

Abstract

The scope of palladium-catalyzed, auxiliary-assisted direct arylation and alkylation of sp2 and sp3 C-H bonds of amine and carboxylic acid derivatives has been investigated. The method employs a palladium acetate catalyst, substrate, aryl, alkyl, benzyl, or allyl halide, and inorganic base in t-amyl alcohol or water solvent at 100-140 °C. Aryl and alkyl iodides as well as benzyl and allyl bromides are competent reagents in this transformation. Picolinic acid auxiliary is used for amine γ-functionalization and 8-aminoquinoline auxiliary is used for carboxylic acid β-functionalization. Some optimization of base, additives, and solvent is required for achieving best results.
1. Introduction
Transition-metal-catalyzed functionalization of carbon-hydrogen bonds is becoming an important synthetic tool that allows to create carbon-carbon bonds efficiently.1 Regioselective, intermolecular arylation and alkylation of heterocycles and other arenes can be efficiently accomplished by employing first- and second-row transition-metal catalysis.2 A recent report by Ackerman shows that meta-alkylation of 2-phenylpyridine derivatives is feasible.2l In contrast, intermolecular functionalization of unactivated (not benzylic or α to heteroatom) sp3 C-H bonds has attracted less attention.3 Many of the published examples of sp3 C-H bond functionalization involve positions adjacent to quaternary centers.3a-f Fewer reports deal with functionalization of sp3 C-H bonds in systems where β-hydride elimination from metalated intermediates is possible. Ohno has prepared indolines from N-alkyl-2-bromoanilines.3i Yu has developed methods for palladium-catalyzed olefination, carbonylation, and arylation of sp3 C-H bonds by employing perfluoroaniline auxiliaries and utilized pyridine as a directing group in an example of sp3 C-H bond alkylation by alkylboronic acids.3g-l Sanford has reported an aerobic sp3 C-H bond olefination by using a pyridine directing group.3m A direct Pd-catalyzed γ-arylation of amino acid esters bearing a removable N-(2-pyridyl)sulfonyl directing group has been described by Fernández-Ibáñez.3n
In 2005, we reported the β-arylation of carboxylic acid and γ-arylation of amine derivatives by employing an 8-aminoquinoline or picolinic acid auxiliary, catalytic Pd(OAc)2, stoichiometric AgOAc, and an aryl iodide coupling partner.4a Subsequently, a number of auxiliaries were investigated for carboxylic acid β-arylation and it was shown that silver salts can be replaced by simple inorganic bases (Scheme 1).4b Omission of silver allowed catalytic alkylation of sp2 and sp3 C-H bonds. Functionalization regiochemistry is determined by a double five-membered palladacycle intermediate 1 that is formed in C-H bond activation step. The electron-rich dianionic pincer-type ligand on palladium facilitates oxidative addition of aryl halide to the Pd(II) intermediate 1 and stabilizes the presumed high-valent Pd intermediates.4b,6 We have also reported auxiliary-directed synthesis of unnatural amino acids as well as picolinic acid-directed heterocycle formation.4c,d Aminoquinoline and picolinic acid can also direct copper-catalyzed carbon-heteroatom bond formation.4e-g Subsequently, several other groups have used these auxiliaries for new reaction development and synthetic purposes. Corey has used the 8-aminoquinoline auxiliary to arylate sp3 C-H bonds in amino acid derivatives.5a Chen has employed 8-aminoquinoline auxiliary in total synthesis of Celogentin C.5b Synthesis of the Leu-Trp component of the celogentin family of cyclic peptides via C-H bond functionalization methodology has also been disclosed.5c Elegant total syntheses of piperborenines and the proposed structure of pipercyclobutanamide A by using 8-aminoquinoline and 2-thiomethylaniline directing groups have been developed by Baran.5d,e Carbocycles have been constructed by using 8-aminoquinoline directing group.5f Chen has employed picolinic acid directing group for arylation, alkenylation, and alkylation of sp2 and sp3 C-H bonds.5g,h,l,m Quinolinecarboxylic acid naphthylamide arylation has been recently disclosed.3k Picolinamide and 8-aminoquinoline-directed alkynylation of sp2 and sp3 C-H bonds has also been reported.5i,j Furthermore, iron, nickel, copper, and ruthenium catalysis has been used for 8-aminoquinoline-containing carboxamide functionalization.4e-g, 5n-q These examples show that monoanionic, chelating auxiliaries have found wide applications for C-H to C-C bond conversion in a variety of catalytic systems. Significantly, application of these auxiliaries in the construction of complex natural products shows that C-H bond functionalization methodology has been introduced into mainstream organic synthesis. Consequently, further methodological and mechanistic investigations that would increase the scope and understanding of C-H bond functionalization processes are warranted. We report here the scope and limitations of auxiliary-assisted, palladium-catalyzed arylation and alkylation of sp2 and sp3 C-H bonds in amine and carboxylic acid derivatives (Scheme 2).
Scheme 1.

Auxiliary-Assisted Arylation
Scheme 2.

Auxiliary-Assisted, Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 C-H Bonds
2. Results and Discussion
2.1 Picolinamide Arylation Optimization
Based on our previous results with carboxylic acid derivative functionalization, the initial optimization experiments were aimed at replacing silver acetate with other stoichiometric additives for arylation of cumylamine picolylamide (Table 1). Potassium phosphate and cesium carbonate bases were inefficient (entries 1 and 2). Better results were obtained with cesium acetate in t-amyl alcohol (entry 4). Addition of 10 mol % of CuBr2 allowed achieving full conversion to diarylation product (entry 6). Thus, the optimized arylation conditions include 4 equivalents of CsOAc base in t-amyl alcohol solvent, 5 mol % Pd(OAc)2, 10 mol % CuBr2, and 4 equivalents of ArI at 140 °C.
Table 1.
Arylation Optimizationa

| ||
|---|---|---|
|
| ||
| entry | reaction conditions | conv, % |
| 1 | K3PO4, toluene/CH3CN, 140 °C, 16 h |
15 |
| 2 | Cs2CO3, toluene/CH3CN, 140 °C, 16 h |
15 |
| 3 | CsOAc, toluene/CH3CN, 140 °C, 16 h |
30 |
| 4 | CsOAc, t-amyl alcohol, 140 °C, 24 h |
90 |
| 5 | CsOAc, 10 % CuBr2, t-amyl alcohol, 110 °C, 24 h |
75 (70)b |
| 6 | CsOAc, 10 % CuBr2, t-amyl alcohol, 140 °C, 24 h |
>99 (99)b |
conversion measured by GC with internal standard. Please see Experimental Section for details.
Isolated yield.
2.2. Benzylpicolinamide arylation
The silver-free conditions were applied to arylation of a number of benzylpicolinamides (Table 2). Benzylamine derivatives are arylated in excellent yields (entries 1-3). Diarylated products are obtained if unsubstituted benzylamines are employed (entries 1-2). We are interested in synthesis of 8-aryl-1-naphthylamines that could be used in the synthesis of ligands for Brookhart-type transition-metal catalyzed olefin polymerization.7 Consequently, arylation of picolinamide of 1-naphthylamine was investigated in depth (entries 4-12). The reaction is successful both by using AgOAc base and by using CsOAc base. Thus, reaction with 4-iodoanisole gives 98% isolated yield if AgOAc is used, and 73% yield if CsOAc is employed (entry 6). 4-Bromophenylation of 1-naphthylamine picolinamide affords nearly identical yield of product in both cases (entry 10). However, AgOAc conditions allow for a lower Pd(OAc)2 loading (2% vs. 5% for CsOAc base). Three large scale reactions (entries 4, 11, and 12) afforded excellent product yields showing that scale-up to at least 50 mmol is possible. Phenethylamine derivative is arylated in moderate yield, presumably due to requirement for less favorable six-membered palladacycle intermediate (entry 13). In contrast to this result, our previous benzylamine arylation methodology is not applicable to arylation of phenethylamines.8 Alkenylation of sp2 C-H bonds is also possible, and benzylamine picolinamide was reacted with iodostyrene to give coupling product in 86% yield. The ester, chloro, bromo, ether, and trifluoromethyl functionalities are compatible with the arylation conditions. The reaction fails if aryl bromide coupling partners are used. Benzylamine picolinamide was reacted with bromobenzene under conditions of entry 2 and arylation product was not detected in the reaction mixture.
Table 2.
Arylation of Benzylpicolinamidea

| ||||
|---|---|---|---|---|
|
| ||||
| entry | Picolinamide | Aryl Iodide | Arylated Picolinamide | Yield |
| 1b |

|

|
|
99% |
| 2b |
|
PhI |

|
99% |
| 3b |

|
|
|
92% |
| 4c,d |

|
|
|
91% |
| 5c | 3 |
|

|
99% |
| 6 | 3 |
|

|
73%b
98%c |
| 7c | 3 |

|
|
99% |
| 8c3 | 3 |
|

|
92% |
| 9c | 3 |
|
|
98% |
| 10 | 3 |

|

|
98% |
| 11c,e | 3 |

|

|
84% |
| 12c,f | 3 |
|
|
65% |
| 13b |
|

|
|
52% |
| 14b |

|

|
|
86% |
For CsOAc base: 5 mol % Pd(OAc)2, 10 mol% CuBr2 additive, 4 equiv CsOAc, tAmOH solvent, 4 equiv ArI, 1 mmol scale. For AgOAc base: 2 mol% Pd(OAc)2, 2 equiv AgOAc, no solvent, 4 equiv ArI, 0.5 mmol scale. Yields are isolated yields. Please see Experimental Section for details.
CsOAc base.
AgOAc base.
20 mmol scale, 1.5 equiv AgOAc.
50 mmol scale, 2 equiv ArI, 1.5 equiv AgOAc.
35 mmol scale, 3 equiv ArI, 1.5 equiv AgOAc.
Directing groups can be removed by using n-butylamine and AlCl3 in toluene at 90 °C or NaOH in ethanol (Scheme 3).9 Free arylated amines are obtained in good to excellent yields.
Scheme 3.

Directing Group Removal
Synthesis of even more hindered amines is possible. Acylation of 8-(p-tolyl)-1-naphthylamine by propionyl chloride followed by palladium-catalyzed arylation affords 2-(4-carbethoxyphenyl)-8-8-(p-tolyl)-1-naphthylamine derivative in a good yield (Scheme 4).10 Recently, a method for 1-aminonaphthalene quinolinecarboxamide arylation has been reported; however, it requires use of 15 mol% Pd(OAc)2.5k
Scheme 4.

Introduction of Second Aryl Group
2.3 Arylation of Alkylpicolinamides
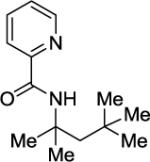
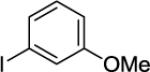
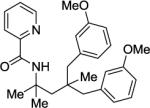
Arylation of unactivated sp3 C-H bonds can be accomplished by employing conditions developed for sp2 C-H bond functionalization (Table 3). Comparison of the arylation yields for propyl (entry 1), s-butyl (entry 2), and 2-(2-methylbutyl) derivatives (entry 3) shows that the reaction is most efficient for the substrates possessing the most α–methyl groups. The increase in yield is likely due to Thorpe-Ingold effect.11 Arylation of secondary aliphatic C-H bonds is also feasible and proceeds in good yield (entry 4). An amide derived from t-octylamine was arylated in modest yield. A mixture of mono- and diarylation products was obtained, with functionalization occurring at δ-positions (entry 5). Sixmembered palladacycle intermediate may be responsible for less efficient arylation. 2-Iodotoluene was unreactive in all reactions tested as shown before for arylations proceeding via high-valent palladium intermediates.4a,b,10
Table 3.
Arylation of Alkyl Picolinamidesa
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Picolinamide | Aryl Iodide | Arylated Picolinamide | Yield |
| 1 |
|
|

|
56% |
| 2 |
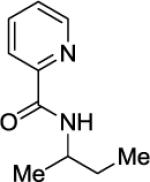
|
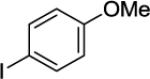
|
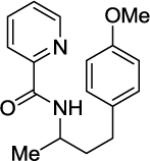
|
75% |
| 3 |
|
|
|
91% |
| 4 |
|

|
|
86% |
| 5b |
|
|
|
29% |
Palladium acetate (5 mol %), 10 mol% CuBr2 additive, 4 equiv CsOAc, t-amyl alcohol solvent, 4 equiv ArI, 1 mmol scale. Yields are isolated yields. Please see Experimental Section for details.
Palladium acetate (10 mol %), 20 mol % CuBr2, and K2CO3 base used. Monoarylation product also isolated in 13% yield.
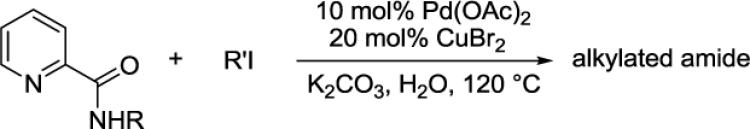
2.4 Picolinamide alkylation
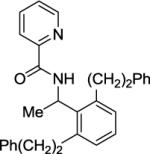
The alkylation of picolinamide C-H bonds is presented in Table 4. Short optimization showed that the best results are obtained by employing potassium carbonate base in conjunction with water solvent. α-Methylbenzylamine derivatives can be alkylated by various alkyl iodides such as butyl iodide (entry 1), 4,4,4-trifluorobutyl iodide (entry 2), isobutyl iodide (entry 3), and 2-phenethyl iodide (entry 4) in good yields. However, if butyl iodide was replaced with butyl bromide, no product was obtained. Benzylation can be performed by employing benzyl iodide (entry 5). α,α-Dimethylbenzylamine picolinamide reaction with n-butyl iodide gave the dialkylation product in a good yield (entry 6). Unexpectedly, secondary alkyl iodides are also reactive. Cyclohexylation of benzyl picolinamide affords a 20% yield of monoalkylation product in addition to 14% of dialkylation (entry 7). Similarly, alkylation of a 2-methoxybenzylamine derivative gives the product in 14% yield (entry 8). 1-Naphthylamine derivative is alkylated by n-octyl iodide in moderate yield (entry 6). The alkylation of unactivated sp3 C-H bonds is inefficient. Reaction of picolinic acid 2-(2-methyl)butylamide with n-amyl iodide yielded only 27% of the product (entry 10). Chen has recently reported method for picolinamide sp3 C-H bond alkylation.51 We have previously shown that α-methylbenzylamines do not racemize under palladium-catalyzed arylation conditions.4h
Table 4.
Alkylation of aryl and alkylpicolinamidesa
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Picolinamide | Aryl Iodide | Arylated Picolinamide | Yield |
| 1b |

|
nBuI |

|
99% |
| 2b | 11 | CF3(CH2)3I |
|
79% |
| 3c | 11 | (CH3)2CHCH2I |

|
84% |
| 4 | 11 | PhCH2CH2I |
|
86% |
| 5b | 11 | BnI |

|
85% |
| 6b,c,d |
|
nBuI |

|
54% |
| 7e |
|

|
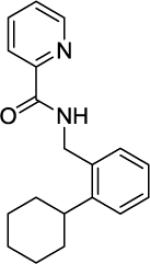
|
20% |
| 8 |
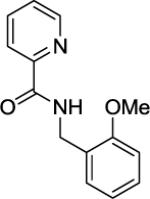
|

|

|
14% |
| 9b,f |
|
nOctylI |

|
49% |
| 10g |

|
nAmylI |

|
27% |
Picolinamide (1 mmol), RI (4 mmol), Pd(OAc)2 (10 mol%), CuBr2 (20 mol%), K2CO3 (4 mmol), H2O solvent, 24 h at 120 °C. Yields are isolated yields. Please see Experimental Section for details.
Palladium acetate (5 mol%), CuBr2 (10 mol%).
Six equiv RI.
Monoalkylation product also isolated (14 %).
Dialkylation product also isolated (11 %).
t-Amyl alcohol solvent, CsOAc base, 140 °C.
Pivalic acid additive, t-amyl alcohol solvent, 110 °C.
2.5 Alkylation of 8-Aminoquinoline Benzamides
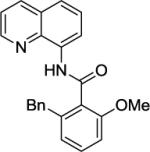
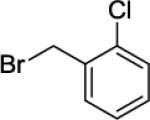
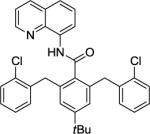
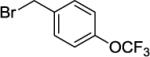
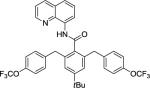
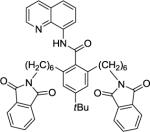
Our initial conditions reported in 2005 that use AgOAc for iodide removal were not successful for C-H bond alkylation. Silver acetate reacts with alkyl iodides competing with C-alkylation. The new silver-free conditions, developed in 2010, are successful since the competitive destruction of alkyl iodides is slow.4b We have determined that the optimal auxiliary for C-H bond alkylation of benzamide derivatives is 8-aminoquinoline. As reported earlier, alkylation conditions involve heating of the substrate with alkyl iodide or benzyl bromide to 100 - 110° C in t-amyl alcohol in the presence of K2CO3 base and catalytic amount of pivalic acid. We have previously reported 3 examples of 8-aminoquinoline benzamide alkylation.4b Table 5 shows the alkylation scope and functional group tolerance. Benzylation of alkoxy- (entries 1, 5 and 6), bromo- (entry 2), and trifluoromethyl-substituted (entry 4) benzoic acid amides occurs with good to excellent yields. The reaction cleanly affords dialkylation products if benzamide is substituted at 4-position or possesses a small substituent at 3-position (entry 6). The alkylation of 4-t-butylbenzoic acid derivative can be accomplished by employing ethyl iodide (entry 7), i-butyl iodide (entry 8), 2-phenethyl iodide (entry 9), and ethyl-7-iodoheptanoate (entry 10). Phthaloyl-protected 6-amino-1-iodohexane is also reactive (entry 15). Furthermore, a variety of benzyl bromides can be employed in the alkylation. Thus, reaction is successful with benzyl bromides possessing chloro (entry 11), ester (entry 12), trifluoromethoxy (entry 13), and nitro (entry 14) substituents, attesting to the functional group tolerance of C-H bond alkylation methodology. Allylation is also possible by employing 1-bromo-3-methylbut-2-ene, although the isolated yield of the product is low (entry 16).
Table 5.
Alkylation of 8-Aminoquinoline Benzamides
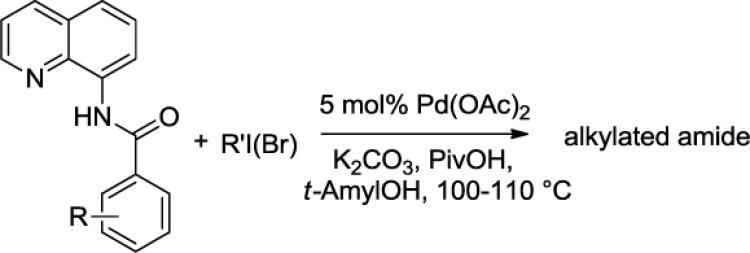
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Amide | Alkyl Halide | Product | Yield |
| 1 |
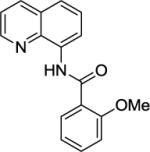
|
BnBr |
|
61% |
| 2 |
|
BnBr |
|
76% |
| 3 |
|
BnBr |
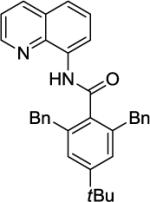
|
88% |
| 4 |
|
BnBr |

|
71% |
| 5 |
|
BnBr |
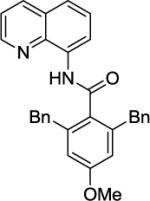
|
58% |
| 6 |
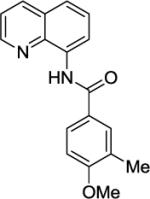
|
BnBr |

|
74% |
| 7 |

|
EtI |
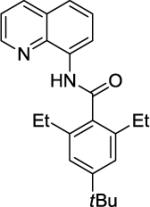
|
90% |
| 8 | 12 | iBuI |
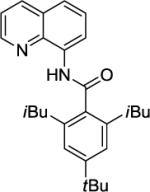
|
75% |
| 9 | 12 | Ph(CH2)2I |

|
92% |
| 10 | 12 | EtO2C(CH2)6I |

|
77% |
| 11 | 12 |
|
|
70% |
| 12 | 12 |
|

|
94% |
| 13 | 12 |
|
|
90% |
| 14 | 12 |
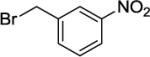
|

|
86% |
| 15 | 12 |

|
|
74% |
| 16 | 12 |

|

|
25% |
a Pd(OAc)2 (5 mol %), K2CO3 (2.5 equiv), substrate (0.74 mmol), pivalic acid (2 mol %), alkyl bromide or iodide (3–4 equiv), t-amyl–OH solvent, 12–96 h at 110– °C. Yields are isolated yields. Please see Experimental section for details.
2.6 Alkylation of 8-Aminoquinoline Amide sp3 C-H Bonds
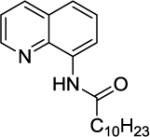
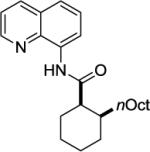
We have previously determined that the optimal auxiliary for carboxamide C-H bond alkylation is 8-aminoquinoline. Two examples of 8-aminoquinoline propionylamide α-alkylation were published.4b Alkylation conditions involve heating of the substrate with 5% Pd(OAc)2 and alkyl iodide or benzyl bromide to 100 - 110° C in t-amyl alcohol in the presence of K2CO3base and catalytic amount of pivalic acid (Table 6). 8-Aminoquinoline propionamide can be alkylated with simple alkyl iodides such as ethyl (entry 1), butyl (entry 2), octyl (entry 3), and phenetyl iodide (entry 4). Allylation by 1-bromo-3-methylbut-2-ene is also possible, affording the product in a low yield (entry 5). Isobutyl iodide is reactive and the alkylation proceeds in a good yield (entry 6). Benzylation with 2-bromobenzyl bromide is successful and product is obtained in 60% yield (entry 7). 2-Methylbutyric acid derivative is selectively alkylated in the α-methyl group (entry 8). Flurbiprofen12 amide can also be alkylated in a moderate yield (entry 9). Finally, alkylation of a secondary C-H bond proceeds in a low yield (entry 10).
Table 6.
Alkylation of 8-Aminoquinoline Amide sp3 C-H Bonds
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Amide | Alkyl Halide | Product | Yield |
| 1 |

