

Background: MGP inhibits tissue calcification, but underlying mechanisms are understudied.
Results: In MGP null mice, TG2 ablation prevents calcifying cartilaginous vascular lesions but does not affect elastocalcinosis and elastin fragmentation associated with increased elastase adipsin.
Conclusion: MGP acts via two distinct mechanisms.
Significance: Our study identifies TG2 and adipsin as potential therapeutic targets in vascular disease linked to MGP deficiency.
Keywords: Calcification, Cardiovascular Disease, Elastin, Transgenic Mice, Vascular Smooth Muscle Cells, Adipsin, Matrix Gla Protein, Transglutaminase 2
Abstract
Mutations in matrix Gla protein (MGP) have been correlated with vascular calcification. In the mouse model, MGP null vascular disease presents as calcifying cartilaginous lesions and mineral deposition along elastin lamellae (elastocalcinosis). Here we examined the mechanisms underlying both of these manifestations. Genetic ablation of enzyme transglutaminase 2 (TG2) in Mgp−/− mice dramatically reduced the size of cartilaginous lesions in the aortic media, attenuated calcium accrual more than 2-fold, and doubled longevity as compared with control Mgp−/− animals. Nonetheless, the Mgp−/−;Tgm2−/− mice still died prematurely as compared with wild-type and retained the elastocalcinosis phenotype. This pathology in Mgp−/− animals was developmentally preceded by extensive fragmentation of elastic lamellae and associated with elevated serine elastase activity in aortic tissue and vascular smooth muscle cells. Systematic gene expression analysis followed by an immunoprecipitation study identified adipsin as the major elastase that is induced in the Mgp−/− vascular smooth muscle even in the TG2 null background. These results reveal a central role for TG2 in chondrogenic transformation of vascular smooth muscle and implicate adipsin in elastin fragmentation and ensuing elastocalcinosis. The importance of elastin calcification in MGP null vascular disease is highlighted by significant residual vascular calcification and mortality in Mgp−/−;Tgm2−/− mice with reduced cartilaginous lesions. Our studies identify two potential therapeutic targets in vascular calcification associated with MGP dysfunction and emphasize the need for a comprehensive approach to this multifaceted disorder.
Introduction
Vascular calcification is a known risk factor for cardiovascular disease and a cause of mortality. Molecular mechanisms contributing to this pathology include a decrease in circulating and local inhibitors of mineralization, especially in dialysis patients with chronic kidney disease, as well as osteogenic and chondrogenic transformations of vascular smooth muscle cells (VSMCs)2 that support calcification in the vascular wall via processes thought to recapitulate physiological ossification.
Matrix Gla protein (MGP), a 10-kDa polypeptide containing calcium-binding γ-carboxyglutamic acid (Gla) residues and three phosphoserines, is expressed in vascular tissue by both VSMCs and endothelium (1, 2). In patients, MGP polymorphisms have been linked to coronary artery calcification (3, 4), and in the arteries of mice, genetic loss of MGP leads to extensive calcification of cartilaginous lesions in the tunica media (5). Calcified cartilaginous lesions in the MGP null vasculature originate from phenotypically transformed VSMCs (6) and/or from endothelial progenitor cells (7) and cause rupture of the aortic wall and premature death by 8 weeks of age (5). Restoring circulating levels of MGP in Mgp−/− mice does not rescue the vascular phenotype, but re-establishment of MGP expression in VSMCs does prevent both cartilaginous lesions and calcium accrual (8), identifying VSMC-derived MGP as a potent local inhibitor of matrix mineralization and chondrogenic dedifferentiation of VSMCs in the arterial wall context.
Several biological activities of MGP have been proposed to mediate its protective effects. Because MGP is an extracellular binder of calcium phosphate (9–11), it has been hypothesized that when MGP is absent, unbound calcium phosphate precipitates in the tissue. In addition, MGP is able to sequester BMP2/4 proteins and thus inhibit vascular BMP signaling (12), a potent stimulator of osteochondrogenic differentiation. However, reports citing the limited effects of activated (13) or inhibited (14, 15) BMP signaling on osteogenic differentiation of VSMCs in vitro indicate that additional BMP-independent mechanisms may be involved in the MGP-mediated inhibition of phenotypic transformation in VSMCs.
Because of the dramatic phenotype of calcifying chondrogenic lesions, MGP null vascular disease has been described primarily as a product of ectopic chondrogenesis. However, elastocalcinosis is also a prominent feature in the MGP-deficient vascular wall, and elastin haploinsufficiency has been shown to impede progression of calcification in this tissue (16). However, the mechanisms underlying elastocalcinosis in Mgp−/− arteries and its role in the formation of cartilaginous lesions remain unclear because elastocalcinosis has been considered as both a possible cause (17) and a consequence (6) of chondrogenic transformation. Previous research has shown that calcification of elastin often associates with its degradation (18), and elastin fragmentation points represent preferential sites for hydroxyapatite crystal nucleation (19). Moreover, is it possible that elastin degradation is a common cause of calcification because protease inhibitors have efficiently attenuated aortic calcification induced by diverse stimuli including kidney disease, vitamin D3, warfarin, and peri-adventitial application of CaCl2 (20–22). Nevertheless, elastin degradation occurring in uremic mice on a normal phosphate diet is not sufficient to induce arterial medial calcification (18), and elastocalcinosis induced by warfarin treatment is not associated with significant elastin fragmentation,3 indicating potential mechanistic differences between vascular calcification of different etiologies.
Enzyme transglutaminase 2 (TG2) accumulates in the calcifying aortae of Mgp−/− mice (23). TG2 regulates osteochondrogenic differentiation in committed skeletal cells (24–28) and is a critical regulator of the phenotypic transformation of VSMCs into osteogenic cells (29–31). Therefore, we hypothesize that the elevated levels of TG2 in MGP null arterial tissue (23) may contribute to the phenotypic trans-differentiation of VSMCs into chondrocyte-like cells (6). In this study, we tested the hypothesis that genetic ablation of TG2 will prevent the formation of calcifying cartilaginous lesions in Mgp−/− mice. In the generated Mgp−/−;Tgm2−/− double knock-out mice, we observed a discontinuity between cartilaginous lesions and elastocalcinosis, with both contributing to overall calcium accrual in the MGP null aortae. Elastocalcinosis was preceded by fragmentation of the elastic lamellae that associated with elevated serine elastase activity. Biochemical analysis identified adipsin protease as a major contributor of the elevated elastase activity in MGP null VSMCs. These data demonstrate that both chondrogenic transformation and elastin fragmentation contribute significantly to MGP null vascular disease and suggest that MGP has an unanticipated regulatory function during arterial development, controlling the expression of adipsin and stabilizing the structure of the vascular elastic lamellae.
EXPERIMENTAL PROCEDURES
Reagents were purchased from Sigma-Aldrich unless otherwise noted.
