

Abstract
Background
Despite malignant glioma vascularity, anti-angiogenic therapy is largely ineffective. We hypothesize that efficacy of the antiangiogenic agent cediranib is synergistically enhanced in intracranial glioma via combination with the late-stage autophagy inhibitor quinacrine.
Methods
Relative cerebral blood flow and volume (rCBF, rCBV), vascular permeability (Ktrans), and tumor volume were assessed in intracranial 4C8 mouse glioma using a dual-bolus perfusion MRI approach. Tumor necrosis and tumor mean vessel density (MVD) were assessed immunohistologically. Autophagic vacuole accumulation and apoptosis were assessed via Western blot in 4C8 glioma in vitro.
Results
Cediranib or quinacrine treatment alone did not alter tumor growth. Survival was only marginally improved by cediranib and unchanged by quinacrine. In contrast, combined cediranib/quinacrine reduced tumor growth by >2-fold (P < .05) and increased median survival by >2-fold, compared with untreated controls (P < .05). Cediranib or quinacrine treatment alone did not significantly alter mean tumor rCBF or Ktrans compared with untreated controls, while combined cediranib/quinacrine substantially reduced both (P < .05), indicating potent tumor devascularization. MVD and necrosis were unchanged by cediranib or quinacrine treatment. In contrast, MVD was reduced by nearly 2-fold (P < .01), and necrosis increased by 3-fold (P < .05, one-tailed), in cediranib + quinacrine treated vs untreated groups. Autophagic vacuole accumulation was induced by cediranib and quinacrine in vitro. Combined cediranib/quinacrine treatment under hypoxic conditions induced further accumulation and apoptosis.
Conclusion
Combined cediranib/quinacrine treatment synergistically increased antivascular/antitumor efficacy in intracranial 4C8 mouse glioma, suggesting a promising and facile treatment strategy for malignant glioma. Modulations in the autophagic pathway may play a role in the increased efficacy.
Keywords: antiangiogenesis, autophagy, cediranib, glioma, MRI
Malignant gliomas are highly aggressive vascular tumors for which prognosis remains extremely poor and for which novel therapeutic modalities are urgently required. The morbidity and mortality of malignant gliomas, especially the most common type, glioblastoma multiforme, can be attributed in part to their robust angiogenesis.1–5 Hence, anti-angiogenic therapy is potentially promising. However, anti-angiogenic monotherapy largely fails to induce durable responses with malignant glioma, with tumors often developing treatment resistance.2,3,5–7 Resistance can involve circumvention of angiogenic blockade, via activation of alternative angiogenic pathways or by increased invasion with existing vessel co-option.2,3,6 Additionally, various mechanisms are activated in tumor cells that enable adaption to the hypoxic stress exacerbated by anti-angiogenic treatment.8–11 As anti-angiogenic receptor tyrosine kinase (RTK) inhibitors also have direct cytotoxic effects on tumor cells that extend beyond angiogenic blockade, drug resistance to these agents can involve a number of other mechanisms as well.7 Recent studies have shown that autophagy, a cellular degradation pathway, is an important cytoprotective and drug resistance mechanism.12–15 During autophagy, cellular proteins and organelles are sequestered within characteristic double-layered autophagic vacuoles that subsequently fuse with lysosomes for bulk degradation. Activated during metabolic stress, autophagy provides energy and biosynthetic substrates and removes potentially toxic damaged proteins and organelles. However, while autophagy can function as a survival promoting pathway, the incomplete degradation of autophagic vacuoles, and their excessive accumulation, can trigger autophagic cell death, a cell death mechanism exhibiting significant crosstalk with apoptotic signaling.12,14–17 Known to play a role in autophagic cell death is lysosomal membrane permeabilization, which releases catabolic enzymes that can trigger cell death via caspase-dependent or -independent death pathways or through necrosis. The involvement of autophagy in cell death can be therapeutically exploited, as the implementation of late-stage autophagic inhibition when autophagic flux is stimulated can induce cell death.17–21 Antimalarial agents such as chloroquine and quinacrine effectively inhibit late-stage autophagy, through their lysosomotropic action, which enables them to selectively enter and concentrate within autophagic vacuoles.16,19–23 While chloroquine and its analog hydroxychloroquine are more commonly employed in cancer treatment, quinacrine is an FDA-approved drug for malaria that is well tolerated and has superior blood–brain barrier penetration to that of chloroquine/hydroxychloroquine, a key issue with CNS diseases.16,24 Various cancer therapeutics have been shown to activate autophagy, and a number of recent studies in different cancer models have demonstrated increased efficacy via combined autophagy inhibition.17–21,23,25 However, the combination of anti-angiogenic agents with autophagy inhibitors is a relatively new concept.8 Efficacy of the small molecule anti-angiogenic agent cediranib, a potent inhibitor of vascular endothelial growth factor (VEGF) receptors 1, 2, and 3, platelet derived growth factor (PDGF) receptors (α and β), and c-Kit,26 has not been investigated in combination with an autophagy inhibitor. We have recently hypothesized that the efficacy of cediranib will be synergistically enhanced in intracranial glioma via combination with the autophagy inhibitor quinacrine.27 Because tumor growth and angiogenesis involve interaction between tumor and host, they must ultimately be evaluated in vivo. For this study, we have employed the syngeneic intracranial mouse 4C8 glioma model, which fosters a seamless tumor–host interaction in immunocompetent mice.28,29 We have utilized a comprehensive perfusion MRI approach that quantifies key vascular biomarkers noninvasively in this model, measuring both vascular flow and permeability while also monitoring tumor growth.29 To accomplish this, we employed dynamic contrast-enhanced (DCE) MRI, producing high resolution maps of Ktrans, an index of vascular permeability that is a key biomarker of tumor neovasculature. Subsequently, dynamic susceptibility contrast (DSC) MRI was used to determine cerebral blood flow (CBF) and cerebral blood volume (CBV). This dynamic imaging approach provides functional vascular parameters difficult to obtain histologically and has revealed evidence that cediranib works synergistically with the autophagy inhibitor quinacrine to induce antivascular/antitumor efficacy and improve survival of treated mice. Combined histological analysis provided further evidence for the unique efficacy of the combined treatment. Measurements of autophagic and apoptotic markers in 4C8 glioma cell cultures supported the concept that the combinatorial treatment induces modulations in the autophagic pathway that are associated with tumor cell lethality.
