins, the relevant bands were excised and analyzed by tandem mass spectrometry. All of the regacin-affected proteins (EspA, EspB, EspD, Tir, Aap, and KfcC) we identified are known secreted effectors or surface proteins encoded by genes located on the LEE or other prophage elements whose expression is directly or indirectly activated by RegA (Fig. 4). These results are in agreement with those from the β-gal assays which demonstrated that regacin at 100 μm inhibited the transcription from the promoters of aap, LEE4 (encodes EspA, EspB, EspD) and LEE5 (encodes Tir) (data not shown).
Regacin Does Not Affect RegA Dimerization
As with other AraC-like regulators, RegA functions as a dimer (40). To assess whether regacin interferes with RegA dimerization, we carried out a sedimentation velocity study. Because RegA, like many other AraC-like proteins, is highly insoluble, we examined the quaternary structure of RegA by using a fully functional fusion protein in which the N terminus of RegA was linked to the C terminus of E. coli maltose-binding protein (MBP::RegA) (4, 40).
Continuous size (c(s)) distribution analysis of MBP::RegA in solution in the absence and presence of 30 μm regacin yielded excellent fits under the conditions tested. The quality of the distribution of best-fits was demonstrated by the random distribution of residuals obtained for each analysis (Fig. 5A). The data showed that the MBP::RegA protein exists as a dimer in solution in the absence or presence of 30 μm regacin with a sedimentation coefficient (s20,w) of 6.2 S (Fig. 5B) and indicate that regacin does not affect the dimeric state of functional RegA.
FIGURE 5.
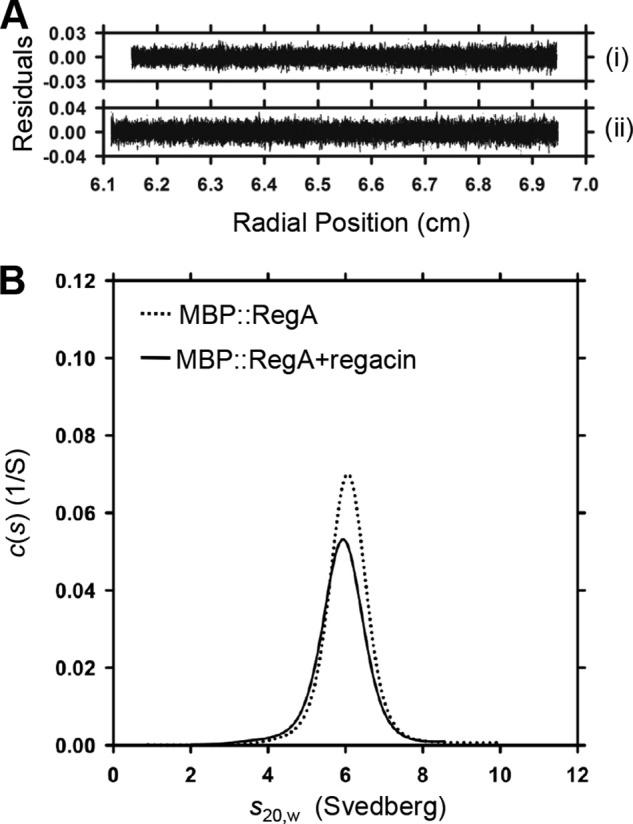
Determination of the effect of regacin on RegA dimerization by analytical ultracentrifugation. A, residuals were plotted as a function of radial position (cm) from the axis of rotation MBP::RegA alone (i) and MBP::RegA plus 30 μm regacin (ii). B, continuous size (c(s)) distribution plotted as a function of s20,w (Svedberg) at a resolution of 300, with Smin = 0.1 and Smax = 12 and a confidence level (F-ratio) of 0.95. Together, these data indicate that regacin does not interfere with RegA dimer formation.
Regacin Inhibits the DNA Binding Capacity of RegA
To determine if regacin inhibited the DNA binding activity of RegA, we carried out an EMSA. The fusion protein MBP::RegA and a 32P-labeled kfc-promoter DNA fragment that contains a known RegA binding site were used for this assay (27). MBP::RegA (200 nm) was incubated with the labeled DNA fragment in the presence of RegA cofactor, sodium bicarbonate (45 mm), and with 0, 5, 10, or 30 μm regacin for 20 min at 30 °C, after which the samples were analyzed on native polyacrylamide gels.
In the absence of regacin, a distinct band, which represented a MBP::RegA-DNA complex, was formed (Fig. 6A). This retarded band faded in the presence of 5 and 10 μm regacin and disappeared completely in the presence of 30 μm regacin, indicating that regacin interferes with the binding of RegA to its DNA target.
FIGURE 6.