Animals
Five-week-old C57BL/6J mice (WT), matrix Gla Protein-deficient (Mgp−/−, KO) mice, and transglutaminase 2 (Tgm2−/−) mice (all transgenic mice on C57BL/6J genetic background) were used in this study. Mgp+/− mice were courteously provided by Gerard Karsenty (Columbia University, New York, NY) (5). Tgm2−/− mice were a kind gift from Robert Graham, Victor Chang Cardiovascular Institute, New South Wales, Australia. To study the effect of deletion of TG2 in the Mgp−/− mouse model, we generated mice deficient in both MGP and TG2 (Mgp−/−;Tgm2−/−). For this, animals heterozygous for MGP (Mgp+/−) were crossed with homozygous TG2 null mice (Tgm2−/−) to generate MGP and TG2 heterozygotes (Mgp+/−;Tgm2+/− ). These F1 mice were used as breeders to generate mice carrying different genotypes (F2), including 1) Mgp−/−;Tgm2−/−, 2) Mgp−/−;Tgm2+/+, and 3) Mgp+/+;Tgm2+/+. F2 mice were genotyped using standard PCR for Mgp and Tgm2 genes. All procedures were approved by the institutional animal care and use committee at the University of Maryland Medical School and were conducted in compliance with National Institutes of Health guidelines for the care and use of laboratory animals.
Histology
Frozen 10-μm sections of freshly dissected aortas, non-perfusion-fixed in 4% paraformaldehyde, were stained for proteoglycan deposition and calcified matrix using the Alcian blue and von Kossa silver nitrate methods, respectively, according to standard protocols (32). Elastic lamellae were stained using eosin Y and visualized by fluorescence microscopy at 488 nm. Images were collected using a Leica DMIL inverted microscope equipped with a SPOT RT3 real time CCD camera (Diagnostic Instruments).
Morphologic Analysis
For morphologic analyses, serial sections spaced 100 μm apart along a 1-mm length of descending aorta from Mgp+/+;Tgm2+/+ (WT), Mgp−/−;Tgm2+/+ (KO), and Mgp−/−;Tgm2−/− (DKO) animals were analyzed for chondrogenic lesions and thickness of tunica media. For each animal, the average value of four or five serial sections was used. Mean and standard error values were calculated using the average values from each animal. All of the values were normalized to WT animals (set at 100%).
Calcium Determination
Aortas were dried at 55 °C overnight and then dissolved in 0.1 n HCl overnight. Calcium content was determined biochemically using the Calcium (CPC) LiquiColor kit as measured with ο-Cresolphthalein (Stanbio). Calcium content of the aorta was normalized to dry weight of the tissue.
Cell Culture
Primary VSMCs from C57BL/6J (WT), Tgm2−/− (TGKO), or Mgp−/− (KO) mice were obtained by a modification of the explant method originally described by Ross (33). For all experiments, cells were used between passages 3 and 4 and were stained using a rabbit anti-α-smooth muscle actin antibody (1:250; Abcam) visualized by fluorescence microscopy using an Alexa 488-conjugated goat anti-rabbit secondary (Invitrogen). To induce chondrogenic transformation, VSMCs were seeded as high density micromasses (2.5 × 105 cells in 10 μl of volume) in chondrogenic medium (DMEM supplemented with 10−7 mmol/liter dexamethasone, 0.1 mmol/liter ascorbic acid (Wako Chemicals), 1% insulin, transferrin, selenium (ITS) premix (BD Biosciences, NJ), 1 mmol/liter sodium pyruvate, 0.35 mmol/liter l-proline, 4 mmol/liter l-glutamine (Invitrogen), 1% penicillin-streptomycin (Invitrogen), and 10 ng/ml TGF-β3 (ProSpec, NJ). The medium was changed twice a week for 8 days. Sulfated glycosaminoglycan (GAG) synthesized by cultured VSMCs was detected by staining with 1% Alcian blue (8GX) dissolved in 0.1 n HCl. For quantitative analysis, Alcian blue was extracted with 4 m guanidine hydrochloride, and absorbance at 590 nm was measured using an Optima spectrophotometer (PolarStar). Cell density was estimated using water-soluble tetrazolium salts substrate (Dojindo, CA).
Elastase Activity
Elastase activity was measured in aortic tissue and primary VSMCs from WT and KO animals according to the published protocol (34), in the presence or absence of 5 mmol/liter EDTA, 1 mmol/liter PMSF, or an equal volume of ethanol (EtOH) vehicle. Absorbance of degraded elastin substrate was measured at 410 nm in a PolarStar Optima spectrophotometer. A standard curve for degradation was prepared using porcine pancreatic elastase. The results are expressed as defined units, where 1 unit is equivalent to the cleavage of 1 μg of substrate in 24 h by 1 mg of total protein in cell or tissue lysate (determined by Pierce micro BCA protein quantitation kit). For adipsin depletion studies, lysates were preincubated with 4 μg of anti-adipsin antibody (Santa Cruz Biotechnology) and protein A/G beads (Millipore) in elastase buffer without Me2SO and elastin substrate, overnight at 4 °C. Me2SO and elastin substrate were then added to the resulting adipsin-deficient supernatant, and elastase activity was measured as described above.
Quantitative Real Time PCR
Quantitative real time PCR was performed according to the standard protocol with EVA green chemistry in a CFX96 thermocycler (Bio-Rad) using the primers in Table 1. Relative change in gene expression was calculated by the ΔΔCt method using Microsoft Excel. mRNA was isolated using the RNeasy kit (Qiagen), and first strand synthesis was performed using the Maxima RT kit (Thermo-Fisher) according to the manufacturer's instructions in a DYAD thermocycler (MJ Research).
TABLE 1.
Primer sequences for mouse genes analyzed by real time PCR
| Gene | Accession no. | Forward primer | Reverse primer |
|---|---|---|---|
| Serine proteases | |||
| cfd (adipsin) | NM_013459 | ggtatgatgtgcagagtgtagtg | caacgaggcattctgggatag |
| cela1 | NM_010703 | tcactgtcaggcgacacttc | tgaggcttcacgtgcattag |
| cela2a | NM_007919 | gtgaccctgagcaagaacat | gcctgtgacatagcagacatag |
| cela3b | NM_026419 | gcacctaccaggttgtactt | ccacagctcacacacataga |
| prss1 | NM_053243 | cccgcatccaagtgagatt | tgtcattgttcagggtcttcc |
| prss2 | NM_009430 | gctttccctgtggatgatga | agtggtagccagcatttagg |
| prtn3 | NM_011178 | acggtggtcaccttcctat | ggagtccactccatgaagaatg |
| ctsg | NM_007800 | gcgagaagacttcgtcctaac | atgagttgctgggtcctttc |
| try4 | NM_011646 | ctgggagagcacaacatcaa | gttgttcagggtcctgctatt |
| elane | NM_015779 | atggctccgctaccattaac | actgggtgatggtctgtttg |
| Housekeeping | |||
| Ribosomal protein L19 (Rpl 19) | NM_009078 | aagaggaagggtactgccaatgct | tgaccttcaggtacaggctgtgat |
Western Blot
Aortic tissue was cut into small pieces on dry ice and lysed by freeze-thaw cycles in radioimmune precipitation assay buffer containing EDTA-free protease and phosphatase inhibitors (Thermo Fisher). Denatured 40-μg protein samples were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad), and Western blot was performed using the standard protocol. Primary antibody used was rabbit anti-adipsin (1:1,000; Santa Cruz Biotechnology). Proteins were detected using HRP-conjugated secondary goat anti-rabbit antibody (1:5000; Abcam). As a loading control, membranes were incubated in HRP-conjugated mouse anti-GAPDH (1:35,000). Signal was visualized with SuperSignal West Pico chemiluminescent substrate (Thermo Fisher).
