

Abstract
Mutations in FBN1 cause a range of overlapping but distinct conditions including Marfan syndrome (MFS), Weill-Marchesani syndrome (WMS), familial thoracic aortic aneurysms/dissections (FTAAD), acromicric dysplasia (AD), and geleophysic dysplasia (GD). Two forms of acromelic dysplasia, AD and GD, characterized by short stature, brachydactyly, reduced joint mobility, and characteristic facies, result from heterozygous missense mutations occurring in exons 41 and 42 of FBN1; missense mutations in these exons have not been reported to cause MFS or other syndromes. Here we report on probands with MFS and WMS who have heterozygous FBN1 missense mutations in exons 41 and 42, respectively. The proband with WMS has ectopia lentis, short stature, thickened pinnae, tight skin, striae atrophicae, reduced extension of the elbows, contractures of the fingers and toes, and brachydactyly and has a missense mutation in exon 42 of FBN1 (c.5242T>C ;p.C1748R). He also experienced a previously unreported complication of WMS, an acute thoracic aortic dissection. The second proband displays classic characteristics of MFS, including ectopia lentis, skeletal features and aortic root dilatation, and has a missense mutation in exon 41 of FBN1 (c.5084G>A; p.C1695Y). These phenotypes provide evidence that missense mutations in exons 41 and 42 of FBN1 lead to MFS and WMS in addition to AD and GD and also suggest that all individuals with pathogenic FBN1 mutations in these exons should be assessed for thoracic aortic disease and ectopia lentis. Further studies are necessary to elucidate the factors responsible for the different phenotypes associated with missense mutations in these exons of FBN1.
Keywords: geleophysic dysplasia, acromicric dysplasia, aortic aneurysm, aortic dissection, ectopia lentis
INTRODUCTION
Mutations in FBN1, encoding fibrillin-1, are responsible for various distinct yet overlapping inherited disorders, including Marfan syndrome (MFS, [OMIM 154700]), Weill-Marchesani syndrome (WMS, [OMIM 227600]), acromicric dysplasia (AD, [OMIM 102370]) and geleophysic dysplasia (GD, [OMIM 231050]) [Faivre et al., 2003b; Le et al., 2011; Milewicz et al., 1996; Pyeritz, 2000]. The most common inherited disease caused by FBN1 mutations is Marfan syndrome, an autosomal dominant connective tissue disease primarily affecting the cardiovascular, ocular, and skeletal systems. The cardiovascular manifestations of MFS are characterized by thoracic aortic aneurysms primarily involving the aortic root that lead to acute aortic dissections. A wide array of skeletal findings is observed in individuals with MFS and includes chest wall deformities, scoliosis, arachnodactyly, and disproportionate overgrowth of the long bones, often resulting in tall stature. The major ocular feature observed in at least half of individuals with MFS is ectopia lentis. FBN1 was identified as the causal gene for MFS in 1991, and FBN1 mutations were subsequently found to be the underlying etiology in a spectrum of MFS-related conditions, including ectopia lentis syndrome (ELS) and familial thoracic aortic aneurysms and dissections (FTAAD) [Ades et al., 2004; Milewicz et al., 1996].