|
EtI |

|
78% |
| 2 | 13 | nBuI |

|
|
| 3 | 13 | nOctI |
|
52% |
| 4 | 13 | Ph(CH2)2I |
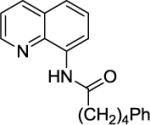
|
64% |
| 5 | 13 |

|

|
29% |
| 6 | 13 | iBuI |

|
78% |
| 7 | 13 |

|

|
60% |
| 8 |
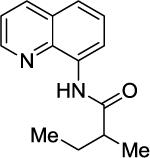
|
nOctI |
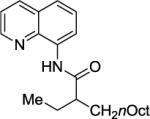
|
40% |
| 9 |

|
nOctI |

|
45% |
| 10 |
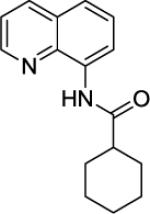
|
nOctI |
|
15% |
a Pd(OAc)2 (5 mol %), K2CO3 (2.5 equiv), substrate (0.74 mmol), pivalic acid (2 equiv), alkyl bromide or iodide (4 equiv), t-amyl alcohol solvent, 24 h at 110 °C. Yields are isolated yields. Please see Experimental section for details.
3. Summary
In this paper, we report the scope and limitations of auxiliary-assisted, palladium-catalyzed arylation and alkylation of sp2 and sp3 C-H bonds in amine and carboxylic acid derivatives. The method employs a palladium acetate catalyst, substrate, aryl, alkyl, benzyl, or allyl halide, and inorganic base in t-amyl alcohol or water solvent at 100-140 °C. Aryl and alkyl iodides as well as benzyl and allyl bromides are competent reagents in this transformation. Picolinic acid auxiliary is used for amine γ-functionalization and 8-aminoquinoline auxiliary is used for carboxylic acid β-functionalization. Some optimization of base, additives, and solvent is required for achieving best results. The arylation is possible for both secondary and primary sp3 C-H bonds; however, alkylation of secondary sp3 C-H bonds in aminoquinoline derivatives and primary C-H bonds in picolinamides is low-yielding.
4. Experimental Section
General considerations
Flash chromatography was performed on 60 Å silica gel. Preparative TLC was performed on TLC plates, 20 x 20 cm, 2000 μm thick, with fluorescent indicator. GC analyses were performed on a Restek column (Rtx®-5, 15 m, 0.25 mm ID). Residual solvent peaks were used as reference in 1H NMR and 13C NMR spectra. Melting points are uncorrected. The following starting materials were obtained from commercial sources and were used without further purification: picolinic acid, triethylamine, dichloromethane, ethyl chloroformate, MgSO4, hexanes, ethyl acetate, cumylamine, α-methylbenzylamine, benzylamine, 1-naphthylamine, pyridine, triphenylphosphite, H2SO4, 2-methoxybenzylamine, 3,4-dimethoxyphenethylamine, tert-pentylamine, 2-aminobutane, 1-propylamine, cyclohexylamine, tert-butylamine, palladium (II) acetate, copper (II) bromide, cesium acetate, tert-amyl alcohol, iodo-4-methylbenzene, iodobenzene, 1-iodo-4-methoxybenzene, 1-bromo-4-iodobenzene, ethyl 4-iodobenzoate, iodoethane, 1,1,1-trifluoro-4-iodobutane, 1-iodo-2-methylpropane, (2-iodoethyl)benzene, benzyl bromide, octyl iodide, iodobutane, iodocyclohexane, iodopentane, aluminum chloride. Mass spectra were performed on a Micromass Ultima Magnetic Sector.
Synthesis of starting materials
General procedure for the preparation of the picolinamides from amines13
Picolinic acid (35 mmol, 4.3 g) and triethylamine (70 mmol, 9.70 mL) were dissolved in dry dichloromethane (80 mL). The solution was cooled to 0 °C followed by addition of ethyl chloroformate (35 mmol, 3.30 mL). The mixture was subsequently stirred for 30 minutes in ice bath. The amine (20 mmol) was added dropwise via a syringe and the suspension was stirred for 1 hour. The solution was warmed to room temperature and stirred for 24 hours. After that, water (100 mL) was added to the reaction mixture and the layers were separated. The aqueous layer was extracted with dichloromethane (2 x 100 mL). The organic layers were combined, dried with MgSO4, concentrated. The residue was purified by a silica gel column chromatography using hexanes/ethyl acetate eluent.
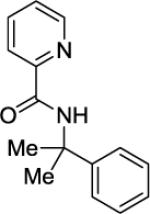
N-(2-Phenylpropan-2-yl)picolinamide (SM01)

Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.30 mL), and cumylamine (20 mmol, 2.7 g). After chromatography (hexanes/ethyl acetate 70/30), white crystalline material was obtained (4.44 g, 93 %). Rf = 0.40 (hexanes/ethyl acetate 70/30), mp = 87–88 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.55–8.54 (m, 1H), 8.48 (br s, 1H), 8.15–8.13 (m, 1H), 7.84–7.80 (m, 1H), 7.48–7.45 (m, 2H), 7.44–7.40 (m, 1H), 7.36–7.31 (m, 2H), 7.25–7.21 (m, 1H), 1.85 (s, 6H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.3, 150.6, 147.9, 146.9, 137.5, 128.5, 126.8, 126.1, 124.9, 122.0, 55.7, 29.3. FT-IR (neat, cm−1) ν 3381, 1682, 15.13, 1570, 1436, 1384, 1365, 1280. Anal. Calcd. for C15H16N2O (240.30 g/mol): C, 74.97; H, 6.71; N, 11.66; Found: C, 75.02; H, 6.71; N, 11.62.
N-(1-Phenylethyl)picolinamide (SM02, 11)
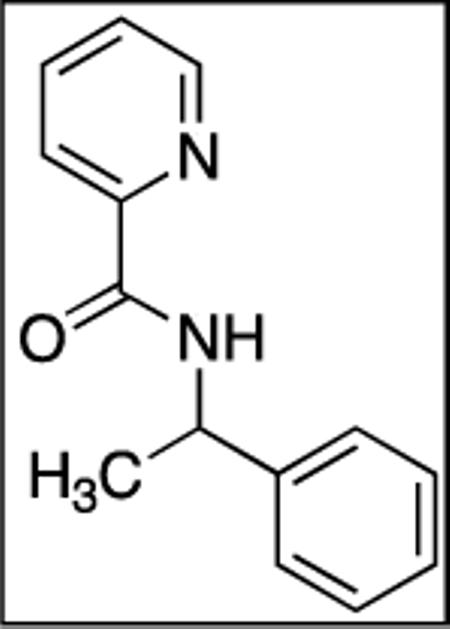
Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.30 mL), and α-methylbenzylamine (20 mmol, 2.60 mL). After chromatography (hexanes/ethyl acetate 70/30), white crystals were obtained (4.35 g, 96 %). Rf = 0.31 (hexanes/ethyl acetate 70/30). This compound is known.141H NMR (400 MHz, CDCl3, ppm) δ 8.54–8.52 (m, 1H), 8.32 (d, J = 6.31H), 8.20–8.18 (m, 1H), 7.84–7.80 (m, 1H), 7.41–7.38 (m, 3H), 7.36–7.31 (m, 2H), 7.28–7.23 (m, 1H), 5.38–5.26 (m, 1H), 1.62 (d, J=6.9 Hz, 3H).
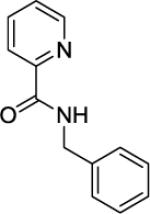
N-Benzylpicolinamide (SM03)

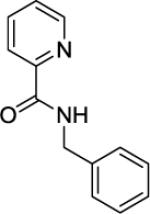
Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.30 mL), and benzylamine (20 mmol, 2.18 mL). After chromatography (hexanes/ethyl acetate 60/40), white crystals were obtained (3.81 g, 90 %). Rf = 0.36 (hexanes/ethyl acetate 60/40). This compound is known.151H NMR (400 MHz, CDCl3, ppm) δ 8.53–8.51 (m, 1H), 8.37 (br s, 1H), 8.23 (d, J=7.8 Hz, 1H), 7.87–7.83 (m, 1H), 7.43–7.40 (m, 1H), 7.38–7.32 (m, 4H), 7.30–7.26 (m, 1H), 4.67 (d, J=6.0 Hz, 2H).
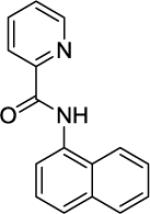
N-(Naphthalen-1-yl)picolinamide (SM04, 3)
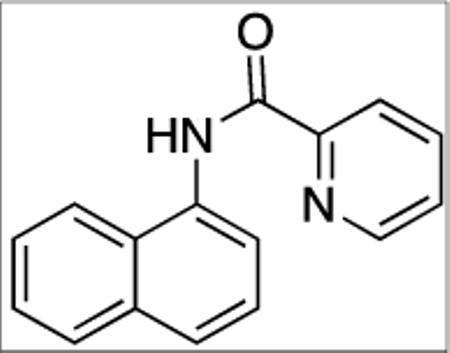
1-Naphthylamine (7.2 g, 50 mmol) in pyridine (10 mL) was added dropwise in 15 minutes to a stirred solution of picolinic acid (6.2 g, 50 mmol) in pyridine (14 mL) at 50 °C. Triphenylphosphite (13 mL, 50 mmol) was added to the resulting mixture followed by stirring at 110 °C for 4 hours. The mixture was cooled to room temperature followed by addition of distilled water (50 mL) and dichloromethane (50 mL). The mixture was placed in a 500 mL Erlenmeyer flask and aqueous H2SO4 (150 mL; concentrated H2SO4/water 1/1 v/v) was added. The mixture was shaken and the layers were separated. The organic layer was washed with aqueous H2SO4 (2 × 100 ml). The acidic aqueous layers were combined and neutralized with solid sodium bicarbonate. The tan solids formed were filtered and washed thoroughly with distilled water, then recrystallized from methanol to afford tan needles (10.9 g, 87 %). This compound is known.161H NMR (400 MHz, CDCl3, ppm) δ 10.77 (s, 1H), 8.70 (d, J = 8.2 Hz, 1 H), 8.36 (d, J = 8.2 Hz, 1 H), 8.36 (d, J = 7.8 Hz, 1 H), 8.09 (d, J = 8.2 Hz, 1 H), 7.95–7.88 (m, 2 H), 7.70 (d, J = 8.2 Hz, 1 H), 7.61–7.50 (m, 4H).
N-(2-Methoxybenzyl)picolinamide(SM05)
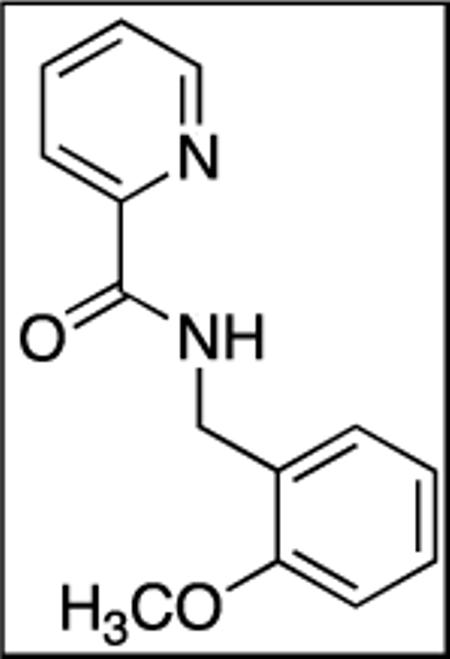
Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.30 mL), and 2-methoxybenzylamine (20 mmol, 2.74 g). After chromatography (hexanes/ethyl acetate 60/40), white powder was obtained (3.2 g, 71 %). Rf = 0.34 (hexanes/ethyl acetate 60/40). This compound is known.171H NMR (400 MHz, CDCl3, ppm) δ 8.53–8.52 (m, 1H), 8.45 (br s, 1H), 8.21–8.19 (m, 1H), 7.85–7.77 (m, 1H), 7.35 (dd, J = 7.45, 1.7 Hz, 2H), 7.27–7.24 (m, 2H), 6.93–6.88 (m, 2H), 4.67 (d, J=6.3 Hz, 2H), 3.88 (s, 3H).
N-(3,4-Dimethoxyphenethyl)picolinamide(SM06)
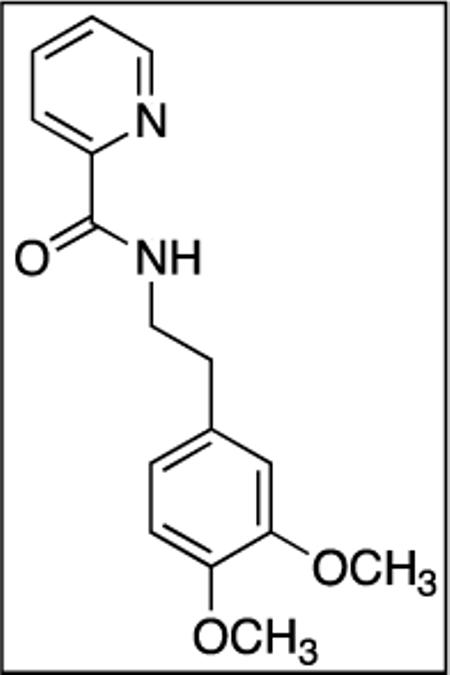
Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.7 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.3 mL), and 3,4-dimethoxyphenethylamine (20 mmol, 3.4 mL). After chromatography (hexanes/ethyl acetate 60/40), white powder was obtained (5.56 g, 97 %). Rf = 0.30 (hexanes/ethyl acetate 60/40). This compound is known.181H NMR (400 MHz, CDCl3, ppm) δ 8.52–8.51 (m, 1H), 8.21–8.16 (m, 2H), 7.87–7.82 (m, 1H), 7.43–7.39 (m, 1H), 6.84–6.78 (m, 3H), 3.87–3.85 (m, 6H), 3.74–3.69 (m, 2H), 2.91–2.87 (m, 2H).
N-(tert-Pentyl)picolinamide(SM07)

Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.3 mL), and tert-pentylamine (20 mmol, 2.19 mL). After chromatography (hexanes/ethyl acetate 70/30), colorless liquid was obtained (3.77 g, 98 %). Rf = 0.55 (hexanes/ethyl acetate 70/30). This compound is known.5l 1H NMR (400 MHz, CDCl3, ppm) δ 8.52-8.51 (m, 1H), 8.19–8.16 (m, 1H), 7.97 (br s, 1H), 7.85–7.81 (m, 1H), 7.41–7.38 (m, 1H), 1.87 (q, J=7.3 Hz, 2H), 1.45 (s, 6H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.4, 150.8, 147.8, 137.4, 125.9, 121.7, 53.7, 33.0, 26.4, 8.5.
N-(sec-Butyl)picolinamide(SM08)
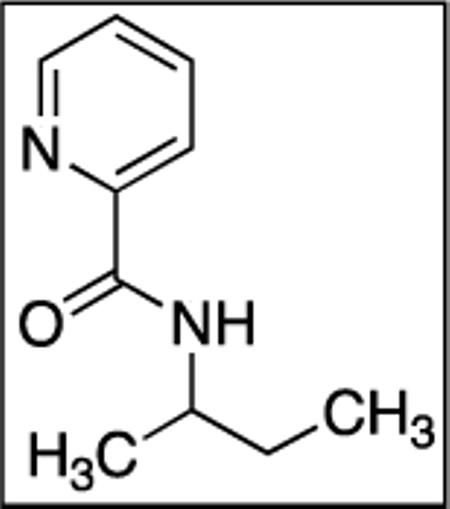
Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.3 mL), and 2-aminobutane (20 mmol, 2.01 mL). After chromatography (hexanes/ethyl acetate 70/30), white powder was obtained (3.27 g, 92 %). Rf = 0.35 (hexanes/ethyl acetate 70/30). This compound is known.4a1H NMR (400 MHz, CDCl3, ppm) δ 8.56–8.55 (m, 1H), 8.21 (d, J=7.8 Hz, 1H), 7.87–7.83 (m, 2H), 7.35–7.40 (m, 1H), 4.15–4.08 (m, 1H), 1.65–1.58 (m, 2H), 1.26 (d, J=6.4 Hz, 3H), 0.97 (t, J=7.5 Hz, 3H).
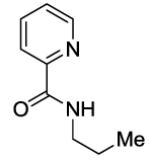
N-Propylpicolinamide (SM09)

Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.3 mL), and 1-propylamine (20 mmol, 1.64 mL). After chromatography (hexanes/ethyl acetate 70/30), colorless liquid was obtained (3.11 g, 95 %). Rf = 0.31 (hexanes/ethyl acetate 70/30). This compound is known.4a1H NMR (400 MHz, CDCl3, ppm) δ 8.55–8.53 (m, 1H), 8.21 (d, J=7.8 Hz, 1H), 8.15 (br s, 1H), 7.86–7.82 (m, 1H), 7.35–7.40 (m, 1H), 3.46 (q, J=6.4 Hz, 2H), 1.71–1.64 (m, 2H), 0.99 (t, J = 7.3 Hz, 3H).
N-Cyclohexylpicolinamide(SM10)

Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.70 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.3 mL), and cyclohexylamine (20 mmol, 2.29 mL). After chromatography (hexanes/ethyl acetate 70/30), white needles were obtained (4.30 g, 98 %). Rf = 0.32 (hexanes/ethyl acetate 70/30). This compound is known.191H NMR (400 MHz, CDCl3, ppm) δ 8.55–8.53 (m, 1H), 8.22–8.20 (m, 1H), 7.96 (s, J=3.7 Hz, 1H), 7.86–7.82 (m, 1H), 7.43–7.40 (m, 1H), 4.01–3.94 (m, 1H), 2.04–2.00 (m, 2H), 1.80–1.75 (m, 2H), 1.68–1.63 (m, 1H), 1.49–1.19 (m, 5H).
N-(2,4,4-Trimethylpentan-2-yl)picolinamide(SM11)
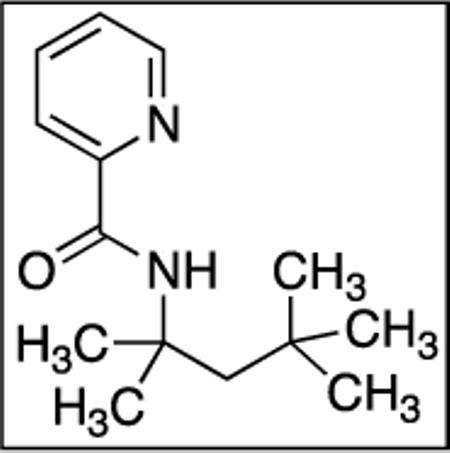
Picolinic acid (35 mmol, 4.3 g), triethylamine (70 mmol, 9.7 mL), dichloromethane (80 mL), ethyl chloroformate (35 mmol, 3.3 mL), and tert-octylamine (20 mmol, 3.2 mL). After column chromatography (hexanes/ethyl acetate 70/30), colorless oil was obtained (3.2 g, 62 %). Rf = 0.56 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.52 (d, J=4.0 Hz, 1H), 8.18 (d, J=8.0, Hz, 1H), 8.12 (br s, 1H), 7.84–7.81 (m, 1H), 7.40–7.38 (m, H), 1.87 (s, 2H), 1.56 (s, 6H), 1.03 (s, 9H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.1, 151.0, 148.9, 137.4, 125.8, 121.7, 54.7, 52.0, 31.8, 31.6, 29.2. FT-IR (neat, cm−1) ν 2956, 1681, 1522, 1464, 1432, 1365, 1228. Anal. Calcd. for C14H22N2O (234.34 g/mol): C, 71.76; H, 9.46; N, 11.95; Found: C, 71.46; H, 9.29; N, 11.86.
General procedure for the preparation of the 8-aminoquinoline amides
A round-bottom flask was charged with 8-aminoquinoline and triethylamine in dichloromethane. The respective benzoyl chloride was added as a solution in dichloromethane (35 mL). The mixture was stirred overnight at room temperature. The reaction mixture was transferred into separatory funnel and washed with water (2x35 mL). The water layer was extracted with dichloromethane (3x30 mL). Organic layers were combined, washed with saturated aqueous NaHCO3 (30 mL), and dried over MgSO4. Filtration and evaporation under reduced pressure gave the product. Amides not reported are known.4b
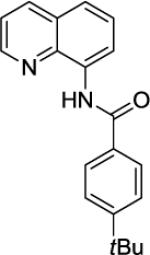
4-tert-Butyl-N-(quinolin-8-yl)benzamide (SM12, 12)
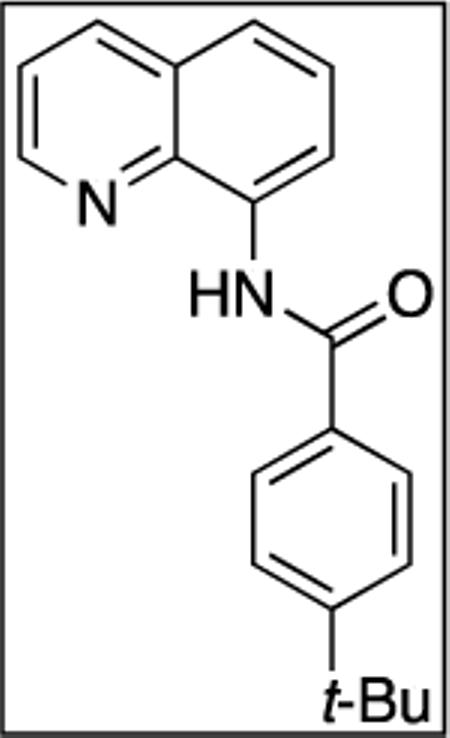
8-Aminoquinoline (13.1 mmol, 1.9 g) and Et3N (15.8 mmol, 2.2 mL) in dichloromethane (35 mL), followed by 4-tert-butylbenzoyl chloride (13.1 mmol, 2.6 g) in dichloromethane (25 mL). The mixture was stirred for 24 h at room temperature. After chromatography (hexanes/ethyl acetate 7/1), tan crystalline compound (6.92 g, 99 % yield) was obtained. Rf = 0.33 (hexanes/ethyl acetate 7/1), mp = 93–94 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 10.73 (s, 1H), 8.95 (dd, J = 7.4, 1.1 Hz, 1H), 8.85 (dd, J = 4.3, 1.6 Hz, 1H), 8.19 (dd, J = 8.3, 1.4 Hz, 1H), 8.05–8.01 (m, 2H), 7.62–7.52 (m, 4H), 7.50–7.46 (m, 1H), 1.38 (s, 9H). 13C NMR (100 MHz, CDCl3, ppm) δ 165.5, 155.4, 148.3, 138.8, 136.4, 134.8, 132.4, 128.0, 127.5, 127.2, 125.8, 121.8, 121.6, 116.5, 35.1, 31.3. FT-IR (neat, cm-1) ν 3349, 1665, 1531, 1485. Anal. Calcd. for C20H20N2O (304.35 g/mol): C, 78.92; H, 6.62; N, 9.20. Found: C, 78.88; H, 6.68; N, 9.22.
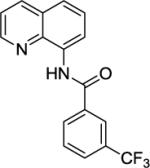
N-(Quinolin-8-yl)-3-(trifluoromethyl)benzamide (SM13)

