

Abstract
A relationship between status epilepticus (SE) and oxidative stress has recently begun to be recognized. To explore whether the flavonoids extracted from licorice (LFs) have any protective effect on kainate (KA)-induced seizure in mice, we treated mice with LFs before and after KA injection. In KA-treated mice, we found that superoxide dismutase (SOD) activity decreased immediately after the onset of seizure at 1 h and then increased at 6 h. It returned to baseline 1 d after seizure and then increased again at 3, 7, and 28 d, while malondialdehyde (MDA) content remained at a high level at 1 h, 6 h, 3 d, 7 d, and 28 d, indicating a more oxidized status related to the presence of more reactive oxygen species (ROS). Treatment with LFs before KA injection reversed the seizure-induced change in SOD activity and MDA content at 1 h, 6 h, 3 d, 7 d, and 28 d. Treatment with LFs after seizure decreased KA-induced SOD activity and MDA content at 7 and 28 d. Also, LF pre- and post-KA treatments decreased seizure-induced neuronal cell death. Subsequently, Morris water maze tests revealed that the escape latency was significantly decreased and the number of target quadrant crossings was markedly increased in the LF-treated groups. Thus, our data indicate that LFs have protective effects on seizure-induced neuronal cell death and cognitive impairment through their anti-oxidative effects.
Keywords: Seizure, Kainate, Flavonoid, Licorice, Antioxidant, Malondialdehyde (MDA), Superoxide dismutase (SOD)
1. Introduction
Epilepsy, one of the most common neurological disorders, is characterized by recurrent highly synchronized discharges of neurons and leads to mortality and morbidity in patients. Most available treatments, however, suppress seizure only symptomatically, due to the lack of a clear understanding of the underlying mechanisms. Most patients have to take antiepileptic drugs (AEDs) for their whole life. These produce severe side effects such as cognitive impairment and psychiatric problems. Accordingly, new AEDs with fewer adverse effects and higher efficacy are needed (Temkin, 2001; Löscher and Schmidt, 2006; Elger and Schmidt, 2008).
A relationship between status epilepticus (SE) and oxidative stress has recently begun to be recognized both in animal models (Ashrafi et al., 2007; Shin et al., 2008; Lehtinen et al., 2009) and in patients (Ben-Menachem et al., 2000; Shiihara et al., 2006). It has been established that blood flow, energy, and oxygen are increased during seizure and that SE induces the production of redundant reactive oxygen species (ROS). Compared with other organs, the brain uses the highest amount of oxygen and contains a high concentration of polyunsaturated fatty acids that are easily peroxidated, which makes it particularly susceptible to oxidative stress (Floyd and Carney, 1992). Studies have revealed that prolonged seizure activity initiates calcium influx via voltage-gated and N-methyl-D-aspartate-dependent ion channels and subsequently triggers biochemical cascades, which result in neuronal cell death and the production of ROS (Bruce and Baudry, 1995; Frantseva et al., 2000). Similarly, increased oxidative stress contributes to seizure-induced brain injury and subsequently results in epilepsy. In turn, ROS may be a contributing factor in the generation of epileptic seizures in animal models (Ashrafi et al., 2007; Aguiar et al., 2012; Ryan et al., 2012) and in patients (Shiihara et al., 2006; Martinc et al., 2012). In various experimental studies, it has been demonstrated that SE induces changes in superoxide dismutase (SOD) and malondialdehyde (MDA) levels, as well as in mitochondrial DNA. Treatments with antioxidants such as curcumin, baicalin, and Capparis ovata prevent the excitotoxicity in animal models (Tejada et al., 2007; Golechha et al., 2011; Nazıroğlu et al., 2013). Thus, antioxidants may have a potential role in preventing excitotoxicity-induced brain damage and possible epileptogenesis.