Materials and Methods
Cell Culture and Tumor Inoculation
Mouse studies were conducted with the approval of the Oregon Health and Science University Institutional Animal Care and Use Committee and under the supervision of the OHSU Department of Comparative Medicine. The 4C8 mouse glioma cells (provided by Dr G. Yancey Gillespie of the University of Alabama–Birmingham) were grown in Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 50/50 Mix (Invitrogen), supplemented with 7% fetal bovine serum (FBS; Hyclone) and 1% l-glutamine (Sigma-Aldrich). Female C57BL/6 × DBA/2 F1 hybrid mice (B6D2F1) were purchased from Charles River Laboratories. Brain tumors were induced by the intracerebral injection of 0.5–1 × 106 4C8 cells, suspended in DMEM/F12 (5–10 μL) using a stereotaxic frame as previously described.8 To assess cell viability in response to treatment, MTS solution (CellTiter 96 AQueous MTS Reagent) and phenazine methosulfate solution were added to cells, and absorbance measured using an ELX800 Micro Plate Reader (Bio-Tek Instruments) at 490 nm.30
MRI Procedures
Starting at 10 days after tumor cell inoculation, mice were routinely imaged using contrast-enhanced (Magnevist i.p.) T1-weighted MRI to determine when tumor growth initiates. When tumors reached a cross-sectional area of ∼2 mm2, (generally at 2–3 wk post-inoculation), pretreatment perfusion MRI studies were implemented, after which mice were randomized into 4 treatment groups, with treatments starting within 24 h: (i) untreated, n = 6 (vehicle: 1% Tween in phosphate buffered saline [PBS], oral gavage, daily); (ii) cediranib, n = 6 (AZD2171, 6 mg/kg in 1% Tween/PBS, daily, oral gavage; Selleck Chemicals); (iii) quinacrine, n = 5 (50 mg/kg in PBS, daily, oral gavage; Sigma-Aldrich); and (iv) cediranib + quinacrine, n = 6 (daily). Biweekly perfusion MRI experiments were discontinued when right brain displacement by the tumor approached maximum or when mice showed neurological/behavorial changes or signs of physical deterioration, such as excessive weight loss or skull deformation, at which point mice were sacrificed and their brains were harvested and stored in 10% formalin for histology. For perfusion MRI, mice were initially anesthetized with a ketamine/xylazine mixture (15 mg xylazine/kg, 100 mg ketamine/kg) and one of the lateral tail veins was cannulated using a 30 gauge needle attached to polyethylene (PE-10) tubing, filled with sterile saline containing 15 U heparin/mL. MRI experiments employed a Bruker-Biospin 11.75 T small animal MR system with a Paravision 4.0 software platform, 9 cm inner diameter gradient set (750 mT/m), and a mouse head (20 mm inner diameter) quadrature radiofrequency transceiver coil (M2M Imaging). The mice were positioned with their heads immobilized with a specially designed head holder with adjustable ear pieces. Body temperature of the mice was monitored and maintained at 37°C using a warm air temperature control system (SA Instruments). Isoflurane (0.5%–2%) in 100% oxygen was administered and adjusted while respiration was monitored.
A coronal 25-slice T1-weighted image set was obtained using a spoiled gradient echo sequence (Paravision FLASH, 256 × 256 matrix, 98 μm in-plane resolution, 0.5 mm slice width, repetition time [TR] = 500 ms, echo time [TE] = 1.4 ms, flip angle [FA] = 60°, 1 average) to determine the perfusion MRI slice position within the tumor, which was matched to that used in any previous imaging sessions using brain/skull anatomical features. A T2-weighted image set (Paravision rapid acquisition with relaxation enhancement [RARE] spin echo sequence, same spatial geometry as for multislice T1 weighted, TR = 4000 ms, TEeffective = 32 ms, RARE factor 8, 1 average) was obtained to assess tumor volume. A fully relaxed (M0) spoiled gradient echo precontrast image (Paravision FLASH, TR = 6000 ms, TE = 1.2 ms, FA = 90°, 128 × 128 matrix, 1 slice, 1 mm slice thickness, 195 μm in-plane resolution) was obtained at the position of the subsequent DCE T1-weighted image series (450 images), which used the same parameters except with TR = 15.6 ms and FA = 20° (2.0 s/image), and with injection of gadopentetate dimeglumine (i.v., 10× diluted, 3.0 μL/g, 0.15 mmol/kg; Magnevist [Bayer Healthcare Pharmaceuticals]). DSC-MRI was then implemented (150 T2*-weighted images, Paravision FLASH, TR = 8, TE = 4.2 ms, FA = 5°, 128 × 128 matrix, 1 slice, 1 mm slice thickness, 195 μm in-plane resolution, 1 s/image) employing Feridex, a superparamagnetic iron oxide agent (4× diluted, 2.4 μL/g, 26.9 μg iron/g; Bayer Healthcare Pharmaceuticals). Both injections occurred at 30 s after initiation of the image series, at 1 mL/min, using a 150-μL saline/heparin chase.
Image Processing
DCE-MRI parameters were computed voxelwise using the Extended Tofts-Kety model31,32 and custom pharmacokinetic modeling software (developed by M. C. Schabel) written in MATLAB (MathWorks). Fully relaxed M0 image intensities were used to compute pre-injection longitudinal relaxation time, enabling quantitative concentration measurement and Ktrans (in min−1) estimates.29,33 The Monte Carlo blind estimation algorithm was used to determine the arterial input function directly from measured tumor curves.34,35 Calculation of DSC-MRI perfusion parameters followed the model-independent method described by Ostergaard et al36 using the Jim software package (Xinapse Systems). The arterial input function was determined from 12 nontumor brain pixels identified by an automatic scanning and selection routine that targeted brain parenchymal arterial microvessels.29 DSC data analysis was restricted to the first 50 postcontrast images in the series to bracket the susceptibility bolus intensity changes. Parametric maps were generated for CBF (in mL blood/100 g tissue/min), CBV (in blood volume percentage of total tissue volume), and mean transit time (MTT; in seconds). Analysis of the DCE/DSC-MRI parametric maps and T2 multislice images employed the Jim software package. Tumor volumes were calculated from the multislice T2-weighted images. The DSC-MRI parameters (relative [r]CBF, rCBV, rMTT) were reported relative to contralateral values to reduce measurement error and minimize effects from alterations in intracranial pressure, blood pressure, and depth of anesthesia.