8-Amino-quinoline (13.1 mmol, 1.9 g) and Et3N (15.8 mmol, 2.2 mL) in dichloromethane (35 mL), followed by 3-(trifluoromethyl)benzoyl chloride (23.4 mmol, 3.4 g) in dichloromethane (25 mL). The mixture was stirred for 16 h at room temperature. After recrystallization (ethanol/water), tan crystalline compound (4.9 g, 96 % yield) was obtained. Rf = 0.25 (toluene/ethyl acetate 50/1), mp = 86–87 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) 10.84(s, 1H), 8.93 (dd, J = 7.4, 1.4 Hz, 1H), 8.90 (dd, J = 4.3, 1.5 Hz, 1H), 8.37 (s, 1H), 8.33–8.28 (m, 2H), 7.87–7.84 (m, 1H), 7.74–7.62 (m, 3H), 7.59–7.55 (m, IH). 13C NMR (100 MHz, CDCl3, ppm) δ 164.0, 148.5, 138.7, 136.6, 136.0, 134.2, 131.5 (q, JC-F = 32.8 Hz), 130.3, 129.5, 128.5 (q, JC-F = 3.6 Hz), 128.0, 127.5, 124.6 (q, JC-F = 3.7 Hz), 122.4, 122.2, 121.9, 116.9. F NMR (376 MHz, CDCl3, ppm) δ-62.5 (s). Anal. Calcd for C17H11F3N2O (316.28 g/mol): C, 64.56; H, 3.51; N, 8.86. Found: C, 64.58; H, 3.45; N, 8.80.
4-Methoxy-3-methyl-N-(quinolin-8-yl)benzamide (SM14)
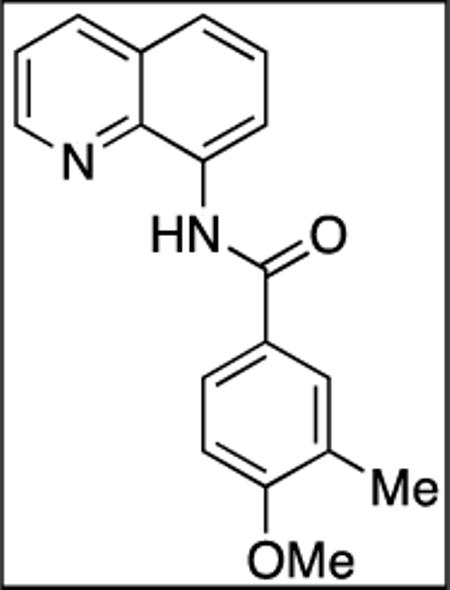
8-Amino-quinoline (10.9 mmol, 1.6 g) and Et3N (14.7 mmol, 2.0 mL) in dichloromethane (20 mL), followed by 4-methoxy-3-methylbenzoyl chloride (12 mmol, 2.2 g) in dichloromethane (15 mL). The mixture was stirred for 16 h at room temperature. After recrystallization (ethanol/water), tan crystalline compound (3.18 g, 99 % yield) was obtained, mp = 121-123 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 10.64 (s, IH) 8.97 (dd, J = 7.4, 1.3 Hz, 1H), 8.83 (dd, J = 4.4, 1.5 Hz, 1H), 8.14 (dd, J = 8.3, 1.8 Hz, 1H), 7.95–7.86 (m, 2H), 7.60–7.47 (m, 2H), 7.46–7.42 (m, 1H), 6.92 (d, J = 8.5 Hz, 1H), 1.55 (s, 3H), 0.92 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) 165.4, 160.8, 148.2, 138.8, 136.5, 134.9, 129.9, 128.1, 127.6, 127.1, 126.9, 126.7, 121.7, 121.4, 116.4, 109.5, 55.6, 16.5. Anal. Calcd for C18H16N2O2 292.33 g/mol): C, 73.95; H, 5.52; N, 9.58. Found: C, 74.00; H, 5.50; N, 9.53.
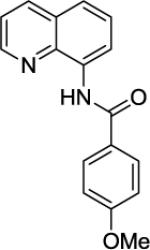
4-Methoxy-N-(quinolin-8-yl)benzamide (SM15)

8-Aminoquinoline (20.8 mmol, 3.0 g) and Et3N (25 mmol, 3.50 mL), in dichloromethane (35 mL), followed by 4-methoxybenzoyl chloride (21.8 mmol, 3.7 g) in dichloromethane (20 mL). The mixture was stirred for 24 h at room temperature. A crystalline compound (5.6 g, 98 % yield) was obtained, mp = 117–118 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 10.67 (s, IH), 8.92 (dd, J = 7.3, 1.4 Hz, 1H), 8.84 (dd, J = 4.3, 1.5 Hz, 1H), 8.15 (dd, J = 8.3, 1.6 Hz, 1H), 8.08–8.03 (m, 2H), 7.60–7.49 (m, 2H), 7.48-7.43 (m, 1H), 7.05–7.01 (m, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 165.0, 162.5, 148.3, 138.8, 136.4, 134.8, 129.2, 128.0, 127.6, 127.5, 121.7, 121.5, 116.4, 114.0, 55.5. Anal. Calcd for C17H14N2O2 (278.31 g/mol): C, 73.37; H, 5.07; N, 10.07. Found: C, 73.35; H, 5.09; N, 9.95.
2-(2-Fluoro-[1,1'-biphenyl]-4-yl)-N-(quinolin-8-yl)propan-amide (SM16)
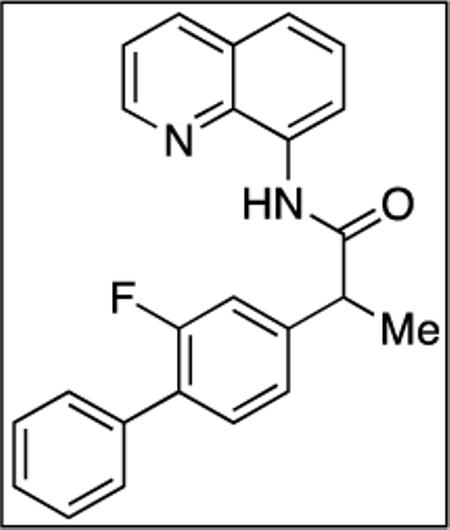
8-Aminoquinoline (7.4 mmol, 1.1 g) and Et3N (8.2 mmol, 1.2 mL) in dichloromethane (25 mL), followed by 2-(2-fluorobiphenyl-4-yl)propanoyl chloride (8.2 mmol, 2.1 g) in dichloromethane (20 mL). The mixture was stirred for 16 h at room temperature. After chromatography (toluene/ethyl acetate 25/1), tan crystalline compound (2.6 g, 84 % yield) was obtained. Rf = 0.40 (toluene/ethyl acetate 25/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.98 (s, 1H), 8.81–8.69 (m, 2H), 8.11 (dd, J = 8.3, 0.9 Hz, 1H), 7.57–7.27 (m, 11H), 4.00–3.92 (m, 1H), 1.72 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm; list of signals, C-F coupling not assigned) δ 172.1, 161.2, 158.7, 147.4, 142.6, 142.5, 138.5, 136.4, 135.6, 134.5, 131.3, 131.2, 129.1, 129.0, 128.6, 128.4, 128.1, 128.0, 127.8, 127.4, 123.8, 123.7, 121.8, 127.7, 116.5, 115.6, 115.4, 48.2, 18.7. 19F NMR (376 MHz, CDCl3, ppm) δ -117.3 (m). HRMS electrospray (m/z): [M+ + Na] calcd for C24H19FN2O, 393.13792; found, 393.13782, error=1.16 ppm.
Optimization of reaction conditions for arylation ofN-(2-phenylpropan-2-yl)picolinamide
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), N-(2-phenylpropan-2-yl)picolinamide (1.0 mmol, 246 mg), and 1-iodo-4-methylbenzene (4.0 mmol, 896 mg). The reactants and solvent were added to this mixture (Table 7). The resulting suspension was stirred in an oil bath at the specified temperature. After the designated time, the reaction mixture was cooled, diluted with dichloromethane (4 mL) and analyzed by GC-MS.
Table 7.
Optimization of Picolinamide Arylationa

| ||
|---|---|---|
|
| ||
| Entry | Reaction conditions | % GC Yield (isolated) |
| 1 | 4 eq NaOAc, 2 mL MeCN, 60 °C, 16 h | 0 |
| 2 | 4 eq NaOAc, 2 mL toluene, 140 °C, 16 h | 10 |
| 3 | 4 eq CsOAc, 1.6 mL toluene, 0.4 mL MeCN, 140 °C, 16 h | 30 |
| 4 | 2 eq K3PO4, 1.6 mL toluene, 0.4 mL MeCN, 140 °C, 16 h | 15 |
| 5 | 2 eq Cs2CO3, 1.6 mL toluene, 0.4 mL MeCN, 140 °C, 16 h | 15 |
| 6 | 3 eq K3PO4, 1.6 mL t-amyl alcohol, 0.4 mL H2O, 90 °C, 16 h | 70 |
| 7 | 3 eq K2CO3, 2 mL t-amyl alcohol, 90 °C, 20 h | 80 |
| 8 | 4 eq K2CO3, 2 mL t-amyl alcohol, 110°C, 24 h | 90 (86) |
| 9 | 4 eq K2CO3, 2 mL t-amyl alcohol, 140°C, 24 h | 80 |
| 10 | 4 eq CsOAc, 1 mL t-amyl alcohol, 110°C, 24 h | 85 |
| 11 | 4 eq CsOAc, 1 mL t-amyl alcohol, 140°C, 24 h | 90 |
| 12 | 10 % CuBr2, 4 eq CsOAc, 1 mL t-amyl alcohol, 110°C, 24 hr | 75 (70) |
| 13 | 10 % CuBr2, 4 eq CsOAc, 1 mL t-amyl alcohol, 140°C, 24 h | 99 (99) |
a Dodecane as internal standard; amide (0.5 mmol), ArI (4 mmol).
Determination of the GC conversion using internal standard
The GC conversion for the optimization experiments was calculated based on an internal standard (dodecane) as described here. First, a 1:1 molar mixture of dodecane and the pure target compound was dissolved in ethyl acetate and injected into GC to determine detector response ratio F = Atc/Ado (Atc: area of target compound peak, Ado: area of dodecane peak). Second, the reaction is set up as usual on 1 mmol scale with the addition of dodecane as internal standard (0.3 mmol). After the completion of reaction, 1 drop of reaction mixture is diluted with CH2Cl2 and injected into GC to determine area of dodecane (Ador) and the target compound (Atcr). The amount of target compound in reaction mixture can be calculated by the following equation: ntcr = 0.3.Atcr/(Ador.F) (mmol). The conversion is derived based on the amount of starting material added (nsm): C = (ntcr/nsm)*100%.
Attempted synthesis ofN-((2,2"-dimethyl-[1,1':3',1"-terphenyl]-2'-yl)methyl)picolinamide

A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-benzylpicolinamide (1.0 mmol, 224 mg), 2-iodotoluene (4.0 mmol, 872 mg), CsOAc (4.0 mmol, 794 mg), and tert-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. An aliquot of the reaction mixture was diluted with ethyl acetate and passed though a short silica plug. GC-MS analysis indicated that no product was formed.
Solvent optimization for the alkylation ofN-(1-phenylethyl)picolinamide
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 6 mg), N-(1-phenylethyl)picolinamide (0.5 mmol, 134 mg), iodobutane (2 mmol, 367 mg), and K3PO4 (2 mmol, 424 mg). The solvent (0.50 mL) was added to this mixture (Table 8). The resulting suspension was stirred in an oil bath at 140 °C. After 24 hours, the reaction mixture was cooled and diluted with dichloromethane (4 mL) followed by analysis with GCMS using dodecane as an internal standard as described earlier.
Table 8.
Alkylation of Picolinamides – Optimization of Solventa

