

Abstract
The use of adjuvant molecules that have the ability to restore the susceptibility of multi-drug resistant bacteria, such as MRSA, to clinically available antibiotics is a promising alternative to the development of novel antimicrobials. We report an extremely potent small molecule that, at sub-MIC levels, lowers the MIC of oxacillin against a number of MRSA strains by up to 512-fold. Preliminary mechanistic investigations indicate that the VraSR two-component system plays a role in the activity of this compound.
Keywords: antibiotic resistance, MRSA, two-component system, 2-aminoimidazole, adjuvant
The emergence of resistance to multiple antimicrobial agents in pathogenic bacteria is a significant global public health threat and causes considerable patient mortality and morbidity. In the United States, methicillin resistant Staphylococcus aureus (MRSA) accounts for 80% of all hospital-acquired S. aureus infections.[1] In 2005, almost 95,000 people acquired MRSA infections in the United States, of which nearly 19,000 people died - more than die annually from HIV/AIDS, emphysema, Parkinson’s disease, and homicide combined.[2] In addition, MRSA infections, traditionally only observed among hospitalized patients, have now become prevalent outside of the hospital setting, with the emergence of community associated MRSA (CA-MRSA).[3] In the USA, the USA300 clone is the most prevalent CA-MRSA clone.[3] β-Lactam antibiotics have typically been the most effective drugs for the treatment of infections caused by staphylococci; however increasing occurrence of resistance means they are often no longer efficacious.
Although the design of new antibiotics could address the threat of multi-drug resistance, the sobering fact is that there have been very few novel classes of antibiotics marketed in the last four decades.[4] Furthermore, bacteria inevitably develop resistance to all microbicidal agents that are introduced into the clinic.[5] An orthogonal approach to the development of new antibiotic entities is the use of small molecule adjuvants.[6] Recently, our group and others, have been exploring the use of small molecules that are able to render MDR bacteria sensitive to the effects of conventional antibiotics.[7–10] One of the key features of this approach is that by targeting pathways within the bacteria that, by themselves, are not essential for bacterial growth the rate of resistance acquisition may be significantly reduced.
We have developed a class of 2-aminoimidazole/triazole conjugates (2-AIT) that are able to inhibit and disperse biofilms of several pathogenic bacteria.[11,12] Some members of this class of compounds also possess the ability to suppress the resistance of planktonic bacteria to β-lactam antibiotics.[7] We recently reported (2-AIT) 1 (Scheme 1), which lowers the minimum inhibitory concentration (MIC) of oxacillin against an Iberian MRSA clone when co-dosed at sub-MIC levels.[13] Herein, we report that this compound exhibits similar activity against a USA300 MRSA clone. Analogue synthesis resulted in the identification of a significantly more active derivative that reduced oxacillin MICs up to 512-fold, taking the MIC significantly below the breakpoint for clinical resistance. Finally, screening this compound against a number of USA300 MRSA mutant strains indicates that the VraSR two-component system (TCS) plays a role in the activity of this compound.
Scheme 1.