In contrast to bone overgrowth and tall stature in individuals with MFS and Marfan-related conditions, heterozygous FBN1 mutations also have been described as the underlying etiology in three heritable short stature syndromes including: WMS, AD, and GD. WMS is an autosomal dominant or autosomal recessive connective tissue disease characterized by proportionate short stature, brachydactyly, joint stiffness, and abnormalities of the ocular lens, including microspherophakia, ectopia lentis, and glaucoma. Cardiac anomalies such as mitral valve prolapse, aortic and pulmonary valve stenosis, ventricular septal defects, and QTc prolongation have each been described in individuals with WMS [Faivre et al., 2003a; Kojuri et al., 2007; van de Woestijne et al., 2004]. However, thoracic aortic disease has not been reported as a feature of WMS. Mutations in ADAMTS10 are responsible for the autosomal recessive cases of WMS [Dagoneau et al., 2004]. More recently, FBN1 mutations were found to be causal in two types of acromelic dysplasias, AD and GD [Le et al., 2011]. Both dysplasias have phenotypes overlapping with WMS and are characterized by short stature, reduced joint mobility, skin thickening, and brachydactyly. These syndromes can be differentiated from WMS by other clinical manifestations, such as characteristic facies, premature death due to cardiac valve thickening, or tracheal narrowing leading to dyspnea or pulmonary infection in patients with GD, or distinct radiological findings in patients with AD [Faivre et al., 2001;Maroteaux et al., 1986]. Mutations in ADAMTSL2 have been reported in a subset of patients with autosomal recessive GD, but more recently FBN1 mutations were identified in patients with autosomal dominant AD and GD. Interestingly, all of the reported heterozygous FBN1 mutations for AD and GD are missense mutations located in exons 41 and 42 in the TGF-β binding-protein-like domain 5 (TB5) domain of FBN1. Therefore, these data suggest that the short stature phenotypes of AD and GD are due to FBN1 mutations in exons 41 and 42, but missense mutations in these exons have not been described in patients with either WMS or MFS. Here, we describe two families, one with typical MFS and the other with WMS complicated by thoracic aortic disease, who harbor heterozygous FBN1 missense mutations in exons 41 and 42, respectively, indicating that mutations in these exons can also cause these phenotypes.
CLINICAL REPORTS
Patient 1
The proband, a 41-year-old African American male, was born as the second child to a then 27-year-old father and 24-year-old mother. He weighed 3.18 kg at birth, the product of a normal gestation and full term pregnancy. His developmental and motor milestones were achieved on time, but he had short stature and performed poorly at school, repeating the first grade. He was first placed in eyeglasses at age 4 years when he was found to have high myopia and spherophakia in each eye with a superior temporal subluxation of each lens. At age 7.5 years, he had an uncomplicated pars plana vitrectomy and lensectomy of his left eye. At age 9 years, he was admitted to the hospital with a seven-day history of cough, pleuritic chest pain, and progressive shortness of breath, and was subsequently diagnosed with congestive heart failure and pleural effusion. He did not respond to medical therapy and pericardiocentesis, and thus a pericardiopleural window was created surgically with attendant drainage of about 400cc of sterile fluid.
At age 9, he was 109 cm. tall (<5th centile) and 20.5 kg. (<5th centile) with a normal head circumference (53.5 cm = 60th centile). He was disproportionately short-statured with a depressed glabella, thickened pinnae, and short, thick hands which did not fully extend or flex. His protuberant abdomen had a small umbilical hernia. His ocular examination was normal except for iridodonesis with superotemporal subluxation of the right lens and surgical absence of the left lens. Urine amino acids, chemical metabolic screening tests, and a G-banded karyotype were normal. Radiographs showed shortened metacarpals and metatarsals and his bone age was retarded by approximately two years. During his teen years, he underwent two hernia repairs. As an adult, he developed asthma and systemic hypertension.
At 38 years of age, he presented to an emergency department with a three-week history of shortness of breath and retrosternal “tightness”. A chest CT scan demonstrated a sub-acute aortic dissection extending from the aortic root to the left subclavian artery of the thoracic aortic arch (Stanford type A dissection) and showed dilatation of the aortic root and ascending aorta (Fig 1A). An intraoperative transesophageal echocardiogram documented pre-operative measurements of 2.1 cm at the aortic annulus, 3.6 cm as the sinus of Valsalva, 4.4 cm at the sinotubular junction, and 4.5 cm at the proximal ascending aorta. The patient underwent resection and graft replacement of both the ascending aorta and the proximal transverse aortic arch, reconstruction of the aortic root, and resuspension of the tri-leaflet aortic valve.