| |||
|---|---|---|---|
|
| |||
| Entry | Solvent, 0.50 mL | % GC Yield |
|
| A | B | ||
| 1 | CF3CH2OH | 26 | 19 |
| 2 | CF3CH(OH)CF3 | 52 | 13 |
| 3 | CH3COOH | 36 | 13 |
| 4 | Piv-OH | 20 | 5 |
| 5 | H2O, m-xylene | 50 | 24 |
| 6 | H2O | 47 | 23 |
| 7 | H2O (0.15 mL) | 50 | 14 |
Dodecane internal standard, amide (1 mmol), BuI (4 mmol)
Optimization of the additive used for the alkylation ofN-(1-phenylethyl)picolinamide
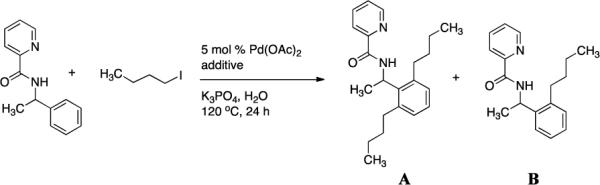
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 6 mg), N-(1-phenylethyl)picolinamide (0.5 mmol, 134 mg), iodobutane (2 mmol, 367 mg), and K3PO4 (2.0 mmol, 424 mg). The additive (10 mol %) was added to this mixture (Table 9). The resulting suspension was stirred in an oil bath at 120 °C. After 24 hours, the reaction mixture was cooled and diluted with dichloromethane (4 mL) followed by GC-MS analysis using dodecane as an internal standard as described earlier.
Table 9.
Alkylation of Picolinamides – Optimization of Additivesa
| |||
|---|---|---|---|
|
| |||
| Entry | Additive | % GC Yield |
|
| A | B | ||
| 1 | No additive | 45 | 15 |
| 2 | 10 % CuBr2 | 95 | 5 |
| 3 | 20 % CuBr2 | 76 | 14 |
| 4 | 10 % Cu(NO3)2 | 65 | 17 |
| 5 | 20 % Cu(NO3)2 | 95 | 4 |
| 6 | 10 % CuCl2 | 55 | 20 |
| 7 | 10 % CuCO3 | 58 | 19 |
| 8 | 10 % CuOAc | 84 | 10 |
| 9 | 10 % MnO2 | 70 | 17 |
Dodecane internal standard; amide (0.5 mmol), BuI (4 mmol).
Optimization of the base used for the alkylation ofN-(1-phenylethyl)picolinamide
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 6 mg), CuBr2 (10 mol %, 11 mg) N-(1-phenylethyl)picolinamide (0.5 mmol, 134 mg), iodobutane (2 mmol, 367 mg), base (2 mmol), and water (0.30 mL). The resulting suspension was stirred in oil bath at 120 °C. After 24 hours, the reaction mixture was cooled and diluted with dichloromethane (4 mL) followed by analysis with GC-MS using dodecane as an internal standard as described earlier.
General procedure for the arylation of sp2 C-H bonds of picolinamides
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), picolinamide (1 mmol), aryl iodide (4 mmol), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 hours. The reaction mixture was then extracted with dichloromethane (3x4 mL). The extracts were combined, filtered through pad of cotton, concentrated, and then loaded onto a chromatography column with hexanes/ethyl acetate mixture as an eluent and subjected to column chromatography. After concentration of the fractions containing the product, the residue was dried under reduced pressure.
General procedure for the arylation ofN-(naphthalen-1-yl)picolinamide using Pd(OAc)2 catalyst and AgOAc base
A 2-dram screw-cap via was charged with Pd(OAc)2 (2 mol%, 4.4 mg), AgOAc (166 mg, 1 mmol), aryl iodide (2 mmol), and N-(naphthalen-1-yl)picolinamide (0.5 mmol, 125 mg). The resulting solution was stirred at 140 °C for 24 hours. The reaction mixture was then diluted with dichloromethane (2 mL), filtered through pad of celite, concentrated, then loaded on a chromatography column with hexane/ethyl acetate mixture as an eluent.
N-(2-(4,4"-Dimethyl-[1,1':3',1"-terphenyl]-2'-yl)propan-2-yl)-picolinamide(Table 2, Entry 1)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol%, 11 mg), CuBr2 (10 mol %, 22 mg), N-(2-phenylpropan-2-yl)picolinamide (1 mmol, 246 mg), 1-iodo-4-methylbenzene (4 mmol, 896 mg), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), tan powder (425 mg, 99 % yield) was obtained. Rf = 0.45 (hexanes/ethyl acetate 70/30), mp=164–165 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.21–8.20 (m, 1H), 7.99–7.97 (m, 1H), 7.76–7.72 (m, 1H), 7.58 (br s, 1H), 7.31-7.28 (m, 1H), 7.17–7.13 (m, 5H), 7.02 (d, J=7.3 Hz, 2H), 6.90 (d, J=7.3 Hz, 2H), 2.25 (s, 6H), 1.60 (s, 6H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 150.7, 147.1, 143.4, 141.8, 141.7, 136.9, 135.7, 132.8, 128.7, 128.2, 125.4, 124.9, 121.5, 57.5, 33.4, 21.2. FT-IR (neat, cm−1) ν 3369, 1679, 1527, 1444, 1224, 1042. Anal. Calcd for C29H28N2O (420.55 g/mol): C, 82.82; H, 6.71; N, 6.66; Found: C, 82.47; H, 6.69; N, 6.55.
N-([1,1':3',1"-Terphenyl]-2'-ylmethyl)picolinamide(Table 2, Entry 2)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-benzylpicolinamide (1 mmol, 212 mg), iodobenzene (4 mmol, 816 mg), CsOAc (4 mmol, 794 mg), and t-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), white needles (360 mg, 99 % yield) were obtained. Rf = 0.34 (hexanes/ethyl acetate 70/30), mp = 119–120 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.44–8.43 (m, 1H), 7.98–7.96 (m, 1H), 7.78–7.72 (m, 2H), 7.43–7.29 (m, 14H), 4.49 (d, J = 5.1 Hz, 2H) 13C NMR (100 MHz, CDCl3, ppm) δ 162.9, 149.8, 147.9, 143.9, 141.2, 137.1, 132.7, 129.8, 129.1, 128.3, 127.5, 127.4, 125.9, 122.0, 39.4. FT-IR (neat, cm−1) ν 3378, 1678, 1510, 1464, 1435, 1000. Anal. Calcd for C25H20N2O (364.44 g/mol): C, 82.39; H, 5.53; N, 7.69; Found: C, 82.50; H, 5.45; N, 7.68. When bromobenzene (4 mmol, 628 mg) was used instead of iodobenzene, product was not detected by GC-MS.
Ethyl 3'-methoxy-2'-(picolinamidomethyl)-[1,1'-biphenyl]-4-carboxylate(Table 2, Entry 3)
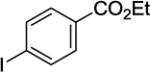
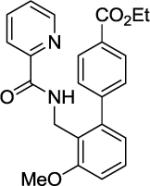
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(2-methoxybenzyl)picolinamide (1 mmol, 247 mg), ethyl-4-iodobenzoate (4 mmol, 1104 mg), CsOAc (4 mmol, 794 mg), and t-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 60/40), white needles (353 mg, 92 % yield) were obtained. Rf = 0.33 (hexanes/ethyl acetate 60/40), mp=163–164 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.50–8.48 (m, 1H), 8.33–8.36 (m, 1H), 8.16–8.13 (m, 1H), 8.10–8.07 (m, 2H), 7.81–7.76 (m, 1H), 7.43–7.41 (m, 2H), 7.38–7.31 (m, 2H), 6.96 (d, J=7.8 Hz, 1H), 6.89 (dd, J = 7.8, 0.9 Hz, 1H), 4.58 (d, J = 5.5 Hz, 2H), 4.37 (q, J = 7.3 Hz, 2H), 3.94 (s, 3H), 1.38 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 166.6, 163.4, 158.8, 150.3, 148.1, 145.3, 143.0, 137.3, 129.6, 129.5, 128.7, 126.0, 123.6. 122.5, 122.3, 110.2, 61.0, 56.0, 38.6, 14.5. FT-IR (neat, cm−1) ν 3396, 1709, 1668, 1584. 1512, 1462, 1271, 1176, 1023. Anal. Calcd for C23H22N2O4 (390.43 g/mol): C, 70.75; H, 5.68; N, 7.17; Found: C, 70.86; H, 5.65; N, 7.16.
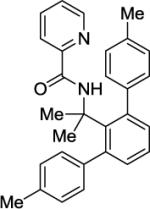
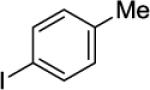
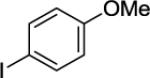
N-(8-p-Tolylnaphthalen-1-yl)picolinamide (Table 2, Entry 4)
N-(Naphthalen-1-yl)picolinamide (5.1 g, 20.5 mmol), 4-iodotoluene (17.5 g, 80.3 mmol), AgOAc (5.1 g, 30.5 mmol), and Pd(OAc)2 (101 mg, 0.45 mmol). The resulting suspension was stirred at 140 °C for 24 h. After column chromatography (hexanes/ethyl acetate 90/10 then hexanes/ethyl acetate 65/35), the solvent was evaporated to give light brown crystals (6.45 g, 91 % yield). Rf = 0.50 (hexanes/ethyl acetate 65/35), mp=123-124 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.61 (s, 1H), 8.23 (dd, J = 7.7, 1.5 Hz, 1H), 8.18–8.16 (m, 1H), 8.10–8.08 (m, 1H), 7.86 (dd, J = 8.4, 1.5 Hz, 1H), 7.80–7.74 (m, 2H), 7.58–7.54 (m, 1H), 7.48–7.44 (m, 1H), 7.32–7.24 (m, 4H), 6.96 (d, J = 7.7 Hz, 2H), 2.80 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 150.0, 147.4, 139.9, 137.8, 137.0, 136.6, 135.6, 133.0, 130.5, 129.2, 128.9, 128.6, 126.5, 126.0, 125.7, 125.1, 125.0, 122.6, 121.9, 21.2. FT-IR (neat, cm−1) ν 1689, 1493, 1433. Anal. Calcd for C23H18N2O (388.4 g/mol): C, 81.63; H, 5.36; N, 8.28; Found: C, 81.54; H, 5.35; N, 8.23.
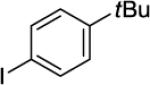
N-(8-(4-tert-Butylphenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 5)
N-(Naphthalen-1-yl)picolinamide (124, 0.5 mmol), 4-t-butyliodobenzene (mg, 2 mmol), AgOAc (166 mg, 1.0 mmol), and Pd(OAc)2 (4.4 mg, 0.01 mmol). After column chromatography (hexanes/ethyl acetate 80/20), the solvent was evaporated to give white powder (190 mg, 99 % yield). Rf = 0.34 (hexanes/ethyl acetate 80/20), mp=136-137 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.60 (s, 1H), 8.32–8.18 (m, 2H), 8.11–8.10 (m, 1H), 7.88 (dd, J = 8.2, 0.9 Hz, 1H), 7.81–7.73 (m, 1H), 7.59–7.55 (m, 2H), 7.50–7.47 (m, 2H), 7.36–7.34 (m, 3H), 7.31–7.28 (m, 1H), 7.21–7.17 (m, 1H), 1.06 (s, 9H). 13C NMR (100 MHz, CDCl3, ppm) δ 161.7, 149.8, 149.7, 147.6, 139.6, 137.8, 137.1, 135.6, 133.1, 130.7, 128.9, 128.4, 126.3, 126.0, 125.9, 125.2, 124.8, 122.3, 121.9, 34.3, 31.3. Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 1690, 1521, 1495. HRMS (m/z): [M+] calcd for C26H24N2O, 380.1889; found, 380.1885, error=−1.1 ppm.
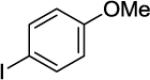
N-(8-(4-Methoxyphenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 6)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(naphthalen-1-yl)picolinamide (1.0 mmol, 247 mg), 1-iodo-4-methoxybenzene (4.0 mmol, 936 mg), CsOAc (4.0 mmol, 794 mg), and tert-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexanes/ethyl acetate 50/50), a beige powder (260 mg, 73 % yield) was obtained. Rf = 0.31 (hexane/ethyl acetate 50/50), mp = 107–108 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.71 (s, 1H), 8.30–8.28 (dd, J = 7.8, 1.4 Hz, 1H), 8.20–8.19 (d, J = 5.0, 1H), 8.11 (d, J = 7.8, 1H), 7.86 (dd, J = 8.2, 0.9 Hz, 1H), 7.86 (dd, J = 8.2, 0.9 Hz, 1H), 7.80–7.56 (m, 2H), 7.59–7.55 (m, 1H), 7.49–7.45 (m, 1H), 7.34–7.30 (m, 4H), 6.75–6.67 (m, 2H), 3.59 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 158.9, 150.1, 147.5, 137.4, 137.0, 135.5, 135.1, 133.1, 130.7, 130.4, 128.6, 126.4, 126.0, 125.8, 125.0, 122.3, 122.0, 113.6, 55.0. Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 1683, 1494, 1515, 1433, 1243, 1176, 1036. Anal. Calcd for C23H18N2O2 (354.40 g/mol): C, 77.95; H, 5.12; N, 7.90; Found: C, 77.68; H, 5.09; N, 7.78.
N-(8-(4-Methoxyphenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 6)
N-(Naphthalen-1-yl)picolinamide (127 mg, 0.5 mmol), 4-iodoanisole (468 mg, 2 mmol), AgOAc (166 mg, 1.0 mmol), and Pd(OAc)2 (4.4 mg, 0.01 mmol). The resulting suspension was stirred at 140 °C for 24 h. After column chromatography (hexanes/ethyl acetate 70/30), the solvent was evaporated to give white powder (176 mg, 98 % yield). 1H NMR (400 MHz, CDCl3, ppm) δ 9.71 (s, 1H), 8.30–8.28 (dd, J = 7.8, 1.4 Hz, 1H), 8.20–8.19 (d, J = 5.0, 1H), 8.11 (d, J = 7.8, 1H), 7.86 (dd, J = 8.2, 0.9 Hz, 1H), 7.86 (dd, J = 8.2, 0.9 Hz, 1H), 7.80–7.56 (m, 2H), 7.59–7.55 (m, 1H), 7.49–7.45 (m, 1H), 7.34–7.30 (m, 4H), 6.75–6.67 (m, 2H), 3.59 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 158.9, 150.1, 147.5, 137.4, 137.0, 135.5, 135.1, 133.1, 130.7, 130.4, 128.6, 126.4, 126.0, 125.8, 125.0, 122.3, 122.0, 113.6, 55.0. Signal for one carbon could not be located. Anal. Calcd for C23H18N2O2 (354.4 g/mol): C, 77.95; H, 5.12; N, 7.90; Found: C, 77.68; H, 5.09; N, 7.78.
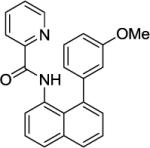
N-(8-(3-Methoxyphenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 7)
N-(Naphthalen-1-yl)picolinamide (125 mg, 0.5 mmol), 3-iodoanisole (468 mg, 2 mmol), AgOAc (166 mg, 1.0 mmol), and Pd(OAc)2 (4.4 mg, 0.01 mmol). After column chromatography (hexanes/ethyl acetate 70/30), the solvent was evaporated to give white powder (178 mg, 99 % yield). Rf = 0.30 (hexanes/ethyl acetate 70/30), mp=99–100 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.60 (s, 1H), 8.23–8.18 (m, 2H), 8.10–8.08 (m, 1H), 7.88 (dd, J = 8.2, 1.4 Hz, 1H), 7.82–7.75 (m, 2H), 7.60–7.56 (m, 1H), 7.47 (dd, J = 8.2 Hz, 7.3 Hz, 1H), 7.34–7.31 (m, 2H), 7.07–7.04 (m, 2H), 6.91–6.93 (m, 2H), 6.52–6.49 (m, 1H), 3.67 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.2, 159.4, 149.9, 147.5, 144.2, 137.6, 137.1, 135.5, 132.9, 130.3, 129.2, 128.9, 126.6, 126.0, 125.9, 125.3, 125.0, 123.1, 121.9, 121.7, 114.0, 113.3, 55.2. FT-IR (neat, cm−1) ν 1682, 1521, 1577, 1498, 1427, 1215, 1160, 1041. Anal. Calcd for C23H18N2O2 (354.4 g/mol): C, 77.95; H, 5.12; N, 7.90; Found: C, 77.69; H, 5.10; N, 7.83.
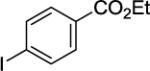
Ethyl 4-(8-(picolinamido)naphthalen-1-yl)benzoate (Table 2, Entry 8)
N-(Naphthalen-1-yl)picolinamide (122 mg, 0.5 mmol), ethyl 4-iodobenzoate (122 mg, 2 mmol), AgOAc (166 mg, 1.0 mmol), and Pd(OAc)2 (4.4 mg, 0.01 mmol). After column chromatography (hexanes/ethyl acetate 70/30), the solvent was evaporated to give white powder (179 mg, 92 % yield). Rf = 0.28 (hexanes/ethyl acetate 70/30), mp=82–83 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.35 (s, 1H), 8.12–8.10 (m, 2H), 8.06 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.83–7.80 (m, 3H), 7.76–7.70 (m, 1H), 7.60–7.56 (m, 1H), 7.50–7.46 (m, 1H), 7.43 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 7.4 Hz, 1H), 7.24–7.22 (m, 1H), 4.29 (q, J = 7.3, 2H), 1.32 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 166.3, 162.0, 149.6, 147.7, 147.5, 137.1, 136.8, 135.5, 132.5, 130.2, 129.4, 129.2, 128.8, 126.9, 126.2, 125.9, 125.4, 125.0, 123.8, 122.0, 60.8, 14.5. Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 1710, 1682, 1495, 1266, 1102. Anal. Calcd for C25H20N2O3 (396.4 g/mol): C, 75.74; H, 5.08; N, 7.07; Found: C, 75.63; H, 5.05; N, 7.00.
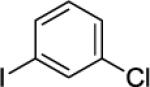
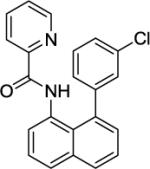
N-(8-(3-Chlorophenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 9)
N-(Naphthalen-1-yl)picolinamide (135 mg, 0.5 mmol), 1-chloro-3-iodobenzene (476 mg, 2 mmol), AgOAc (166 mg, 1.0 mmol), and Pd(OAc)2 (4.4 mg, 0.01 mmol). After column chromatography (hexanes/ethyl acetate 80/20), the solvent was evaporated to give white powder (190 mg, 98 % yield). Rf = 0.30 (hexanes/ethyl acetate 80/20), mp=120–121 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.50 (s, 1H), 8.30 (d, J = 4.6 Hz, 1H), 8.18 (dd, J = 7.8, 0.9 Hz, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.89 (dd, J = 8.2, 1.4 Hz, 1H), 7.82–7.76 (m, 2H), 7.60–7.56 (m, 1H), 7.49–7.47 (m, 2H), 7.37–7.34 (m, 1H), 7.28 (dd, J = 7.4, 1.4 Hz, 1H), 7.18–7.15 (m, 1H), 7.01–6.96 (m, 1H), 6.92–6.90 (m, 1H). 13C NMR (100 MHz, CDCl3, ppm) δ 161.9, 149.6, 147.5, 144.7, 137.2, 136.2, 135.5, 134.2, 132.6, 130.4, 129.3, 129.1, 129.0, 127.8, 126.8, 126.2, 126.1, 125.2, 125.0, 123.3, 122.0. Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 1684, 1526, 1498, 1432. Anal. Calcd for C22H15ClN2O (358.8 g/mol): C, 73.64; H, 4.21; N, 7.81; Found: C, 73.89; H, 4.09; N, 7.76.
N-(8-(4-Bromophenyl)naphthalen-1-yl)picolinamide (Table 2, entry 10)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(naphthalen-1-yl)picolinamide (1.0 mmol, 251 mg), 1-bromo-4-iodobenzene (4.0 mmol, 1.13 g), CsOAc (4.0 mmol, 794 mg), and tert-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 80/20), light brown powder (342 mg, 84 % yield) was obtained. Rf = 0.33 (hexanes/ethyl acetate 80/20), mp = 134–135 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ9.56 (s, 1H), 8.32–8.30 (m, 1H), 8.21–8.20 (m, 1H), 8.12 (d, J = 7.8 Hz, 1H), 7.91 (dd, J = 8.3, 0.9 Hz, 1H), 7.84–7.80 (m, 2H), 7.61–7.57 (m, 1H), 7.50–7.46 (m, 1H), 7.42–7.39 (m, 1H), 7.32–7.28 (m, 5H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 149.5, 147.9, 141.9, 137.2, 136.4, 135.6, 132.6, 131.3, 130.9, 130.5, 129.2, 129.7, 126.3, 126.2, 125.0, 123.2, 122.0, 121.4. Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 1687, 1498, 1433, 1009. Anal. Calcd for C22H15BrN2O (403.3 g/mol): C, 65.52; H, 3.75; N, 6.95; Found: C, 65.10; H, 3.54; N, 6.82.
N-(8-(4-Bromophenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 10)
N-(Naphthalen-1-yl)picolinamide (121 mg, 0.5 mmol), 1-bromo-4-iodobenzene (564 mg, 2 mmol), AgOAc (166 mg, 1.0 mmol), and Pd(OAc)2 (4.4 mg, 0.01 mmol). The resulting suspension was stirred at 140 °C for 24 h. After column chromatography (hexanes/ethyl acetate 70/30), the solvent was evaporated to give white powder (161 mg, 82 % yield). 1H NMR (400 MHz, CDCl3, ppm) δ 9.56 (s, 1H), 8.32–8.30 (m, 1H), 8.21–8.20 (m, 1H), 8.12 (d, J = 7.8 Hz, 1H), 7.91 (dd, J = 8.3, 0.9 Hz, 1H), 7.84–7.80 (m, 2H), 7.61–7.57 (m, 1H), 7.50–7.46 (m, 1H), 7.42–7.39 (m, 1H), 7.32–7.28 (m, 5H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 149.5, 147.9, 141.9, 137.2, 136.4, 135.6, 132.6, 131.3, 130.9, 130.5, 129.2, 129.7, 126.3, 126.2, 125.0, 123.2, 122.0, 121.4. Signal for one carbon could not be located. Anal. Calcd for C22H15BrN2O (403.3 g/mol): C, 65.52; H, 3.75; N, 6.95; Found: C, 65.10; H, 3.54; N, 6.82.
Large Scale Synthesis of N-(8-(4-(Trifluoromethyl)phenyl)naphthalen-1-yl)picolinamide (Table 2, Entry 11)
N-(Naphthalen-1-yl)picolinamide (12.4 g, 50 mmol), 4-iodobenzotrifluoride (27.2 g, 100 mmol), AgOAc (12.45 g, 75 mmol), and Pd(OAc)2 (224 mg, 1.0 mmol). The flask was sealed with rubber septum and then heated with stirring at 140 °C for 24 hours. After the reaction was complete, the mixture was cooled and 150 mL of ethyl acetate was added. The mixture was filtered and the filtrate was washed with brine (150 mL). The layers were separated and the aqueous solution was extracted with ethyl acetate (2×50 mL). The organic layers were combined, dried with MgSO4, and concentrated. The residue was subjected to column chromatography (hexanes/ethyl acetate 65/35), and the solvent was evaporated to give light orange crystals (16.3 g, 84 % yield). Rf = 0.48 (hexanes/ethylacetate 70/30), mp = 155–156 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.34 (s, 1H), 8.15 (d, J = 4.1 Hz, 1H), 8.09–8.07 (m, 2H), 7.93 (dd, J = 8.2, 2.3 Hz, 1H), 7.84 (dd, J = 8.2, 1.3 Hz, 1H), 7.79–7.75 (m, 1H), 7.62–7.58 (m, 1H), 7.52–7.48 (m, 3H), 7.39 (d, J = 7.8 Hz, 2H), 7.34–7.30 (m, 2H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.0, 149.3, 147.6, 146.7, 137.3, 136.3, 135.6, 132.4, 130.5, 129.5, 129.1, 128.7, 127.0, 126.3, 125,5, 125.1, 124.9, 124.8 (q, JC-F = 3.8 Hz), 124.1, 122.7, 121.9. FT-IR (neat, cm−1) ν 1670, 1491, 1320, 1185, 1141, 1111, 1070, 1058, 1018. Anal. Calcd for C23H15F3N2O (392.4 g/mol): C, 70.40; H, 3.85; N, 7.14; Found: C, 70.59; H, 3.65; N, 7.14.
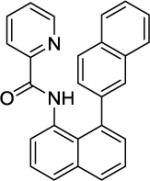
Large Scale Synthesis of N-(1,2'-Binaphthyl-8-yl)picolinamide (Table 2, Entry 12)
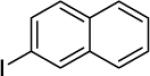
N-(Naphthalen-1-yl)picolinamide (8.68 g, 35 mmol), 2-iodonaphthalene (26.7 g, 105 mmol), AgOAc (8.71 g, 52.5 mmol), and Pd(OAc)2 (392 mg, 1.75 mmol). The flask was sealed with rubber septum and then heated with stirring at 140 °C for 24 hours. After the reaction was complete, the mixture was cooled and ethyl acetate (150 mL) was added. The mixture was filtered and the filtrate was washed with brine (150 mL). The layers were separated and the aqueous solution was extracted with ethyl acetate (2x50 mL). The organic layers were combined, dried with MgSO4, and concentrated. The residue was subjected to column chromatography (dichloromethane/ethyl acetate 50/50), and the solvent was evaporated and the residue obtained was recrystallized from methanol to give light brown crystals (8.5 g, 65 % yield). Rf = 0.32 (dichloromethane/ethyl acetate 50/50), mp = 155–156 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.56 (s, 1H), 8.31 (dd, J = 7.8, 1.4 Hz, 1H), 8.03 (s, 1H), 7.94–7.87 (m, 3H), 7.81 (dd, J = 8.3, 0.9 Hz, 1 H), 7.61–7.47 (m, 6H), 7.41–7.34 (m, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.1, 149.4, 146.8, 140.7, 137.6, 136.6, 135.5, 133.8, 133.1, 132.7, 130.7, 129.0, 128.1, 128.0, 127.7, 127.6, 127.4, 126.4, 126.3, 126.1, 125.8, 125.7, 125.1, 125.0, 122.3, 121.5. FT-IR (neat, cm−1) ν 1692, 1496. Anal. Calcd for C26H18N2O (374.4 g/mol): C, 83.40; H, 4.85; N, 7.48; Found: C, 83.19; H, 4.88; N, 7.39.