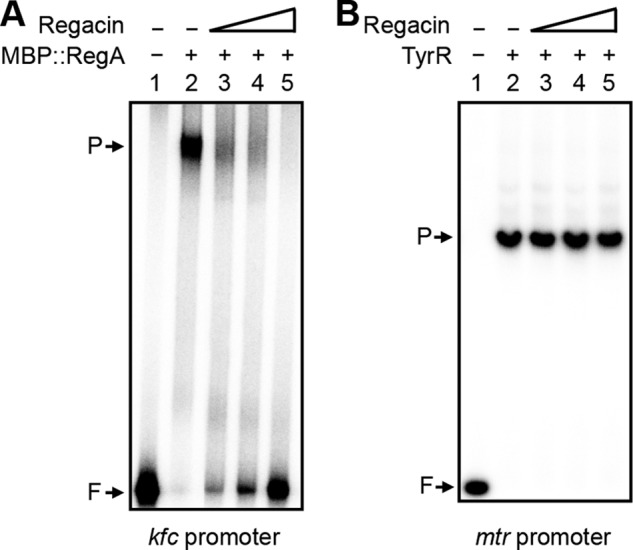
Determination of the effect of regacin on the DNA binding activity of RegA and TyrR by EMSA. A, a 32P-labeled 233-bp DNA fragment containing the regulatory region of kfc was mixed with 200 nm MBP::RegA and 45 mm NaHCO3 in the absence or presence of varying concentrations of regacin. B, a 32P-labeled 157-bp DNA fragment containing the regulatory region of mtr was mixed with 200 nm TyrR, 50 μm ATP, and 100 μm tyrosine in the absence or presence of varying concentrations of regacin. After incubation at 30 °C for 20 min, the samples were separated by electrophoresis on a native polyacrylamide gel. Lane 1, DNA fragment alone (control); lanes 2–5, DNA fragment mixed with 200 nm MBP::RegA (panel A) or TyrR (panel B) in the presence of 0, 5, 10, and 30 μm regacin, respectively. Free DNA (F) and the protein-DNA complex (P) are indicated.
In a control experiment, 200 nm TyrR protein was incubated with the 32P-labeled mtr-promoter DNA fragment in the presence of its cofactors, ATP (50 μm) and tyrosine (100 μm), and with 0, 5, 10, or 30 μm regacin for 20 min at 30 °C. Electrophoretic analysis showed that under these conditions TyrR caused a complete shift of its target promoter and that up to 30 μm regacin had no effect on the DNA binding activity of TyrR (Fig. 6B).
Determination of the Site of Action of Regacin
In the absence of a crystal or NMR structure of the RegA protein, we probed the point of interaction of regacin by using a genetic approach to identify inhibitor-resistant mutants of regA. A random regA-mutant plasmid library was generated that was transformed into the recipient E. coli strain MC4100(kfc-lacZ). Cells were plated on X-gal indicator plates in the presence of saturating levels of regacin (100 μm). After screening >50,000 colonies, we identified one that was darker than the pale blue colonies of the regacin-inhibited RegA strains, suggesting that this isolate expressed a regacin-resistant form of RegA. β-Gal analysis showed that in the absence of regacin the mutant RegA protein activated the kfc promoter to the same degree as that of the wild-type protein. In contrast, unlike the wild-type RegA, the mutant protein exhibited strong but incomplete resistance to 100 μm regacin (Fig. 7A).
FIGURE 7.

Characterization of the RegA I222T mutant. A, β-gal analysis. E. coli strains MC4100(kfc-lacZ) carrying plasmids pACYC184-regA (RegA wild type) or pACYC184-regA-mutant (RegA I222T) were grown in LB broth with NaHCO3 (45 mm) in the absence or presence of 100 μm regacin to mid-log phase (A600 ∼ 0.6), after which their β-gal activities were measured. The data show that regacin completely inhibited the function of the wild-type RegA protein but only partially reduced that of the mutant RegA protein (I222T). Data are the mean ± S.D. of three independent assays. B, amino acid sequence of the RegA DNA binding domain (16). The positions of residues (188–262) are shown, and the two helix-turn-helix motifs are underlined. Three residues, Trp-188, Ile-222, and Arg-223, which were mutated, are shown in boldface type and marked with triangles.
Sequence analysis revealed that this RegA mutant carried a single thymine to cytosine change at nucleotide position 615 of the regA coding region, resulting in an isoleucine-to-threonine missense mutation at position 222 of the RegA protein (I222T). This mutation is located in the region between the two helix-turn-helix DNA binding motifs in the C-terminal region of RegA (Fig. 7B). Given this location, the most likely explanation for the phenotype of the mutation is that the isoleucine residue lies within the binding pocket for regacin and that the mutation altered its local structure in a way that partially prevented regacin from interacting with RegA.