Figure 1. Clinical features and pedigree of Weill-Marchasani family, Patient 1.
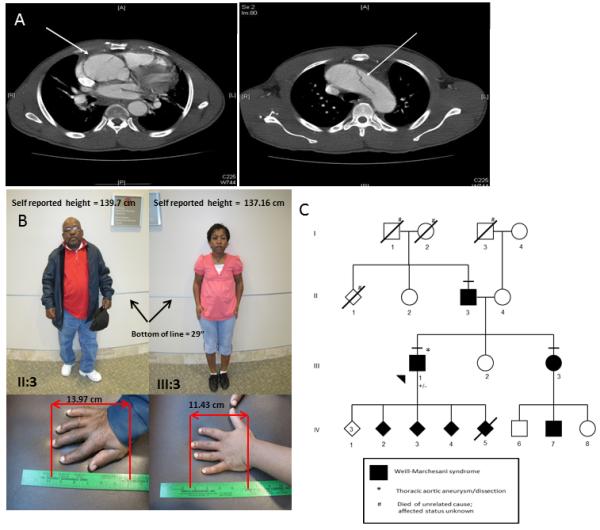
(A) Aneurysm and dissection of the ascending thoracic aorta. The left panel show a Chest CT with contrast of the 38-year-old proband in the WMS family (Person III-1) demonstrating an aneurysm of the ascending aorta measuring 47 mm. The right panel is a Chest CT with contrast of the WMS proband (person III-1) showing the transverse aortic arch with a luminal flap visualized, demarcating the aortic dissection. (B) Clinical features observed in the WMS family. Short stature observed in the proband’s father (Person II-3) and sister (Person III-3) and brachydactyly observed in the proband’s father (Person II-3) and sister (Person III-3). Written consent was obtained for publication of these images. (C) Pedigree of WMS family with p.C1748R mutation in exon 42 of FBN1. The disease status of individuals is indicated in the figure legend. The horizontal line above the symbol indicates an examination by a medical geneticist. The presence (+/−) of the FBN1 mutation is indicated below the symbol.
Following the proband’s aortic repair, physical examination showed iridodonesis in the right eye, a high palate, a crumpled right ear helix, tight skin, striae atrophicae, reduced extension of the elbows, contractures of the toes, and brachydactyly. The proband’s height was 149 cm (<5th centile). A four-generation pedigree showed that the proband’s father (Person II-3), sister (Person III-3), and four of his seven children have short stature and brachydactyly (Fig 1B, C). In addition, the proband’s electrocardiograms showed prolonged QTc, with calculated QTc ranging between 486 and 531ms. Molecular analysis of the FBN1 gene in the proband identified a heterozygous missense mutation c.5242T>C (p.C1748R) in exon 42. The proband and his family declined further clinical and genetic evaluations.
The proband’s older sister had been examined at age 13 (Person III-3) and was noted to be short-statured and stocky for her age. At age 7, she had been found to have high myopia (spherical equivalent of ~−14 D. O.U.) with spherophakia and superotemporal subluxation O.U. At age 13, she stood 129.5 cm. (> 4 S.D below the mean), weighed 34 kg (<5th centile), but had a normal head circumference (55 cm = 75th centile). She had a depressed glabella, short palms and fingers, restricted movements of all joints of her upper extremities, lumbar lordosis, protuberant abdomen, and a small umbilical hernia.
The father (Person II-3) of both children was examined at age 34 years. He had worn an astigmatic correction for approximately four years. He was142 cm. tall (>6 S.D. below the mean), but had a normal head circumference. He was stocky and muscular with limited flexion and extension of his thick but stubby fingers. Although his corrected acuity was normal (with ~3.00 D astigmatism), his ocular examination showed minimal iridodonesis, poor iris dilation in response to mydriatic agents, and subtle superotemporal subluxation of each lens. Gonioscopy revealed a flat iris insertion, poor definition of the trabeulum, and no iris processes. Radiographic studies were characteristic of WMS.