Licorice root has been widely used in China as herbal medicine and food. Flavonoids, such as liquiritin, glabridin, and isoliquiritigenin, the major bioactive components of licorice root, have shown various antioxidant, anti-tumor, and antivirus biochemical activities (Sun et al., 2010; Asha et al., 2013; Wang et al., 2013). However, the effect of licorice flavonoids (LFs) on seizures has not been studied. Therefore, in this study, the protective effect of LFs in a mice seizure model induced by kainate (KA) was studied by examining their effects on SOD, MDA, neuronal cell death, and cognitive impairment.
2. Materials and methods
2.1. Reagents
Fluoro-Jade B (FJB) was purchased from Sigma (St. Louis, MO, USA). KA was obtained from Nanocs Inc. (New York, NY, USA). SOD and MDA detection kits were provided by Jiancheng Bioengineering Institute of Nanjing (Jiangsu, China).
2.2. Preparation of LFs
The preparation of LFs was conducted as previously reported (Xie et al., 2009). Briefly, the air-dried roots of Glycyrrhiza uralensis were crushed and extracted twice with H2O. The water extract was further extracted by methanol and chromatographed on silica gel followed by C18 column chromatography. The final contents of liquiritin, liquiritin apioside, and liquiritigenin were 3.81%, 1.38%, and 0.52%, respectively.
2.3. Animal model and drug treatment
The animal protocol described here is compliant with the guidelines of the Animal Care and Use Committee of Zhejiang University. Male Imprinting Control Region (ICR) mice of six weeks of age were purchased from Shanghai Slac Laboratory Animal Corporation (certificate: SCXK 2007-0005) and were kept at (24±1) °C with 40%–60% humidity and 12 h: 12 h light:dark. Mice were injected intraperitoneally once with KA at a dose of 25 mg/kg body weight (BW) to induce SE. Seizure severity was scored as previously reported (Zeng et al., 2009). Briefly, category 1=immobility and facial twitch; category 2=head nodding; category 3=forelimb clonus; category 4=rearing; and category 5=rearing and falling. The onset of SE was defined as the beginning of stages 4–5 seizure. If the animals did not develop stage 4 or 5 seizure, they were not used for further experiment.
The paradigm for LF treatment is shown in Fig. 1. For pretreatment, LFs were administered intragastrically at a dose of 10 mg/kg BW once a day for 7 d prior to KA injection. For post-treatment, LFs were administered once a day 24 h after KA-induced SE and continued for 7 d. The control group received the same volume of vehicle simultaneously (n=12–15 mice/group).
Fig. 1.
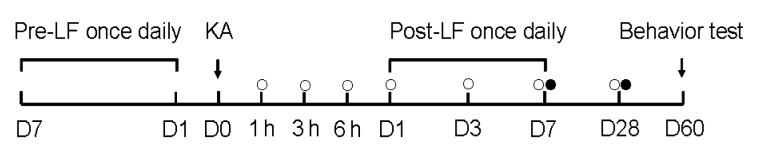
Paradigm of animal treatment and drug administration
○: the time points for analyses of SOD and MDA in the pre-LF treatment; ●: the time points for analyses of SOD and MDA in the post-LF treatment. D0–D60: Day 0–Day 60
2.4. FJB staining
FJB staining was performed to detect neuronal cell death as described previously (Zeng et al., 2009). Briefly, mice sacrificed with 10% chloral hydrate (1 g/kg BW, i.p.) were intracardially perfused with 0.1 mol/L phosphate buffered saline (PBS) followed by 0.04 g/ml paraformaldehyde (PFA) 7 d after SE. The brains were fixed in 4% PFA overnight followed by 0.3 g/ml sucrose solution. Coronary sections of 50 μm thickness were cut using a vibratome (VT 1000S, Leika, Nussloch, Germany). Five sections selected from a one-in-six series were collected from each animal at the same level of the hippocampus, starting at 2.8 mm posterior to bregma. Floating sections were mounted on gelatin-coated slides and dried at room temperature. Sections were re-hydrated in 0.01 g/ml NaOH/80% ethanol for 5 min, and then sequentially in 70% ethanol, 50% ethanol, and distilled water for 2 min. After incubation in 0.6 g/L potassium permanganate solution for 10 min, the sections were rinsed in distilled water and stained with 0.0004% FJB in 0.1% acetate for 20 min in the dark. Images of 920 μm×920 μm fields were acquired using a Carl Zeiss LSM PASCAL confocal microscope. The number of FJB-positive cells per image field in the hippocampal CA1, CA3, and hilus was counted in each of the five sections per animal and the counts averaged (n=8–10 mice/group).