Histology
At the termination of longitudinal MRI studies, mice were sacrificed and brains were stored in 10% formalin. Fixed tissue samples were paraffin-embedded, and 7-μm sections were mounted on slides. The slides were then processed and stained with hemotoxylin and eosin (H&E; Sigma-Aldrich) using standard methodologies. For each tumor sample (n = 4 tumor samples/treatment group), a total of 3 image fields (4× magnification) were positioned randomly across a minimum of 2 stained tumor sections and photographed using an Olympus BX50 microscope mounted with a Leica camera (DFC320) equipped with Leica Firecam v3.4.1. Necrotic regions within the image fields were quantified using the region-of-interest analysis tool of the Jim software package under the supervision of an experienced neuropathogist (R.L.W.) and averaged. In adjacent sections from 3 mice per treatment group, immunohistochemical vascular staining was performed after deparaffinized sections were subjected to antigen retrieval (5 min treatment at room temperature with 95% formic acid followed by incubation at 85–90°C in citrate buffer, pH 6.0, for 30 min), using mouse monoclonal primary antibody to CD31, clone JC70A (Dako), with hematoxylin counterstain. Slides were prepared using the Mouse-On-Mouse (M.O.M) Peroxidase Kit (Vector Labs) to prevent nonspecific background, and the presence of CD31 was investigated using the avidin-biotin complex method (Vector Labs), with diaminobenzidine. Slides were studied at 40× magnification, and the number of CD31 stained vessels was counted in 10 random fields across 2 sections for each tumor and averaged to obtain mean vessel density (MVD).
Western Blot Analysis
Cells were plated at a density of 4.8 × 104 cells per well in 6-well plates and treated with vehicle or drugs at the concentrations and times indicated in the legend for Fig. 7A and C. After the indicated time of treatment, cells were lysed by brief sonication in whole-cell lysis buffer (62.5 mM Tris, 10% glycerol, 1% sodium dodecyl sulfate, pH 6.8), and cellular proteins were collected in the supernatant fraction after centrifugation at 14 000 g for 10 min. Proteins were reduced by heating at 100°C for 5 min in 50 mM dithiothreitol with 0.05 bromphenol blue, electrophoresed in 4%–20% Tris-HCl gels, and transferred onto nitrocellulose using the NuPage electrophoresis system (Invitrogen). The membranes were probed with antibodies to microtubule-associated protein 1 light chain 3 (LC3) and cleaved caspase-3 (Cell Signaling) and actin (Santa Cruz Biotechnology). Membranes were treated with appropriate secondary antibody, exposed to enhanced chemiluminescence solutions (Invitrogen), and visualized using the Molecular Imager Gel Documentation System.
Fig. 7.
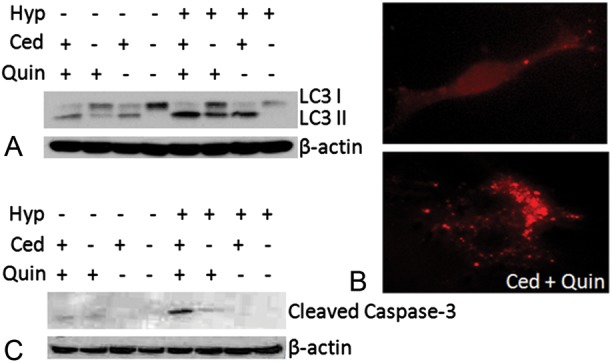
(A) Representative LC3 Western blots obtained from 4C8 cells grown under normoxic (20% O2) or hypoxic (0.5% O2) conditions for 10 h while untreated or exposed to cediranib (3 µM), quinacrine (0.8 µM), or combined cediranib/quinacrine. (B) RFP-LC3–expressing 4C8 cells were visualized using EVOS fl microscopy, grown under hypoxic conditions for 24 h while untreated or exposed to cediranib (2.5 µM), quinacrine (2.5 µM), or combined cediranib/quinacrine. (C) Representative cleaved caspase-3 Western blots obtained from 4C8 cells grown under normoxic (20% O2) or hypoxic (0.5% O2) conditions for 72 h while untreated or exposed to cediranib (2 µM), quinacrine (2 µM), or combined cediranib/quinacrine.
Microscopy for Red Fluorescent Protein–LC3 Expression
Cells were transduced with lentiviral particles (EMD Millipore) to stably express the red fluorescent protein (RFP)–LC3 fusion protein. Cells were then treated with the cediranib/quinacrine combination at the concentrations indicated in the legend for Fig. 7B for 24 h. Cells expressing RFP-LC3 were then visualized using an EVOS fl fluorescence microscope (Advanced Microscopy Group).
Statistics
Values are expressed as mean ± SEM. For perfusion MRI and CD31 immunohistochemistry results, the 2-tailed unpaired Student's t-test (NCSS Statistical Software) was employed to test for differences at 0, 3 ± 1, 6 ± 1, and 11 ± 1 days after tumor growth initiation versus the untreated group (time points corresponding to ≥50% untreated-group survival). Differences in median survival were tested using the log-rank test. The 1-tailed paired Student's t-test (tumors paired to tumor cell injection batch) was used for analysis of percent necrosis (MATLAB).
Results
The T2-weighted MR images depicted in Fig. 1 were obtained from mice at 14 days after tumor growth initiation and show clearly delineated tumors. Figure 1A–C indicates similar cross-sectional areas for untreated tumor in comparison with single-agent treatment with cediranib or quinacrine. In contrast, Fig. 1D indicates that the tumor from the combined cediranib/quinacrine treatment group was substantially smaller. Figure 2A shows tumor growth curves obtained from the MRI evaluations for the different treatment groups, with tumor volume plotted versus the time from tumor growth initiation. Rapid tumor growth was observed for untreated mice, increasing to over 70 mm3 over an 11-day period. The growth curves indicated that tumor growth was essentially unaltered by cediranib or quinacrine treatment. In contrast, tumor growth was markedly reduced with combined cediranib/quinacrine treatment throughout the treatment period (P < .05 vs untreated group), with tumor volume at 11 days of growth, indicating a 60% lower tumor volume than the untreated group. Consistent with this, exponential curves fit to the individual tumor volume versus time data provided mean exponential growth rate constants (in days−1) and indicated a substantially lower growth rate constant for combined cediranib/quinacrine (0.12 ± 0.01) versus no treatment (0.26 ± 0.03 P < .0014), with values for quinacrine (0.21 ± 0.017334) and cediranib (0.18 ± 0.017) that were not significantly different from those in the untreated group. The Kaplan–Meier survival curves in Fig. 2B indicate that no increase in median survival over the untreated group (9.5 ± 1.1) occurred with quinacrine-treated mice (10.0 ± 1.8). Cediranib-treated mice indicated only a modest increase in survival (14 ± 0.5, P < .05 vs untreated). In contrast, median survival was substantially extended with combined cediranib/quinacrine treatment (25.5 ± 1.9, P < .0001) versus the untreated group.