The mother and the younger sister of the proband had normal stature for their respective ages and normal physical and ophthalmologic examinations.
Patient 2
The proband, a 28-year-old African American female, presented to our medical genetics clinic after her brother died of an acute Stanford type A aortic dissection. She was diagnosed with MFS at the age of 5 years based on ectopia lentis, tall stature, arachnodactyly, and joint hypermobility. Previous aortic imaging via transthoracic echocardiogram were reported to show stable, mild aortic root dilatation (echocardiograms not available for review). Physical examination of the proband found a high palate, tall stature, arachnodactyly, striae atrophicae, joint hypermobility, arm span to height ratio of 1.08, a reduced upper to lower segment ratio, and characteristic facial features of dolichocephaly, enophthalmos, and malar hypoplasia (Fig 2A). Based on the revised Ghent Criteria for the diagnosis of MFS, the proband’s systemic score was 6 out of 20 points (a score of ≥7 qualifies for systemic involvement)[Loeys et al., 2010]. Transthoracic echocardiography showed an aortic root measurement of 3.91 cm at the level of the sinus of Valsalva (Z-score +3.4) and a normal proximal ascending aorta measuring 3.0 cm. Subsequent molecular analysis of the FBN1 gene revealed a heterozygous missense mutation in exon 41, c.5084G>A (p.C1695Y), a novel FBN1 mutation not previously reported.
Figure 2. Clinical features and pedigree of the MFS family, Patient 2.
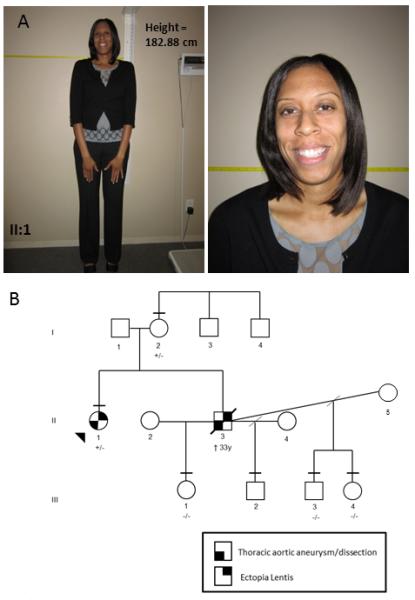
(A) Tall stature observed in proband with MFS (Person II-1) and facial features, which include dolichocephaly, enopthalmos and mild malar hypoplasia. Written consent was obtained for publication of these images. (B) Pedigree of MFS family with p.C1695Y mutation in exon 41 of FBN1. The disease status of individuals is as indicated in the figure legend. The horizontal line above the symbol indicates an examination by a medical geneticist. Age of death is documented below the symbol. The presence (+/−) or absence (−/−) of the FBN1 mutation is indicated below the symbol.
The proband’s brother was also diagnosed with MFS after presenting at age two years with bilateral ectopia lentis, scoliosis, pectus excavatum, and joint hypermobility (Fig 2B, person II-3). He underwent serial aortic imaging until the age of 22 years but then was lost to follow-up. He presented at the age of 33 years with an acute type A aortic dissection and underwent an emergent aortic repair. His aortic root was noted to be 5.7 cm in diameter at the time of surgery. He did not survive and DNA was not available from him. The FBN1 mutation was also present in the proband’s 57-year-old mother, who had not been diagnosed with MFS (Fig 2B, Person I-2). She had a history of hip dislocations and joint hypermobility beginning at age 7 years and had a total hip replacement at 22 years of age. At age 54, her transthoracic echocardiogram reportedly demonstrated a normal aortic root measurement of 3.4 cm (Z-score −0.66; echocardiograph was not available for review). On physical examination, she had a high palate, straie atrophicae, pes planus and a fifth finger joint contracture. She presented with similar facial features as her daughter, including dolichocephaly, enophthalmos, and malar hypoplasia.
