

Difluoromethyl ethers are found increasingly in pharmaceuticals, agrochemicals, and materials.[1] Aryl difluoromethyl ethers are found in medicinally important compounds that include enzyme inhibitors,[2] anti-HIV agents[3] and atimicrobial agents.[4] Pantoprazole (Protonix®), a proton-pump inhibitor, is among the top 100 pharmaceuticals and contains a difluoromethyl ether.[5]
However, current syntheses of difluoromethyl ethers require the ozone-depleting compound HCF2Cl (Freon 22) that is difficult to handle because it is a gas (Scheme 1).[6] Non-ozone-depleting sources have been reported for the formation of difluoromethyl ethers from phenols,[7] but the reactions with these reagents often require high-temperatures, long reaction times, and have only been demonstrated to work with simple substrates.
Scheme 1.

Conventional synthesis of difluoromethyl ethers.
We report a procedure for the difluoromethylation of phenols and thiophenols that occurs with broad scope starting with difluoromethyltriflate (HCF2OTf), a non-ozone-depleting liquid. The fast rates, tolerance for additional functionality and tolerance of byproducts formed by prior reactions made possible the development of one-pot protocols for the conversion of aryl halides, aryl boronic acids, and even arenes, to difluoromethyl ethers.
Difluoromethyltriflate is an attractive source of a difluoromethyl unit because it can be prepared in multi-gram scale from readily available, non-ozone-depleting reagents. The reaction between TMSCF3 (the Ruppert-Prakash reagent) and triflic acid with catalytic TiCl4 at room temperature provides difluoromethyltriflate (HCF2OTf) in good yield (eq. 1).[8] HCF2OTf is an air-stable liquid which makes handling the reagent easier than gaseous HCF2Cl.
| (1) |
Reaction conditions were examined for the difluoromethylation of 4-butylphenol with HCF2OTf (Table 1). Initial results showed that reactions conducted with aqueous base occurred in significantly higher yields than reactions conducted under anhydrous conditions. Reactions conducted with aqueous KOH formed the desired product in higher yield than those conducted with LiOH or NaOH (Table 1, entries 12 and 13). Reactions with MeCN were found to give higher yields than those in other co-solvents examined. Under the reaction conditions shown in Table 1, the difluoromethylation of 4-butylphenol was complete within minutes at room temperature and formed minimal side-products. The reactions are trivial to perform; they simply involve the addition of HCF2OTf to a solution of phenol in 1:1 MeCN/6M KOH at ambient temperature (entry 6).
Table 1.
Screen of reaction conditions for the difluoromethylation of 4-butylphenol with HCF2OTf and KOH.

| ||||
|---|---|---|---|---|
|
| ||||
| Entry | KOH (equiv) | Co-Solvent | ArOCF2H (%) | ArOTf (%) |
| 1 | 12 | DMF | 43 | 34 |
| 2 | 12 | DMSO | 59 | 5 |
| 3 | 12 | Dioxane | 54 | 16 |
| 4 | 12 | THF | 62 | 7 |
| 5 | 12 | Water | 5 | 2 |
|
| ||||
| 6 | 12 | MeCN | 75 | 12 |
|
| ||||
| 7 | 8 | MeCN | 59 | 6 |
| 8 | 10 | MeCN | 70 | 19 |
| 9 | 16 | MeCN | 54 | 11 |
| 10 | 20 | MeCN | 61 | 7 |
| 11 | 24 | MeCN | 56 | 7 |
| 12 | 12 | MeCN | 38 | 11[b] |
| 13 | 12 | MeCN | 61 | 10[c] |
Reactions were performed on a 0.1 mmol scale and the yields were determined by GC with 1-bromo-4-fluorobenzene as an internal standard
The reaction was performed with LiOH in place of KOH
The reaction was performed with NaOH in place of KOH.
The reaction conditions identified for the difluoromethylation of 4-butylphenol (Table 1, entry 6) were evaluated for the synthesis of difluoromethyl ethers from a range of phenols (Table 2). Electron-rich, electron-deficient and sterically hindered phenols reacted under the standard conditions. The short reaction time and mild conditions were tolerant of esters, amides, ketones, acetals, nitriles, aldehydes, aryl halides, and heterocycles. In each reaction, the only by-products observed were unreacted phenol and varying amounts of aryl-triflate.[9] Stable enols(1s, 1t)also underwent the difluoromethylation reaction in high yield. Capsaicin and estrone reacted to form the difluoromethyl ethers 2w and 2x in modest yield. The same reaction conditions for the difluoromethylation of phenols also led to difluoromethylsulfides 2u and 2v from the corresponding thiophenols. The substrate scope and generality demonstrated here is unrivaled for the synthesis of difluoromethyl ethers.
Table 2.
Difluoromethylation of phenols with HCF2OTf.