Fig. 1.

Representative T2-weighted coronal mouse brain MR images obtained at 14 days after tumor growth initiation for the 4 treatment groups: (A) untreated, (B) quinacrine-treated, (C) cediranib-treated, and (D) cediranib/quinacrine treated.
Fig. 2.
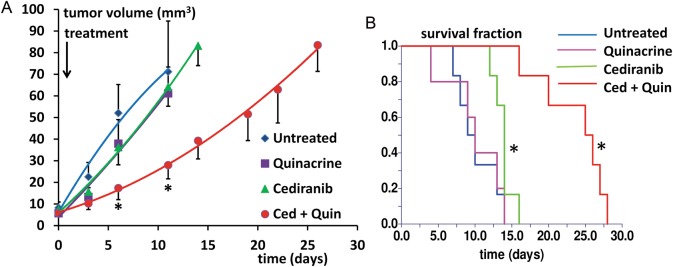
(A) Tumor growth curves are shown for the 4 treatment groups, in tumor volume vs time after tumor growth initiation. Curves are indicated for time points corresponding to ≥50% survival. *P < .05 vs untreated, indicated for time points corresponding to ≥50% untreated group survival. (B). Kaplan–Meier survival curves for the 4 treatment groups (in days from tumor growth initiation) *P < .05 vs untreated.
Tumor vascularity was evaluated with the dual bolus-tracking DCE/DSC perfusion MRI approach that we previously validated with intracranial 4C8 mouse glioma.29 Figure 3 indicates typical rCBF and Ktrans parametric maps obtained during both the first week after tumor growth initiation and the final week of MRI evaluation for representative tumors from each treatment group. While negligible Ktrans values were observed in nontumor regions due to the intact blood–brain barrier, Fig. 3A indicates that relatively high and heterogeneously distributed Ktrans values were observed in untreated tumor. High values of rCBF were also observed, with local “hot spots” exceeding flow in contralateral regions by more than 5-fold, also with a heterogeneous distribution. This pattern, observed throughout the tumor growth period, is consistent with the robust yet heterogeneously distributed and permeable vasculature typical of malignant glioma.1,3,9 Figure 3B shows that similar characteristics are observed in a quinacrine-treated mouse tumor. Figure 3C shows parametric maps obtained from a cediranib-treated mouse tumor. Consistent with cediranib's anti-angiogenic mode of action was the development of a very poorly perfused tumor core. Notably, however, the tumor clearly retained a well-vascularized rim. Persistence of an angiogenic tumor rim, in a continually expanding tumor, demonstrates resilience of gliomas in the face of angiogenic blockade, congruent with clinical observations.2,3,6,7 While poorly perfused tumor core regions were consistently observed with cediranib treatment, persistence of regions with robust tumor vascularity was also a consistent finding. In contrast, Fig. 3D indicates that with combined cediranib/quinacrine treatment, there was a dramatic reduction in tumor rCBF and Ktrans in comparison with untreated tumors, starting at the early treatment stage and continuing to the end of treatment. At the latter stage of treatment, the rCBF map indicates an extensive poorly perfused tumor core region, involving a much larger tumor fraction than was observed with cediranib alone. The vascularized rim is also noticeably thinner than that observed following single-agent cediranib treatment, with greatly reduced Ktrans and CBF values compared with those observed in cediranib-treated or untreated tumors. The marked reduction in these key flow and permeability vascular biomarkers indicates greatly increased antivascular efficacy with combined treatment. Figure 4 displays mean tumor rCBF, rCBV, rMTT, and Ktrans values plotted versus the time from tumor growth initiation for the 4 different treatment groups. Figure 4A indicates that pretreatment rCBF levels were uniformly high (∼2.8) in the different groups. Consistent with the parametric maps shown in Fig. 3, in the untreated group, as well as in cediranib-treated and quinacrine-treated groups, mean tumor rCBF remained relatively unchanged at this level during the following period of tumor growth. In contrast, with combined cediranib/quinacrine treatment, large and sustained decreases in mean rCBF occurred starting at early treatment stages (P < .05 vs untreated). Unlike rCBF, rCBV increased substantially (Fig. 4B) throughout the period of tumor growth in the untreated group. The relatively robust increase in rCBV, in comparison with that of rCBF, suggests the development of a tumor vascular network that is increasingly inefficient, consistent with our previous findings with 4C8 glioma.29 Quinacrine treatment did not substantially alter tumor rCBV in comparison with the untreated group. Cediranib treatment moderately attenuated the increase in rCBV, indicating lower values in comparison with the untreated group at 11 days after initiation of tumor growth. Combination of cediranib with quinacrine produced a substantially greater effect, however, with rCBV decreasing rather than increasing, to values significantly below those of the untreated group throughout the treatment period. Figure 4C indicates that rMTT, which is inversely related to vascular efficiency, increased substantially with tumor growth for the untreated group, consistent with the robust increase in rCBV in comparison with rCBF. Quinacrine did not substantially alter rMTT in comparison with no treatment. Consistent with its effects on rCBV, cediranib tended to attenuate rMTT values below those observed in the untreated group (nonsignificant vs untreated). Combined cediranib/quinacrine treatment entirely prevented increases