DISCUSSION
FBN1 missense mutations in exons 41 and 42 cause autosomal dominantly inherited forms of AD and GD[Le et al., 2011]. Here we describe that missense mutations in these exons can also cause MFS and WMS. Exons 41 and 42 encode the fifth of seven domains in fibrillin-1 that contain eight cysteines and have homology to a domain found in latent TGF-β binding proteins, thus termed TGF-β binding-protein-like domain (TB5). The mutations reported here are both novel mutations that disrupt cysteines in this domain and therefore are predicted to disrupt the structure of the domain. Patient 2 had classic features of MFS with ectopia lentis and Patient 1 had dominantly inherited WMS. Therefore, both individuals were expected to have FBN1 mutations. The entire FBN1 gene was sequenced in both cases, and these missense mutations were the only alterations identified. Patient 1′s family was not available to verify segregation of the mutation with WMS in the family. Patient 2′s mother carried the mutation and had several systemic features of MFS but did not have either a dilated aortic root or lens dislocation, possibly due to either reduced expressivity or somatic mosaicism for the mutation. It is notable that there are six mutations in exon 41 and 42 reported in the FBN1 mutation database [Faivre et al., 2007]; five nonsense mutations are predicted to lead to nonsense mediated decay of the transcript in MFS patients and one is a missense mutation, c.5162 G>A (p.C1721Y), but the clinical phenotype of this patient is not described. Therefore, we conclude that missense mutations in FBN1 exon 41 and 42 can cause both MFS and WMS.
Both AD and GD are characterized by severe short stature, short extremities, and stiff joints. Patients with GD have facial features that include full cheeks, shortened nose, hypertelorism, and thin upper lip vermillion, and die early from either cardiac valve thickening and dysfunction or tracheal narrowing leading to dyspnea or pulmonary infections [Le and Cormier-Daire, 2012]. Individuals with AD exhibit a milder phenotype without cardiac involvement or early death. Neither syndrome is reported to be associated with lens dislocation or thoracic aortic disease. It is notable that FBN1 missense mutations in exons 41 and 42 causing AD and GD include a subset that disrupts cysteines.
WMS similarly manifests as short stature, short fingers, and stiff joints, but these patients also develop ectopia lentis. Both autosomal dominant and recessive transmissions of WMS have been described. The autosomal recessive form is caused by mutations in the ADAMTS10 gene [Dagoneau et al., 2004] and the dominant form by FBN1 mutations [Faivre et al., 2003b], which suggests that ADAMTS10 and fibrillin-1 may interact in a common pathway [Kutz et al., 2011]. A 24 bp in-frame deletion in exon 41 of FBN1 in four individuals of a three-generation WMS family and a 7895 bp genomic deletion of FBN1 with boundaries in intron 8 and intron 11, leading to an in-frame putative deletion of 160 amino acids, have been reported in unrelated WMS patients [Faivre et al., 2003b; Kutz et al., 2011].
Aortic disease has not been described in patients with WMS. The most commonly reported echocardiographic abnormality is mitral valve prolapse and congenital valvular aortic stenosis [Kojuri et al., 2007]. Subvalvular fibromuscular aortic stenosis, mitral stenosis, and pulmonic valve stenosis have been described in WMS patients, findings that overlap the valvular thickening in AD and GD patients [Faivre et al., 2003a; Ferrier et al., 1980; Le et al., 2011; Megarbane et al., 2000; van de Woestijne et al., 2004]. A review of the cardiovascular findings in six patients with WMS found prolonged QTc interval in three patients [Kojuri et al., 2007] and Patient 1 reported here also has prolonged QTc intervals.