Statistical Analysis
The data are presented as means ± S.E. For experiments containing two groups, significance was determined by comparison using a Wilcoxon-Mann-Whitney test. For experiments containing more than two groups, Levene's test was used to determine equality of variance (homoscedasticity) followed by one-way analysis of variance and Tukey-Kramer post hoc analysis for comparison between groups. A p value of <0.05 was considered to be statistically significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Survival curves were analyzed using the Kaplan-Meier method, and p values were determined by Mantel-Cox log-rank test.
RESULTS
Genetic Deletion of TG2 Attenuates Chondrogenic Transformation of Primary VSMCs
To determine whether TG2 can be involved with chondrogenic transformation of VSMCs, we employed the in vitro model of TGF-β-induced chondrogenesis in high density micromasses using primary mouse WT and Tgm2−/− (TGKO) VSMCs. The VSMC nature of the isolated cells was confirmed by the presence of α-smooth muscle actin (Fig. 1A). After 8 days in culture, both types of VSMCs formed chondrogenic nodules and deposited cartilaginous sulfated GAG-rich matrix, detectable with Alcian blue stain. An equal number of cells were plated as control cells and cultured in serum-containing medium in the absence of TGF-β. These control cells did not deposit GAG-positive matrix and rapidly proliferated (Fig. 1). In contrast, proliferation was ceased in the micromasses undergoing chondrogenic transformation, resulting in a similar number of WT and TGKO VSMCs after 8 days of culture (Fig. 1B). Quantitative analysis of the extracted Alcian blue dye revealed a ∼50% decrease in GAG deposition by TGKO VSMC cultures compared with WT micromasses (Fig. 1C), suggesting a role for TG2 in chondrogenic dedifferentiation in VSMCs.
FIGURE 1.
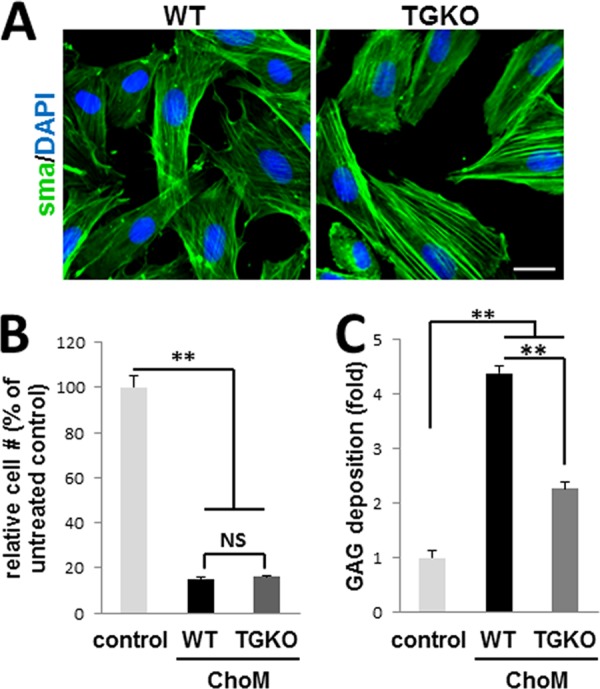
Genetic ablation of TG2 attenuates chondrogenic transformation of VSMCs. A, immunostain for smooth muscle actin (green) (nuclei counterstained with DAPI (blue)) in primary cultures of WT and Tgm2−/− (TGKO) VSMCs prior to micromass culture. Scale bar, 25 μm. B, relative cell numbers in WT and TGKO micromasses cultured in chondrogenic medium (ChoM) compared with cells cultured in normal growth medium (control) (n = 4). C, GAG deposition, detected by Alcian blue stain, in primary WT and TGKO VSMCs induced to undergo spontaneous chondrogenesis by TGFβ3 in high density micromass culture compared with control cells (n = 4).
Genetic Deletion of TG2 Attenuates Chondrogenic Lesions in Mgp−/− Aortae
Previous studies have shown that TG2 is elevated in Mgp−/− aortic tissue (23) and that VSMC-derived cells constitute almost 97% of the chondrogenic cells in MGP null neoplasia (6). We therefore hypothesized that ablation of TG2 will attenuate cartilaginous lesions in the MGP null model. To test this hypothesis, we generated Mgp−/−;Tgm2−/− double knock-out (DKO) animals by crossing Mgp+/− and Tgm2−/− mice. Histological analysis revealed a significant reduction of cartilaginous lesions and an improvement in the gross morphology of the aortic wall in 3.5-week-old DKO mice compared with age-matched Mgp−/− (KO) animals (Fig. 2, n = 6). Quantitative analysis of serial sections spaced 100 μm apart along a 1-mm segment of aorta from each animal (Fig. 2A) showed that although cartilaginous lesions in KO mice comprised 49.9 ± 5.5% of the total circumference of vessel wall, the proportion of these lesions in the DKO mice was significantly reduced to 19.2 ± 2.7% (n = 6, p < 0.001) (Fig. 2B). Because chondrogenic lesions thicken vessel walls, the effects of TG2 deletion included an ∼30% reduction in arterial wall thickness (51.6 ± 2.5 μm in DKO mice compared with 88.2 ± 6.3 μm in KO animals; p < 0.01), rendering this parameter similar to the wild-type aortae (42.2 ± 2.2 μm) (Fig. 2C, WT, n = 3). These results demonstrate that TG2 is central in the formation of cartilaginous lesions in the MGP null arterial wall.
FIGURE 2.