in rMTT with tumor growth, resulting in rMTT values substantially lower than those observed in the untreated group. Figure 4D indicates that mean tumor Ktrans values gradually increased with tumor growth, consistent with development of permeable tumor neovasculature. Quinacrine-treated mice indicated a similar pattern, while the cediranib-treated group exhibited a tendency toward decreased Ktrans values in the later stages of tumor growth (nonsignificant vs untreated), likely due at least in part to decreased vascular permeability, consistent with cediranib's VEGF receptor inhibition. With combined cediranib/quinacrine treatment, marked decreases in Ktrans occurred, to levels substantially below those in all the other groups. As Ktrans is, under most conditions, proportional to the permeability surface area product per unit volume of tissue, the marked decrease is likely to be due primarily to decreased vessel surface area in conjunction with tumor devascularization, given the substantial reductions in tumor rCBF and rCBV that also occurred, suggesting a greatly reduced vascular network.37 Taken together, the perfusion MRI results indicate that combined cediranib/quinacrine treatment has a profound effect on tumor vasculature, inducing a synergistic increase in antivascular/antitumor efficacy and effectively slowing tumor growth and extending survival in mouse 4C8 glioma. Histological studies performed after completion of the longitudinal MRI studies confirmed the perfusion MRI results and provided further information regarding tumor pathology. Representative histological tumor sections with CD31 vascular staining are shown in Fig. 5A–D and indicate clearly demarcated tumor vessels (see arrows). Consistent with the perfusion MRI results, we generally observed a greater prevalence of vessels at tumor borders. Multiple vessels are indicated in Fig. 5A–C, in sections from untreated, quinacrine-treated, and cediranib-treated tumors. In contrast, in Fig. 5D, a section from a cediranib/quinacrine-treated tumor shows substantial necrosis (indicated by nonspecific staining) and fewer vessels. In Fig. 5E–H, representative H&E sections from the various treatment groups are shown and clearly indicate regions of tumor necrosis. While similar levels of necrosis were observed in the sections from untreated, quinacrine-treated, and cediranib-treated tumors, necrosis was comparatively increased in sections from cediranib/quinacrine-treated tumors. Figure 6 indicates mean values of MVD and percent necrosis that were carefully quantified from multiple CD31 and H&E stained sections, respectively. Figure 6A indicates that the MVD values were very similar for the untreated, cediranib, and quinacrine groups. In contrast, MVD was reduced in the cediranib/quinacrine-treated group by nearly 2-fold in comparison with the other groups (P < .01 vs untreated). Figure 6B indicates that similar levels of tumor necrosis were observed in the untreated, quinacrine, and cediranib groups. In contrast, necrosis was markedly elevated, by ∼3-fold, in the cediranib/quinacrine group (P < .05, one-tailed paired t-test). The immunohistological vascular staining and MVD quantification shown in Figs 5 and 6 are very consistent with the perfusion MRI data in Figs 3 and 4, indicating an absence of antivascular efficacy with single-agent treatment and markedly improved efficacy with combined cediranib/quinacrine treatment. That the marked devascularization and reduced tumor growth shown in Figs 1–6 occurs in the context of greatly increased tumor necrosis further documents the marked antitumor efficacy of the combined cediranib/quinacrine treatment.
Fig. 3.

rCBF and Ktrans (min−1) parametric maps (nonbrain regions masked) obtained from representative tumors from each treatment group both during the first week after tumor growth initiation and during the final week of MRI evaluation (CBF color scale is relative: contralateral hues set to ∼unity).
Fig. 4.

Mean tumor (A) rCBF, (B) rCBV, (C) rMTT, and (D) Ktrans (min−1) values plotted vs the time from tumor growth initiation for the 4 different treatment groups. Curves are indicated for time points corresponding to ≥50% survival. *P < .05 vs untreated, indicated for time points corresponding to ≥50% untreated group survival.
Fig. 5.
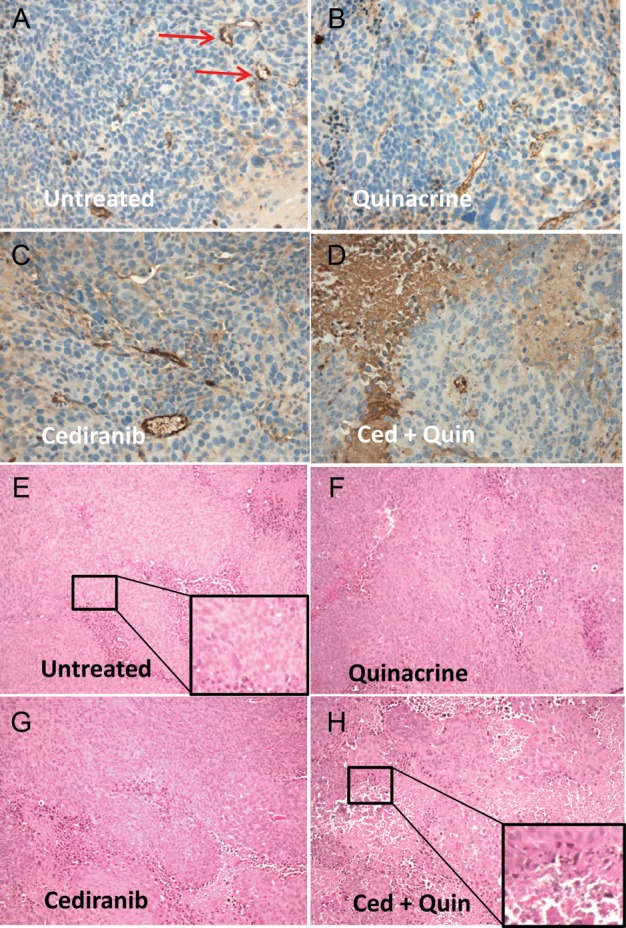
(A–D) Representative histological tumor sections with CD31 vascular staining (brown) and hematoxylin nuclear counterstain (blue) from the 4 treatment groups. Discrete staining is associated with vascular endothelial cells, whereas more diffuse and variable staining is nonspecific and associated with tumor necrosis. Microvessel examples are marked by arrows. (E–H) Representative H&E stained sections from the 4 treatment groups, indicating areas of necrosis, which appear as condensed cells in areas of vacuolated tissue. Insets indicate enlarged areas of the section as indicated.
Fig. 6.

(A) MVD quantified from multiple CD31 stained sections for the 4 treatment groups. *P < .01 vs untreated. (B) Percent necrosis, quantified from multiple H&E stained sections, for the 4 treatment groups. *P < .05 vs untreated (1-tailed paired t-test).