To investigate this hypothesis we generated a putative three-dimensional model of RegA based on the crystal structure of the ToxT protein of V. cholerae (35). Potential ligand binding pockets within RegA were identified by using SiteID in SYBYL-X 1.2 (Tripos). Of the nine possible pockets identified, only three (designated red, green, and yellow, Fig. 8A) were predicted to interfere with DNA binding and be influenced by an I222T mutation. Docking regacin into the three alternative sites using the Surflex-Dock program (38) indicated that the green pocket was an unlikely site for regacin binding due to a significantly lower value of the scoring function (and hence lower protein-ligand binding energy) when compared with the other two pockets (Fig. 8B). In the red pocket, regacin was predicted to form two hydrogen bonds with the side chain of Asn-16, but eliminating this side chain by making an alanine substitution did not reduce the inhibitory activity of regacin (Fig. 8C), suggesting that the red pocket is not the site of regacin binding.
FIGURE 8.

Analysis of the mode of binding of regacin to the DNA binding domain of RegA. A, docking model showing the interaction of regacin with the DNA binding domain of RegA. The homology model of RegA was generated from the crystal structure of ToxT of V. cholerae. The predicted RegA dimerization helix (cyan) and the three potential regacin binding pockets (red, green, and yellow) are shown. In the enlargement, regacin is docked into the yellow pocket, and the putative hydrogen bonding of regacin with Trp-188 (2.9, 2.0, and 3.0 Å) and Arg-223 (3.2 Å) is shown. B, CScore values for the top-docked solution of regacin. C, effect of various mutations on RegA activity in the presence or absence of regacin. E. coli strain MC4100(kfc-lacZ) carrying either pACYC184-regA (wild type), pACYC184-regA-mutant (N16A), pACYC184-regA-mutant (W188A), or pACYC184-regA-mutant (R223A) was grown in LB broth with 45 mm NaHCO3 in the absence or presence of 100 μm regacin to log phase (A600 ∼ 0.6), after which β-gal activity was measured. Data are the mean ± S.D. of three independent assays.
The yellow pocket is situated directly in what is assumed to be part of the DNA binding groove of RegA. Docking regacin into the yellow pocket gave one cluster of solutions that hydrogen-bonded the inhibitor to the backbone of Trp-188 and the side chain of Arg-223 (Fig. 8A). Consistent with this docking option is the prediction that the change of isoleucine to threonine at position 222 (I222T) would cause the side chain of the adjacent Arg-223 to rotate toward the threonine side chain, thus reducing the potential of this residue to hydrogen bond with regacin. Mutational analysis confirmed that the side chains of both Trp-188 and Arg-223 are critical for the function of RegA, as an alanine replacement at either position (W188A and R223A) completely destroyed the ability of the resultant RegA mutant to activate transcription of its target promoter (Fig. 8C).
Regacin Reduces Intestinal Colonization by C. rodentium in Mice
Previously we reported that a regA mutant of C. rodentium showed markedly reduced virulence for mice and that reduced virulence corresponded with reduced intestinal colonization (16, 17). Because regacin-treated wild-type C. rodentium phenotypically resembled a regA mutant in vitro (Fig. 4), we examined whether regacin can reduce the virulence of the wild-type to the level of a regA mutant in vivo. Regacin (50 mg/kg) was administered orally to C57BL/6 mice, which 15 min later were infected by gavage with wild-type C. rodentium strain ICC169. After infection, mice continued to receive regacin twice daily for 5 days. In control experiments, mice were infected with the wild-type C. rodentium strain without regacin or with a regA-mutant strain, EMH1, with or without regacin using the same protocol as described above.
The colonization efficiency of wild-type C. rodentium for mice given regacin was significantly lower than that in mice that were not treated (Student's t test, two-tailed, p < 0.005 days 3 and 4, p < 0.0005 day 5; Fig. 9) but was indistinguishable from that of the regA mutant (Fig. 9). These results indicate that regacin caused a highly significant attenuation of intestinal colonization by the mouse pathogen. The results also showed that regacin acts on RegA in vivo and not via a host factor, as there was no significant difference in colonization levels between the regA mutant with or without regacin throughout the course of infection (Student's t test, two-tailed, p > 0.2 days 1–5; Fig. 9).
FIGURE 9.