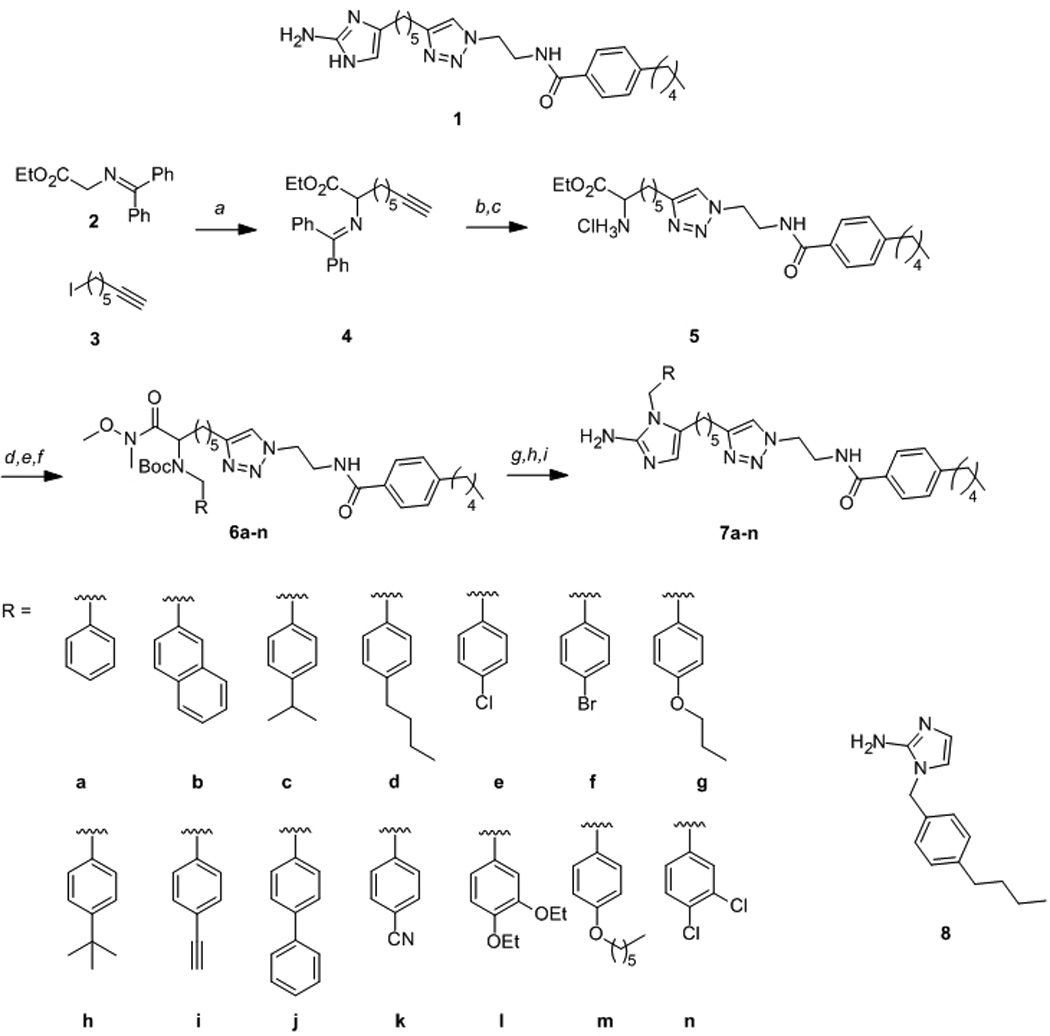
Synthesis of N1-substituted 2-AITs. Reagents and conditions: a) tBuOK, THF, −78 °C–rt 72 h; b) N-(2-azidoethyl)-4-pentylbenzamide, CuSO4, sodium ascorbate, H2O/EtOH/CH2Cl2, rt 3 h; c) 2 M HCl/Et2O, rt, 2 h; d)i RCHO, LiOH.H2O, MeOH, rt 2 h, ii NaBH4, rt, 1 h; e) Boc2O, TMAH, CH3CN, rt, 16 h; f) HN(OMe)Me.HCl, iPrMgCl, THF, −20 °C–rt, 18 h; g) DIBAL-H, THF, −78 °C, 2 h; h) 9:1 CH2Cl2/TFA, rt, 15 min; i) H2O/EtOH, pH 4.3, H2NCN, 95 °C, 3 h.
The MIC of compound 1 against a USA300 MRSA strain (ATCC BAA-1556) was determined to be 50 µM (23.7 µg/mL) using the CLSI broth microdilution protocol[14]. We subsequently determined the oxacillin MIC in the absence and presence of 40% of the MIC of compound 1 (20 µM, 9.5 µg/mL). Compound 1 lowered the oxacillin MIC by eight-fold at this concentration, from 32 µg/mL to 4 µg/mL. With the aim of developing a compound with increased activity, a series of analogues of compound 1 in which substituents were placed at the N1-position of the 2-aminoimidazole were synthesized using an approach that we had previously developed.[15]
Reaction of α-amino ester 48 with N-(2-azidoethyl)-4-pentylbenzamide[13] under Huisgen cycloaddition conditions and subsequent deprotection delivered the key intermediate 5 (Scheme 1). Diversity was introduced through reductive amination of 5 with a variety of commercially available benzaldehydes. Boc-protection of the resulting secondary amines and subsequent conversion of the ester to the corresponding Weinreb amide generated intermediates 6a-n. Finally, each Weinreb amide was reduced to the corresponding α-amino aldehyde with DIBAL-H, which following Boc-deprotection and cyclization with cyanamide afforded the 1,5-substituted 2-aminoimidazole/triazole conjugates 7a-n. Following purification, each compound was converted to the corresponding HCl salt for biological screening.