We employed 4C8 glioma cells in culture to evaluate the possible effects of the various treatment conditions on autophagy and apoptosis. Cediranib and quinacrine decreased 4C8 glioma cell viability, with half-maximal inhibitory concentration values of 2.7 ± 0.1 µM and 3.2 ± 0.2 µM, respectively. Figure 7A indicates a representative Western blot of LC3 obtained from 4C8 cells grown under normoxic (20% O2) as well as hypoxic (0.5% O2) conditions38 and exposed to cediranib, quinacrine, or combined cediranib/quinacrine treatment. LC3 comprises both the cytosolic form, LC3-I, and its lipidated form, LC3-II, which is converted from LC3-I and is a well-accepted biomarker of autophagic vacuoles. LC3-II increases are generally indicative of increased autophagic flux and/or late-stage autophagy inhibition. Such increases are clearly observed in response to cediranib, strongly suggesting that it activates autophagic flux. The increase in response to cediranib was potentiated by the presence of hypoxia, consistent with the known activation of autophagic flux by hypoxia.8–11,39 Quinacrine also induced an increase in LC3-II accumulation under normoxic conditions, likely through inhibition of autophagic vacuole degradation, and as with cediranib, the increase was potentiated by hypoxia. Combined cediranib/quinacrine resulted in a strong accumulation of LC3-II and notably in the presence of hypoxia induced the highest levels that were observed. Figure 7B shows representative fluorescence microscopy images of 4C8 cells expressing RFP-LC3 under hypoxic conditions, with and without exposure to combined cediranib/quinacrine. While the untreated cells indicate a diffuse RFP-LC3 distribution indicative of cytosolic LC3-I, cells exposed to combined cediranib/quinacrine clearly indicate a punctate distribution, indicative of LC3-II, confirming the increased presence of autophagic vacuoles. Figure 7C indicates representative cleaved caspase-3 Western blots, which revealed that apoptosis was markedly increased by combined cediranib/quinacrine under hypoxic conditions in comparison with all other treatment groups. As these conditions were also associated with the highest LC3-II levels, the observations are consistent with the concept that cediranib in combination with hypoxia can substantially stimulate autophagic flux in 4C8 glioma cells, leading to a toxic accumulation of autophagic vacuoles in the presence of late-stage autophagy inhibition.
Discussion
Malignant glioma remains a frustratingly difficult disease to treat. Approximately 12 000–15 000 cases per year are diagnosed in the United States alone; and with current treatment options, the disease has a prognosis of 25% survival after 2 years, with only a 14-month average survival for the most common malignant glioma, glioblastoma multiforme.1–4 Malignant glioma is characterized by robust angiogenesis, proliferation, and invasion.1–5 It often exhibits marked treatment resistance, and despite its elevated vascularity, anti-angiogenic monotherapy is of limited utility.2,3,5–7 The current study employed the syngeneic intracranial 4C8 mouse glioma model, which is a clone of the MOCH-1 tumor that arose spontaneously in the brain of a B6D2F1 mouse transgenic for myelin basic protein promoter–driven c-neu. The model employs immunocompetent wild-type F1 hybrid mice and is characterized by aggressive tumor growth with robust neovasculature and core necrosis.28,29 A unique perfusion MRI approach was employed with the 4C8 glioma model in combination with immunohistological approaches to investigate the effects of separate and combined treatment with cediranib and quinacrine. Our perfusion MRI results demonstrated that while single-agent treatment with cediranib in the 4C8 glioma model did not decrease mean tumor rCBF in comparison with untreated tumors, poorly perfused tumor core regions were prevalent in cediranib-treated mice, in conjunction with robustly vascularized tumor rim regions. These findings were corroborated by immunohistological determinations of MVD, averaged across the tumor, which were found to be unchanged with cediranib treatment alone. Mean rCBV increases were attenuated with cediranib, in conjunction with a tendency to attenuate mean rMTT increases. In combination with the absence of mean rCBF reduction, this may suggest an augmented vascular efficiency with cediranib, as has been proposed to occur at early stages of anti-angiogenic monotherapy, due to normalization of abnormal and dilated tumor vasculature.3,40–42 The time window of effective vascular normalization is reportedly brief in orthotopic rodent brain tumor models, however, and improvements in overall perfusion should not be inferred given the heterogeneous vascularity we observed in cediranib-treated tumors, particularly at later treatment stages.40–42 Consistent with this, Kamoun et al41 reported a transient vascular normalization in intracranial mouse glioma, maximizing at day 2 of cediranib treatment, after which tumor core microvascular density significantly decreased. In the current study, cediranib-treated mice also indicated a tendency to exhibit decreased Ktrans in the later stages of tumor growth, consistent with a decreased vascular permeability and with cediranib's inhibition of VEGF receptors.26,41,43,44 As has been previously suggested, this effect may contribute to the modest increases in survival that we and others have observed with cediranib in glioma models in the absence of altered tumor growth, related to reduction of peritumoral edema.41 While our study is consistent with previous studies reporting reduced rCBV and vascular permeability in cediranib-treated glioma, to our knowledge the current study is the first to report mean tumor rCBF measurements in a cediranib-treated preclinical glioma model.41,43,45 Our results clearly demonstrated, with both perfusion MRI and immunohistological approaches, that while cediranib may have limited perfusion in localized regions of the tumor core, it did not prevent a vigorous neovascularization process in the 4C8 mouse glioma model. Cediranib had little effect on tumor growth and only modest effects on survival, consistent with previous preclinical and clinical studies indicating that cediranib and other small molecule anti-angiogenic RTK inhibitors with similar molecular targets have limited efficacy as single-agent cancer therapeutics, particularly with malignant glioma.2,3,5–7,41,46,47 Our study is the first to document that in vivo single-agent treatment with quinacrine induces no measureable effects on mean rCBF, rCBV, Ktrans, tumor growth, or survival in malignant glioma. Similarly it did not alter tumor necrosis or MVD. In contrast, our study provides dramatic evidence for a strong synergy between cediranib and quinacrine in terms of both antivascular and antitumor efficacy. Combined cediranib and quinacrine induced markedly reduced tumor perfusion, consistent with the sharply reduced histological determinations of MVD. Consistent with this, tumor necrosis was found to be 3-fold greater in the combined cediranib/quinacrine than in untreated or single agent–treated tumors. Tumor growth was slowed by more than a factor of 2, resulting in substantially increased survival. The effects reveal a potentially important therapeutic avenue for treatment of malignant glioma, a disease for which prognosis is extremely poor and therapeutic options are limited. The similarity of cediranib's molecular targets to those of other recently developed RTK anti-angiogenic agents7 suggests that the novel and effective combination tested in the current study could represent an important new combinational paradigm, which could greatly increase the utility of this class of agent.