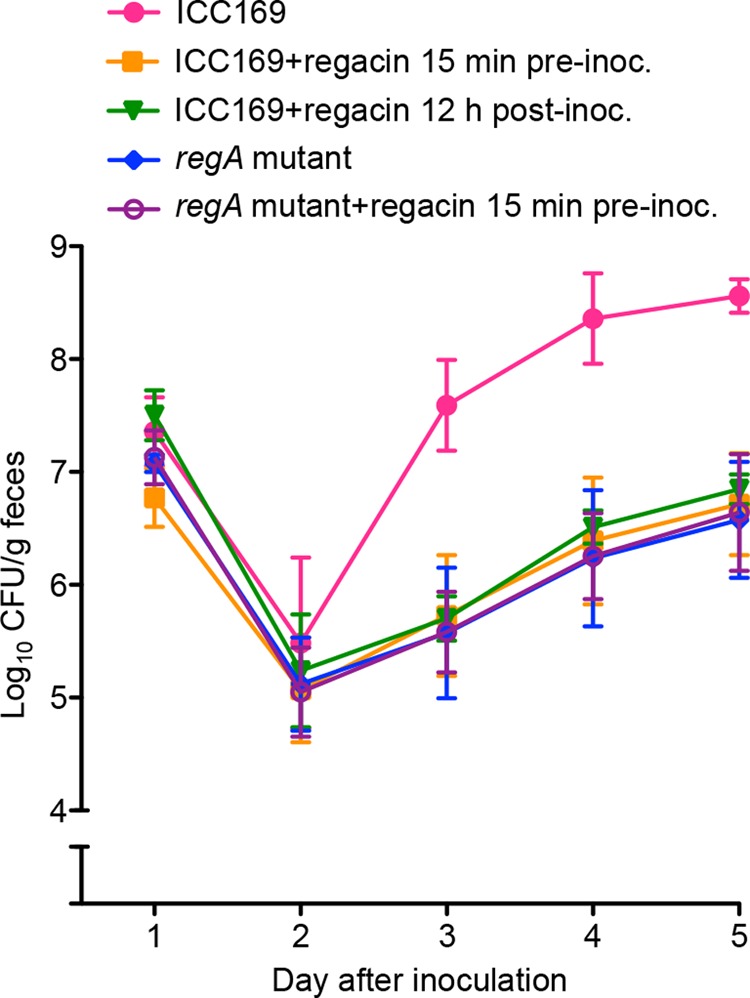
Suppression of C. rodentium virulence by regacin. C57BL/6 mice in groups of five were inoculated with 2 × 109 cfu of either C. rodentium wild-type strain, ICC169 (four groups), or its isogenic regA mutant (two groups). Regacin was administered perorally to one group of mice 15 min before inoculation with wild-type C. rodentium ICC169, to another group of mice 15 min before inoculation with the regA mutant, and 12 h after inoculation with C. rodentium ICC169 to a third group of mice. All mice in these three groups then received regacin twice daily for the duration of the experiment. Regacin was not given to the remaining two groups. Data are the mean ± S.D. cfu/g of feces from mice on days 1–5.
To determine if regacin can inhibit C. rodentium infection after colonization has occurred, we also administered regacin to mice 12 h after inoculation with C. rodentium and twice daily thereafter for the duration of the experiment. The data showed that administration of regacin 12 h after inoculation also resulted in a significant inhibition of bacterial colonization (Fig. 9; Student's t test, two-tailed, p < 0.05 days 3 and 4, p < 0.005 day 5).
Analysis of the Effect of Regacin on Other AraC-like Regulators
We next examined the effect of regacin on the ability of four RegA-related virulence regulatory proteins, Rns, RegR, AggR, and ToxT, to activate their cognate promoter targets, cooB, sefA, aggR, and tcpA. To this end, E. coli strain MC4100 derivatives carrying plasmid pairs (cooB-lacZ/pACYC184-rns, cooB-lacZ/pACYC184, sefA-lacZ/pACYC184-regR, sefA-lacZ/pACYC184, aap-lacZ/pACYC184-aggR, aap-lacZ/pACYC184, and tcpA-lacZ/pACYC184-toxT, tcpA-lacZ/pACYC184) were grown in the absence or presence of regacin (100 μm) to mid-log phase, and the β-gal activities of the samples were measured. The results showed that under the assay conditions regacin significantly inhibited the ability of Rns and RegR to activate the transcription of their target promoters but had little or no effect on the activity of AggR and ToxT (Fig. 10).
FIGURE 10.

β-Galactosidase analysis of Rns-, RegR-, AggR-, and ToxT-mediated activation in the presence or absence of regacin. The E. coli MC4100 strains used in this analysis all contain a pair of plasmids comprising a promoter-lacZ transcriptional fusion with either pACYC184 (Control) or pACYC184 carrying a relevant regulatory gene (Test). β-Gal assays were performed to evaluate the effect of regacin on transcriptional activation of the cooB promoter by the Rns protein of enterotoxigenic E. coli (A), the sefA promoter by the RegR protein of enteropathogenic E. coli (B), the aap promoter by the AggR protein of enteroaggrevative E. coli (C), and the tcpA promoter by the ToxT protein of V. cholerae (D). Data are the mean ± S.D. of three separate determinations.
DISCUSSION
The profligate use of conventional antimicrobial agents has led to the selection and spread of resistance-encoding genes, which are readily transmissible from commensal bacteria to pathogens as well as between pathogens (41). Because the intestinal microbiota is a major reservoir of resistance genes, treatment of gastrointestinal infections with antibiotics is discouraged. Furthermore, in the case of EHEC, antibiotic therapy is contraindicated due to the possibility of antibiotic-induced synthesis and release of Shiga toxins, which are major contributors to the life-threatening complications associated with EHEC infections (42). For these reasons, there is an urgent need to develop novel approaches to treat intestinal and other infections (43–45).