|
Reactions were performed on a 0.1 mmol scale to determine yields by 19F NMR spectroscopy with PhCF3 as an internal standard. Isolated yields are shown in parenthesis for reactions performed on a 0.5 mmol scale.
Reactions were performed on a 0.1 mmol scale with HCF2ONf in place of HCF2OTf and yields were determined by 19F NMR spectroscopy.
The aryl difluoromethyl ether products are stable and were isolated by silica gel chromatography. Difluoromethyl ethers 2a, 2f, 2g, and a 2p, which contain an electron-withdrawing group were obtained in analytically pure form after an aqueous workup. Isolated yields of the reactions performed with 0.5 mmol of substrate were comparable to the yields determined by 19F NMR spectroscopy for reactions performed on a 0.1 mmol scale. The volatility of some products prevented their isolation in high yield, and the yields determined by 19F NMR spectroscopy are given in those cases.
The aryl-triflate side products are most prevalent in the reactions of phenol substrates bearing electron-donating groups. It was proposed that nucleophilic attack at the sulfur atom of HCF2OTf would be inhibited by the use of a bulkier sulfonate group. Thus, we prepared difluoromethylnonaflate (HCF2ONf) according to the literature procedure by the reaction of nonafluorobutanesulfonic acid (NfOH) with TMSCF3 (eq 1).[8] Phenols 1c, 1e, 1i, and 1x, which formed significant amounts of ArOTf in the reactions with HCF2OTf, gave measurably higher yields of the difluoromethyl ethers when HCF2ONf was used as the difluoromethyl source (Table 2). It is important to note that the aryl triflate from reactions with HCF2OTf can be recycled to the starting phenol by basic hydrolysis (eq 2).[10]
 |
(2) |
Because these reactions occur rapidly under mild conditions, we considered that they would tolerate byproducts from the synthesis of phenols. If so, then the difluoromethylation could be used in combination with several processes that form phenols from common precursors, such as arylboronates, aryl halides and arenes themselves.
Phenols can be prepared by oxidation of aryl boronic acids with aqueous hydrogen peroxide. We proposed that aryl boronic acids could be transformed to aryl difluoromethyl ethers by first forming the phenol in-situ. Indeed, the reaction between arylboronic acids in MeCN with 30% aqueous H2O2 for 15 minutes, followed by the addition of KOH and HCF2OTf provided difluoromethyl ethers from aryl boronic acids (Table 3). The two-step, one-pot reaction sequence was tolerant of ketones, aldehydes and amides.[11]
Table 3.
One-pot difluoromethoxylation of arylboronic acids.
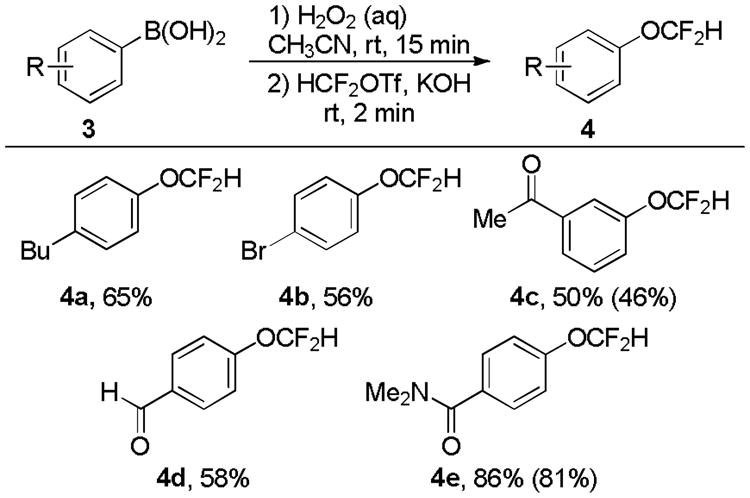
|
Reactions were performed on a 0.1 mmol scale to determine yields by 19F NMR spectroscopy with PhCF3 as an internal standard added after the reaction. Isolated yields are shown in parenthesis for reactions performed on a 0.5 mmol scale.
Phenols can also be prepared by the hydroxylation of aryl halides catalyzed by transition-metal complexes. We envisioned a two-step sequence for the conversion of aryl halides to difluoromethoxyarenes based on the palladium-catalyzed conversion of aryl halides to phenols and in-situ conversion of the resulting phenoxides with HCF2OTf. Indeed, we found that the phenols formed in the Pd-catalyzed hydroxylation were readily transformed into difluoromethyl ethers (Table 4).[12] The phenols formed in the first step were used without purification. Dilution of the crude reaction with MeCN and additional aqueous KOH, and treatment of this solution with HCF2OTf, gave the difluoromethyl ether products in good yield. Aryl bromides and aryl chlorides both underwent the two step process in good yield.[13]
Table 4.
One-pot difluoromethoxylation of aryl halides.