The goal of the current study was to investigate the in vivo antivascular and antitumor efficacy of a novel treatment combination. We clearly demonstrated that the combination of cediranib and quinacrine administration exhibited a potent synergy in this regard, in terms of tumor devascularization, necrosis, growth, and increased survival. While the underlying mechanisms were not fully characterized in this initial study, we obtained evidence suggesting that modulation of autophagy could play a key role. We employed controlled cell culture conditions in order to carefully investigate the interaction between hypoxia and the induction of autophagic vacuole accumulation and apoptosis via the single and combined administration of cediranib and quinacrine. These experiments demonstrated that cediranib and hypoxia each induced substantially increased levels of the autophagic vacuole marker LC3-II in 4C8 glioma, and increased it further when applied in combination. The presence of quinacrine further increased LC3-II levels with cediranib or hypoxia, with the highest levels being observed with combined cediranib/quinacrine treatment under hypoxic conditions. Fluorescent microscopy confirmed the increased presence of autophagic vacuoles under this condition. Importantly, combined cediranib/quinacrine treatment under hypoxic conditions not only induced maximal autophagic vacuole accumulation but was also associated with maximal activation of apoptosis. Demonstration of these cytotoxic effects in glioma cell culture provides evidence that despite cediranib's known anti-angiogenic capacity, the therapeutic efficacy may substantially derive from the direct targeting of glioma cells. The results strongly suggest that the in vivo therapeutic efficacy derives from more than anti-angiogenic effects and are consistent with the concept that cediranib and hypoxia each drive autophagic flux, which in combination with quinacrine's well-documented ability to induce late-stage autophagy inhibition promotes autophagic vacuole accumulation and tumor cell death.8,12–23 As autophagy regulation primarily occurs through signaling of mammalian target of rapamycin (mTOR) signaling, a possible mechanism by which autophagy activation with cediranib may occur is via its known inhibition of VEGF and PDGF receptor signaling, which could have downstream inhibitory effects on the phosphatidylinositol-3 kinase/Akt/mTOR signaling axis.48,49 Autophagy is activated strongly by hypoxic/nutrient stress, and hence the exacerbation of hypoxia in vivo through cediranib's anti-angiogenic effects represents another possible avenue for autophagy activation.8 Although mean tumor rCBF did not decrease with single-agent cediranib treatment, considerable perfusion heterogeneity was observed in cediranib-treated tumors, suggesting that hypoxia could be increased in local regions, particularly in the tumor core regions. However, while cediranib's exacerbation of hypoxia may contribute to autophagic flux stimulation, it may not be a required element, as it is well documented that a state of chronic hypoxia exists in most tumors.1,4,9,10 This is particularly evident in malignant glioma, which is characterized by focal hypoxic regions with pseudopalisading necrosis.1,4
In summary, using a novel treatment combination with the intracranial 4C8 mouse glioma model, we documented a synergistic increase in antivascular/antitumor efficacy, using a comprehensive DCE/DSC perfusion MRI approach and immunohistology. The antiangiogenic RTK inhibitor cediranib in combination with the autophagy inhibitor quinacrine produced a powerful antivascular and antitumor effect that far exceeded treatment with either agent alone, with markedly decreased tumor perfusion, increased tumor necrosis, decreased tumor growth, and increased survival. In vitro studies revealed a direct modulatory effect of the drug combination on the autophagic pathway, which was potentiated by hypoxia and accompanied by apoptotic cell death. The data strongly suggest that the combination offers a new and promising treatment avenue for malignant glioma.
Conflict of interest statement. None declared.
Funding
This work was supported by NIH/NCI grant 1R21 CA114279–01A2 (M.M.P.) and NIH/NCI grant 1R21 CA167302–01 (M.M.P.).
Acknowledgments
We gratefully thank Mary Stenzel-Poore, PhD for generously providing cell culture facilities; Peter Kurre, MD for generously providing a hypoxic chamber; and Kerri Forquer for expert assistance with immunohistology.
References
- 1.Gladson CL, Prayson RA, Liu WM. The pathobiology of glioma tumors. Annu Rev Pathol. 2010;5:33–50. doi: 10.1146/annurev-pathol-121808-102109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol. 2009;5(11):610–620. doi: 10.1038/nrneurol.2009.159. [DOI] [PubMed] [Google Scholar]
- 3.Rahman R, Smith S, Rahman C, Grundy R. Antiangiogenic therapy and mechanisms of tumor resistance in malignant glioma. J Oncol. 2010;2010:251231. doi: 10.1155/2010/251231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65(6):529–539. doi: 10.1097/00005072-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Tabatabai G, Stupp R. Primetime for antiangiogenic therapy. Curr Opin Neurol. 2009;22(6):639–644. doi: 10.1097/WCO.0b013e328332ba28. [DOI] [PubMed] [Google Scholar]
- 6.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13(1):1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773–1783. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7(2):134–153. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rouschop KM, Wouters BG. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med. 2009;9(4):417–424. doi: 10.2174/156652409788167131. [DOI] [PubMed] [Google Scholar]
- 11.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 12.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6(3):322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge P, Luo Y, Fu S, Ji X, Ling F. Autophagy: a strategy for malignant gliomas' resistance to therapy. Med Hypotheses. 2009;73(1):45–47. doi: 10.1016/j.mehy.2008.11.047. [DOI] [PubMed] [Google Scholar]
- 14.Hoyer-Hansen M, Jaattela M. Autophagy: an emerging target for cancer therapy. Autophagy. 2008;4(5):574–580. doi: 10.4161/auto.5921. [DOI] [PubMed] [Google Scholar]
- 15.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 16.Geng Y, Kohli L, Klocke BJ, Roth KA. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol. 2010;12(5):473–481. doi: 10.1093/neuonc/nop048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaza N, Kohli L, Roth KA. Autophagy in brain tumors: a new target for therapeutic intervention. Brain Pathol. 2012;22(1):89–98. doi: 10.1111/j.1750-3639.2011.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amaravadi R. Autophagy can contribute to cell death when combining targeted therapy. Cancer Biol Ther. 2009;8(21):130–133. doi: 10.4161/cbt.8.21.10416. [DOI] [PubMed] [Google Scholar]