One such approach is to block bacterial virulence, including inhibiting adhesion to host cells, inactivating toxic proteins, disrupting protein secretion, and disabling virulence regulation (2). To date, however, only a few examples of small molecule inhibitors targeting virulence regulators have been reported (46–51), and in most cases the mechanism of inhibition is unknown. In this study we developed a simple but effective screening assay that allowed us to identify two inhibitors of RegA, DCTTA and regacin.
AraC-like virulence regulatory proteins share common functional and structural features. For example, the basic functional unit of these proteins is a dimer, and they all possess two helix-turn-helix DNA binding motifs usually at their C-terminal end (4). Thus, any chemical compound that disrupts dimerization or DNA binding of these regulators would be expected to inhibit their activity. Indeed, a small molecule compound, virstatin, inhibits the regulatory action of the AraC-like, ToxT protein from V. cholerae by preventing its dimerization (47, 52). By contrast, regacin is the first specific virulence inhibitor shown to interact with the double helix-turn-helix domain, resulting in a loss of binding affinity of RegA for its DNA targets. A major attraction of this target is that the two residues, Trp-188 and Arg-223, which are predicted to interact with regacin, are conserved in several RegA homologs including ToxT, AggR, RegR, and Rns. As with RegA, these residues are also critical for ToxT activity (53), indicating that they are either required to maintain the correct tertiary structure of their DNA binding domains or are directly involved in DNA binding. The fact that these residues are conserved makes them an ideal target in computer-aided drug design for antimicrobials of a variety of intestinal pathogens. Indeed, regacin can significantly inhibit the activity of Rns and RegR but has little or no effect on the activity of AggR and ToxT. The different effects of regacin on Rns, RegR, AggR, and ToxT are probably due to differences in amino acid residues within the putative ligand binding pockets. For these reasons, regacin will need to be modified to produce inhibitors that exert strong inhibition of other AraC-like regulatory proteins. Our data, which showed that the minor modification to the original hit (DCTTA) that led to regacin, which is almost four times more potent (Fig. 2), indicates how leads can be adapted in the future to develop compounds that block AraC-like virulence regulators of human pathogens.
Unlike conventional antibiotics, which disrupt the intestinal microbiota and encourage overgrowth of pathogens such as Clostridium difficile (54), small molecule inhibitors that specifically target virulence regulatory pathways should not cause a growth disadvantage of the bacterial pathogen or commensal bacteria, which would be left intact to resist further intrusions by pathogens (55). Indeed, despite being a strong inhibitor of RegA with an IC50 of 1.7 μm, regacin did not inhibit the growth of C. rodentium even at the concentration of 100 μm (Fig. 3A).
Proteomic analysis of C. rodentium grown with regacin showed that regacin is RegA-specific in that it inhibited the production of those proteins encoded by genes or operons regulated by RegA (Fig. 4). These proteins are homologs of known virulence factors of intestinal pathogens, including several putative colonization factors, such as Aap (dispersin from EAEC) (56), KfcC (a key component of the K99 pilus of ETEC) (57), and Tir (the translocated intimin receptor from EHEC and EPEC) (58). The down-regulation of these factors is expected to interfere with bacterial virulence. Indeed, regacin was highly effective when administered perorally to mice starting either 15 min before or 12 h after inoculation with C. rodentium and reduced colonization by wild-type C. rodentium to the level of a regA mutant (Fig. 9). This is the first time that a virulence inhibitor of this type has been shown to disrupt infection with a bacterial pathogen in its natural host infected by the natural route.
Our in vivo findings suggest that inhibition of RegA homologs, such as Rns, PerA, ToxT, and AggR, will similarly reduce the infectivity of their respective pathogens for humans. Although targeting virulence genes will not necessarily rid the host of the pathogen, preventing the expression of these genes will limit or perhaps even prevent colonization entirely. Even if the bacteria persist in the intestine after treatment ceases, their ability to cause disease will be severely reduced if they cannot colonize their preferred niche in the intestine. For example, the enterotoxins of ETEC cause diarrhea by interfering with water and electrolyte transport in the small intestine (59), and EPEC causes disease in adult volunteers when administered by mouth but not when given per rectum (60). Furthermore, we and others (5, 61) have shown that many different intestinal pathogens respond to specific environmental signals in the small intestine, such as bicarbonate ions, to activate the expression of their virulence genes. Accordingly, once these bacteria are located in the large intestine they are unlikely to become virulent unless they are re-ingested. Asymptomatic colonization of the large intestine by these bacteria may even confer an advantage by inducing an immune response, which protects against infection with the same pathogen in future. Small molecule inhibitors of virulence may be particularly useful for the prevention of travelers diarrhea caused by enterovirulent strains of E. coli or in controlling outbreaks such as the recent outbreak of infection with Shiga-toxin producing E. coli O104:H4 in Germany (62).