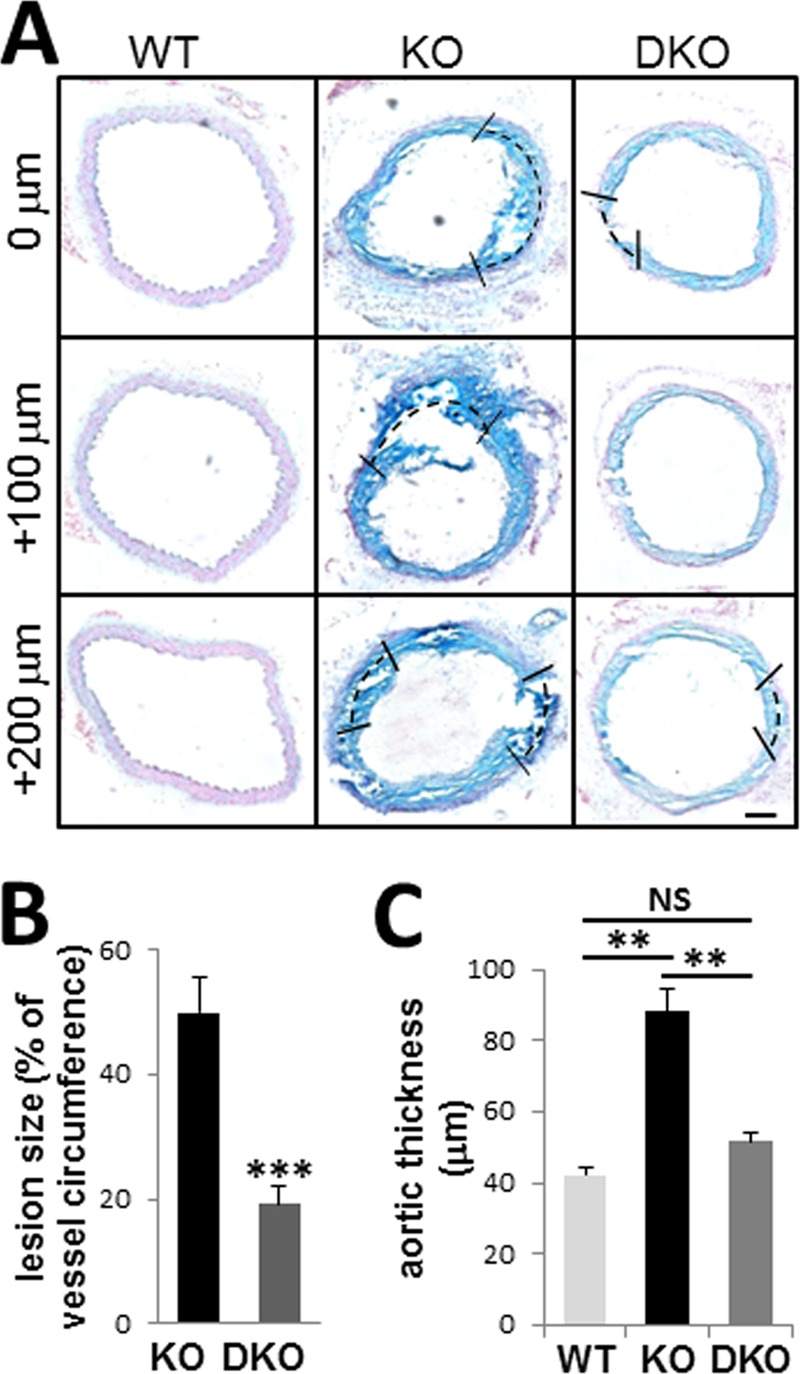
Genetic ablation of TG2 reduces cartilaginous lesions and improves vessel morphology of Mgp−/− mice. A, Alcian blue stain for GAG deposition in Mgp−/− (KO) and Mgp−/−;Tgm2−/− (DKO) aortic tissue. Tissue sections separated by 100 μm (denoted as 0 μm, +100 μm, and +200 μm) along a segment of aorta from each animal were examined. Scale bar, 50 μm. B and C, quantitative analysis of vessel morphology of aortae from 3.5-week-old KO and DKO animals. Shown are percentages of vessel circumference occupied by chondrogenic lesions (B) and quantitation of cross-sectional thickness of tunica media (C) (n = 3, WT; n = 6, KO; n = 6, DKO).
Persistent Elastocalcinosis in Mgp/Tgm2 Double Knock-out Animals
Cartilaginous lesions in MGP null arteries rapidly calcify, and this process contributes to early mortality caused by vessel rupture (5). Eradication of ectopic chondrogenesis in MGP-deficient arteries by TG2 ablation was associated with a significant 48.1 ± 8.0% (p < 0.05) reduction in total calcium content of aortic tissue (Fig. 3A) and significantly attenuated the early mortality characteristic of MGP null mice (5) (Fig. 3B; p < 0.005). Nevertheless, despite the improved longevity, DKO mice still died prematurely, indicating that cartilaginous lesions may not be the only life-threatening complication of MGP deficiency. Indeed, the remaining calcium levels in the aortae of DKO mice were still significantly higher than that in wild-type control littermates (126.99 ± 21.86 μg calcium/mg of dry weight in DKO animals compared with 0.39 ± 0.01 μg calcium/mg of dry weight in WT animals; p < 0.001). To examine this phenomenon further, localization of the calcium phosphate deposits in the KO and DKO arterial walls was detected with von Kossa staining. Although the massive calcified zones corresponding to cartilaginous lesions in KO aortae were no longer observed in the DKO mice (Fig. 3C, upper panels), calcium phosphate deposits aligning with elastic lamellae were still present (Fig. 3C, lower panels), indicating that MGP null disease involves two discrete vascular calcification mechanisms, including calcification of the TG2-dependent cartilaginous lesions and TG2-independent elastocalcinosis.
FIGURE 3.
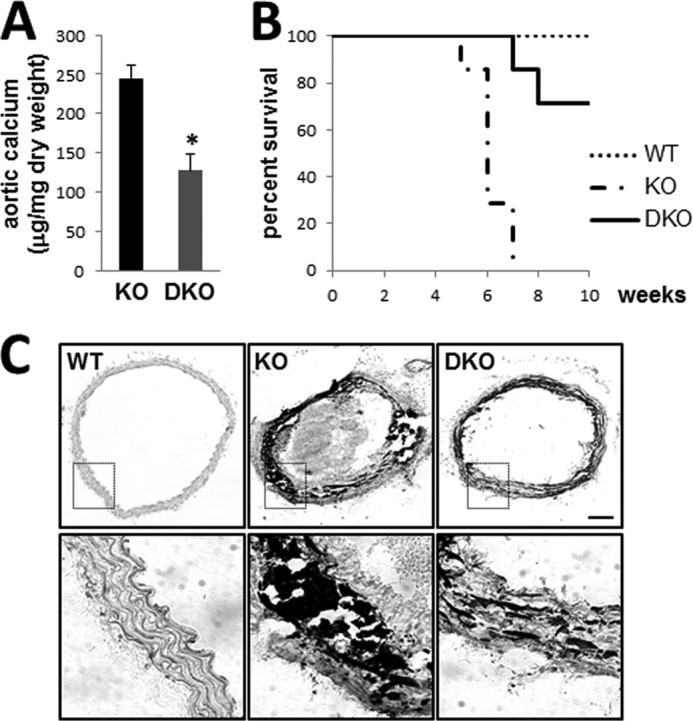
Genetic ablation of TG2 reduces calcium accrual and attenuates mortality of MGP-deficient mice. A, quantitation of total aortic calcium in Mgp−/− (KO) (n = 5) and Mgp−/−;Tgm2−/− (DKO) (n = 4) mice normalized to dry weight of the tissue shows significant reduction in calcium content in DKO aortae. B, Kaplan-Meier survival curve for WT (n = 11), KO (n = 7), and DKO (n = 7) animals showing percentage of animals living each week from birth to 10 weeks old (p < 0.005, log-rank test). C, von Kossa stain for calcified matrix deposition in KO and DKO aortic tissue (upper panels). Scale bar, 50 μm. The lower panels show higher magnification of calcium deposits in the vessel wall.
Elastin Fragmentation in Mgp Null Aortae
Because previous studies have established that calcification of the elastic lamellae can be promoted by their degradation (18), we analyzed the integrity of the aortic elastic lamellae detected by eosin staining. A significant decrease in the length of continuous elastin layers was observed in KO compared with WT littermates (Fig. 4A). Genetic ablation of TG2 did not prevent fragmentation of the elastic lamellae in the DKO animals, in agreement with elastin fragmentation contributing to its calcification. To clarify the possible causative relationship between these two phenomena, we analyzed the dynamics of both processes at earlier developmental stages. In newborn Mgp−/− mice, elastocalcinosis was absent, and the length of the continuous elastic lamellae was the same as in the heterozygous littermates (Fig. 4B). In 1-week-old Mgp−/− mice, elastin fragmentation is pronounced in all animals (Fig. 4C), whereas elastocalcinosis was observed with von Kossa staining in only two of the four animals analyzed (Fig. 4C). Importantly, fragmentation was not different between the calcifying vessels and those that have not yet calcified (Fig. 4C), supporting a model in which elastin degradation precedes its calcification in the MGP null vasculature.
FIGURE 4.