As with 2-AIT 1, the MIC of each compound against MRSA ATCC BAA-1556 was first established. The MIC of oxacillin in the presence of 40% of the MIC of each compound was then determined (Table 1). Previously we had shown that the introduction of an N1-substituent resulted in increased antibiotic activity relative to the parent compound[15] As expected, this trend was followed in the generation of this library. Most compounds exhibited a significantly reduced or abrogated ability to lower the oxacillin MIC. Three compounds however (7d, 7k, and 7l), displayed a marked increase in activity, lowering the oxacillin MIC by 128-fold, 64-fold and 64-fold respectively. The ability of the pilot library to suppress oxacillin resistance in another CA-MRSA USA300 strain (JE2) was next investigated (Table 1) and activity for each compound against this strain was virtually identical to BAA-1556.
Table 1.
MIC values and oxacillin resistance suppression activity against MRSA USA300 strains.
| Compound | 2-AIT concentration (µM) |
Oxacillin MIC (µg/mL) | |
|---|---|---|---|
| ATCC BAA- 1556 |
NARSA JE2 | ||
| - | 32 | 32 | |
| 1 | 20 | 4 | 4 |
| 7a | 10 | 32 | 32 |
| 7b | 5 | 32 | 16 |
| 7c | 5 | 32 | 32 |
| 7d | 5 | 0.25 | 0.5 |
| 7e | 5 | 32 | 32 |
| 7f | 5 | 16 | 16 |
| 7g | 5 | 32 | 32 |
| 7h | 5 | 4 | 16 |
| 7i | 5 | 16 | 32 |
| 7j | 5 | 4 | 8 |
| 7k | 40 | 0.5 | 0.5 |
| 7l | 20 | 0.5 | 0.5 |
| 7m | 10 | 32 | 16 |
| 7n | 20 | 4 | 2 |
| 8 | 50 | 32 | 32 |
2-AIT concentration represents 40% of the MIC up to a limit of 50 µM.
Compound 7d, which possess a 4-butylbenzyl substituent was selected as the lead compound for further study owing to the considerably lower concentration at which it displayed activity compared to 7k and 7l (5 µM compared to 40 µM and 20 µM respectively). A control compound, 8, which possesses only the N-1 substituent of compound 7d was synthesized in the same manner as the pilot library and shown to be devoid of bactericidal activity (MIC > 200 μM) and resistance suppression activity at concentrations as high as 50 µM. Time-kill curves were constructed for strain JE2 cultured in the presence of combinations of oxacillin and compound 7d (Figure 1). Compound 7d, when dosed alone at 5 µM (3.1 µg/mL), is bactericidal at early time points (up to 8 h); however bacterial growth is similar to that of the control by the 24 h time point. When bacteria are cultured in the presence of combinations of oxacillin and compound 7d, a large reduction in the number of colony forming units (CFU) compared to treatment with oxacillin alone is observed. A considerable synergistic effect can be observed at the 24 h time point. Compound 7d alone, at 5 µM, effected a 1.08 log reduction in CFU after 24 h, and oxacillin effected less than 0.4 log reduction at concentrations of 16 µg/mL and below. Combining 7d (5 µM) and oxacillin resulted in log CFU reductions of 6.41, 5.54 and 4.38 for oxacillin concentrations of 16, 4, and 1 µg/mL respectively. Finally, we tested the ability of lead compound 7d at 5 µM to suppress oxacillin resistance in eight additional MRSA isolates obtained from the ATCC (supporting information) and observed reduction in MIC values of 4 - 512 fold.
Figure 1.