In summary, the findings of this study indicate that inhibiting AraC-mediated activation of transcription is a promising and effective strategy to combat intestinal infections in humans and animals. This proof-of-principle study lays the foundation for the computational aided design of drugs that target similar virulence regulators from other bacterial pathogens.
This work was supported by grants from the Australian National Health and Medical Research Council (NHMRC), the NHMRC Independent Research Institutes Infrastructure Support Scheme, and the Victorian State Government Operational Infrastructure Support Program to St. Vincent's Institute and the Murdoch Childrens Research Institute.
- EPEC
- enteropathogenic E. coli
- ETEC
- enterotoxigenic E. coli
- EAEC
- enteroaggregative E. coli
- EHEC
- enterohemorrhagic E. coli
- regacin
- 5-[(4-chlorobenzyl)thio]-1,3,4-thiadiazol-2-amine
- LEE
- locus for enterocyte effacement
- MBP
- maltose-binding protein
- DCTTA
- 5-[(2,5-dichlorobenzyl)thio]-1,3,4-thiadiazol-2-amine
- X-gal
- 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside.
REFERENCES
- 1. Petri W. A., Jr., Miller M., Binder H. J., Levine M. M., Dillingham R., Guerrant R. L. (2008) Enteric infections, diarrhea, and their impact on function and development. J. Clin. Invest. 118, 1277–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rasko D. A., Sperandio V. (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug. Discov. 9, 117–128 [DOI] [PubMed] [Google Scholar]
- 3. Clatworthy A. E., Pierson E., Hung D. T. (2007) Targeting virulence. A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3, 541–548 [DOI] [PubMed] [Google Scholar]
- 4. Gallegos M. T., Schleif R., Bairoch A., Hofmann K., Ramos J. L. (1997) Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61, 393–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang J., Tauschek M., Robins-Browne R. M. (2011) Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends Microbiol. 19, 128–135 [DOI] [PubMed] [Google Scholar]
- 6. Dorman C. J. (2004) H-NS. A universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2, 391–400 [DOI] [PubMed] [Google Scholar]
- 7. Bender J. K., Praszkier J., Wakefield M. J., Holt K., Tauschek M., Robins-Browne R. M., Yang J. (2012) Involvement of PatE, a prophage-encoded AraC-like regulator, in the transcriptional activation of acid resistance pathways of enterohemorrhagic Escherichia coli strain EDL933. Appl. Environ. Microbiol. 78, 5083–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hacker J., Kaper J. B. (2000) Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679 [DOI] [PubMed] [Google Scholar]
- 9. Gómez-Duarte O. G., Kaper J. B. (1995) A plasmid-encoded regulatory region activates chromosomal eaeA expression in enteropathogenic Escherichia coli. Infect. Immun. 63, 1767–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Srikhanta Y. N., Hocking D. M., Praszkier J., Wakefield M. J., Robins-Browne R. M., Yang J., Tauschek M. (2013) RegR virulence regulon of rabbit-specific enteropathogenic Escherichia coli strain E22. Infect. Immun. 81, 1078–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murphree D., Froehlich B., Scott J. R. (1997) Transcriptional control of genes encoding CS1 pili. Negative regulation by a silencer and positive regulation by Rns. J. Bacteriol. 179, 5736–5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nataro J. P., Yikang D., Yingkang D., Walker K. (1994) AggR, a transcriptional activator of aggregative adherence fimbria I expression in enteroaggregative Escherichia coli. J. Bacteriol. 176, 4691–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DiRita V. J., Parsot C., Jander G., Mekalanos J. J. (1991) Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 88, 5403–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mundy R., MacDonald T. T., Dougan G., Frankel G., Wiles S. (2005) Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706 [DOI] [PubMed] [Google Scholar]
- 15. Deng W., Puente J. L., Gruenheid S., Li Y., Vallance B. A., Vázquez A., Barba J., Ibarra J. A., O'Donnell P., Metalnikov P., Ashman K., Lee S., Goode D., Pawson T., Finlay B. B. (2004) Dissecting virulence. Systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U.S.A. 101, 3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang J., Tauschek M., Hart E., Hartland E. L., Robins-Browne R. M. (2010) Virulence regulation in Citrobacter rodentium. The art of timing. Microb. Biotechnol. 3, 259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hart E., Yang J., Tauschek M., Kelly M., Wakefield M. J., Frankel G., Hartland E. L., Robins-Browne R. M. (2008) RegA, an AraC-like protein, is a global transcriptional regulator that controls virulence gene expression in Citrobacter rodentium. Infect. Immun. 