|
Reactions were performed on a 0.5 mmol scale to determine yields by 19F NMR spectroscopy with PhCF3 as an internal standard added after the reaction.
Finally, we considered that a one-pot route could be developed for the conversion of arenes to aryl difluoromethyl ethers by sequential C-H borylation, oxidation, and difluoromethylation.Tandem reactions involving initial C-H borylation are useful for preparing diversely functionalized arenes,[14] due in part to the high selectivity of the borylation reaction for the least-hindered C-H bond.[15]The results for the overall conversion of Ar-H to Ar-OCF2H are shown in Table 5. Synthetically useful yields of the difluoromethyl ether were obtained with substrates containing amides, ketones and aryl halides. The aryl boronate esters formed in the first step were used without purification. However, a change in solvent from THF to MeCN was necessary after the borylation reaction. Thus, the three-step sequence reported here provides an unusual conversion of arenes to 3,5-disubsituted aryl difluoromethyl ethers.
Table 5.
One-pot difluoromethoxylation of arenes through Ir-catalyzed C-H borylation.

|
Reactions were performed on a 0.1 mmol scale to determine yields by 19F NMR spectroscopy with PhCF3 as an internal standard added after the reaction. Isolated yields are shown in parenthesis for reactions performed on a 0.5 mmol scale.
The mechanism of the reactions of phenols with HCF2OTf was studied experimentally. All reactions that have been reported for the difluoromethylation of phenols are proposed to occur through initial formation of difluorocarbene.[16] To determine if the reaction of phenols with HCF2OTf proceeds through the formation of difluorocarbene or by nucleophilic displacement of the triflate of HCF2OTf by phenol, we performed reactions with D2O. If the reaction with phenol occurs by nucleophilic displacement of triflate, than the unlabeled product ArOCF2H would be expected to form. However, if the reaction proceeds by nucleophilic addition to difluorocarbene, then the deuterium-labeled product ArOCF2D would be expected to form by protonation of the intermediate ArOCF2[−] with D2O. These labeling experiments reflect the reaction pathway because no H-D exchange occurs to generate DCF2OTf in the presence of D2O and KOH, and the difluoromethyl ether product does not undergo H-D exchange under the reaction conditions. In the event, the reaction with D2O gave 91% incorporation of deuterium at the difluoromethyl group of the ether (eq 3).[17]
 |
(3) |
We further evaluated whether difluorocarbene is formed under the reaction conditions by conducting the reaction of HCF2OTf with an alkene under the same conditions as the difluoromethylation of phenol. The reaction of tetramethylethylene with HCF2OTf and KOH in CH3CN/H2O provided the difluorocyclopropane product in 22% yield, as determined by 19F NMR spectroscopy (eq 4). The difluorocarbene formed undergoes competing hydrolysis with water to form formate and fluoride ions, which accounts for the low yield of the difluorocyclopropane. Nevertheless, the observation of the cyclopropane further supports the formation of difluorocarbene from the reaction of KOH with HCF2OTf. These results are consistent with a mechanism for the formation of a difluoromethyl ether by reaction of the phenol (or phenolate) with difluorocarbene, not by nucleophilic displacement of the triflate of HCF2OTf by phenoxide.
 |
(4) |
In summary, we have developed a simple method for the difluoromethylation of phenols and thiophenols with a readily available and non-ozone-depleting liquid reagent, HCF2OTf. This method allows difluoromethyl ethers and sulfides to be prepared within minutes at room temperature in aqueous solvent. The broad functional group tolerance and mild conditions of this reaction make possible the difluoromethylation of a wide range of complex phenols, including phenols generated in-situ by a series of catalytic and oxidation processes. One-pot procedures have been developed for the difluoromethyoxylation of aryl boronic acids, aryl halides and arenes. The direct conversion of arenes, boronic acids, and aryl halides to difluoromethoxyarenes has been challenging, in part, because of the instability of the [-]OCF2H anion.[18] A series of mechanistic studies show that the difluoromethylation of phenols reported here proceeds through initial formation of difluorocarbene and subsequent nucleophilic addition of the phenolate or thiophenolate anion to difluorocarbene.
Supplementary Material
Scheme 2.