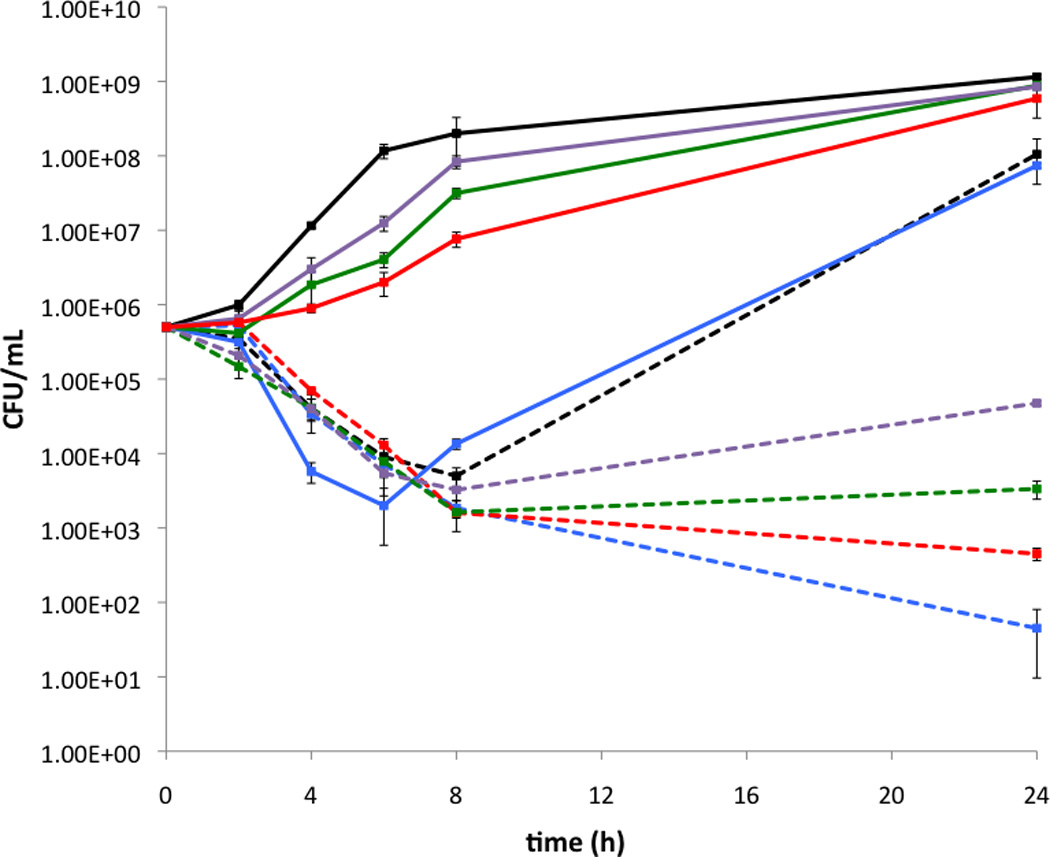
Time-kill curves for USA300 MRSA strain JE2. Solid lines: no 2-AIT, broken lines: 5 µM 7d. Black: no oxacillin, blue: 64 µg/mL oxacillin, red: 16 µg/mL oxacillin, green: 4 µg/mL oxacillin, purple: 1 µg/mL oxacillin.
As the molecules are amphipathic, we first investigated the effect of the compounds on cell membrane integrity. The ability of compound 7d to permeabilize the bacterial cell membrane was quantified using the BacLight assay.[16] After exposure of strain JE2 to compound 7d for one hour, the ratio of intact/damaged cells was measured and compared to control (DMSO only treated) bacteria. At 4x the MIC, 96% of cells were damaged, while at 1x, 0.4x, and 0.25x the MIC, only 33%, 21%, and 9% of cells were damaged respectively. An inactive compound (7e) was found to be com-parable, with 83%, 24%, 23%, and 16% of cells damaged at 4x, 1x, 0.4x, and 0.25x the MIC respectively, suggesting that cell membrane permeabilization is not the mechanism by which compound 7d suppresses resistance to oxacillin. Importantly for a potential antibiotic adjuvant, 7d exhibited little effect on eukaryotic cell membranes, as determined by measuring the hemolytic activity against mechanically difibrinated sheep blood[9]. At its active resistance suppression concentration (5 µM), less than 1% lysis was observed compared to triton×positive control, while only 5.6% lysis was observed as high as 50 µM.