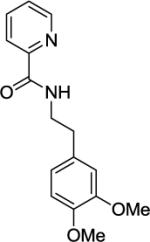
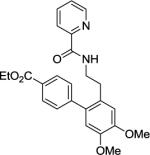
Ethyl 4',5'-dimethoxy-2'-(2-(picolinamido)ethyl)-[1,1'-biphenyl]-4-carboxylate(Table 2, entry 13)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(3,4-dimethoxyphenethyl)picolinamide (1.0 mmol, 265 mg), ethyl 4-iodobenzoate (4.0 mmol, 1.10 g), CsOAc (4.0 mmol, 794 mg), and tert-amyl alcohol (1.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 40/60), white powder (207 mg, 52 % yield) was obtained. Rf = 0.39 (hexanes/ethyl acetate 40/60), mp = 133–134 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.50–8.49 (m, 1H), 8.14–8.11 (m, 1H), 8.06–8.03 (m, 2H), 7.97–7.95 (m, 1H), 7.84–7.80 (m, 1H), 7.42–7.37 (m, 3H), 6.86 (s, 1H), 6.72 (s, 1H), 4.34 (q, J=7.3 Hz, 3.87 (s, 3H), 3.86 (s, 3H), 3.52 (q, J=7.3 Hz, 2H), 2.85 (t, J=7.3 Hz, 2H), 1.41 (t, J=7.3 Hz, 2H). 13C NMR (100 MHz, CDCl3, ppm) δ 166.6, 164.2, 149.9, 148.7, 148.0, 147.4, 146.2, 137.5, 133.6, 129.6, 129.1, 128.5, 126.3, 122.3, 113.1, 112.7, 61,1, 56.1, 56.0, 40.6, 32.6, 14.5. Signal for one carbon could not be located. δ. FT-IR (neat, cm−1) ν 3364, 1706, 1666, 1520, 1502, 1440, 1272, 1237, 1212, 1139, 1097, 1032. Anal. Calcd for C25H26N2O5 (434.48 g/mol): C, 69.11; H, 6.03; N, 6.45; Found: C, 69.08; H, 5.95; N, 6.44.
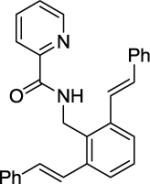
N-(2,6-Di((E)-styryl)benzyl)picolinamide (Table 2, Entry 14)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-benzylpicolinamide (1 mmol, 194 mg), (E)-(2-iodovinyl)benzene (4 mmol, 0.92 g), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 h. After chromatography (hexanes/ethyl acetate 70/30), tan needles (269 mg, 86 % yield) were obtained. Rf = 0.35 (hexanes/ethyl acetate 70/30), mp=145–146 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ8.43-8.41 (m, 1H), 8.23–8.21 (m, 1H), 8.14–8.12 (m, 1H), 7.82–7.78 (m, 1H), 7.61–7.53 (m, 8H), 7.40–7.34 (m, 6H), 7.27–7.24 (m, 2H), 7.02 (d, J=16.0 Hz, 2H), 4.98 (d, J=5.5 Hz, 2H). 13C NMR (100 MHz, CDCl3, ppm) φ 163.9, 149.8, 148.1, 138.3, 137.4, 137.3, 132.5, 132.4, 128.7, 128.5, 127.9, 126.9, 126.2, 126.0, 125.9, 122.4, 37.4. FT-IR (neat, cm−1) ν 3395, 1677, 1515. Anal. calcd for C29H24N2O (416.51g/mol): C, 83.63; H, 5.81; N, 6.73; Found: C, 83.89; H, 5.70; N, 6.57.
General procedure for the hydrolysis of the arylated picolinamides
The N-(8-arylnaphthalen-1-yl)picolinamide was dissolved in ethanolic NaOH solution (NaOH in EtOH/H2O 10/1) and refluxed for 6 hours. The reaction mixture was cooled and diluted with an equal volume of water. The product was extracted with dichloromethane (3×60 mL). The combined organic layers were combined, dried with MgSO4, and concentrated. The crude compound was subjected to column chromatography and the fractions containing the product were combined and the solvent was evaporated to give pure 8-arylnaphthalen-1-amines.
8-p-Tolylnaphthalen-1-amine (6)
N-(8-p-tolylnaphthalen-1-yl)picolinamide (10.1 g, 30 mmol), ethanolic NaOH solution (12 g NaOH, 300 mmol in EtOH/H2O 10/1 v/v, 120 mL). After chromatography (hexane/ethyl acetate/triethylamine 94/5/1), beige crystals were obtained (7.0 g, quantitative yield). Rf = 0.16 (hexane/ethyl acetate/triethylamine 94/5/1), mp=73–74 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 7.75 (d, J = 8.1 Hz, 1H), 7.38−7.22 (m, 7H), 7.13 (d, J = 7.0 Hz, 1H), 6.60 (d, J = 7.3 Hz, 1H), 3.74 (s, 2H), 2.42 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 143.8, 140.6, 138.4, 137.3, 135.9, 129.2, 128.8, 128.6, 128.4, 126.6, 124.7, 121.0, 119.1, 111.4, 21.4. FT-IR (neat, cm−1) ν 3490, 3393. 1615, 1579, 1522. Anal. Calcd for C17H15N 233.3 g/mol): C, 87.52; H, 6.48; N, 6.00; Found: C, 87.44; H, 6.42; N, 5.96.
8-(4-(Trifluoromethyl)phenyl)naphthalen-1-amine (7)
N-(8-(4-(Trifluoromethyl)-phenyl)-naphthalene-1-yl)picolinamide (16.3 g, 42 mmol), NaOH (16.8 g, 420 mmol) in EtOH/H2O (10/1 v/v, 200 mL). After chromatography (hexane/ethyl acetate/triethylamine 94/5/1), beige crystals were obtained (9.0 g, 75%). Rf = 0.21 (hexane/ethyl acetate/triethylamine 94/5/1), mp=108–109 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 7.80 (dd, J = 8.2, 0.9 Hz, 1H), 7.68 (d, J = 7.8 Hz, 2H), 7.56 (d, 7.8 Hz, 2 H), 7.41–7.28 (m, 3H), 7.12 (dd, J = 8.7, 1.4, 1H), 6.64 (dd, J = 8.7, 1.4 Hz, 1H), 3.56 (s, 2 H).13C NMR (100 MHz, CDCl3, ppm) δ 147.3, 143.4, 136.9, 135.9, 129.8, 129.5, 128.5, 126.9, 125.7, 125.0 (q, JC-F = 3.8 Hz), 124.7, 123.0, 120.4, 119.4, 111.8. FT-IR (neat, cm−1) ν 3707, 3618, 2973, 2922, 2865, 2844, 1323, 1057, 1032, 1015. Anal. Calcd for C17H12F3N (287.3 g/mol): C, 71.07; H, 4.21; N, 4.88; Found: C, 71.23; H, 4.12; N, 4.82.
1,2'-Binaphthyl-8-amine (8)
N-(1,2'-Binaphthyl-8-yl)picolinamide (8.23 g, 22 mmol), NaOH (8.8 g, 220 mmol) in EtOH/H2O (10/1 v/v, 100 mL). After chromatography (hexane/dichloromethane 50/50), light brown crystals were obtained (5.2 g, 88% yield). Rf = 0.26 (hexane/dichloromethane 50/50), mp=113–114 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 7.91–7.84 (m, 4H), 7.81–7.79 (m, 1H), 7.57–7.51 (m, 3H), 7.41–7.26 (m, 3H), 7.22–7.20 (m, 1H), 6.60 (dd, J = 7.3, 1.4 Hz, 1H), 3.71 (s, 2H). 13C NMR (100 MHz, CDCl3, ppm) δ 143.9, 141.3, 138.3, 136.0, 132.9, 132.6, 128.9, 128.7, 128.2, 128.0, 127.9, 127.6, 127.5, 126.8, 126.7, 126.4, 124.7, 120.9, 119.1, 111.3. FT-IR (neat, cm−1) ν 3707, 3681, 2972, 2922, 2865, 2844, 1055, 1032, 1014. Anal. Calcd for C20H15N (269.2 g/mol): C, 89.19; H, 5.61; N, 5.20; Found: C, 8.39; H, 5.56; N, 5.18.
Cleavage of the picolinic acid auxiliary: [1,1':3',1″-Terphenyl]-2'-ylmethanamine (4)
A known procedure was followed.20N-([1,1’:3’,1”-Terphenyl]-2’-ylmethyl)picolinamide (0.5 mmol, 182 mg), (0.5 mmol, 67 mg), n-butylamine (5 mmol, 0.5 mL), and toluene (1.5 mL) were mixed in a 2-dram vial inside glovebox. The mixture was shaken until the contents dissolved. Anhydrous AlCl3 (0.5 mmol, 67 mg) was then added to the mixture. The vial was capped, taken outside the glovebox, heated and stirred at 90 °C for 24 h. After the reaction was complete, water (2 mL) was added to the reaction mixture. The mixture was extracted with ethyl acetate (3×5 mL). The organic layers were combined, concentrated, and purified by column chromatography in hexanes/ethyl acetate 60/40. The fractions containing the product were combined, concentrated and the solvent was evaporated to give white crystals (118 mg, 91 % yield). Rf = 0.12 (hexanes/ethyl acetate 60/40), mp=70–72 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 7.44–7.30 (m, 12 H), 7.23–7.22 (m, 1H), 3.71 (s, 2H), 1.01 (br s, 2H). 13C NMR (100 MHz, CDCl3, ppm) δ 142.6, 141.8, 138.7, 129.8, 129.3, 128.4, 127.2, 126.5, 40.8. FT-IR (neat, cm−1) ν 3060, 3031, 2937, 1603, 1580, 1498, 1454, 1443, 1185, 1157, 1074, 1031. Anal. Calcd for C19H17N (259.34 g/mol): C, 87.99; H, 6.61; N, 5.40; Found: C, 87.79; H, 6.70; N, 5.36.
Installation of the propanamide auxiliary: N-(8-p-Tolylnaphthalen-1-yl)propionamide (9)
8-p-Tolylnaphthalen-1-amine 6 (2.02 g, 8.7 mmol) and triethylamine (1.34 mL, 9.57 mol) were dissolved in dichloromethane (35 mL). The resulting mixture was cooled in ice bath. Propionyl chloride (1.55 mL, 17.4 mmol) in dichloromethane (10 mL) was added dropwise. The reaction mixture was warmed to room temperature and stirred for 24 h. The reaction mixture was diluted with water (25 mL) and the layers were separated. The organic layer was dried with MgSO4, concentrated and subjected to column chromatography (hexane/ethyl acetate 75/25) to give 2.50 g (99 % yield) of a white powder. Rf = 0.29 (hexane/ethyl acetate 75/25), mp=134-135 °C (hexanes).1H NMR (400 MHz, CDCl3, ppm) δ 8.14 (d, J = 7.1 Hz, 1H), 7.85 (dd, J = 8.2, 1.4 Hz, 1H), 7.81 (d, J =7.8 Hz, 1H), 7.50–7.42 (m, 2H), 7.33–7.28 (m, 4H), 7.26–7.25 (m, 1H), 7.15 (s, 1H), 2.45 (s, 3H), 1.57 (q, J = 7.3 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 171.5, 140.6, 137.7, 136.8, 135.2, 133.3, 129.9, 129.4, 129.0, 126.1, 125.7, 124.7, 124.0, 121.2, 30.5, 21.3, 9.4. Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 1651, 1378, 1219. HRMS electrospray (m/z): [M+ + Na] calcd for C20H19NO, 312.13644; found, 312.13589, error=0.87 ppm.
Arylation of naphthyl propanamide: Ethyl 4-(1-propionamido-8-p-tolylnaphthalen-2-yl)benzoate (10)
A 2-dram screw-cap via was charged with Pd(OAc)2 (5 mol %, 6 mg) AgOAc (166 mg, 1 mmol), ethyl 4-iodobenzoate (0.52 g, 2 mmol), N-(8-p-tolylnaphthalen-1-yl)propionamide (149.9 mg, 0.5 mmol), and trifluoroacetic acid (0.5 mL). The resulting solution was stirred at 110 °C for 3 h. The reaction mixture was diluted with dichloromethane (2 mL), filtered through pad of Celite® and concentrated. Purification by chromatography (hexanes/ethyl acetate 80/20) gave white powder (180 mg, 80 % yield). Rf = 0.09 (hexanes/ethyl acetate 80/20), mp=259-260 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.00 (d, J = 8.2 Hz, 2H), 7.94–7.89 (m, 2H), 7.50–7.45 (m, 2H), 7.40 (d, J = 8.2 Hz, 2H), 7.30 (dd, J = 7.2, 0.7 Hz, 1H), 7.26–7.21 (m, 4H), 6.38 (s, 1H), 4.36 (q, J =7.3 Hz, 2H), 2.40 (s, 3H), 1.39 (t, J = 7.3 Hz, 3H), 1.22 (q, J = 7.4 Hz, 2H), 0.57 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 171.6, 166.6, 145.7, 142.0, 138.6, 138.4, 136.6, 135.1, 131.0, 129.4, 129.3, 129.1, 129.0, 128.9, 128.8, 128.7, 128.6, 128.4, 127.9, 125.4, 61.0, 29.1, 21.2, 14.5, 9.0. FT-IR (neat, cm−1) ν 3710, 3680, 2956, 2844, 1716, 1662, 1266, 1055, 1033, 1014. HRMS electrospray (m/z): [M+ + Na] calcd for C29H27NO3Na, 460.18831; found, 460.18870, error=0.84 ppm.
General procedure for the arylation of sp3 C-H bonds of picolinamides
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), picolinamide (1 mmol), aryl iodide (4 mmol), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 hours. The reaction mixture was extracted with dichloromethane (3×4 mL). The extracts were combined, filtered through pad of cotton, concentrated, and then loaded onto a chromatography column with hexanes/ethyl acetate mixture as eluent and subjected to column chromatography. After concentration of the fractions containing the product, the residue was dried under reduced pressure.
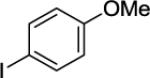
N-(3-(4-Methoxyphenyl)propyl)picolinamide(Table 3, Entry 1)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-propylpicolinamide (1 mmol, 199 mg), 1-iodo-4-methoxybenzene (4 mmol, 936 mg), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), yellowish oil (168 mg, 56 % yield) was obtained. Rf = 0.19 (hexanes/ethyl acetate 70/30). This compound is known.4a1H NMR (400 MHz, CDCl3, ppm) δ 8.51 (d, J=4.6 Hz, 1H), 8.18 (d, J=8.0 Hz, 1H), 8.09 (br s, 1H), 7.82–7.80 (m, 1H), 7.40–7.38 (m, 1H), 7.11 (d, J=8.0 Hz, 2H), 6.81 (d, J=8.0 Hz, 2H), 3.76 (s, 3H), 3.50–3.46 (m, 2H), 2.68–2.64 (m, 2H), 1.97–1.94 (m, 2H).
N-(4-(4-Methoxyphenyl)butan-2-yl)picolinamide (Table 3, Entry 2)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(2-methylpropan-2-yl)picolinamide (1 mmol, 221 mg), 1-iodo-4-methoxybenzene (4 mmol, 936 mg), K2CO3 (4 mmol, 794 mg), and tert-amyl alcohol (2.0 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), pale yellow oil (255 mg, 75 % yield) was obtained. Rf= 0.27 (hexanes/ethyl acetate 70/30). This compound is known.4a1H NMR (400 MHz, CDCl3, ppm) δ 8.49–8.47 (m, 1H), 8.17–8.16 (m, 1H), 7.92 (br d, J=8.7 Hz, 1H), 7.79–7.75 (m, 1H), 7.36–7.33 (m, 1H), 7.09–7.05 (m, 2H), 6.78–6.75 (m, 2H), 4.25–4.15 (m, 1H), 3.70 (s, 3H), 2.61 (t, J=8.2 Hz, 2H), 1.91–1.75 (m, 1H), 1.25 (d, J=6.9 Hz, 3H).
N-(4-(4-Methoxyphenyl)-2-methylbutan-2-yl)picolinamide (Table 3, Entry 3)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(2-methylpropan-2-yl)picolinamide (1 mmol, 198 mg), 1-iodo-4-methoxybenzene (4 mmol, 936 mg), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), pale yellow oil (278 mg, 91 % yield) was obtained. Rf = 0.26 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.50–8.49 (m, 1H), 8.17–8.15 (m, 1H), 8.00 (br s, 1H), 7.82–7.78 (m, 1H), 7.39–7.36 (m, 1H), 7.12–7.09 (m, 2H), 6.80–6.76 (m, 2H), 3.73 (s, 3H), 2.61–2.56 (m, 2H), 2.16–2.12 (m, 2H), 1.50 (s, 6H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.4, 157.8, 150.7, 147.9, 137.4, 134.4, 129.4, 126.0, 121.8, 113.8, 55.3, 53.5, 42.4, 29.9, 27.2. FT-IR (neat, cm−1) ν 2963, 1675, 1510, 1464, 1247, 1178, 1033. Anal. Calcd for C18H22N2O2 (298.38 g/mol): C, 72.46; H, 7.43; N, 9.39; Found: C, 72.15; H, 7.31; N, 9.37.
Ethyl 4-(3-(picolinamido)cyclohexyl)benzoate (Table 3, Entry 4)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol%, 11 mg), CuBr2 (10 mol %, 22 mg), N-cyclohexylpicolinamide (1 mmol, 194 mg), ethyl 4-iodobenzoate (4 mmol, 1.10 g), CsOAc (4 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 hr. After chromatography (hexane/ethyl acetate 70/30), light yellow powder (269 mg, 86 %) was obtained. Rf=0.35 (hexanes/ethyl acetate 70/30), mp=117–118 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 8.45–8.43 (m, 1H), 8.13–8.11 (m, 1H), 7.97 (br d, J=8.7 Hz, 1H), 7.91 (d, J=8.2 Hz, 2H), 7.76–7.72 (m, 1H), 7.34–7.30 (m, 1H), 7.20 (d, J=8.2 Hz, 2H), 4.27 (q, J=6.9 Hz, 2H), 4.12–4.02 (m, 1H), 2.75–2.67 (m, 1H), 2.20 (d, J=12.4 Hz, 1H), 2.07 (d, J=12.4 Hz, 1H), 1.91–1.80 (m, 2H), 1.57–1.26 (m. 7H). 13C NMR (100 MHz, CDCl3, ppm) δ 167.7, 163.5, 151.4, 150.0, 148.1, 137.5, 129.8, 128.5, 126.9, 126.2, 122.3, 60.9, 48.7, 43.3, 40.4, 33.1, 32.7, 25.2, 14.4. FT-IR (neat, cm−1) ν 3371, 1713, 1656, 1519, 1276, 1110. Anal. Calcd for C21H24N2O3 (352.43 g/mol): C, 71.57; H, 6.86; N, 7.95; Found: C, 71.31; H, 6.69; N, 7.73.
N-(4-(3-Methoxybenzyl)-5-(3-methoxy-phenyl)-2,4-dimethylpentan-2-yl)picolinamide (A) and N-(5-(3-methoxyphenyl)-2,4,4-trimethylpentan-2-yl)picolinamide (B) (Table 3, Entry 5)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol%, 22 mg), CuBr2 (20 mol %, 44 mg), N-(2,4,4-trimethylpentan-2-yl)picolinamide (1 mmol, 245 mg), 1-iodo-4-methoxybenzene (4 mmol, 936 mg), K2CO3 (6 mmol, 794 mg), and tert-amyl alcohol (0.5 mL). The resulting suspension was stirred at 140 °C for 24 hr. The following products were obtained after column chromatography in hexanes/ethyl acetate 70/30.
Product A was obtained as a light yellow oil (138 mg, 29 % yield). Rf = 0.69 (hexanes/ethyl acetate 70/30), 1H NMR (400 MHz, CDCl3, ppm) δ 8.49 (m, 1H), 8.17–8.15 (m, 2H), 7.82–7.79 1H), 7.38–7.35 (m, 1H), 7.17–7.14 (m, 2H), 6.76–6.72 (m, 4H), 6.68–6.67 (m, 2H), 3.76 (s, 6H), (d, J=13.0 Hz, 2H), 2.60 (d, J=13.0, 2H), 2.10 (s, 2H), 1.56 (s, 6H), 1.08 (s, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.3, 159.1, 150.8, 147.9, 140.5, 137.5, 134.4, 128.6, 125.9, 123.7, 121.7, 116.9, 111.3, 55.2, 54.7, 48.3, 48.0, 39.0, 29.9, 24.8. FT-IR (neat, cm−1) ν 2955, 1679, 1582, 1521, 1488, 1263, 1154, 1043. Anal. Calcd for C28H34N2O3 (446.58 g/mol): C, 75.31; H, 7.67; N, 6.27; Found C, 74.96; H, 7.67; N, 6.22.
Product B was obtained as a colorless oil (46 mg, 13 %). Rf = 0.64 (hexane/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.51–8.49 (m, 1H), 8.18–8.16 (m, 1H), 8.14 (br s, 1H), 7.83–7.79 (m, 1H), 7.38–7.35 (m, 1H), 7.17–7.14 (m, 1H), 6.75–6.71 (m, 2H), 6.68–6.72 (m, 1H), 3.77 (s, 3H), 2.58 (s, 2H), 1.97 (s, 2H), 1.57 (s, 6H), 1.01 (s, 6H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.2, 159.0, 150.8, 147.9, 140.62, 137.4, 128.5, 125.9, 123.5, 121.7, 116.9, 111.1, 55.2, 54.7, 51.2, 51.1, 35.5, 29.6, 27.8. FT-IR (neat, cm−1) ν 2916, 1679, 1583, 1520, 1488, 1463, 1264, 1045. Anal. Calcd for C21H28N2O2 (340.46 g/mol): C, 74.08; H, 8.29; N, 8.23; Found: C, 73.79; H, 8.28; N, 8.11.
General procedure for the alkylation of sp2 and sp3 C–H bonds of picolinamides
A Kontes flask or a 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol %, 22 mg), CuBr2 (20 mol %, 44 mg), picolinamide (1 mmol), alkyl iodide (4–6 mmol), K2CO3 (4 mmol, 794 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 h. The reaction mixture was diluted with dichloromethane (4 mL) and filtered through a pad of cotton. The residue was then washed with dichloromethane (2×4 mL). The organic solvents were combined, concentrated, and then loaded onto a chromatography column with hexanes/ethyl acetate mixture as eluent and subjected to column chromatography. After concentration of the fractions containing the product, the residue was dried under reduced pressure.
N-(1-(2,6-Dibutylphenyl)ethyl)picolinamide (Table 4, Entry 1)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol%, 11 mg), CuBr2 (20 mol %, 22 mg), N-(1-phenylethyl)picolinamide (1 mmol, 239 mg), n-butyl iodide (4 mmol, 736 mg), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 h. After chromatography (hexanes/ethyl acetate 70/30), light yellow oil (336 mg, 99 % yield) was obtained. Rf = 0.60 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.62 (br d, J=7.8 Hz, 1H), 8.52–8.50 (m, 1H), 8.19–8.17 (m, 1H), 7.80 (td, J=7.5, 1.37 Hz, 1H), 7.40–7.36 (m, 1H), 7.14–7.10 (m, 2H), 7.06–7.04 (m, 1H), 5.76 (q, J=7.3, 1H), 2.98–2.90 (m, 2H), 2.80–2.73 (m, 2H), 1.70–1.60 (m, 7H), 1.52–1.43 (m, 4H), 0.93 (t, J=7.3 Hz, 1H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.4, 150.1, 148.0, 141.0, 138.3, 137.4, 128.6, 127.1, 126.1, 122.2, 45.1, 34.5, 34.2, 23.2, 22.2, 14.1. FT-IR (neat, cm−1) ν 2956, 1678, 1511, 1432, 1462, 1374, 1206. Anal. Calcd for C22H30N2O (338.49g/mol): C, 78.06; H, 8.93; N, 8.28; Found: C, 77.84; H, 8.92; N, 8.19.
When n-butyl bromide (4 mmol, 548 mg) was used as an alkylating agent, no product was detected by GC-MS.
N-(1-(2,6-Bis(4,4,4-trifluorobutyl)phenyl)ethyl)picolinamide (Table 4, Entry 2)
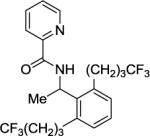
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(1-phenylethyl)picolinamide (1 mmol, 229 mg), 1,1,1-trifluoro-4-iodobutane (4 mmol, 948 mg), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 h. After chromatography (hexanes/ethyl acetate 70/30), light yellow oil (356 mg, 79 % yield) was obtained. Rf = 0.52 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.54–8.51 (m, 2H), 8.17–8.14 (m, 1H), 7.84–7.80 (m, 1H), 7.42–7.39 (m, 1H), 7.16 (t, J = 7.8 Hz, 1H), 7.06 (d, J=7.8 Hz, 1H), 5.69 (q, J = 7.3 Hz, 3.10–3.01 (m, 1H), 2.88–2.80 (m, 2H), 2.29–2.16 (m, 4H), 1.95–1.88 (m, 4H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.4, 149.7, 148.1, 139.3, 138.8, 137.6, 129.1, 127.5, 127.2 (q, J = 276.1 Hz), 126.4, 122.2, 45.1, 33.8 (q, J = 28.8 Hz), 33.1, 24.1, 22.1. 19F NMR (376 MHz, CDCl3, ppm) δ 66.1. FT-IR (neat, cm−1) ν 1678, 1512, 1465, 1434, 1388, 1251, 1132, 1005. Anal. Calcd for C22H24F6N2O (446.43g/mol): C, 59.19; H, 5.42; N, 6.27; Found: C, 59.28; H, 5.48; N, 6.24.