Proposed mechanism for the reaction of phenols with HCF2OTf.
Footnotes
The authors thank the NIH-NIGMS (R29-55382) for support of this work
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Hiyama T. Organofluorine Compounds : Chemistry and Applications. Springer; Berlin; New York: 2000. [Google Scholar]; b) Kirsch P. Modern Fluoroorganic Chemistry : Synthesis, Reactivity, Applications. Wiley-VCH; Weinheim; Great Britain: 2004. [Google Scholar]; c) Hu JB, Zhang W, Wang F. Chem Commun. 2009:7465–7478. doi: 10.1039/b916463d. [DOI] [PubMed] [Google Scholar]; d) Kirsch P, Bremer M. Angew Chem Int Ed. 2000;39:4217–4235. doi: 10.1002/1521-3773(20001201)39:23<4216::AID-ANIE4216>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; e) Bégué JP, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. John Wiley & Sons; Hoboken, N.J.: 2008. [Google Scholar]
- 2.Chauret N, Guay D, Li C, Day S, Silva J, Blouin M, Ducharme Y, Yergey JA, Nicoll-Griffith DA. Bioorg Med Chem Lett. 2002;12:2149–2152. doi: 10.1016/s0960-894x(02)00349-9. [DOI] [PubMed] [Google Scholar]
- 3.Ohmine T, Katsube T, Tsuzaki Y, Kazui M, Kobayashi N, Komai T, Hagihara M, Nishigaki T, Iwamoto A, Kimura T, Kashiwase H, Yamashita M. Bioorg Med Chem Lett. 2002;12:739–742. doi: 10.1016/s0960-894x(02)00003-3. [DOI] [PubMed] [Google Scholar]
- 4.Takahata M, Mitsuyama J, Yamashiro Y, Yonezawa M, Araki H, Todo Y, Minami S, Watanabe Y, Narita H. Antimicrobial Agents and Chemotherapy. 1999;43:1077–1084. doi: 10.1128/aac.43.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheer SM, Prakash A, Faulds D, Lamb HM. Drugs. 2003;63:101–132. doi: 10.2165/00003495-200363010-00006. [DOI] [PubMed] [Google Scholar]
- 6.Miller TG, Thanassi JW. J Org Chem. 1960;25:2009–2012. [Google Scholar]
- 7.a) Zafrani Y, Sod-Moriah G, Segall Y. Tetrahedron. 2009;65:5278–5283. [Google Scholar]; b) Zhang LJ, Zheng J, Hu JB. J Org Chem. 2006;71:9845–9848. doi: 10.1021/jo061799l. [DOI] [PubMed] [Google Scholar]; c) Zheng J, Li Y, Zhang LJ, Hu JB, Meuzelaar GJ, Federsel HJ. Chem Commun. 2007:5149–5151. doi: 10.1039/b713156a. [DOI] [PubMed] [Google Scholar]; d) Chen QY, Wu SW. J Fluorine Chem. 1989;44:433–440. [Google Scholar]; e) Chen QY, Wu SW. J Org Chem. 1989;54:3023–3027. [Google Scholar]
- 8.Levin VV, Dilman AD, Belyakov PA, Struchkova MI, Tartakovsky VA. J Fluor Chem. 2009;130:667–670. [Google Scholar]
- 9.The aryl triflate is formed by nucleophilic attack of the phenoxide on the sulfur atom of HCF2OTf.
- 10.Ohgiya T, Nishiyama S. Tet Lett. 2004;45:6317–6320. [Google Scholar]
- 11.The yield of the difluoromethylation step of this squence was slightly lower than that of the separate reaction of a phenol due to the presence of boric acid.
- 12.Anderson KW, Ikawa T, Tundel RE, Buchwald SL. J Am Chem Soc. 2006;128:10694–10695. doi: 10.1021/ja0639719. [DOI] [PubMed] [Google Scholar]
- 13.The yield of the difluoromethylation step of this squence was slightly lower than that of the separate reaction of a phenol due to the presence of dioxane.
- 14.Hartwig JF. Acc Chem Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 15.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 16.Hine J, Porter JJ. J Am Chem Soc. 1957;79:5493–5496. [Google Scholar]
- 17.The protons in KOH, ArOH, and HCF2OTf limit the maximum deuterium incorporation to 93% based on the amount of O-H and O-D bonds in the reaction.
- 18.Difluoromethoxide ion undergoes rapid elimination to formyl fluoride (CHOF) and fluoride ion. See references 1a and 1b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.