To further delineate the mechanism by which these 2-AIT conjugates are able to lower the oxacillin MIC against MRSA, we obtained a number of mutant strains belonging to the Nebraska Transposon Mutant Library from the Network on Antimicrobial Resistance in S. aureus (NARSA). These mutants are all derived from JE2, allowing us to probe non-essential pathways that may be involved in suppression of oxacillin resistance. For this screen, we focused largely on mutants of non-essential TCS. Bacterial TCS, consisting of a membrane-bound histidine kinase and a response regulator, regulate adaptation to environmental changes and have been shown to play a role in resistance to certain antibiotics[17–19] as well as being master regulators of biofilm formation[20,21]. Fur-thermore, biotinylated analogues of related 2-AI anti-biofilm compounds employed in pull down assays bind to response regulators involved in biofilm formation (unpublished work). Therefore, we posited that these 2-AIT derivatives, which also have anti-biofilm activity, might also target other response regulators involved in antibiotic resistance.
We first established the MICs of oxacillin and 7d against each mutant strain (Table 2, see supporting information for gene descriptions). As expected, the MIC of 7d was fairly consistent against all mutant strains (either 6.25 or 12.5 µM), while a majority of the strains examined, including several response regulator mutants (strains NE958, NE481, NE262, and NE49), histidine kinase mutants (strains NE218, NE147, NE618, NE873, NE820, NE116, and NE423), and a MecR1 regulatory protein mutant (strain NE839), did not exhibit a greater than two-fold difference in oxacillin MIC compared to the parent strain. However, three of the strains tested exhibited considerably lower oxacillin MIC values: NE481, an unidentified DNA-binding response regulator mutant, NE554 (vraR mutant), and NE823 (vraS mutant) exhibited oxacillin MICs that were reduced 16-fold, eight-fold, and eight-fold respectively. These results are in line with previous studies that show expression of VraSR contributes to oxacillin resistance.[22] The ability of 7d to lower the oxacillin MIC against the mutant strains was then examined in an identical manner to that used for the parent strain (at 40% of the MIC). Of the mutants that exhibited altered oxacillin MIC values compared to the parent, compound 7d failed to lower the MIC of both the VraSR two-component system mutant strains NE554 and NE823, suggesting the mode of action of oxacillin resistance suppression activity of compound 7d involves VraSR. Interestingly, compound 7d also failed to lower the oxacillin MIC by more than two-fold against strains NE116 and NE49, suggesting that compound 7d may have some interaction with the pathways controlled by the disrupted genes of these two mutants. NE116 is a putative histidine kinase mutant, while NE49 is an AraC family response regulator mutant. AraC family proteins are known to play a role in antibiotic resistance and stress responses,[23] however as these two mutant strains did not exhibit oxacillin MICs that differed from the parent strain, these pathways most likely do not relate to the mechanism of oxacillin resistance suppression by compound 7d.
Table 2.
MIC values and oxacillin resistance suppression activity of compound 7d against strains from the Nebraska Transposon Mutant Library.