- 19.Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183(1):101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirzoeva OK, Hann B, Hom YK, et al. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl) 2011;89(9):877–889. doi: 10.1007/s00109-011-0774-y. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu S, Takehara T, Hikita H, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2011;131(3):548–557. doi: 10.1002/ijc.26374. [DOI] [PubMed] [Google Scholar]
- 22.Marceau F, Bawolak MT, Bouthillier J, Morissette G. Vacuolar ATPase-mediated cellular concentration and retention of quinacrine: a model for the distribution of lipophilic cationic drugs to autophagic vacuoles. Drug Metab Dispos. 2009;37(12):2271–2274. doi: 10.1124/dmd.109.028480. [DOI] [PubMed] [Google Scholar]
- 23.Gupta A, Roy S, Lazar AJ, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST) Proc Natl Acad Sci U S A. 2010;107(32):14333–14338. doi: 10.1073/pnas.1000248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yung L, Huang Y, Lessard P, et al. Guglielmo BJ, Pharmacokinetics of quinacrine in the treatment of prion disease. BMC Infect Dis. 2004;4:53. doi: 10.1186/1471-2334-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12(12):1395–1403. doi: 10.1634/theoncologist.12-12-1395. [DOI] [PubMed] [Google Scholar]
- 26.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65(10):4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 27.Lobo MRHE, Kurre P, Schabel MC, Gillespie GY, Pike MM. Autophagy inhibitor quinacrine synergistically enhances anti-angiogenic efficacy of cediranib in intracranial mouse glioma [abstract]. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31–Apr 4; Chicago, IL. Philadelphia (PA). AACR. Cancer Research; 2012. 72(8 Suppl) [Google Scholar]
- 28.Dyer CA, Philibotte T. A clone of the MOCH-1 glial tumor in culture: multiple phenotypes expressed under different environmental conditions. J Neuropathol Exp Neurol. 1995;54(6):852–863. doi: 10.1097/00005072-199511000-00012. [DOI] [PubMed] [Google Scholar]
- 29.Pike MM, Stoops CN, Langford CP, Akella NS, Nabors LB, Gillespie GY. High-resolution longitudinal assessment of flow and permeability in mouse glioma vasculature: Sequential small molecule and SPIO dynamic contrast agent MRI. Magn Reson Med. 2009;61(3):615–625. doi: 10.1002/mrm.21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Samadi AK, Roby KF, Timmermann B, Cohen MS. Inhibition of cell growth and induction of apoptosis in ovarian carcinoma cell lines CaOV3 and SKOV3 by natural withanolide Withaferin A. Gynecol Oncol. 2011;124(3):606–612. doi: 10.1016/j.ygyno.2011.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tofts PS, Brix G, Buckley DL, et al. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: standardized quantities and symbols. J Magn Reson Imaging. 1999;10(3):223–232. doi: 10.1002/(sici)1522-2586(199909)10:3<223::aid-jmri2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 32.Sourbron SP, Buckley DL. Tracer kinetic modelling in MRI: estimating perfusion and capillary permeability. Phys Med Biol. 2012;57(2):R1–R33. doi: 10.1088/0031-9155/57/2/R1. [DOI] [PubMed] [Google Scholar]
- 33.Schabel MC, Parker DL. Uncertainty and bias in contrast concentration measurements using spoiled gradient echo pulse sequences. Phys Med Biol. 2008;53(9):2345–2373. doi: 10.1088/0031-9155/53/9/010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schabel MC, DiBella EV, Jensen RL, Salzman KL. A model-constrained Monte Carlo method for blind arterial input function estimation in dynamic contrast-enhanced MRI: II. In vivo results. Phys Med Biol. 2010;55(16):4807–4823. doi: 10.1088/0031-9155/55/16/012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schabel MC, Fluckiger JU, DiBella EV. A model-constrained Monte Carlo method for blind arterial input function estimation in dynamic contrast-enhanced MRI: I.: simulations. Phys Med Biol. 2010;55(16):4783–4806. doi: 10.1088/0031-9155/55/16/011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ostergaard L, Weisskoff R, Chesler D, Gyldensted C. High resolution measurement of cerebral blood flow using intravascular tracer bolus passages. Part I: Mathematical approach and statistical analysis. MRM. 1996;36:715–725. doi: 10.1002/mrm.1910360510. [DOI] [PubMed] [Google Scholar]
- 37.Barrett T, Brechbiel M, Bernardo M, Choyke PL. MRI of tumor angiogenesis. J Magn Reson Imaging. 2007;26(2):235–249. doi: 10.1002/jmri.20991. [DOI] [PubMed] [Google Scholar]
- 38.Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4(7):423–436. doi: 10.1158/1541-7786.MCR-06-0150. [DOI] [PubMed] [Google Scholar]
- 39.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goel S, Wong AH, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med. 2012;2(3):a006486. doi: 10.1101/cshperspect.a006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamoun WS, Ley CD, Farrar CT, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009;27(15):2542–2552. doi: 10.1200/JCO.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6(6):553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 43.Bradley DP, Tessier JJ, Lacey T, et al. Examining the acute effects of cediranib (RECENTIN, AZD2171) treatment in tumor models: a dynamic contrast-enhanced MRI study using gadopentate. Magn Reson Imaging. 2009;27(3):377–384. doi: 10.1016/j.mri.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 44.Farrar CT, Kamoun WS, Ley CD, et al. Sensitivity of MRI tumor biomarkers to VEGFR inhibitor therapy in an orthotopic mouse glioma model. PLoS One. 2011;6(3):e17228. doi: 10.1371/journal.pone.0017228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farrar CT, Kamoun WS, Ley CD, et al. Sensitivity of MRI tumor biomarkers to VEGFR inhibitor therapy in an orthotopic mouse glioma model. PLoS One. 2011;6(3):e17228. doi: 10.1371/journal.pone.0017228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorensen AG, Emblem KE, Polaskova P, et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012;72(2):402–407. doi: 10.1158/0008-5472.CAN-11-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeuchi H, Kanzawa T, Kondo Y, Kondo S. Inhibition of platelet-derived growth factor signalling induces autophagy in malignant glioma cells. Br J Cancer. 2004;90(5):1069–1075. doi: 10.1038/sj.bjc.6601605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gomez-Rivera F, Santillan-Gomez AA, Younes MN, et al. The tyrosine kinase inhibitor, AZD2171, inhibits vascular endothelial growth factor receptor signaling and growth of anaplastic thyroid cancer in an orthotopic nude mouse model. Clin Cancer Res. 2007;13(15 Pt 1):4519–4527. doi: 10.1158/1078-0432.CCR-06-2636. [DOI] [PubMed] [Google Scholar]