Although the proband with WMS has features overlapping AD and GD, including short stature, joint limitations, facial features, and stiff skin, WMS may be a more appropriate clinical diagnosis, given the ectopia lentis and the absence of cardiac valvular thickening, tracheal narrowing, and bronchopulmonary infections. According to the revised Ghent nosology for Marfan syndrome, Patient 1 meets criteria for MFS due to the findings of aortic dilation/dissection, ectopia lentis, and an FBN1 mutation [Loeys et al., 2010]. Interestingly, the dislocation of the lens in this family was in the superior temporal direction, similar to what is observed in MFS, rather than inferior nasal dislocation typical of WMS [Evereklioglu et al., 1999; Maumenee, 1981]. However, the revised nosology emphasizes the importance of additional diagnostic considerations, particularly if they segregate with disease in the family or if they are suggestive of a specific alternative diagnosis. As an illustration of the overlap of phenotypes due to probable FBN1 mutations, there is a report of a large, multi-generation family with classic MFS in which one family member had ectopia lentis and skeletal features consistent with WMS rather than MFS [Bowers, 1959].
In summary, the clinical phenotypes of MFS, WMS, AD, and GD are the spectrum that result from missense mutations in exons 41 and 42 of FBN1. Based on the aortic dissection in our WMS Patient 1, we recommend that all patients with pathogenic FBN1 mutations in these exons undergo cardiovascular assessment for both congenital valvular disease and thoracic aortic enlargement. The frequency of aortic imaging should be tailored individually until more information on prevalence and outcomes is available, but regular monitoring of these patients with thoracic aortic disease risk factors, such as systemic hypertension or pregnancy in women, is warranted. As with other populations with short stature and a predisposition to aortic disease, such as individuals with Turner syndrome, body surface area must be considered in both diagnosis and treatment of aortic disease. Ultimately, prevention of untimely death from thoracic aortic dissections depends on the early identification of individuals predisposed to thoracic aortic disease, careful monitoring of the diameter of the thoracic ascending aorta, and timely surgical repair of diseased segments [Finkbohner et al., 1995]. The two cases reported here also suggest that individuals with AD and GD have an increased risk to develop aortic disease and ectopia lentis due to the underlying FBN1 mutation and should be assessed for these conditions. Finally, these cases illustrate the overlap between syndromes resulting from FBN1 mutations.
ACKNOWLEDGMENTS
We are grateful to the patients and their families for participating in this study. The following sources provided resources or funding for these studies: RO1 HL62594 (D.M.M.), UL1 RR024148 (CTSA), Vivian L. Smith Foundation (D.M.M.), and the Richard T. Pasani Funds (D.M.M.). Dr. Lewis is a Senior Scientific Investigator of Research to Prevent Blindness, New York, that provided unrestricted funds for part of this study.
REFERENCES
- Ades LC, Holman KJ, Brett MS, Edwards MJ, Bennetts B. Ectopia lentis phenotypes and the FBN1 gene. Am J Med Genet A. 2004;126:284–289. doi: 10.1002/ajmg.a.20605. [DOI] [PubMed] [Google Scholar]
- Bowers D. Marfan’s syndrome and the Weill-Marchesani syndrome in the S. family. Ann Intern Med. 1959;51:1049–1070. doi: 10.7326/0003-4819-51-5-1049. [DOI] [PubMed] [Google Scholar]
- Dagoneau N, oist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, Cormier-Daire V. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75:801–806. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evereklioglu C, Hepsen IF, Hamdi ER. Weill-Marchesani syndrome in three generations. Eye (Lond) 1999;13(Pt 6):773–777. doi: 10.1038/eye.1999.226. [DOI] [PubMed] [Google Scholar]
- Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, rslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Ades LC, Biggin A, Benetts B, Brett M, Holman KJ, De BJ, Coucke P, Francke U, De PA, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Dollfus H, Lyonnet S, Alembik Y, Megarbane A, Samples J, Gorlin RJ, Alswaid A, Feingold J, Le MM, Munnich A, Cormier-Daire V. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet A. 2003a;123A:204–207. doi: 10.1002/ajmg.a.20289. [DOI] [PubMed] [Google Scholar]
- Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, Le Merrer M, Collod-Beroud G, Boileau C, Munnich A, Cormier-Daire V. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003b;40:34–36. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Le MM, Baumann C, Polak M, Chatelain P, Sulmont V, Cousin J, Bost M, Cordier MP, Zackai E, Russell K, Finidori G, Pouliquen JC, Munnich A, Maroteaux P, Cormier-Daire V. Acromicric dysplasia: long term outcome and evidence of autosomal dominant inheritance. J Med Genet. 2001;38:745–749. doi: 10.1136/jmg.38.11.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier S, Nussle D, Friedli B, Ferrier PE. [Marchesani’s syndrome (spherophakia-brachymorphism)] Helv Paediatr Acta. 1980;35:185–198. [PubMed] [Google Scholar]
- Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM. Marfan syndrome. Long-term survival and complications after aortic aneurysm repair. Circulation. 1995;91:728–733. doi: 10.1161/01.cir.91.3.728. [DOI] [PubMed] [Google Scholar]
- Kojuri J, Razeghinejad MR, Aslani A. Cardiac findings in Weill-Marchesani syndrome. Am J Med Genet A. 2007;143A:2062–2064. doi: 10.1002/ajmg.a.31861. [DOI] [PubMed] [Google Scholar]
- Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, Sakai LY, Keene DR, Apte SS. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem. 2011;286:17156–17167. doi: 10.1074/jbc.M111.231571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le GC, Cormier-Daire V. From tall to short: the role of TGFbeta signaling in growth and its disorders. Am J Med Genet C Semin Med Genet. 2012;160C:145–153. doi: 10.1002/ajmg.c.31337. [DOI] [PubMed] [Google Scholar]
- Le GC, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, Zylberberg L, Collod-Beroud G, Bonnet D, Alanay Y, Brady AF, Cordier MP, Devriendt K, Genevieve D, Kiper PO, Kitoh H, Krakow D, Lynch SA, Le MM, Megarbane A, Mortier G, Odent S, Polak M, Rohrbach M, Sillence D, Stolte-Dijkstra I, Superti-Furga A, Rimoin DL, Topouchian V, Unger S, Zabel B, Bole-Feysot C, Nitschke P, Handford P, Casanova JL, Boileau C, Apte SS, Munnich A, Cormier-Daire V. Mutations in the TGFbeta binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. doi: 10.1016/j.ajhg.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De BJ, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- Maroteaux P, Stanescu R, Stanescu V, Rappaport R. Acromicric dysplasia. Am J Med Genet. 1986;24:447–459. doi: 10.1002/ajmg.1320240307. [DOI] [PubMed] [Google Scholar]
- Maumenee IH. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc. 1981;79:684–733. [PMC free article] [PubMed] [Google Scholar]
- Megarbane A, Mustapha M, Bleik J, Waked N, Delague V, Loiselet J. Exclusion of chromosome 15q21.1 in autosomal-recessive Weill-Marchesani syndrome in an inbred Lebanese family. Clin Genet. 2000;58:473–478. doi: 10.1034/j.1399-0004.2000.580608.x. [DOI] [PubMed] [Google Scholar]
- Milewicz DM, Michael K, Fisher N, Coselli JS, Markello T, Biddinger A. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation. 1996;94:2708–2711. doi: 10.1161/01.cir.94.11.2708. [DOI] [PubMed] [Google Scholar]
- Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- van de Woestijne PC, rk-Jan Ten HA, Bogers AJ. Two patients with Weill-Marchesani syndrome and mitral stenosis. Interact Cardiovasc Thorac Surg. 2004;3:484–485. doi: 10.1016/j.icvts.2004.04.004. [DOI] [PubMed] [Google Scholar]