| Strain | Concentration of 7d (µM) |
Oxacillin MIC (µg/mL) |
Oxacillin MIC w/7d (µg/mL)[a] |
|---|---|---|---|
| JE2 | 5 | 32 | 0.5 |
| NE218 | 2.5 | 32 | 8 |
| NE147 | 2.5 | 32 | 0.25 |
| NE958 | 5 | 32 | 0.25 |
| NE481 | 5 | 2 | 0.25 |
| NE262 | 5 | 32 | 0.5 |
| NE618 | 5 | 32 | 1 |
| NE554 | 2.5 | 4 | 4 |
| NE823 | 2.5 | 4 | 4 |
| NE873 | 5 | 32 | 0.25 |
| NE210 | 5 | 32 | 0.5 |
| NE820 | 5 | 32 | 0.25 |
| NE839 | 5 | 32 | 0.5 |
| NE49 | 2.5 | 32 | 16 |
| NE116 | 2.5 | 32 | 32 |
| NE95 | 5 | 16 | 0.5 |
| NE423 | 2.5 | 16 | 4 |
Oxacillin MIC values recorded in the presence of 40% MIC of 7d
As compound 7d exhibited a lower MIC against a number of mutant strains than the parent strain (and was therefore screened for resistance suppression at a lower concentration, 2.5 µM, 1.55 µg/mL), we wanted to ensure that the lack of resistance suppression activity was not simply a result of lower bactericidal activity of the compound. A time kill curve was therefore constructed for NE554 in the presence of 2.5 µM 7d (40% MIC) and compared to the time kill curve of JE2 in the presence of 5 µM 7d (supporting information). The bactericidal activity of compound 7d, was in fact slightly higher against strain NE554 than JE2 at the concentrations used in the resistance suppression assay, suggesting that the lack of activity against NE554 is due to the absence of VraR, rather than altered bactericidal activity.
VraSR has been proposed to be a ‘sentinel’ system that is capable of sensing perturbation of cell wall synthesis and coordinating a response involving expression of a number of genes involved in antibiotic resistance.[22] Expression of VraSR is induced upon exposure to cell wall-acting antibiotics including β-lactams, glycopeptides, daptomycin, and bacitracin,[24,25] and it has been shown that VraSR mutants are treatable with an oxacillin regimen in vivo[26]. To further establish disruption of VraSR signaling in the mechanism of 7d, we tested the ability of 7d to lower the MIC of vancomycin. The MIC of vancomycin against JE2 was established as 1 µg/mL, while in the presence of 5 µM 7d this is lowered to 0.25 µg/mL. The MIC of vancomycin is 0.5 µg/mL against both NE554 and NE823, and this remained unchanged in the presence of 2.5 µM 7d. Furthermore, compound 7d had little or no effect on the MICs of streptomycin or chloramphenicol (supporting information), non-cell wall acting antibiotics that do not activate the VraSR TCS. The fact that compound 7d did not lower the MIC of these latter antibiotics against the parent strain further suggests that the reduction in oxacillin MIC brought about by this compound is not simply due to a combined microbicidal effect, but due to disruption of the VraSR TCS pathway.
In conclusion, we have developed a compound that is able to suppress resistance to oxacillin in several diverse MRSA strains, achieving MIC suppressions upwards of 512-fold. We have shown that this activity is not dependent upon membrane disruption, while preliminary screening of USA300 mutants indicates that VraSR plays an important role in the activity of this compound. Given the pressing need for new strategies to deal with the threat of multi-drug resistant pathogenic bacteria, the identification of molecules that restore the efficacy of approved antibiotics by interfering with bacterial TCS represents a potential avenue for the development of antibiotic adjuvants.
Supplementary Material
Acknowledgments
The authors thank the DOD DMRDP program (W81XWH-11-2-0115) for support of this work. The DMRDP program is administered by the Department of Army; The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014 is the awarding and administering office. The content of this manuscript does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. NARSA isolates were obtained through the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) program, supported by NIAID/NIH.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Eiland EH, Gatlin DJ., III Pharmacy Practice. 2008;21:313–318. [Google Scholar]