N-(1-(2,6-Diisobutylphenyl)ethyl)picolinamide (Table 4, Entry 3)
A 10-mL Kontes flask was charged with Pd(OAc)2 (10 mol %, 22 mg), CuBr2 (20 mol %, 44 mg), N-(1-phenylethyl)picolinamide (1 mmol, 239 mg), 1-iodo-2-methylpropane (6 mmol, 1.10 g), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 h. After chromatography (hexane/ethyl acetate 70/30), light yellow oil (301 mg, 84 % yield) was obtained. Rf = 0.39 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.61 (br d, J=8.0 Hz, 1H) 8.52–8.51 (m, 1H), 8.19–8.17 (m, 1H), 7.79 (td, J7.5, 1.7 Hz, 1H), 7.39–7.36 (m, 1H), 7.12–7.10 (m, 1H), 7.05–7.04 (m, 2H), 5.76 (q, J=8.0 Hz, 1H), 2.91 (dd, J=13.8, 6.87 Hz, 2H), 2.62 (dd, J=13.8, 5.7 Hz, 2H), 2.04 (m, 2H), 1.67 (d, J=6.9 Hz, 3H), 1.00 (d, J=6.9 Hz, 6H), 0.96 (d, J= 6.9 Hz, 6H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.4, 150.1, 148.0, 139.7, 139.2, 137.4, 129.3, 126.4, 126.1, 122.2, 45.2, 43.3, 29.7, 23.0, 22.6, 22.3. FT-IR (neat, cm−1) ν 2954, 1677, 1509, 1464, 1432, 1383. Anal. Calcd for C22H30N2O (338.49 g/mol): C, 78.06; H, 8.93; N, 8.28; Found: C, 78.03; H, 9.11; N, 8.37.
N-(1-(2,6-Diphenethylphenyl)ethyl)picolinamide (Table 4, Entry 4)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol %, 22 mg), CuBr2 (20 mol %, 44 mg), N-(1-phenylethyl)picolinamide (1 mmol, 228 mg), (2-iodoethyl)benzene (4 mmol, 984 mg), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), light yellow oil (375 mg, 86 % yield) was obtained. Rf = 0.33 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.68 (br d, J=7.8 Hz, 1H), 8.45–8.43 (m, 1H), 8.20–8.18 (m, 1H), 7.77 (td, J=7.8, 1.8 Hz, 1H), 7.36–7.27 (m, 9H), 7.22–7.13 (m, 5H), 5.88 (q, J=7.8 Hz, 1H), 3.31–2.99 (m, 6H), 1.68 (d, J=6.87 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ□ 163.6, 150.0, 148.2, 142.1, 140.2, 138.8, 137.6, 129.2, 128.7, 128.6, 127.5, 126.4, 126.2, 122.4, 45.4, 38.5, 36.6, 22.2. FT-IR (neat, cm−1) ν 1676, 1509, 1453, 1432. Anal. Calcd for C30H30N2O (434.57 g/mol): C, 82.91; H, 6.96; N, 6.45; Found: C, 82.71; H, 7.22; N, 6.41.
N-(1-(2,6-Dibenzylphenyl)ethyl)picolinamide (Table 4, Entry 5)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(1-phenylethyl)picolinamide (1 mmol, 228 mg), benzyl iodide (4 mmol, 872 mg), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 hours. After chromatography (hexanes/ethyl acetate 70/30), light yellow oil (349 mg, 85 % yield) was obtained. Rf = 0.43 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, 50 °C, ppm) δ 8.34 (br d, J=6.87 Hz, 1H), 8.30–8.28 (m, 1H), 8.04 (d, J=7.5 Hz, 1H), 7.71 (td, J= 7.5, 1.7 Hz, 1H), 7.28–7.26 (m, 1H), 7.19–7.03 (m, 13H), 5.72 (q, J=7.5 Hz, 1H), 4.45 (d, J=16.0 Hz, 2H), 4.21 (d, J=16.0 Hz, 2H), 1.24 (d, J=7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.6, 149.8, 147.9, 141.2, 139.9, 138.9 (br), 137.1, 130.7 (br), 129.0, 128.4, 127.3, 125.9, 125.8, 121.9, 45.6, 40.1 (br), 20.6. FT-IR (neat, cm−1) ν 3381, 1676, 1497, 1462, 1431. Anal. Calcd for C28H26N2O (406.52 g/mol): C, 82.73; H, 6.45; N, 6.89; Found: C, 82.54; H, 6.44; N, 6.79.
N-(2-(2,6-Dibutylphenyl)propan-2-yl)picolinamide (A) and N-(2-(2-butylphenyl)-propan-2-yl)picolinamide (B) (Table 4, Entry 6)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol %, 11 mg), CuBr2 (10 mol %, 22 mg), N-(2-phenylpropan-2-yl)picolinamide (1 mmol, 224 mg), iodobutane (6 mmol, 1.10 g), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), two products were obtained.
Product A was obtained as a light yellow oil (178 mg, 54 % yield). Rf = 0.44 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.54 (br s, 1H), 8.51–8.49 (m, 1H), 8.15 (d, J=7.8 Hz, 1H), 7.80 (td, J=9.2, 1.4 Hz, 1H), 7.40–7.36 (m, 1H), 7.10–7.02 (m, 3H), 2.90–2.86 (m, 4H), 2.08 (s, 6H), 1.56–1.48 (m, 4H), 1.29–1.20 (m, 4H), 0.74 (t, J=7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.1, 150.9, 147.8, 141.9, 141.5, 137.4, 130.5, 126.5, 125.9, 121.8, 59.1, 36.5, 35.5, 31.0, 23.3, 14.0. FT-IR (neat, cm−1) ν 2956, 1678, 1510, 1463. Anal. Calcd for C23H32N2O (352.51 g/mol): C, 78.36; H, 9.15; N, 7.95; Found: C, 78.12; H, 9.31; N, 7.83.
Product B was obtained as a light yellow oil (38 mg, 14 % yield). Rf = 0.31 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.51–8.47 (m, 2H), 8.14–8.12 (m, 1H), 7,82–7.79 (m, 1H), 7.50–7.48 (m, 1H), 7.41–7.37 (m, 1H), 7.23–7.15 (m, 3H), 2.82–2.78 (m, 2H), 1.92 (s, 6H), 1.47–1.40 (m, 2H), 1.18–1.12 (m, 2H), 0.61 (t, J=7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.8, 150.7, 147.8, 143.2, 140.8, 137.4, 131.6, 127.4, 127.2, 126.0, 125.7, 121.8, 56.3, 34.8, 33.8, 29.0, 23.4, 13.9 Signal for one carbon could not be located. FT-IR (neat, cm−1) ν 2930, 1678, 1511, 1463, 1432. HRMS (m/z): [M+] calcd for C19H24N2O, 296.1889; found, 296.1883, error=−2.0 ppm.
N-(2,6-Dicyclohexylbenzyl)picolinamide (A) and N-(2-cyclohexylbenzyl)picolinamide (B) (Table 4, Entry 7)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol %, 22 mg), CuBr2 (20 mol %, 44 mg), N-benzylpicolinamide (1 mmol, 223 mg), iodocyclohexane (4 mmol, 840 mg), K2CO3 (4 mmol, 552 mg), and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 h. After chromatography (hexane/ethyl acetate 80/20), two products were obtained.
Product A was obtained as a light yellow oil (42 mg, 11 % yield). Rf = 0.31 (hexanes/ethyl acetate 80/20). 1H NMR (400 MHz, CDCl3, ppm) δ 8.45–8.44 (m. 1H), 8.24 (d, J=7.8 Hz, 1H), 7.89–7.82 (m, 2H), 7.40–7.37 (m, 1H), 7.30–7.27 (m, 1H), 7.18–7.16 (m, 1H), 4.73 (d, J=4.6 Hz, 2H), 2.83–2.75 (m, 2H), 1.80–1.69 (m, 12H), 1.49–1.18 (m, 10H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.7, 149.9, 148.7, 147.5, 137.4, 131.4, 128.3, 126.2, 124.2, 122.2, 40.5, 36.6, 35.0, 27.1, 26.3. FT-IR (neat, cm−1) ν 2925, 2850, 1673, 1521, 1568, 1433, 1242. Anal. Calcd for C25H32N2O (376.53 g/mol): C, 79.75; H, 8.57; N, 7.44; Found: C, 79.89; H, 8.21; N, 7.48.
Product B was obtained as a light yellow oil (62 mg, 20 % yield). Rf = 0.50 (hexanes/ethyl acetate 80/20). 1H NMR (400 MHz, CDCl3, ppm) δ 8.50–8.48 (m, 1H), 8.25–8.19 (m, 2H), 7.86–7.82 (m, 1H), 7.42–7.38 (m, 1H), 7.33–7.30 (m, 1H), 7.19–7.15 (m, 1H), 4.71 (d, J=5.5 Hz, 2H), 2.81–2.74 (m, 1H), 1.81–1.70 (m, 5H), 1.49–1.19 (m, 5H. 13C NMR (100 MHz, CDCl3, ppm) δ 164.9, 149.9, 148.2, 146.6, 137.4, 134.7, 129.3, 128.2, 126.6, 126.2, 126.0, 122.3, 41.5, 39.7, 34.5, 27.0, 26.3. FT-IR (neat, cm−1) ν 2926, 2851, 1674, 1568, 1522, 1241. Anal. Calcd for C19H22N2O (294.39 g/mol): C, 77.52; H, 7.53; N, 9.52; Found: C, 77.13; H, 7.67; N, 9.40.
N-(2-Cyclohexyl-6-methoxybenzyl)picolinamide (Table 4, Entry 8)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol %, 22 mg), CuBr2 (20 mol %, 44 mg), N-(2-methoxybenzyl)-picolinamide (1 mmol, 217 mg), iodocyclohexane (4 mmol, 840 mg), K2CO3 (4 mmol, 552 mg) and water (0.30 mL). The resulting suspension was stirred at 120 °C for 24 h. After chromatography (hexanes/ethyl acetate 70/30), light yellow oil (47 mg, 14 % yield) was obtained. Rf = 0.39 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) δ 8.49–8.47 (m, 1H), 8.23–8.20 (m, 1H), 7.81 (td, J=9.52, 2.00 Hz, 1H), 7.38–7.35 (m, 1H), 7.27–7.23 (m, 1H), 6.93–6.91 (m, 1H), 6.77–6.74 (m, 1H), 4.76 (d, J=5.49 Hz, 2H), 3.87 (s, 3H), 3.02–2.97 (m, 1H), 1.78–1.72 (m, 5H), 1.51–1.36 (m, 4H), 1.27–1.21 (m, 1H). 13C NMR (100 MHz, CDCl3, ppm) δ 163.7, 158.3, 150.4, 148.4, 148.1, 137.3, 128.7, 125.9, 123.5, 122.3, 118.8, 108.0, 55.7, 39.9, 34.6, 34.4, 27.0, 26.3. FT-IR (neat, cm−1) ν 2926, 1674, 1582, 1518, 1464, 1249, 1136, 1096. Anal. Calcd for C20H24N2O2 (324.42 g/mol): C, 74.04; H, 7.46; N, 8.64; O, 9.86 Found: C, 73.76; H, 7.50; N, 8.49.
N-(8-Octylnaphthalen-1-yl)picolinamide (Table 4, Entry 9)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (5 mol%, 11 mg), CuBr2 (10 mol %, 22 mg), N-(naphthalen-1-yl)picolinamide (1 mmol, 217 mg), octyl iodide (4 mmol, 960 mg), CsOAc (3 mmol, 594 mg), and tert-amyl alcohol (0.50 mL). The resulting suspension was stirred at 140 °C for 24 hours. After chromatography (hexane/ethyl acetate 70/30), light yellow oil (153 mg, 49 % yield) was obtained. Rf = 0.33 (hexanes/ethyl acetate 70/30). 1H NMR (400 MHz, CDCl3, ppm) γ 10.56 (s, 1H), 8.64–8.62 (m, 1H), 8.39–8.36 (m, 1H), 8.02 (d, J=7.8 Hz, 1H), 7.89 (td, J=7.6, 1.6 Hz, 1H), 7.77–7.72 (m, 2H), 7.52–7.45 (m, 2H), 7.38–7.30 (m, 2H), 3.30–3.26 (m, 2H), 1.68–1.61 (m, 2H), 1.27-1.13 (m, 10H), 0.86 (t, J=7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, ppm) δ 162.5, 150.3, 148.1, 137.9, 137.8, 136.3, 132.7, 126.9, 128.0, 127.9, 127.5, 126.6, 125.6, 125.2, 122.8, 37.8, 32.9, 31.9, 29.8, 29.6, 29.4, 22.8, 14.3. FTIR (neat, cm−1) ν 2926, 1686, 1522, 1498, 1431, 1339. Anal. Calcd for C24H28N2O (360.49 g/mol): C, 79.96; H, 7.83; N, 7.77; Found: C, 79.78; H, 7.93; N, 7.79.
N-(2-Methylnonan-2-yl)picolinamide(Table 4, Entry 10)
A 2-dram screw-cap vial was charged with Pd(OAc)2 (10 mol %, 11 mg), CuBr2 (20 mol %, 22 mg), N-(tert-pentyl)picolinamide (1 mmol, 192 mg), iodopentane (4 mmol, 792 mg), K2CO3 (4 mmol, 552 mg), pivalic acid (2 mmol, 202 mg), and tert-amyl alcohol (0.7 mL). The resulting suspension was stirred at 110 °C for 24 h. After chromatography (hexanes/ethyl acetate 80/20), colorless oil (67 mg, 27 % yield) was obtained. Rf = 0.31 (hexanes/ethyl acetate 80/20), 1H NMR (400 MHz, CDCl3, ppm) γ 8.53–8.52 (m, 1H), 8.19–8.16 (m, 1H), 7.97 (br s, 1H), 7.83 (td, J=9.6, 1.8 Hz, 1H), 7.41–7.38 (m, 1H), 1.88–1.79 (m, 2H), 1.46 (s, 6H), 1.34-1.26 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) 13C NMR (100 MHz, CDCl3, ppm) γ 163.3, 150.9, 147.8, 137.4, 125.9, 121.7, 53.6, 40.7, 32.0, 30.1, 29.4, 26.9, 24.3, 22.7, 14.2. FT-IR (neat, cm−1) ν 2926, 1681, 1520, 1464, 1432, 1363, 1287. Anal. Calcd for C16H26N2O (262.39 g/mol): C, 73.24; H, 9.99; N, 10.68; Found: C, 73.04; H, 10.04; N, 10.38.
General procedure for the alkylation of sp2 C–H bonds of 8-aminoquinoline amides
A 2– dram screw—capped vial was charged with Pd(OAc)2 (5 mol%), K2CO3 (2.5 equiv), substrate, pivalic acid (20 mol%), and alkyl bromide or iodide (3–4 equiv). The t-amyl alcohol (0.7–3.0 mL) solvent was added and the resulting mixture was stirred at 100–110 °C for 12–96 h. The reaction mixture was allowed to cool to room temperature and diluted with ethyl acetate (10 mL), followed by washing with water (10 mL). The aqueous layer was extracted with ethyl acetate (3x10 mL). The organic layers were combined, dried over MgSO4, and filtered. The filtrate was concentrated under vacuum. The crude product was purified flash column chromatography.
2-Benzyl-6-methoxy-N-(quinolin-8-yl)benzamide (Table 5, Entry 1)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 2-methoxy-N-(quinolin-8-yl)benzamide (206 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.148 mmol), and benzyl bromide (380 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 70/1), 166 mg (61 % yield) of a crystalline material was obtained. Rf = 0.19 (toluene/ethyl acetate 70/1), mp = 137–139 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.98 (s, 1H), 8.99-8.95 (dd, 1H, J = 7.7, 1.3 Hz), 8.71-8.68 (dd, 1H, J = 4.0, 1.3 Hz), 8.17–8.14 (dd, 1H, J = 8.3, 1.2 Hz), 7.61–7.57 (m, 1H), 7.55–7.52 (m, 1H), 7.44–7.41 (m, 1H), 7.33–7.29 (m, 1H), 7.21–7.17 (m, 2H), 7.13–7.09 (m, 2H), 7.04–7.00 (m, 1H), 6.89–6.81 (m, 2H), 4.13 (s, 2H), 3.84 (s, 3H). 13C NMR (500 MHz, CDCl3, ppm) γ 166.4, 156.5, 148.1, 140.5, 140.4, 138.5, 136.2, 134.7, 130.3, 129.2, 128.3, 128.0, 127.5, 127.2, 126.0, 122.6, 121.7, 121.5, 116.8, 109.0, 55.9, 38.9. FT-IR (neat, cm−1) ν 3330, 1670, 1525, 1483. Anal. calcd. for C24H20N2O2 (368.43 g/mol): C, 78.24; H, 5.47; N, 7.60. Found: C, 78.27; H, 5.54; N 7.46.
2,6-Dibenzyl-4-bromo-N-(quinolin-8-yl)benzamide (Table 5, Entry 2)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-bromo-N-(quinolin-8-yl)benzamide (242 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and benzyl bromide (380 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 12 h. After column chromatography (toluene), 285 mg (76 %) of a crystalline material was obtained. Rf = 0.27 (toluene), mp = 118–119 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.74 (s, 1H), 8.91–8.87 (dd, 1H, J = 7.4, 1.5 Hz), 8.63–8.60 (dd, 1H, J = 4.3, 1.5 Hz), 8.17–8.13 (dd, 1H, J = 8.6, 1.7 Hz), 7.61–7.53 (m, 2H), 7.44–7.40 (m, 1H), 7.22 (s, 2H), 7.18–7.11 (m, 8H), 7.06–7.02 (m, 2H), 4.07 (s, 4H). 13C NMR (400 MHz, CDCl3, ppm) δ 167.5, 148.2, 140.4, 139.3, 138.4, 136.8, 136.2, 134.0, 131.0, 129.21, 128.6, 127.9, 127.3, 126.4, 123.5, 122.2, 121.7, 116.8, 39.0. FT-IR (neat, cm−1) ν 3358, 1677, 1524, 1485. Anal. calcd. for C30H23BrN2O (507.42 g/mol): C, 71.01; H, 4.57; N; 5.52. Found: C, 71.19; H, 4.52; N 5.48.
2,6-Dibenzyl-4-tert-butyl-N-(quinolin-8-yl)benzamide (Table 5, Entry 3)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and benzyl bromide (380 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 100 °C for 20 h. After column chromatography (toluene/ethyl acetate 100/1 to 50/1), 318 mg (88 % yield) of a crystalline material was obtained. Rf = 0.22 (toluene/ethyl acetate 70/1), mp = 136–137 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.64 (s, 1H), 8.93–8.90 (dd, J = 7.4, 1.3 Hz, 1H), 8.57–8.54 (dd, J = 4.1, 1.7 Hz, 1H), 8.14–8.11 (dd, J = 8.6, 1.6 Hz, 1H), 7.59–7.51 (m, 2H), 7.40–7.37 (m, 1H), 7.17–7.12 (m, 6H), 7.10–7.05 (m, 4H), 7.01–6.97 (m, 2H), 4.10 (s, 4H), 1.25 (s, 9H). 13C NMR (400 MHz, CDCl3, ppm) δ 168.7, 152.2, 148.0, 140.5, 138.4, 137.6, 136.1, 135.4, 134.4, 129.1, 128.3, 127.9, 127.4, 125.9, 125.4, 121.9, 121.5, 116.7, 39.6, 34.7, 31.3. FT-IR (neat, cm−1) ν 3363, 1676, 1521, 1485. Anal. calcd. for C34H32N2O (507.42 g/mol): C, 84.26; H, 6.66; N, 5.78. Found: C, 84.26; H, 6.68; N 5.82.
2-Benzyl-N-(quinolin-8-yl)-5-(trifluoromethyl)benzamide(Table 5, Entry 4)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)-3-(trifluoromethyl)benzamide (234 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and benzyl bromide (380 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (hexanes/ethyl acetate 10/1), 212 mg (71 %) of crystalline material was obtained. Rf = 0.27 (hexanes/ethyl acetate 10/1), mp = 124-126 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 10.11 (s, 1H), 8.91–8.87 (d, J = 7.1 Hz, 1H), 8.75–8.72 (dd, J = 4.1, 1.5 Hz, 1H), 8.21–8.17 (dd, J = 8.5, 1.2 Hz, 1H), 7.90 (s, 1H), 7.67–7.57 (m, 3H), 7.48–7.45 (m, 1H), 7.40–7.37 (m, 1H), 7.21–7.18 (m, 4H), 7.12–7.08 (m, 1H), 4.34 (s, 2H). 13C NMR (400 MHz, CDCl3, ppm) δ 196.8, 148.4, 143.8, 139.6, 138.5, 137.6, 136.4, 134.3, 131.5, 129.3, 128.9 (q, JC–F = 32.0 Hz), 128.6, 128.5, 128.0, 127.0 (q, JC–F = 3.5 Hz), 126.5, 124.3 (q, JC–F = 3.5 Hz), 123.9 (q, JC–F = 271.5 Hz), 122.4, 121.8, 116.9, 38.9. 19F NMR (376 MHz, CDCl3, ppm) δ-62.3 (s). FT-IR (neat, cm−1) ν 3361, 1674, 1525, 1485. Anal. calcd. for C24H17F3N2O (406.40 g/mol): C, 70.93; H, 4.22; N, 6.89. Found: C, 70.83; H, 4.33; N 6.69.
2,6-Dibenzyl-4-methoxy-N-(quinolin-8-yl)benzamide (Table 5, Entry 5)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-methoxy-N-(quinolin-8-yl)benzamide (206 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and benzyl bromide (380 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 ºC for 15 h. After column chromatography (toluene/ethyl acetate 40/1), 198 mg (58 %) of crystalline material was obtained. Rf = 0.33 (toluene/ethyl acetate 40/1), mp = 137-138 ºC (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.74 (s, 1H), 8.92 (dd, J = 7.5, 1.1 Hz, 1H), 8.61 (dd, J = 4.1, 1.3 Hz, 1H), 8.14 (dd, J = 8.2, 1.4 Hz, 1H), 7.60-7.51 (m, 2H), 7.42-7.38 (m, 1H), 7.20-7.17 (m, 4H), 7.14-7.10 (m, 4H), 7.06-7.01 (m, 2H), 6.59 (s, 2H), 4.09 (s, 4H), 3.70 (s, 3H). 13C NMR (400 MHz, CDCl3, ppm) δ 168.5, 159.9, 148.0, 140.1, 140.0, 138.4, 136.1, 134.4, 131.1, 129.2, 128.4, 127.9, 127.4, 126.1, 121.8, 121.5, 116.6, 113.5, 55.2, 39.4. FT-IR (neat, cm-1) ν 3344, 1669, 1521, 1485. Anal. calcd. for C31H26N2O2 (458.55 g/mol): C, 81.20; H, 5.72; N, 6.11. Found: C, 80.94; H, 5.73; N 6.10.
2,6-Dibenzyl-4-methoxy-3-methyl-N-(quinolin-8-yl)benzamide (Table 5, Entry 6)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-methoxy-3-methyl-N-(quinolin-8-yl)benzamide (216 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and benzyl bromide (380 mg, 2.22 mmol) were dissolved in t-amyl alcohol (1.0 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 40/1), 258 mg (74 %) of crystalline material was obtained. TLC Rf = 0.35 (toluene/ethyl acetate 40/1), mp = 138–139 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.70 (s, 1H), 8.87 (dd, J = 7.5, 1.3 Hz, 1H,), 8.53 (dd, J = 4.3, 1.6 Hz, 1H), 8.09 (dd, J = 8.2, 1.5 Hz, 1H), 7.56–7.46 (m, 2H), 7.39–7.34 (m, 1H), 7.25–7.13 (m, 3H), 7.13–7.08 (m, 5H), 7.07–6.96 (m, 2H), 6.63 (s, 1H), 4.15 (s, 2H), 4.11 (s, 2H), 3.76 (s, 3H), 2.07 (s, 3H).13C NMR (400 MHz, CDCl3, ppm) δ169.2, 158.3, 147.9, 140.6, 140.0, 138.4, 136.6, 136.3, 136.0, 134.4, 132.1, 129.1, 128.4, 128.3, 128.3, 127.8, 127.3, 126.0, 125.6, 124.5, 121.7, 121.4, 116.5, 110.2, 55.5, 39.5, 36.9, 11.9. FT-IR (neat, cm−1) ν 3353, 1671, 1521, 1482. HRMS (m/z): [M+] calcd for C32H28N2O2, 472.2151; found, 472.2152, error=0.2 ppm.
4-tert-Butyl-2,6-diethyl-N-(quinolin-8-yl)benzamide (Table 5, Entry 7)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and iodoethane (346 mg, 2.22 mmol) were dissolved in t-amyl alcohol (1.0 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 50/1), 240 mg (90 %) of crystalline material was obtained. Rf = 0.33 (toluene/ethyl acetate 50/1), mp = 125–127 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.94 (s, 1H), 9.00 (dd, J = 7.5, 1.3 Hz, 1H), 8.72 (dd, J = 4.4, 1.6 Hz, 1H), 8.19–8.15 (dd, J = 8.5, 1.7 Hz, 1H), 7.63–7.54 (m, 2H), 7.45–7.41 (m, 1H), 7.19–7.16 (m, 2H), 2.78–2.71 (m, 4H), 1.35 (s, 9H), 1.25 (t, J = 7.8 Hz, 6H). 13C NMR (400 MHz, CDCl3, ppm) δ 169.3, 152.2, 148.3, 140.4, 138.6, 136.3, 134.6, 134.5, 128.1, 127.5, 123.3, 121.8, 121.7, 116.8, 34.8, 31.4, 26.9, 16.2. FT-IR (neat, cm−1) ν 3341, 1673, 1520, 1487. Anal. calcd. for C24H28N2O (360.49 g/mol): C, 79.96; H, 7.83; N, 7.77. Found: C, 79.81; H, 7.88; N 7.66.
4-tert-Butyl-2,6-diisobutyl-N-(quinolin-8-yl)benzamide (Table 5, Entry 8)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and l-iodo-2-methylpropane (408 mg, 2.22 mmol) were dissolved in t-amyl alcohol (1.0 mL). Resulting mixture was stirred at 110 °C for 40 h. After column chromatography (toluene/ethyl acetate 50/1), 229 mg (75 %) of crystalline material was obtained. Rf = 0.37 (toluene/ethyl acetate 50/1), mp = 114–116 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.86 (s, 1H), 8.98 (dd, J = 7.5, 1.4 Hz, 1H), 8.71 (dd, J = 4.2, 1.6 Hz, 1H), 8.18 (dd, J = 8.4, 1.6 Hz, 1H), 7.64–7.54 (m, 2H), 7.46–7.42 (m, 1H), 7.10 (s, 2H) 2.59 (d, J = 7.3 Hz, 4H), 2.04–1.93 (m, 2H), 1.35 (s, 9H), 0.85 (d, J = 6.7 Hz, 12H). 13C NMR (400 MHz, CDCl3, ppm) δ 169.3, 151.1, 148.2, 138.6, 137.8, 136.3, 135.7, 134.6, 128.1, 127.5, 124.7, 121.7, 121.6, 116.7, 43.0, 34.6, 31.3, 30.1, 22.7. FT-IR (neat, cm−1) ν 3348, 1677, 1519, 1483. Anal. calcd. for C28H36N2O (416.60 g/mol): C, 80.73; H, 8.71; N, 6.72. Found: C, 80.66; H, 8.68; N 6.69.