Elastocalcinosis in Mgp−/− animals. A, eosin stain for elastic lamellae in WT, Mgp−/− (KO), and Mgp−/−;Tgm2−/− (DKO) aorta. Scale bar, 40 μm. Quantitation of the average continuous length of elastic fibers along a random 300-μm segment of tunica media is shown at right (n = 3). B and C, von Kossa stain for calcified matrix and eosin stain for elastic lamellae in adjacent sections of aortic tissue from WT and KO mice at birth (B, d0, n = 4) and at 7 days old (C, d7, n = 4). At 7 days, both noncalcified (KO-nc) and calcified (KO-c) vessels are shown. Quantitation of the average continuous length of elastic fibers along a random 250-μm segment of tunica media is shown at right. Scale bar in B, 20 μm; scale bar in C for von Kossa, 50 μm; and scale bar in C for eosin, 20 μm.
Elevated Elastase Activity in MGP Null Arteries Relates to Induced Adipsin
Elastin degradation is mediated by numerous enzymes, mainly serine proteases or matrix metalloproteinases (MMPs). To determine whether elastin fragmentation in the MGP null arteries correlated with enhanced proteolysis, we performed an assay for elastase activity and found an ∼3-fold increase in aortic tissue from Mgp−/− (KO) mice compared with the WT control (Fig. 5A, light gray bars). Both WT and KO elastase activity was equally sensitive to treatment with EDTA (Fig. 5A, dark gray bars), but only the elevated activity in KO arterial tissue was sensitive to PMSF (Fig. 5A, black bars). We concluded that elevated elastase activity in KO is mediated by a serine protease(s) rather than MMP(s). To determine the cellular origin of increased serine elastase activity in the vasculature, we performed the same assay using cultured primary WT and MGP null (KO) VSMCs (P3–4). The MGP null VSMCs produced significantly more PMSF-sensitive elastase activity than the wild-type VSMCs (Fig. 5B, light gray bars), which was not affected by the vehicle control (Fig. 5B, EtOH). These results identify VSMCs as a cellular source of the increased serine elastase in MGP null aortic tissue.
FIGURE 5.
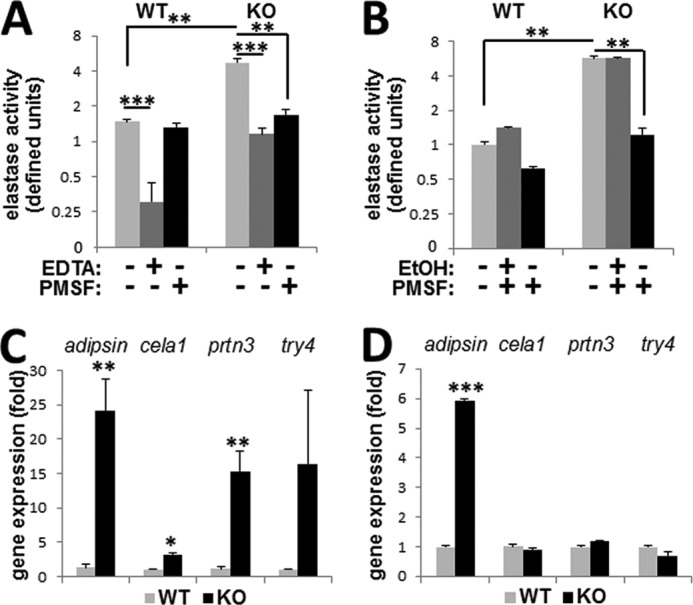
Elastase activity in Mgp−/− animals. A and B, elastase activity in WT and Mgp−/− (KO) aortae from 4.5-week-old mice (A, n = 3) or in primary VSMC cultures (B, two animals from each genotype used to establish primary cultures) in the presence or absence of metalloproteinase inhibitor, EDTA, serine elastase inhibitor, PMSF, or ethanol (EtOH) vehicle control as indicated. C and D, expression of induced serine elastases in WT and KO aortae (C, n = 4) or primary VSMC cultures (D).
To identify candidate serine proteases potentially contributing to increased elastase activity in MGP-deficient VSMCs, we searched the GeneCards database using the keywords “serine elastase” with the score cutoff ≥1.0 and removing the entities that do not have known protease activity. This analysis identified 10 mouse serine elastase genes (Table 2). Quantitative analysis of their transcripts in the wild-type and Mgp−/− arterial tissue by real time RT-PCR identified seven genes expressed at detectable levels in the vasculature, of which four are increased in Mgp−/− arterial tissue (Fig. 5C and Table 2). However, only the expression of adipsin was also significantly up-regulated in primary MGP null VSMCs (Fig. 5D), implicating this protease as a major source of the elevated elastase activity in the MGP null vascular wall.
TABLE 2.
Analysis of candidate serine protease mRNA expression in Mgp−/− versus wild-type aortic tissue
| Gene | Protein | Detectable in aorta | Induced in Mgp−/− |
|---|---|---|---|
| cfd (adipsin) | Complement factor D (adipsin) | Yes | Yesa |
| cela1 | Chymotrypsin-like elastase family, member 1 | Yes | Yesb |
| cela2a | Chymotrypsin-like elastase family, member 2a | No | No |
| cela3b | Chymotrypsin-like elastase family, member 3b | Yes | No |
| prss1 | Protease, serine,1 (trypsin 1) | No | No |
| prss2 | Protease, serine,2 (trypsin 2) | No | No |
| prtn3 | Proteinase 3 | Yes | Yesa |
| ctsg | Cathepsin G | Yes | No |
| try4 | Trypsin 4 | Yes | Yesc |
| elane | Elastase, neutrophil expressed (elastase 2) | Yes | No |
a p = 0.003.
b p = 0.002.
c p = 0.132.
To address this possibility, we first confirmed that protein levels of adipsin are elevated in the Mgp−/− aortae using Western analysis (Fig. 6A). Next, we depleted adipsin from arterial tissue lysates by immunoprecipitation with anti-adipsin antibody. The specific removal of adipsin rendered the elastase activity in the MGP null tissue similar to the levels of activity in wild-type tissue lysates (Fig. 6B), indicating that adipsin is the major active serine elastase induced by genetic loss of MGP. Lastly, we examined adipsin expression in the Mgp−/−;Tgm2−/− DKO animals characterized by elastin degradation similar to Mgp−/− (KO) and found identical levels of expression in the KO and DKO aortae (Fig. 6C).
FIGURE 6.
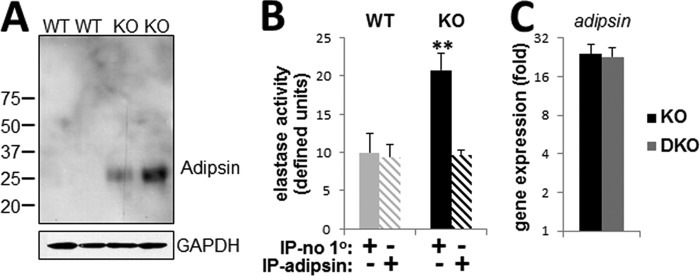
Increased expression and activity of adipsin in Mgp−/− mice. A, Western blot for adipsin protein in aortic tissue from 4.5-week-old WT and Mgp−/− (KO) animals (n = 2). B, elastase activity in supernatant fraction of WT and KO aortic tissue lysates following mock immunoprecipitation (IP-no 1o) or immunoprecipitation with anti-adipsin antibody (IP-adipsin) (n = 3). C, relative expression of adipsin mRNA, detected by real time PCR, in 4.5-week-old KO and DKO aortae compared with wild-type (set at 1) (n = 4).