76, 5247–5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tauschek M., Yang J., Hocking D., Azzopardi K., Tan A., Hart E., Praszkier J., Robins-Browne R. M. (2010) Transcriptional analysis of the grlRA virulence operon from Citrobacter rodentium. J. Bacteriol. 192, 3722–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghaem-Maghami M., Simmons C. P., Daniell S., Pizza M., Lewis D., Frankel G., Dougan G. (2001) Intimin-specific immune responses prevent bacterial colonization by the attaching-effacing pathogen Citrobacter rodentium. Infect. Immun. 69, 5597–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marchès O., Nougayrède J. P., Boullier S., Mainil J., Charlier G., Raymond I., Pohl P., Boury M., De Rycke J., Milon A., Oswald E. (2000) Role of Tir and intimin in the virulence of rabbit enteropathogenic Escherichia coli serotype O103:H2. Infect. Immun. 68, 2171–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evans D. G., Evans D. J., Jr., Pierce N. F. (1973) Differences in the response of rabbit small intestine to heat-labile and heat-stable enterotoxins of Escherichia coli. Infect. Immun. 7, 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nataro J. P., Deng Y., Cookson S., Cravioto A., Savarino S. J., Guers L. D., Levine M. M., Tacket C. O. (1995) Heterogeneity of enteroaggregative Escherichia coli virulence demonstrated in volunteers. J. Infect. Dis. 171, 465–468 [DOI] [PubMed] [Google Scholar]
- 23. Salim A., Lan R., Reeves P. R. (2005) Vibrio cholerae pathogenic clones. Emerg. Infect. Dis. 11, 1758–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Casadaban M. J. (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage λ and μ. J. Mol. Biol. 104, 541–555 [DOI] [PubMed] [Google Scholar]
- 25. Yang J., Ganesan S., Sarsero J., Pittard A. J. (1993) A genetic analysis of various functions of the TyrR protein of Escherichia coli. J. Bacteriol. 175, 1767–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang A. C., Cohen S. N. (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134, 1141–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Hart E., Tauschek M., Price G. D., Hartland E. L., Strugnell R. A., Robins-Browne R. M. (2008) Bicarbonate-mediated transcriptional activation of divergent operons by the virulence regulatory protein, RegA, from Citrobacter rodentium. Mol. Microbiol. 68, 314–327 [DOI] [PubMed] [Google Scholar]
- 28. Yang J., Murakami K., Camakaris H., Fujita N., Ishihama A., Pittard A. J. (1997) Amino acid residues in the α-subunit C-terminal domain of Escherichia coli RNA polymerase involved in activation of transcription from the mtr promoter. J. Bacteriol. 179, 6187–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Argaet V. P., Wilson T. J., Davidson B. E. (1994) Purification of the Escherichia coli regulatory protein TyrR and analysis of its interactions with ATP, tyrosine, phenylalanine, and tryptophan. J. Biol. Chem. 269, 5171–5178 [PubMed] [Google Scholar]
- 30. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., pp. A2.2, Cold Spring Harbour Laboratory Press, Cold Spring Harbour, NY [Google Scholar]
- 31. Miller J. H. (1974) Experiments in Molecular Genetics, Cold Spring Harbour Laboratory Press, Plainview, NY [Google Scholar]
- 32. Laue T. M., Shah B. D., Ridgeway T. M., Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Sciences (Harding S. E., Rowe A. J., Horton J. C., eds) pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 33. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lowden M. J., Skorupski K., Pellegrini M., Chiorazzo M. G., Taylor R. K., Kull F. J. (2010) Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U.S.A. 107, 2860–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sali A., Potterton L., Yuan F., van Vlijmen H., Karplus M. (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23, 318–326 [DOI] [PubMed] [Google Scholar]
- 37. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 38. Jain A. N. (2003) Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 46, 499–511 [DOI] [PubMed] [Google Scholar]
- 39. Pittard J., Camakaris H., Yang J. (2005) The TyrR regulon. Mol. Microbiol. 55, 16–26 [DOI] [PubMed] [Google Scholar]
- 40. Yang J., Dogovski C., Hocking D., Tauschek M., Perugini M., Robins-Browne R. M. (2009) Bicarbonate-mediated stimulation of RegA, the global virulence regulator from Citrobacter rodentium. J. Mol. Biol. 394, 591–599 [DOI] [PubMed] [Google Scholar]
- 41. Levy S. B., Marshall B. (2004) Antibacterial resistance worldwide. Causes, challenges, and responses. Nat. Med. 10, S122–S129 [DOI] [PubMed] [Google Scholar]