- 2.*IDSA Policy Paper d CID. 2011;52(Suppl 5):d S397. [Google Scholar]
- 3.O’Hara FP, Amrine-Madsen H, Mera RM, Brown ML, Close NM, Suaya JA, Acosta CJ. Microb. Drug Resistance. 2012 doi: 10.1089/mdr.2012.0056. [DOI] [PubMed] [Google Scholar]
- 4.Coates ARM, Halls G, Hu Y. Brit. J. Pharmacol. 2011;163:184–194. doi: 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dolgin E. Nat. Med. 2010;16:1054–1054. doi: 10.1038/nm1010-1054a. [DOI] [PubMed] [Google Scholar]
- 6.Kalan L, Wright GD. Expert Rev. Mol. Med. 2011;13:1–17. doi: 10.1017/S1462399410001766. [DOI] [PubMed] [Google Scholar]
- 7.Rogers SA, Huigens RW, III, Cavanagh J, Melander C. Antimicrob. Agents Ch. 2010;54:2112–2118. doi: 10.1128/AAC.01418-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su Z, Peng L, Worthington RJ, Melander C. Chem Med Chem. 2011;6:2243–2251. doi: 10.1002/cmdc.201100316. [DOI] [PubMed] [Google Scholar]
- 9.Worthington RJ, Bunders CA, Reed CS, Melander C. ACS Med. Chem. Lett. 2012;3:357–361. doi: 10.1021/ml200290p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klitgaard JK, Skov MN, Kallipolitis BH, Kolmos HJ. J. Antimicrob. Chemother. 2008;62:1215–1221. doi: 10.1093/jac/dkn417. [DOI] [PubMed] [Google Scholar]
- 11.Rogers SA, Melander C. Angewandte Chemie. 2008;47:5229–5231. doi: 10.1002/anie.200800862. [DOI] [PubMed] [Google Scholar]
- 12.Rogers SA, Bero JD, Melander C. Chem Bio Chem. 2010;11:396–410. doi: 10.1002/cbic.200900617. [DOI] [PubMed] [Google Scholar]
- 13.Su Z, Yeagley AA, Su R, Peng L, Melander C. Chem Med Chem. 2012 doi: 10.1002/cmdc.201200350. [DOI] [PubMed] [Google Scholar]
- 14.Twentieth Informational Supplement. No. 1. Vol. 30. Wayne, PA: Clinical and Laboratory Standards Institute; 2010. Jan, M100-S20. [Google Scholar]
- 15.Harris TL, Worthington RJ, Melander C. Bioorg. Med. Chem. Lett. 2011;21:4516–4519. doi: 10.1016/j.bmcl.2011.05.123. [DOI] [PubMed] [Google Scholar]
- 16.Hilliard JJ, Goldschmidt RM, Licata L, Baum EZ, Bush K. Antimicrob. Agents Ch. 1999;43:1693–1699. doi: 10.1128/aac.43.7.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardete S, Wu SW, Gill S, Tomasz A. Antimicrob. Agents Chemother. 2006;50:3424–3434. doi: 10.1128/AAC.00356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraus D, Herbert S, Kristian SA, Khosravi A, Nizet V, Götz F, Peschel A. BMC Microbiology. 2008;8:85. doi: 10.1186/1471-2180-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shoji M, Cui L, Iizuka R, Komoto A, Neoh H, Watanabe Y, Hishinuma T, Hiramatsu K. Antimicrob. Agents. Chemotherap. 2011;55:3870–3881. doi: 10.1128/AAC.01563-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomaras AP, Flagler MJ, Dorsey CW, Gaddy JA, Actis LA. Microbiology-Sgm. 2008;154:3398–3409. doi: 10.1099/mic.0.2008/019471-0. [DOI] [PubMed] [Google Scholar]
- 21.Parkins MD, Ceri H, Storey DG. Mol. Microbiol. 2001;40:1215–1226. doi: 10.1046/j.1365-2958.2001.02469.x. [DOI] [PubMed] [Google Scholar]
- 22.Belcheva A, Golemi-Kotra D. J. Biol. Chem. 2008;283:12354–12364. doi: 10.1074/jbc.M710010200. [DOI] [PubMed] [Google Scholar]
- 23.Lei MG, Cue D, Roux CM, Dunman PM, Lee CY. J. Bacteriol. 2011;193:5231–5241. doi: 10.1128/JB.05454-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuo M, Kato F, Oogai1 Y, Kawai T, Sugai M, Komatsuzawa H. J. Antimicrob. Chemother. 2010;65:1536–1537. doi: 10.1093/jac/dkq141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dengler V, Stutzmann Meier P, Heusser R, Berger-Bächi B, McCallum N. BMC Microbiol. 2011;11:16. doi: 10.1186/1471-2180-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo DS, Montgomery CP, Yin S, Boyle-Vavra S, Daum RS. Antimicrob. Agents Ch. 2011;55:2818–2823. doi: 10.1128/AAC.01704-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.