DISCUSSION
The present study provides evidence for two mechanisms of arterial calcification in MGP null vascular disease. One mechanism, in which TG2 appears central, promotes the formation of calcifying chondrogenic lesions in VSMCs. In contrast, TG2-independent elastocalcinosis is preceded by elastin fragmentation and associates with induction of the serine elastase adipsin.
Previous data have shown the accumulation of TG2 in the calcifying vascular wall of Mgp−/− mice (23). Consistent with the pro-chondrogenic effects of TG2 previously shown in mesenchymal progenitor cells (28) and committed chondrocytes (27, 35), our study demonstrates a central role for TG2 in the chondrogenic transformation of VSMCs and identifies this enzyme as a key therapeutic target in the calcifying chondrogenic lesions of vascular smooth muscle caused by MGP deficiency. However, despite a significant 50% reduction in total calcium accumulation in the MGP null vessels caused by genetic ablation of TG2, the residual premature mortality in the Mgp/Tgm2 double knock-out animals highlights the importance of elastocalcinosis in MGP null vascular disease.
Elastin is a major extracellular substrate for vascular calcification, and its haploinsufficiency hinders the progression of calcium accumulation in the MGP null vascular wall (16). Our data demonstrate that elastin fragmentation probably brings about elastocalcinosis because visible changes in elastin integrity can be visualized early in development even before detectable levels of calcium phosphate precipitate along the elastic lamellae. It remains to be determined whether mineral accrual occurs merely because of the crystal nucleating activity of elastin fragmentation sites (19) or whether it involves phenotypic changes in VSMCs (23). Indeed, elastin has been characterized as a regulator of arterial development, controlling the proliferation of smooth muscle and stabilizing arterial structure (36), and loss of physical interactions with intact elastin can induce a phenotypic switch in VSMCs (37). In addition, elastin degradation peptides (also called elastokines (38)) can act on vascular cells, amplifying phosphate-driven calcium deposition (39, 40). In any case, loss of continuity of the elastic lamellae is probably a significant, if not the major factor in MGP deficiency-associated elastocalcinosis. Of note, we also detected increased elastase activity in cultured MGP null VSMCs that lose chondrogenic features and revert to muscle phenotype (41), further underscoring the difference between the calcification mechanisms centered on ectopic chondrogenesis and those involving elastin fragmentation.
Taking into account the success of protease inhibition used to attenuate vascular calcification caused by diverse stimuli (20–22), it was of essence to identify the elastase activity induced in MGP null VSMCs. MMP expression is characteristic for arterial tissue (16), and in our study we observed that basal elastase activity in WT aortae is dramatically inhibited by EDTA. A similar degree on inhibition by EDTA was also detected in the MGP null tissue, although elastase activity remained in the presence of the MMP inhibitor. In contrast, serine protease inhibitor PMSF had a profound effect only on MGP null VSMCs, completely abolishing the characteristic increase in elastase activity and reverting it to WT levels. PCR-based expression analysis of known serine elastases identified adipsin as a primary candidate protease, contributing to the increase of total elastase activity in the MGP null vessels. This was confirmed by depleting adipsin from tissue lysates with a specific antibody. Adipsin has been also identified as a source of elevated endogenous vascular elastase activity in pulmonary hypertension in rats (42), indicative of a potential commonality between diverse vascular pathologies.
Aside from the insulted vasculature, adipsin has been detected in the sciatic nerve, bloodstream, and most notably in fat tissue, where it is induced in starvation (43, 44), suggesting that in addition to VSMCs, cells of the perivascular adipose tissue and endothelium may also contribute to the increased adipsin activity in vasculature. Only arterial adipsin has been shown to possess elastolytic activity, and this may be related to post-translational tissue-specific modifications of the enzyme (42). Further biochemical studies on modifications of vascular adipsin may allow its selective targeting in the therapy of cardiovascular disease.
Despite its name, the role of adipsin in fat metabolism appears limited (45), and its major biological role relates to the alternative complement pathway. Thus, adipsin (also known as complement factor D) cleaves factor B (46–48), potentially connecting adipose and complement activation. Accumulating evidence on the involvement of this protease in vascular pathology adds to the complex effects of complement on vasculature, exemplified by the ability of complement proteins C3 and C4 to increase vascular stiffness in atherosclerosis through binding to collagen and elastin (49) and to stimulate adventitial fibroblast migration and differentiation in a rat model of hypertension (50).
Transcriptional up-regulation of adipsin expression in MGP null VSMCs is intriguing. There are very limited data on the transcriptional regulation of adipsin, and we therefore performed a preliminary analysis of the mouse adipsin gene promoter.3 This analysis identified several binding sites for the BMP-regulated Runx1 transcription factor (51). Because BMP signaling is activated in the Mgp−/− vasculature (12, 52), we sought to determine whether the well characterized effect of MGP on BMP signaling is linked with the newly identified mechanism of elastin degradation in MGP null vascular disease. For this, we exposed in vitro rat A10 VSMCs to an elevated BMP2/4 mixture and analyzed elastase activity and adipsin expression. No change in these parameters was detected, nor was there a detectable increase in Runx1 expression. These results suggest that induction of adipsin, and elastin fragmentation and calcification in Mgp−/− aortae are either independent of BMP activity or require a synergistic effect of BMP with as yet unknown regulator(s).
Conventional views assume an important role for MGP γ-carboxylation in the inhibition of vascular calcification (53), based on the importance of this modification in MGP binding to hydroxyapatite crystals (54) and BMP proteins (55) as well as on the seemingly similar pro-calcific effects of MGP deficiency and warfarin treatment (and the ability of warfarin to inhibit vitamin K-dependent protein carboxylation) (10). However, our histologic analysis of the elastic lamellae in calcifying arteries of rats treated with warfarin to induce elastocalcinosis (30, 31, 56) did not reveal overt elastin fragmentation (data not shown) in contrast to MGP null arteries. Further, in warfarin-induced calcification, previous studies identified activation of MMP-9 and no induction of serine elastases (22), whereas in the MGP null arteries, the elevated elastase activity is derived from the serine protease adipsin. Together, these results question the role of MGP carboxylation in the regulation of elastolytic activity in VSMCs.
In conclusion, our results support a model in vascular wall where MGP blocks expression of the serine elastase adipsin and thus supports the stability of elastic lamellae and cell-matrix interactions. Loss of MGP results in adipsin activation and elastin fragmentation, allowing for chondrogenic transformation of VSMCs that leads to formation of cartilaginous lesions in a TG2-dependent manner. This study adds to the growing understanding of the complexity of MGP-mediated effects on vascular tissue. In addition to suppressing BMP (57) and Notch signaling (58), our report identifies TG2 as a therapeutic target in calcifying cartilaginous lesions in the tunica media, and the full spectrum of vascular disease associated with MGP deficiency clearly requires additional approaches that probably include inhibition of the elastase activity of adipsin. With accumulating evidence that therapeutic targets are shared between the manifold variants and manifestations of cardiovascular disease, it is possible that research aimed at developing treatments for MGP deficiency will have a broad and significant impact.
Acknowledgments
We thank Dr. Shobana Shanmugasundaram and Dr. Beatrice Milon for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL099305 (to M. N.) and T32AR007592 (to K. B.).