- 42. Wong C. S., Jelacic S., Habeeb R. L., Watkins S. L., Tarr P. I. (2000) The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 342, 1930–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kollef M. H., Golan Y., Micek S. T., Shorr A. F., Restrepo M. I. (2011) Appraising contemporary strategies to combat multidrug resistant gram-negative bacterial infections. Proceedings and data from the Gram-negative resistance summit. Clin. Infect. Dis. 53, S33–S55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Talbot G. H., Bradley J., Edwards J. E., Jr., Gilbert D., Scheld M., Bartlett J. G. (2006) Bad bugs need drugs. An update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42, 657–668 [DOI] [PubMed] [Google Scholar]
- 45. Stanton T. B. (2013) A call for antibiotic alternatives research. Trends Microbiol. 21, 111–113 [DOI] [PubMed] [Google Scholar]
- 46. Sun F., Zhou L., Zhao B. C., Deng X., Cho H., Yi C., Jian X., Song C. X., Luan C. H., Bae T., Li Z., He C. (2011) Targeting MgrA-mediated virulence regulation in Staphylococcus aureus. Chem. Biol. 18, 1032–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hung D. T., Shakhnovich E. A., Pierson E., Mekalanos J. J. (2005) Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310, 670–674 [DOI] [PubMed] [Google Scholar]
- 48. Kimura K., Iwatsuki M., Nagai T., Matsumoto A., Takahashi Y., Shiomi K., Omura S., Abe A. (2011) A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J. Antibiot. 64, 197–203 [DOI] [PubMed] [Google Scholar]
- 49. Garrity-Ryan L. K., Kim O. K., Balada-Llasat J. M., Bartlett V. J., Verma A. K., Fisher M. L., Castillo C., Songsungthong W., Tanaka S. K., Levy S. B., Mecsas J., Alekshun M. N. (2010) Small molecule inhibitors of LcrF, a Yersinia pseudotuberculosis transcription factor, attenuate virulence and limit infection in a murine pneumonia model. Infect. Immun. 78, 4683–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gauthier A., Puente J. L., Finlay B. B. (2003) Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect. Immun. 71, 3310–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rasko D. A., Moreira C. G., Li de R., Reading N. C., Ritchie J. M., Waldor M. K., Williams N., Taussig R., Wei S., Roth M., Hughes D. T., Huntley J. F., Fina M. W., Falck J. R., Sperandio V. (2008) Targeting QseC signaling and virulence for antibiotic development. Science 321, 1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shakhnovich E. A., Hung D. T., Pierson E., Lee K., Mekalanos J. J. (2007) Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc. Natl. Acad. Sci. U.S.A. 104, 2372–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Childers B. M., Weber G. G., Prouty M. G., Castaneda M. M., Peng F., Klose K. E. (2007) Identification of residues critical for the function of the Vibrio cholerae virulence regulator ToxT by scanning alanine mutagenesis. J. Mol. Biol. 367, 1413–1430 [DOI] [PubMed] [Google Scholar]
- 54. Chang J. Y., Antonopoulos D. A., Kalra A., Tonelli A., Khalife W. T., Schmidt T. M., Young V. B. (2008) Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197, 435–438 [DOI] [PubMed] [Google Scholar]
- 55. Blaser M. (2011) Antibiotic overuse. Stop the killing of beneficial bacteria. Nature 476, 393–394 [DOI] [PubMed] [Google Scholar]
- 56. Sheikh J., Czeczulin J. R., Harrington S., Hicks S., Henderson I. R., Le Bouguénec C., Gounon P., Phillips A., Nataro J. P. (2002) A novel dispersin protein in enteroaggregative Escherichia coli. J. Clin. Invest. 110, 1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee J. H., Isaacson R. E. (1995) Expression of the gene cluster associated with the Escherichia coli pilus adhesin K99. Infect. Immun. 63, 4143–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Deng W., Vallance B. A., Li Y., Puente J. L., Finlay B. B. (2003) Citrobacter rodentium translocated intimin receptor (Tir) is an essential virulence factor needed for actin condensation, intestinal colonization, and colonic hyperplasia in mice. Mol. Microbiol. 48, 95–115 [DOI] [PubMed] [Google Scholar]
- 59. Croxen M. A., Finlay B. B. (2010) Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38 [DOI] [PubMed] [Google Scholar]
- 60. Koya G., Kosakai N., Kono M., Mori M., Fukasawa Y. (1954) Observations on the multiplication of Escherichia coli 0-111 B4 in the intestinal tract of adult volunteers in feeding experiments, the intubation study with Miller-Abbott's double lumen tube. Jpn. J. Med. Sci. Biol. 7, 197–203 [DOI] [PubMed] [Google Scholar]
- 61. Mekalanos J. J. (1992) Environmental signals controlling expression of virulence determinants in bacteria. J. Bacteriol. 174, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Buchholz U., Bernard H., Werber D., Böhmer M. M., Remschmidt C., Wilking H., Deleré Y., an der Heiden M., Adlhoch C., Dreesman J., Ehlers J., Ethelberg S., Faber M., Frank C., Fricke G., Greiner M., Höhle M., Ivarsson S., Jark U., Kirchner M., Koch J., Krause G., Luber P., Rosner B., Stark K., Kühne M. (2011) German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 365, 1763–1770 [DOI] [PubMed] [Google Scholar]