4-tert-Butyl-2,6-diphenethyl-N-(quinolin-8-yl)benzamide (Table 5, Entry 9)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and (2—iodoethyl)benzene (515 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 50/1), 350 mg (92 %) of crystalline material was obtained. Rf = 0.30 (toluene/ethyl acetate 50/1), mp = 160–161 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.98 (s, 1H), 9.04 (dd, J = 7.6, 1.0 Hz, 1H), 8.72 (dd, J = 4.1, 1.7, Hz, 1H), 8.18 (dd, J = 8.4, 1.4 Hz, 1H), 7.66–7.57 (m, 2H), 7.45–7.41 (m, 1H), 7.17–7.13 (m, 4H), 7.10–7.06 (m, 6H), 7.03 (s, 2H), 3.05–2.98 (m, 8H), 1.25 (s, 9H). 13C NMR (400 MHz, CDCl3, ppm) δ 169.0, 151.8, 148.3, 141.8, 138.5, 138.0, 136.4, 135.0, 134.5, 128.6, 128.3, 128.1, 127.5, 125.8, 124.7, 122.0, 121.7, 116.9, 38.3, 36.3, 34.6, 31.3. FT-IR (neat, cm−1) ν 3350, 1670, 1519, 1484. Anal. calcd. for C36H36N2O (512.68 g/mol): C, 84.34; H, 7.08; N, 5.46. Found: C, 84.35; H, 7.10; N 5.56.
Diethyl 7,7'-(5-(tert-butyl)-2-(quinolin-8-ylcarbamoyl)-1,3-phenylene)diheptanoate (Table 5, Entry 10)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and ethyl 7-iodoheptanoate (630 mg, 2.22 mmol) were dissolved in t-amyl alcohol (1.0 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 40/1 to 200/1), 353 mg (77 %) of a yellow oil was obtained. TLC Rf = 0.08 (toluene/ethyl acetate 40/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.90 (s, 1H), 8.97 (dd, J = 7.5, 1.4 Hz, 1H), 8.72 (dd, J = 4.4, 1.6 Hz, 1H), 8.17 (dd, J = 8.3, 1.7 Hz, 1H), 7.63–7.54 (m, 2H), 7.47–7.43 (m, 1H), 7.14 (s, 2H), 4.11–4.04 (m, 4H), 2.72–2.66 (m, 4H), 2.16–2.10 (m, 4H), 1.73–1.64 (m, 4H), 1.53–1.45 (m, 4H), 1.35 (s, 9H), 1.33–1.18 (m, 14H). 13C NMR (400 MHz, CDCl3, ppm) δ 173.8, 169.2, 151.9, 148.3, 139.0, 138.5, 136.3, 134.8, 134.5, 128.1, 127.5, 123.9, 121.8, 121.7, 116.7, 60.1, 34.7, 34.2, 33.8, 31.7, 31.4, 29.3, 28.9, 24.8, 14.3. FT-IR (neat, cm−1) ν 3344, 1734, 1519, 1482. Anal. calcd. for C38H52N2O5 (616.83 g/mol): C, 73.99; H, 8.50; N, 4.54. Found: C, 73.75; H, 8.61; N, 4.43.
4-(tert-Butyl)-2,6-bis(2-chlorobenzyl)-N-(quinolin-8-yl)benzamide (Table 5, Entry 11)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and 1-(bromomethyl)-2-chlorobenzene (456 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 50/1 to 25/1), 286 mg (70 %) of crystalline material was obtained. Rf = 0.38 (toluene/ethyl acetate 50/1), mp = 140–141 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.86 (s, 1H), 8.91 (dd, J = 7.3, 1.7 Hz, 1H), 8.61 (dd, J = 4.3, 1.6 Hz, 1H), 8.14 (dd, J = 8.3, 1.8 Hz, 1H), 7.59–7.50 (m, 2H), 7.42–7.37 (m, 1H), 7.24–7.17 (m, 4H), 7.12–6.99 (m, 6H), 4.25 (s, 4H), 1.20 (s, 9H). 13C NMR (400 MHz, CDCl3, ppm) δ 168.4, 152.3, 148.0, 138.5, 138.2, 136.2, 136.1, 135.6, 134.3, 134.2, 131.4, 129.3, 127.9, 127.6, 127.4, 126.8, 125.3, 121.9, 121.5, 116.8, 36.8, 34.7, 31.1. FT-IR (neat, cm−1) ν 3341, 1670, 1524, 1486. Anal. calcd. for C34H30Cl2N2O (553.52 g/mol): C, 73.78; H, 5.46; N, 5.06;. Found: C, 73.58; H, 5.57; N 5.05.
Dimethyl 3,3'-((5-(tert-butyl)-2-(quinolin-8-ylcarbamoyl)-1,3-phenylene)bis-(methylene))-dibenzoate (Table 5, Entry 12)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and methyl 3-(bromomethyl)benzoate (508 mg, 2.22 mmol) were dissolved in t-amyl alcohol (1.0 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 25/1 to 15/1), 318 mg (94 %) of crystalline material was obtained. Rf = 0.18 (toluene/ethyl acetate 25/1), mp = 110-112 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.43 (s, 1H), 8.87 (dd, J = 7.4, 1.6 Hz, 1H), 8.44 (dd, , J = 4.5, 1.7 Hz, 1H), 8.12 (dd, J = 8.2, 1.5 Hz, 1H), 7.78 (s, 2H), 7.63–7.60 (m, 2H), 7.58–7.50 (m, 2H), 7.39–7.32 (m, 3H), 7.18 (s, 2H), 7.16–7.11 (m, 2H), 4.13 (s, 4H), 3.71 (s, 6H), 1.29 (s, 9H).13C NMR (400 MHz, CDCl3, ppm) δ 168.3, 166.9, 152.5, 147.9, 140.7, 138.2, 137.2, 136.0 (q, Hz=187.8), 135.4, 134.0, 133.6, 130.1, 130.0, 128.4, 127.8, 127.3, 127.2, 125.7, 121.9, 121.5, 116.7, 51.9, 39.5, 34.8, 31.2. FT-IR (neat, cm−1) ν 3328, 1673, 1520, 1484. Anal. calcd. for C38H36N2O5 (600.70 g/mol): C, 75.98; H, 6.04; N, 4.66. Found: C, 75.86; H, 6.08; N 4.67.
4-(tert-Butyl)-N-(quinolin-8-yl)-2,6-bis(4-(trifluoromethoxy)benzyl)benzamide (Table 5, Entry 13)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and 1-(bromomethyl)-4-(trifluoromethoxy)benzene (566 mg, 2.22 mmol) were dissolved in 1.0 ml of t-amyl alcohol. Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 25/1), 433 mg (90 %) of a crystalline material was obtained. Rf = 0.27 (toluene/ethyl acetate 70/1), mp = 118–119 °C (hexanes). 1H NMR (500 MHz, CDCl3, ppm) δ 9.52 (s, 1H), 8.86 (dd, J = 7.1, 1.9 Hz, 1H), 8.50 (dd, J = 4.3, 1.8 Hz, 1H), 8.14 (dd, J = 8.5, 1.6 Hz, 1H), 7.60–7.51 (m, 2H), 7.42–7.37 (m, 1H), 7.19–7.11 (m, 6H), 6.92–6.86 (m, 4H), 4.08 (s, 4H), 1.30 (s, 9H). FT-IR (neat, cm−1) ν 3335, 1670, 1523, 1485. 13C NMR (400 MHz, CDCl3, ppm) δ 168.3, 152.6, 148.1, 147.5, 139.2, 138.1, 137.2, 136.2, 135.4, 134.0, 130.1, 127.9, 127.2, 125.7, 122.1, 121.6, 120.8, 119.1, 116.5, 39.0, 34.8, 31.2. 19F NMR (376 MHz, CDCl3, ppm) δ-57.9. Anal. calcd. for C36H30F6N2O3 (652.63 g/mol): C, 66.25; H, 4.63; N, 4.29. Found: C, 66.51; H, 4.58; N 4.33.
4-(tert-Butyl)-2,6-bis(3-nitrobenzyl)-N-(quinolin-8-yl)benzamide (Table 5, Entry 14)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and 1-(bromomethyl)-3-nitrobenzene (479 mg, 2.22 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 20 h. After column chromatography (toluene/ethyl acetate 40/1 to 30/1), 365 mg (86 %) of a yellow solid was obtained. Rf = 0.31 (toluene/ethyl acetate 40/1), mp = 122–123 °C (hexanes). 1H NMR (400 MHz, CDCl3, ppm) δ 9.39 (s, 1H), 8.76 (q, J = 5.6 Hz, 1H), 8.44 (q, J = 4.2 Hz, 1H), 8.13 (q, J = 8.4 Hz, 1H), 7.97–7.94 (m, 2H), 7.77–7.73 (m, 2H), 7.557.51 (m, 2H), 7.48–7.44 (m, 2H), 7.41–7.37 (m, 1H), 7.26–7.25 (m, 2H), 7.21–7.15 (m, 2H), 4.19 (s, 4H), 1.34 (s, 9H). 13C NMR (500 MHz, CDCl3, ppm) δ 167.8, 153.2, 148.1, 148.1, 142.4, 138.0, 136.5, 136.3, 135.6, 135.2, 133.5, 129.2, 127.8, 127.3, 126.3, 123.7, 122.4, 121.7, 121.2, 116.6, 39.5, 34.9, 31.3. FT-IR (neat, cm−1) ν 3369, 1671, 1517, 1481. Anal. calcd. for C34H30N4O5 (574.63 g/mol): C, 71.07; H, 5.26; N, 9.75. Found: C 71.02, H 5.27, N 9.74.
4-(tert-Butyl)-2,6-bis(6-(1,3-dioxoisoindolin-2-yl)hexyl)-N-(quinolin-8-yl)benzamide (Table 5, Entry 15)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and 2-(6-iodohexyl)isoindoline-l,3-dione (792 mg, 2.22 mmol) were dissolved in t-amyl alcohol (3.0 mL). Resulting mixture was stirred at 110 °C for 40 h. After column chromatography (toluene/ethyl acetate 30/1 to 20/1), 420 mg (74 %) of a yellow oil was obtained. Rf = 0.13 (toluene/ethyl acetate 30/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.88 (s, 1H), 8.96 (dd, J = 7.4, 1.3 Hz, 1H), 8.68 (dd, J = 4.4, 1.6 Hz, 1H), 8.13 (dd, J = 8.4, 1.5 Hz, 1H), 7.82–7.78 (m, 4H), 7.71–7.66 (m, 4H), 7.61–7.51 (m, 2H), 7.39–7.35 (m, 1H), 7.11 (s, 2H), 3.51 (t, J = 7.3 Hz, 4H), 2.63 (t, J = 8.1 Hz, 4H), 1.65 (q, J = 7.8 Hz, 4H), 1.53–1.47 (m, 4H), 1.32 (s, 9H), 1.31–1.19 (m, 8H).13C NMR (400 MHz, CDCl3, ppm) δ 169.2, 168.4, 151.8, 148.2, 138.9, 138.5, 136.3, 134.8, 134.5, 133.8, 132.2, 128.0, 127.5, 124.0, 123.2, 121.8, 121.6, 116.8, 38.0, 34.7, 33.8, 31.8, 31.4, 29.3, 28.5, 26.7. FT-IR (neat, cm−1) ν 3370, 1674, 1519, 1482. Anal. calcd. for C48H50N4O5 (762.93 g/mol): C, 75.57; H, 6.61; N, 7.34. Found: C, 75.29; H, 6.74; N 7.27.
4-(tert-Butyl)-2,6-bis(3-methylbut-2-en-1-yl)-N-(quinolin-8-yl)benzamide (Table 5, Entry 16)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 4-tert-butyl-N-(quinolin-8-yl)benzamide (225 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (14.8 mg, 0.15 mmol), and 1-bromo-3-methylbut-2-ene (330 mg, 2.22 mmol) were dissolved in t-amyl alcohol (1.0 mL). Resulting mixture was stirred at 110 °C for 96 h. After column chromatography (hexanes/ethyl acetate 15/1), 72 mg (22 %) of yellow oil was obtained. R = 0.31 (hexanes/ethyl acetate 15/1). 1f H NMR (500 MHz, CDCl3, ppm) δ 9.89 (s, 1H), 9.02–8.98 (m, 1H), 8.73–8.69 (m, 1H), 8.19–8.14 (m, 1H), 7.61–7.52 (m, 2H), 7.45–7.43 (m, 1H), 7.15 (s, 2H), 5.33–5.27 (m, 2H), 3.45 (d, J = 6.9 Hz, 4H), 1.50 (s, 6H), 1.46 (s, 6H), 1.34 (s, 9H). 13C NMR (400 MHz, CDCl3, ppm) δ 169.1, 152.2, 148.1, 138.5, 138.2, 136.3, 134.9, 134.8, 132.4, 127.9, 127.5, 124.2, 123.3, 121.7, 121.6, 116.7, 34.7, 32.6, 31.3, 25.6, 17.8. FT-IR (neat, cm−1) ν 3339, 1675, 1520, 1482. HRMS (m/z): [M+] calcd for C30H36N2O, 440.2828; found, 440.2827, error=0.2 ppm.
General procedure for alkylation of 8-aminoquinoline amide sp3 C-H bonds
A 2-dram screw—cap vial was charged with Pd(OAc)2 (5 mol%), K2CO3 (3 equiv), substrate, pivalic acid (2 equiv), and alkyl bromide or iodide (4 equiv). t-Amyl alcohol solvent (0.5–1.0 mL) was added and the resulting mixture was stirred and heated at 110 °C for 12–96 h. The conversion was monitored by TLC. After completion of reaction, ethyl acetate was added to reaction mixture followed by extraction with water. Aqueous layer was washed with ethyl acetate (3x5 mL). Combined organic extracts were dried over MgSO4. Filtration and evaporation under reduced pressure followed by purification by flash chromatography gave pure product.
N-(Quinolin-8-yl)pentanamide (Table 6, Entry 1)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and iodoethane (461 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.5 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 20/1), 133 mg (78 %) of a yellow oil was obtained. Rf = 0.20 (toluene/ethyl acetate 20/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.80 (s, 1H), 8.77–8.82 (m, 2H), 8.13 (dd, J = 8.4, 1.6 Hz, 1H), 7.41–7.55 (m, 3H), 2.56 (t, 7.5 Hz, 2H), 1.76–1.85 (m, 2H), 1.41–1.51 (m, 2H), 0.97 (t, J = 7.4 Hz, 2H). 13C NMR (400 MHz, CDCl3, ppm) δ 172.1, 148.2, 138.4, 136.5, 134.7, 128.0, 127.5, 121.7, 121.4, 116.5, 38.1, 27.9, 22.6, 14.0. FT-IR (neat, cm−1) ν 3355, 1688, 1524, 1486. This compound is known.21
N-(Quinolin-8-yl)heptanamide (Table 6, Entry 2)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-iodobutane (544 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.6 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 20/1), 101 mg (53 %) of yellow oil was obtained. Rf = 0.56 (toluene/ethyl acetate 20/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.80 (s, 1H), 8.79–8.77 (m, 2H), 8.13 (dd, J = 8.2, 1.8 Hz, 1H), 7.53–7.41 (m, 3H), 2.54 (t, J = 7.3 Hz, 2H), 1.84–1.77 (m, 2H), 1.44–1.41 (m, 2H), 1.35–1.31 (m, 4H), 0.91–0.87 (m, 3H). This compound is known.22
N-(Quinolin-8-yl)undecanamide (Table 6, Entry 3)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-iodooctane (710 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.5 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 25/1), 126 mg (52 %) of yellow oil was obtained. Rf = 0.46 (toluene/ethyl acetate 20/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.80 (s, 1H), 8.77–8.82 (m, 2H), 8.13 (dd, J = 8.2, 1.5 Hz, 1H), 7.41–7.55 (m, 3H), 2.52–2.59 (t, J = 7.6 Hz, 2H), 1.82 (q, J = 15.0, 7.6 Hz, 2H), 1.19–1.46 (m, 14H), 0.84–0.91 (m, 3H). This compound is known.4b
5-Phenyl-N-(quinolin-8-yl)pentanamide (Table 6, Entry 4)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and (2—iodoethyl)benzene (686 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.5 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 20/1), 138 mg (64 %) of yellow oil was obtained. Rf = 0.45 (toluene/ethyl acetate 20/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.72 (s, 1H), 8.75–8.81 (m, 2H), 8.13 (dd, J = 8.3 ,1.8 Hz, 1H), 7.40–7.54 (m, 3H), 7.14–7.29 (m, 5H), 2.68 (t, J = 7.5 Hz, 2H), 2.58 (t, J = 7.5 Hz, 2H), 1.82–1.92 (m, 2H), 1.71–1.81 (m, 2H). 13C NMR (400 MHz, CDCl3, ppm) δ 171.7, 148.2, 142.3, 138.4, 136.5, 134.61, 128.6, 128.4, 128.0, 127.5, 125.9, 121.7, 121.5, 116.5, 38.2, 35.9, 31.2, 25.5. FT-IR (neat, cm−1) ν 3353, 1687, 1524, 1485. HRMS electrospray (m/z): [M+ + Na] calcd for C20H20N2O, 327.14733; found, 327.14703, error=0.75 ppm.
6-Methyl-N-(quinolin-8-yl)hept-5-enamide (Table 6, Entry 5)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-bromo-3-methylbut-2-ene (441 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.5 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 25/1) 58 mg (29 %) of yellow oil was obtained. Rf = 0.43 (toluene/ethyl acetate 25/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.79 (s, 1H), 8.76–8.81 (m, 2H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 7.42–7.55 (m, 3H), 5.12–5.19 (m, 1H), 2.55 (t, J = 7.6 Hz, 2H), 2.09–2.16 (m, 2H), 1.81–1.91 (m, 2H), 1.69 (d, J = 0.9 Hz, 3H), 1.59 (s, 3H). 13C NMR (400 MHz, CDCl3, ppm) δ 172.1, 148.3, 138.5, 136.5, 134.7, 132.8, 128.1, 127.6, 123.8, 121.7, 121.5, 116.6, 37.8, 27.6, 25.9, 25.9, 17.9. FT-IR (neat, cm−1) ν 3357, 1687, 1524, 1486. HRMS electrospray (m/z): [M+ + Na] calcd for C17H20N2O, 291.14734; found, 291.14693, error=0.48 ppm.
5-Methyl-N-(quinolin-8-yl)hexanamide (Table 6, Entry 6)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and l-iodo-2-methylpropane (544 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.5 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 10/1), 133 mg (78 %) of yellow oil was obtained. Rf = 0.45 (toluene/ethyl acetate 10/1). 1f H NMR (500 MHz, CDCl3, ppm) δ 9.81 (s, 1H), 8.83–8.76 (m, 2H), 8.14 (dd, J = 8.2, 1.6 Hz, 1H), 7.56–7.41 (m, 3H), 2.55 (t, J = 7.6 Hz, 2H), 1.87–1.77 (m, 2H), 1.56 (m, 2H), 1.68 (m, 1H), 1.36–1.27 (m, 2H), 0.94-0.88 (d, J = 6.6 Hz, 6H). This compound is known.4b
4-(2-Bromophenyl)-N-(quinolin-8-yl)butanamide (Table 6, Entry 7)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)propionamide (148 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-bromo-2-(bromomethyl)benzene (739 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.5 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (toluene/ethyl acetate 15/1), 166 mg (60 %) of yellow oil was obtained. Rf = 0.41 (toluene/ethyl acetate 15/1). 1H NMR (400 MHz, CDCl3, ppm) δ 9.81 (s, 1H), 8.82–8.76 (m, 2H), 8.14 (dd, J = 8.2, 1.8 Hz, 1H), 7.55–7.40 (m, 4H), 7.30–7.20 (m, 2H), 7.08–7.02 (m, 1H), 2.87 (t, J = 7.7 Hz, 2H), 2.63 (t, J = 7.3 Hz, 2H), 2.20–2.11 (m, Hz, 2H). 13C NMR (400 MHz, CDCl3, ppm) δ 171.4, 148.3, 140.9, 138.4, 136.5, 134.6, 132.9, 130.7, 128.0, 127.9, 127.6, 127.5, 124.6, 121.7, 121.5, 116.6, 37.4, 35.5, 25.6. FT-IR (neat, cm−1) ν 3344, 1688, 1524, 1486. HRMS electrospray (m/z): [M+ + Na] calcd for C19H17BrN2O, 391.04221; found, 391.04175, error=0.27 ppm.
2-Ethyl-N-(quinolin-8-yl)undecanamide (Table 6, Entry 8)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 2-methyl-N-(quinolin-8-yl)butanamide (169 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-iodooctane (710 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (hexanes/ethyl acetate 12/1), 101 mg (40 %) of yellow oil was obtained. TLC Rf = 0.54 (hexanes /ethyl acetate 12/1). 1H NMR (400 MHz, CDCl3, ppm) δ 9.85 (s, 1H), 8.87–8.78 (m, 2H), 8.15 (dd, J = 8.3,1.8 Hz, 1H), 7.56–7.41 (m, 3H), 2.43–2.34 (m, 1H), 1.87–1.74 (m, 2H), 1.69–1.53 (m, 2H), 1.43–1.12 (m, 14H), 1.03-0.96 (t, J=7.4 Hz, 3H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (400 MHz, CDCl3, ppm) δ 175.1, 148.3, 138.6, 136.5, 134.7, 128.1, 127.6, 121.7, 121.5, 116.6, 51.2, 33.1, 32.0, 29.9, 29.7, 29.6, 29.4, 27.8, 26.4, 22.8, 14.3, 13.0. FT-IR (neat, cm−1) ν 3356, 2926, 2854, 1689, 1524, 1486, 1324. HRMS electrospray (m/z): [M+ + Na] calcd for C24H19FN2O, 393.13792; found, 393.13782, error=1.16 ppm.
2-(2-Fluoro-[1,1'-biphenyl]-4-yl)-N-(quinolin-8-yl)undecanamide (Table 6, Entry 9)
Pd(OAc)2 (8.3 mg, 0.037 mmol), 2-(2-fluoro-[1,1'-biphenyl]-4-yl)-N-(quinolin-8-yl)propanamide (274 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-iodooctane (710 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (hexanes/ethyl acetate 7/1), 162 mg (45 %) of yellow oil was obtained. Rf = 0.29 (hexanes/ethyl acetate 7/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.98 (s, 1H), 8.81–8.74 (m, 2H), 8.13 (dd, J = 8.3, 1.8 Hz, 1H), 7.56–7.29 (m, 11H), 3.77–3.70 (t, J = 7.6 Hz, 1H), 2.37–2.25 (m, 1H), 2.02–1.90 (m, 1H), 1.49–1.15 (m, 14H), 0.89–0.82 (t, J = 6.8 Hz, 3H). 13C NMR (400 MHz, CDCl3, ppm; list of signals, C–F coupling not assigned) δ 171.7, 161.2, 158.7, 148.3, 141.6, 141.5, 138.5, 136.4, 135.7, 134.5, 131.1, 131.0, 129.1, 129.0, 128.5, 128.0, 127.9, 127.7, 127.5, 124.2, 124.1, 121.8, 121.7, 116.5, 115.8, 115.6, 54.6, 33.6, 32.0, 29.7, 29.6, 29.4, 27.9, 22.8, 14.2. 19F NMR (376 MHz, CDCl3, ppm) δ-117.3. FT-IR (neat, cm−1) ν 2927, 2854, 1688, 1525, 1484, 1424, 1325. HRMS electrospray (m/z): [M+ + Na] calcd for C24H19FN2O, 393.13792; found, 393.13782, error=1.16 ppm.
2-Octyl-N-(quinolin-8-yl)cyclohexanecarboxamide (Table 6, Entry 10)
Pd(OAc)2 (8.3 mg, 0.037 mmol), N-(quinolin-8-yl)cyclohexanecarboxamide (188 mg, 0.74 mmol), K2CO3 (256 mg, 1.85 mmol), pivalic acid (151 mg, 1.48 mmol), and 1-iodooctane (710 mg, 2.96 mmol) were dissolved in t-amyl alcohol (0.7 mL). Resulting mixture was stirred at 110 °C for 24 h. After column chromatography (hexanes/ethyl acetate 12/1), 42 mg (15 %) of yellow oil was obtained. TLC Rf = 0.29 (hexanes/ethyl acetate 12/1). 1H NMR (500 MHz, CDCl3, ppm) δ 9.82 (s, 1H), 8.85–8.79 (m, 2H), 8.16 (dd, J = 8.2, 1.7 Hz, 1H), 7.57–7.43 (m, 3H), 2.22–2.12 (m, 1H), 2.06–1.93 (m, 2H), 1.87–1.05 (m, 20 H), 1.04–0.92 (m, 1H), 0.83–0.78 (t, J = 6.9 Hz, 3H). 13C NMR (400 MHz, CDCl3, ppm) δ 175.2, 148.3, 138.6, 136.5, 134.8, 128.1, 127.6, 121.7, 121.4, 116.6, 54.2, 39.5, 35.0, 32.0, 31.3, 31.1, 30.0, 29.7, 29.4, 26.7, 26.1, 26.0, 22.8, 14.3. FT-IR (neat, cm−1) ν 3354, 2924, 2855, 1685, 1523, 1485, 1327, 1160. HRMS electrospray (m/z): [M+ + Na] calcd for C24H34N2O, 389.25689; found, 389.25660, error=0.69 ppm.
Cleavage of the 8-aminoquinoline auxiliary: 5-Phenylpentanoic acid
5-Phenyl-N-(quinolin-8-yl)pentanamide (1.0 mmol, 304 mg), NaOH (2.5 mmol, 100 mg), and ethanol (2.0 mL) were mixed in a 2-dram vial. The vial was capped, heated and stirred at 70 °C for 3 h. After the reaction was complete, the contents of the vial was transferred to flask and water (10 mL) was added to the reaction mixture. The mixture was acidified with HCl (20 % aqueous solution) to pH=1. The product was extracted with ethyl acetate (5x5 mL). The organic layers were combined, concentrated and subjected to column chromatography in hexanes/ethyl acetate 80/20. After concentration of the fractions containing the product, the residue was dried under reduced pressure. Product was obtained as tan crystals (155 mg, 87 % yield). Rf = 0.30 (hexanes/ethyl acetate 80/20). This compound is known.231H NMR (400 MHz, CDCl3, ppm) δ 11.82 (br s, 1H)7.33–7.14 (m, 5H), 2.52–2.40 (t, J = 7.0 Hz, 2H), 2.10–2.04 (t, J = 5.5 Hz, 2H), 1.85-1.54 (m, 4H).
Supplementary Material
Table 10.
Alkylation of Picolinamides – Optimization of Basea