K. E. Beazley, S. Reckard, D. Nurminsky, and M. Nurminskaya, unpublished data.
- VSMC
- vascular smooth muscle cell
- MGP
- matrix Gla protein
- TG2
- transglutaminase 2 protein (alias: TGM2)
- GAG
- glycosaminoglycan
- MMP
- matrix metalloproteinase
- DKO
- double knock-out.
REFERENCES
- 1. Shanahan C. M., Weissberg P. L., Metcalfe J. C. (1993) Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ. Res. 73, 193–204 [DOI] [PubMed] [Google Scholar]
- 2. Boström K., Zebboudj A. F., Yao Y., Lin T. S., Torres A. (2004) Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-β1 activity in endothelial cells. J. Biol. Chem. 279, 52904–52913 [DOI] [PubMed] [Google Scholar]
- 3. Crosier M. D., Booth S. L., Peter I., Dawson-Hughes B., Price P. A., O'Donnell C. J., Hoffmann U., Williamson M. K., Ordovas J. M. (2009) Matrix Gla protein polymorphisms are associated with coronary artery calcification in men. J. Nutr. Sci. Vitaminol. 55, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herrmann S. M., Whatling C., Brand E., Nicaud V., Gariepy J., Simon A., Evans A., Ruidavets J. B., Arveiler D., Luc G., Tiret L., Henney A., Cambien F. (2000) Polymorphisms of the human matrix gla protein (MGP) gene, vascular calcification, and myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 20, 2386–2393 [DOI] [PubMed] [Google Scholar]
- 5. Luo G., Ducy P., McKee M. D., Pinero G. J., Loyer E., Behringer R. R., Karsenty G. (1997) Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386, 78–81 [DOI] [PubMed] [Google Scholar]
- 6. Speer M. Y., Yang H. Y., Brabb T., Leaf E., Look A., Lin W. L., Frutkin A., Dichek D., Giachelli C. M. (2009) Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ. Res. 104, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yao Y., Jumabay M., Ly A., Radparvar M., Cubberly M. R., Boström K. I. (2013) A role for the endothelium in vascular calcification. Circ. Res. 113, 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murshed M., Schinke T., McKee M. D., Karsenty G. (2004) Extracellular matrix mineralization is regulated locally. Different roles of two gla-containing proteins. J. Cell Biol. 165, 625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Price P. A., Thomas G. R., Pardini A. W., Figueira W. F., Caputo J. M., Williamson M. K. (2002) Discovery of a high molecular weight complex of calcium, phosphate, fetuin, and matrix γ-carboxyglutamic acid protein in the serum of etidronate-treated rats. J. Biol. Chem. 277, 3926–3934 [DOI] [PubMed] [Google Scholar]
- 10. Price P. A., Faus S. A., Williamson M. K. (1998) Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler. Thromb. Vasc. Biol. 18, 1400–1407 [DOI] [PubMed] [Google Scholar]
- 11. Chatrou M. L., Reutelingsperger C. P., Schurgers L. J. (2011) Role of vitamin K-dependent proteins in the arterial vessel wall. Hamostaseologie 31, 251–257 [DOI] [PubMed] [Google Scholar]
- 12. Zebboudj A. F., Imura M., Boström K. (2002) Matrix GLA protein, a regulatory protein for bone morphogenetic protein-2. J. Biol. Chem. 277, 4388–4394 [DOI] [PubMed] [Google Scholar]
- 13. Mikhaylova L., Malmquist J., Nurminskaya M. (2007) Regulation of in vitro vascular calcification by BMP4, VEGF and Wnt3a. Calcif. Tissue Int. 81, 372–381 [DOI] [PubMed] [Google Scholar]
- 14. Lomashvili K. A., Wang X., Wallin R., O'Neill W. C. (2011) Matrix Gla protein metabolism in vascular smooth muscle and role in uremic vascular calcification. J. Biol. Chem. 286, 28715–28722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beazley K. E., Eghtesad S., Nurminskaya M. V. (2013) Quercetin attenuates warfarin-induced vascular calcification in vitro independently from matrix Gla protein. J. Biol. Chem. 288, 2632–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khavandgar Z., Roman H., Li J., Lee S., Vali H., Brinckmann J., Davis E. C., Murshed M. (2013) Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 10.1002/jbmr.2039 [DOI] [PubMed] [Google Scholar]
- 17. El-Maadawy S., Kaartinen M. T., Schinke T., Murshed M., Karsenty G., McKee M. D. (2003) Cartilage formation and calcification in arteries of mice lacking matrix Gla protein. Connect Tissue Res. 44, 272–278 [PubMed] [Google Scholar]
- 18. Pai A., Leaf E. M., El-Abbadi M., Giachelli C. M. (2011) Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am. J. Pathol. 178, 764–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rucker R. B., Ford D., Riemann W. G., Tom K. (1974) Additional evidence for the binding of calcium ions to elastin at neutral sites. Calcif. Tissue Res. 14, 317–325 [DOI] [PubMed] [Google Scholar]
- 20. Qin X., Corriere M. A., Matrisian L. M., Guzman R. J. (2006) Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler. Thromb. Vasc. Biol. 26, 1510–1516 [DOI] [PubMed] [Google Scholar]
- 21. Aikawa E., Aikawa M., Libby P., Figueiredo J. L., Rusanescu G., Iwamoto Y., Fukuda D., Kohler R. H., Shi G. P., Jaffer F. A., Weissleder R. (2009) Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation 119, 1785–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bouvet C., Moreau S., Blanchette J., de Blois D., Moreau P. (2008) Sequential activation of matrix metalloproteinase 9 and transforming growth factor β in arterial elastocalcinosis. Arterioscler. Thromb. Vasc. Biol. 28, 856–862 [DOI] [PubMed] [Google Scholar]
- 23. Kaartinen M. T., Murshed M., Karsenty G., McKee M. D. (2007) Osteopontin upregulation and polymerization by transglutaminase 2 in calcified arteries of Matrix Gla protein-deficient mice. J. Histochem. Cytochem. 55, 375–386 [DOI] [PubMed] [Google Scholar]
- 24. Faverman L., Mikhaylova L., Malmquist J., Nurminskaya M. (2008) Extracellular transglutaminase 2 activates β-catenin signaling in calcifying vascular smooth muscle cells. FEBS Lett. 582, 1552–1557 [DOI] [PubMed] [Google Scholar]
- 25. Nurminskaya M., Magee C., Faverman L., Linsenmayer T. F. (2003) Chondrocyte-derived transglutaminase promotes maturation of preosteoblasts in periosteal bone. Dev. Biol. 263, 139–152 [DOI] [PubMed] [Google Scholar]
- 26. Al-Jallad H. F., Nakano Y., Chen J. L., McMillan E., Lefebvre C., Kaartinen M. T. (2006) Transglutaminase activity regulates osteoblast differentiation and matrix mineralization in MC3T3-E1 osteoblast cultures. Matrix Biol. 25, 135–148 [DOI] [PubMed] [Google Scholar]
- 27. Johnson K. A., van Etten D., Nanda N., Graham R. M., Terkeltaub R. A. (2003) Distinct transglutaminase 2-independent and transglutaminase 2-dependent pathways mediate articular chondrocyte hypertrophy. J. Biol. Chem. 278, 18824–18832 [DOI] [PubMed] [Google Scholar]
- 28. Nurminsky D., Shanmugasundaram S., Deasey S., Michaud C., Allen S., Hendig D., Dastjerdi A., Francis-West P., Nurminskaya M. (2011) Transglutaminase 2 regulates early chondrogenesis and glycosaminoglycan synthesis. Mech Dev 128, 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson K. A., Polewski M., Terkeltaub R. A. (2008) Transglutaminase 2 is central to induction of the arterial calcification program by smooth muscle cells. Circ. Res. 102, 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beazley K. E., Deasey S., Lima F., Nurminskaya M. V. (2012) Transglutaminase 2-mediated activation of β-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscler. Thromb. Vasc. Biol. 32, 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beazley K. E., Banyard D., Lima F., Deasey S. C., Nurminsky D. I., Konoplyannikov M., Nurminskaya M. V. (2013) Transglutaminase inhibitors attenuate vascular calcification in a preclinical model. Arterioscler. Thromb. Vasc. Biol. 33, 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishizeki K., Saito H., Shinagawa T., Fujiwara N., Nawa T. (1999) Histochemical and immunohistochemical analysis of the mechanism of calcification of Meckel's cartilage during mandible development in rodents. J. Anat. 194, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ross R. (1971) The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J. Cell Biol. 50, 172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ashcroft G. S., Greenwell-Wild T., Horan M. A., Wahl S. M., Ferguson M. W. (1999) Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am. J. Pathol. 155, 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson K. A., Terkeltaub R. A. (2005) External GTP-bound transglutaminase 2 is a molecular switch for chondrocyte hypertrophic differentiation and calcification. J. Biol. Chem. 280, 15004–15012 [DOI] [PubMed] [Google Scholar]
- 36. Li D. Y., Brooke B., Davis E. C., Mecham R. P., Sorensen L. K., Boak B. B., Eichwald E., Keating M. T. (1998) Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280 [DOI] [PubMed] [Google Scholar]
- 37. Bunton T. E., Biery N. J., Myers L., Gayraud B., Ramirez F., Dietz H. C. (2001) Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 88, 37–43 [DOI] [PubMed] [Google Scholar]
- 38. Wagenseil J. E., Mecham R. P. (2012) Elastin in large artery stiffness and hypertension. J. Cardiovasc. Transl. Res. 5, 264–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simionescu A., Simionescu D. T., Vyavahare N. R. (2007) Osteogenic responses in fibroblasts activated by elastin degradation products and transforming growth factor-β1. Role of myofibroblasts in vascular calcification. Am. J. Pathol. 171, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hosaka N., Mizobuchi M., Ogata H., Kumata C., Kondo F., Koiwa F., Kinugasa E., Akizawa T. (2009) Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif. Tissue Int. 85, 523–529 [DOI] [PubMed] [Google Scholar]
- 41. Speer M. Y., Li X., Hiremath P. G., Giachelli C. M. (2010) Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell. Biochem. 110, 935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu L., Wigle D., Hinek A., Kobayashi J., Ye C., Zuker M., Dodo H., Keeley F. W., Rabinovitch M. (1994) The endogenous vascular elastase that governs development and progression of monocrotaline-induced pulmonary hypertension in rats is a novel enzyme related to the serine proteinase adipsin. J. Clin. Invest. 94, 1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cook K. S., Min H. Y., Johnson D., Chaplinsky R. J., Flier J. S., Hunt C. R., Spiegelman B. M. (1987) Adipsin. A circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science 237, 402–405 [DOI] [PubMed] [Google Scholar]
- 44. Flier J. S., Cook K. S., Usher P., Spiegelman B. M. (1987) Severely impaired adipsin expression in genetic and acquired obesity. Science 237, 405–408 [DOI] [PubMed] [Google Scholar]
- 45. Xu Y., Ma M., Ippolito G. C., Schroeder H. W., Jr., Carroll M. C., Volanakis J. E. (2001) Complement activation in factor D-deficient mice. Proc. Natl. Acad. Sci. 98, 14577–14582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. White R. T., Damm D., Hancock N., Rosen B. S., Lowell B. B., Usher P., Flier J. S., Spiegelman B. M. (1992) Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J. Biol. Chem. 267, 9210–9213 [PubMed] [Google Scholar]
- 47. Choy L. N., Rosen B. S., Spiegelman B. M. (1992) Adipsin and an endogenous pathway of complement from adipose cells. J. Biol. Chem. 267, 12736–12741 [PubMed] [Google Scholar]
- 48. Rosen B. S., Cook K. S., Yaglom J., Groves D. L., Volanakis J. E., Damm D., White T., Spiegelman B. M. (1989) Adipsin and complement factor D activity. An immune-related defect in obesity. Science 244, 1483–1487 [DOI] [PubMed] [Google Scholar]
- 49. Shields K. J., Stolz D., Watkins S. C., Ahearn J. M. (2011) Complement proteins C3 and C4 bind to collagen and elastin in the vascular wall. A potential role in vascular stiffness and atherosclerosis. Clin. Transl. Sci. 4, 146–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruan C. C., Zhu D. L., Chen Q. Z., Chen J., Guo S. J., Li X. D., Gao P. J. (2010) Perivascular adipose tissue-derived complement 3 is required for adventitial fibroblast functions and adventitial remodeling in deoxycorticosterone acetate-salt hypertensive rats. Arterioscler. Thromb. Vasc. Biol. 30, 2568–2574 [DOI] [PubMed] [Google Scholar]
- 51. Pimanda J. E., Donaldson I. J., de Bruijn M. F., Kinston S., Knezevic K., Huckle L., Piltz S., Landry J. R., Green A. R., Tannahill D., Göttgens B. (2007) The SCL transcriptional network and BMP signaling pathway interact to regulate RUNX1 activity. Proc. Natl. Acad. Sci. U.S.A. 104, 840–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yao Y., Jumabay M., Wang A., Boström K. I. (2011) Matrix Gla protein deficiency causes arteriovenous malformations in mice. J. Clin. Invest. 121, 2993–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schurgers L. J., Spronk H. M., Skepper J. N., Hackeng T. M., Shanahan C. M., Vermeer C., Weissberg P. L., Proudfoot D. (2007) Post-translational modifications regulate matrix Gla protein function. Importance for inhibition of vascular smooth muscle cell calcification. J. Thromb. Haemost. 5, 2503–2511 [DOI] [PubMed] [Google Scholar]
- 54. O'Young J., Liao Y., Xiao Y., Jalkanen J., Lajoie G., Karttunen M., Goldberg H. A., Hunter G. K. (2011) Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 133, 18406–18412 [DOI] [PubMed] [Google Scholar]
- 55. Yao Y., Shahbazian A., Boström K. I. (2008) Proline and γ-carboxylated glutamate residues in matrix Gla protein are critical for binding of bone morphogenetic protein-4. Circ. Res. 102, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 56. Dao H. H., Essalihi R., Graillon J. F., Larivière R., De Champlain J., Moreau P. (2002) Pharmacological prevention and regression of arterial remodeling in a rat model of isolated systolic hypertension. J. Hypertens. 20, 1597–1606 [DOI] [PubMed] [Google Scholar]
- 57. Boström K. I., Jumabay M., Matveyenko A., Nicholas S. B., Yao Y. (2011) Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ. Res. 108, 446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sharma B., Albig A. R. (2013) Matrix Gla protein reinforces angiogenic resolution. Microvasc. Res. 85, 24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]