| |||
|---|---|---|---|
|
| |||
| Entry | Base | % GC Yield |
|
| A | B | ||
| 1 | K3PO4 | 95 | 5 |
| 2 | Na3PO4 | 49 | 20 |
| 3 | K2CO3 | 99 | 0 |
| 4 | Na2CO3 | 54 | 20 |
| 5 | Cs2CO3 | 28 | 10 |
| 6 | CsOAc | 7 | 14 |
| 7 | K2HPO4 | 32 | 10 |
| 8 | NaOAc | 6 | 18 |
Dodecane internal standard; amide (0.5 mmol), BuI (4 mmol).
ACKNOWLEDGMENT
This research was supported by the Welch Foundation (Grant E-1571), NIGMS (Grant R01GM077635), and Camille and Henry Dreyfus Foundation.
Footnotes
Supporting Information Available. Spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. Reviews: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ackermann L. Chem. Rev. 2011;111:1315. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; (c) Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; (f) Nakamura E, Yoshikai N. J. Org. Chem. 2010;75:6061. doi: 10.1021/jo100693m. [DOI] [PubMed] [Google Scholar]; (g) Yeung CS, Dong VM. Chem. Rev. 2011;111:1215. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; (h) Sun C-L, Li B-J, Shi Z-J. Chem. Commun. 2010:677. doi: 10.1039/b908581e. [DOI] [PubMed] [Google Scholar]; (i) Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]; (j) Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem.-Eur. J. 2010;16:2654. doi: 10.1002/chem.200902374. [DOI] [PubMed] [Google Scholar]; (k) Catellani M, Motti E, Della Ca N. Acc. Chem. Res. 2008;41:1512. doi: 10.1021/ar800040u. [DOI] [PubMed] [Google Scholar]; (l) Yamaguchi J, Yamaguchi AD, Itami K. Angew. Chem., Int. Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (m) McMurray L, O'Hara F, Gaunt MJ. Chem. Soc. Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- 2.(a) Neufeldt SR, Sanford MS. Acc. Chem. Res. 2012;45:936. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Messaoudi S, Brion J-D, Alami M. Eur. J. Org. Chem. 2010:6495. [Google Scholar]; (c) Bellina F, Rossi R. Tetrahedron. 2009;65:10269. [Google Scholar]; (d) Engle KM, Mei T-S, Wasa M, Yu J-Q. Acc. Chem. Res. 2012;45:788. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Guo X, Li C-J. Org. Lett. 2011;13:4977. doi: 10.1021/ol202081c. [DOI] [PubMed] [Google Scholar]; (f) Tran LD, Daugulis O. Org. Lett. 2010;12:4277. doi: 10.1021/ol101684u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yao T, Hirano K, Satoh T, Miura M. Chem. - Eur. J. 2010;16:12307. doi: 10.1002/chem.201001631. [DOI] [PubMed] [Google Scholar]; (h) Chen Q, Ilies L, Nakamura E-I. J. Am. Chem. Soc. 2011;133:428. doi: 10.1021/ja1099853. [DOI] [PubMed] [Google Scholar]; (i) Ackermann L, Hofmann N, Vicente R. Org. Lett. 2011;13:1875. doi: 10.1021/ol200366n. [DOI] [PubMed] [Google Scholar]; (j) Ren P, Salihu I, Scopelliti R, Hu X. Org. Lett. 2012;14:1748. doi: 10.1021/ol300348w. [DOI] [PubMed] [Google Scholar]; (k) Xiao B, Liu Z-J, Liu L, Fu Y. J. Am. Chem. Soc. 2013;135:616. doi: 10.1021/ja3113752. [DOI] [PubMed] [Google Scholar]; (l) Hofmann N, Ackermann L. J. Am. Chem. Soc. 2013;135:5877. doi: 10.1021/ja401466y. [DOI] [PubMed] [Google Scholar]; (m) Neufeldt SR, Seigerman CK, Sanford MS. Org. Lett. 2013;15:2302. doi: 10.1021/ol400888r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Tauchert ME, Incarvito CD, Rheingold AL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2012;134:1482. doi: 10.1021/ja211110h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Lee D-H, Kwon K-H, Yi CS. J. Am. Chem. Soc. 2012;134:7325. doi: 10.1021/ja302710v. [DOI] [PubMed] [Google Scholar]; (p) Tredwell MJ, Gulias M, Gaunt Bremeyer N, Johansson CCC, Collins BSL, Gaunt M. J. Angew. Chem., Int. Ed. 2011;50:1076. doi: 10.1002/anie.201005990. [DOI] [PubMed] [Google Scholar]
- 3.(a) Dyker G. Angew. Chem., Int. Ed. Engl. 1994;33:103. [Google Scholar]; (b) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; (c) Chaumontet M, Piccardi R, Audic N, Hitce J, Peglion J-L, Clot E, Baudoin O. J. Am. Chem. Soc. 2008;130:15157. doi: 10.1021/ja805598s. [DOI] [PubMed] [Google Scholar]; (d) Chaumontet M, Piccardi R, Baudoin O. Angew. Chem., Int. Ed. 2009;48:179. doi: 10.1002/anie.200804444. [DOI] [PubMed] [Google Scholar]; (e) Lafrance M, Gorelsky SI, Fagnou K. J. Am. Chem. Soc. 2007;129:14570. doi: 10.1021/ja076588s. [DOI] [PubMed] [Google Scholar]; (f) Liégault B, Fagnou K. Organometallics. 2008;27:484. [Google Scholar]; (g) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (h) Wang D-H, Wasa M, Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]; (i) Watanabe T, Oishi S, Fujii N, Ohno H. Org. Lett. 2008;10:1759. doi: 10.1021/ol800425z. [DOI] [PubMed] [Google Scholar]; (j) Chen X, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:12634. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]; (k) Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Wasa M, Chan KSL, Zhang X-G, He J, Miura M, Yu J-Q. J. Am. Chem. Soc. 2012;134:18570. doi: 10.1021/ja309325e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Stowers KJ, Fortner KC, Sanford MS. J. Am. Chem. Soc. 2011;133:6541. doi: 10.1021/ja2015586. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Rodríguez N, Romero-Revilla JA, Fernández-Ibáñez MÀ, Carretero JC. Chem. Sci. 2013;4:175. [Google Scholar]; (o) Baudoin O, Herrbach A, Gueritte F. Angew. Chem., Int. Ed. 2003;42:5736. doi: 10.1002/anie.200352461. [DOI] [PubMed] [Google Scholar]; (p) Renaudat A.; Ludivine J-G, Rodolphe J, Kefalidis CE, Clot E, Baudoin O. Angew. Chem., Int. Ed. 2010;49:7261. doi: 10.1002/anie.201003544. [DOI] [PubMed] [Google Scholar]; (q) Leskinen MV, Yip K-T, Valkonen A, Pihko PM. J. Am. Chem. Soc. 2012;134:5750. doi: 10.1021/ja300684r. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; (b) Shabashov D, Daugulis O. J. Am. Chem. Soc. 2010;132:3965. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tran LD, Daugulis O. Angew. Chem., Int. Ed. 2012;51:5188. doi: 10.1002/anie.201200731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nadres ET, Daugulis O. J. Am. Chem. Soc. 2012;134:7. doi: 10.1021/ja210959p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tran LD, Popov I, Daugulis O. J. Am. Chem. Soc. 2012;134:18237. doi: 10.1021/ja3092278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tran LD, Roane J, Daugulis O. Angew. Chem., Int. Ed. 2013;52:6043. doi: 10.1002/anie.201300135. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Truong T, Klimovica K, Daugulis O. J. Am. Chem. Soc. 2013;135:9342. doi: 10.1021/ja4047125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lazareva A, Daugulis O. Org. Lett. 2006;8:5211. doi: 10.1021/ol061919b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Reddy BVS, Reddy LR, Corey EJ. Org. Lett. 2006;8:3391. doi: 10.1021/ol061389j. [DOI] [PubMed] [Google Scholar]; (b) Feng Y, Chen G. Angew. Chem., Int. Ed. 2010;49:958. doi: 10.1002/anie.200905134. [DOI] [PubMed] [Google Scholar]; (c) Li BTY, White JM, Hutton CA. Aust. J. Chem. 2010;63:438. [Google Scholar]; (d) Gutekunst WR, Baran PS. J. Am. Chem. Soc. 2011;133:19076. doi: 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gutekunst WR, Gianatassio R, Baran PS. Angew. Chem., Int. Ed. 2012;51:7507. doi: 10.1002/anie.201203897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fang Y, Wang Y, Landgraf B, Liu S, Chen G. Org. Lett. 2010;12:3414. doi: 10.1021/ol101220x. [DOI] [PubMed] [Google Scholar]; (g) Zhao Y, Chen G. Org. Lett. 2011;13:4850. doi: 10.1021/ol201930e. [DOI] [PubMed] [Google Scholar]; (h) Zhao Y, He G, Nack WA, Chen G. Org. Lett. 2012;14:2948. doi: 10.1021/ol301214u. [DOI] [PubMed] [Google Scholar]; (i) Ano Y, Tobisu M, Chatani N. Org. Lett. 2012;14:354. doi: 10.1021/ol203100u. [DOI] [PubMed] [Google Scholar]; (j) Ano Y, Tobisu M, Chatani N. J. Am. Chem. Soc. 2011;133:12984. doi: 10.1021/ja206002m. [DOI] [PubMed] [Google Scholar]; (k) Huang L, Li Q, Wang C, Qi C. J. Org. Chem. 2013;78:3030. doi: 10.1021/jo400017v. [DOI] [PubMed] [Google Scholar]; (l) Zhang S-Y, He G, Nack WA, Zhao Y, Li Q, Chen G. J. Am. Chem. Soc. 2013;135:2124. doi: 10.1021/ja312277g. [DOI] [PubMed] [Google Scholar]; (m) He G, Chen G. Angew. Chem., Int. Ed. 2011;50:5192. doi: 10.1002/anie.201100984. [DOI] [PubMed] [Google Scholar]; (n) Shang R, Ilies L, Matsumoto A, Nakamura E.-i. J. Am. Chem. Soc. 2013;135:6030. doi: 10.1021/ja402806f. [DOI] [PubMed] [Google Scholar]; (o) Rouquet G, Chatani N. Chem. Sci. 2013;4:2201. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (p) Aihara Y, Chatani N. J. Am. Chem. Soc. 2013;135:5308. doi: 10.1021/ja401344e. [DOI] [PubMed] [Google Scholar]; (q) Nishino M, Hirano K, Satoh T, Miura M. Angew. Chem., Int. Ed. 2013;52:4457. doi: 10.1002/anie.201300587. [DOI] [PubMed] [Google Scholar]; (r) Suess AM, Ertem MZ, Cramer CJ, Stahl SS. J. Am. Chem. Soc. 2013;135:9797. doi: 10.1021/ja4026424. [DOI] [PubMed] [Google Scholar]; (s) Aihara Y, Chatani N. Chem. Sci. 2013;4:664. [Google Scholar]
- 6.(a) Canty A. J. Acc. Chem. Res. 1992;25:83. [Google Scholar]; (b) Racowski JM, Dick AR, Sanford MS. J. Am. Chem. Soc. 2009;131:10974. doi: 10.1021/ja9014474. [DOI] [PubMed] [Google Scholar]; (c) Sobanov AA, Vedernikov AN, Dyker G, Solomonov BN. Mendeleev Commun. 2002;1:14. [Google Scholar]; (d) Amatore C, Catellani M, Deledda S, Jutand A, Motti E. Organometallics. 2008;27:4549. [Google Scholar]; (e) Guo R, Portscheller JL, Day VW, Malinakova HC. Organometallics. 2007;26:3874. [Google Scholar]; (f) Powers DC, Geibel MAL, Klein JEMN, Ritter T. J. Am. Chem. Soc. 2009;131:17050. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]; (g) Deprez NR, Sanford MS. J. Am. Chem. Soc. 2009;131:11234. doi: 10.1021/ja904116k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ittel SD, Johnson LK, Brookhart M. Chem. Rev. 2000;100:1169. doi: 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]
- 8.Lazareva A, Daugulis O. Org. Lett. 2006;8:5211. doi: 10.1021/ol061919b. [DOI] [PubMed] [Google Scholar]
- 9.Bon E, Bigg DCH, Bertrand G. J. Org. Chem. 1994;59:4035. [Google Scholar]
- 10.Shabashov D, Daugulis O. J. Org. Chem. 2007;72:7720. doi: 10.1021/jo701387m. [DOI] [PubMed] [Google Scholar]
- 11.Beesley RM, Ingold CK, Thorpe JF. J. Chem. Soc., Trans. 1915;107:1080. [Google Scholar]
- 12.Marsh CC, Schuna AA, Sundstrom WR. Pharmacotherapy. 1986;6:10. doi: 10.1002/j.1875-9114.1986.tb03445.x. [DOI] [PubMed] [Google Scholar]
- 13.Shinji C, Maeda S, Imai K, Yoshida M, Hashimoto Y, Miyachi H. Bioorg. Med. Chem. 2006;14:7625. doi: 10.1016/j.bmc.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Suttsuo F, Kato K, Mukaiyama T. Chem. Lett. 2008;37:506. [Google Scholar]
- 15.Yu H, Ballard CE, Wang B. Tetrahedron Lett. 2001;42:1835. [Google Scholar]
- 16.Nag S, Butcher RJ, Bhattacharya S. Eur. J. Inorg. Chem. 2007:1251. [Google Scholar]
- 17.Arvapalli VS, Chen G, Kosarev S, Tan ME, Xie D, Yet L. Tetrahedron Lett. 2010;51:284. [Google Scholar]
- 18.Hermange P, Lindhardt AT, Taaning RH, Bjerglund K, Lupp D, Skrydstrup T. J. Am. Chem. Soc. 2011;133:6061. doi: 10.1021/ja200818w. [DOI] [PubMed] [Google Scholar]
- 19.Gürbüz N, Özdemir I, Çetinkaya B. Tetrahedron Lett. 2005;46:2273. [Google Scholar]
- 20.Bon E, Bigg DCH, Bertrand G. J. Org. Chem. 1994;59:4035. [Google Scholar]
- 21.Lee JC, Jr., Peris E, Rheingold AL, Crabtree RH. J. Am. Chem. Soc. 1994;116:11014. [Google Scholar]
- 22.Heuer HW, Wehrmann R, Elschner A. Elektrolumineszierende Anordnungen unter Verwendung von Bor-Chelaten von 8-Aminochinolin-Derivaten DE 19829949 A1. 05-01-2000.
- 23.Klemm LH, Bower GM. J. Org. Chem. 1958;23:344. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.