2.5. Detections of SOD activity and MDA content in the brain
Mice were sacrificed at pre-determined time points and hippocampal and neocortical tissues were homogenized in ice-cold saline 1:10 (w:v). Homogenate was then centrifuged at 3 000 r/min for 15 min at 4 °C to obtain the supernatant. Protein concentrations were measured by using BCA (bicinchoninic acid, Pierce, Bradford, IL, USA). Determination of SOD activity (WST-1 Assay kit) was performed immediately according to the manufacturer’s instructions and detected by a spectrometer (TU1901, Purkinje General Limited Corporation, Beijing, China) at 450 nm. One unit of enzyme activity was defined as the quantity of SOD required to inhibit the rate of reduction by 50%. SOD activity is presented as U/mg protein. The content of MDA was measured with a modified thiobarbituric acid (TBA) test as described in the manufacturer’s instructions, and detected by a spectrometer (Thermo Scientific, Waltham, MA, USA) at 532 nm. The results are expressed as nmol MDA/mg protein.
2.6. Morris water maze test
Two months after video monitoring, mice were tested for spatial learning and memory using a Morris water maze test as described previously (Chen et al., 2013). A platform of 10 cm diameter was submerged 1 cm beneath the surface and located at a fixed position throughout the training period. The swimming activity of each mouse was monitored using a video camera (Sony, Tokyo, Japan) mounted overhead and automatically recorded via a video tracking system. Mice were trained for four consecutive days before testing. During the four training days, the mice were first placed on the platform for 10 s, and then randomly placed in one of four different quadrants of the water tank. Recording was stopped 10 s after the mice reached the platform. Mice were directed to the platform if they did not find it within 60 s, and they stayed there for 10 s. Each mouse was given four trials per day and each trial was separated by 1 h. On the fifth day, each mouse was placed in the quadrant diagonally opposite from the previous platform location and the time of reaching the platform was recorded as escape latency. Swimming distance, swimming speed, and the number of the target quadrants were also analyzed (n=13‒20 mice/group).
2.7. Statistics
Results are presented as mean±standard error of the mean (SEM). Differences among experimental groups were compared by one-way analysis of variance (ANOVA) using SPSS software followed by Student-Newman-Keuls test for post-hoc comparisons (Version 16.0, SPSS Inc., Chicago, IL, USA). P<0.05 was considered significant.
3. Results
3.1. KA-induced SE resulted in increased SOD activity and MDA content
Systemic KA administration to ICR mice produced a clear sequence of behavioral changes. Mice started repetitive head nodding about 10 min after KA injection, followed by rearing and falling 30 min later. Eventually, the mice developed generalized tonic-clonic seizures lasting for 3–6 h.
The activity of one of the most important antioxidant enzymes, SOD, had decreased 1 h after KA-induced acute seizure (Fig. 2a). However, SOD activity had increased by 3-fold at 6 h and returned to baseline at 1 d. During the chronic period after the acute seizures stopped, SOD activity increased again at 3, 7, and 28 d, indicating a more oxidized status over one month.
Fig. 2.

Effect of LF pretreatment on KA-induced changes in SOD activity (a) and MDA content (b)
Mice were treated with vehicle or LF for 7 d, and then injected with saline or 25 mg/kg KA to induce seizure. Mice were sacrificed at different time points and SOD activity (a) and MDA content (b) were analyzed in brain homogenate. Data are expressed as mean±SEM (n=12‒15 mice/group). * P<0.05 compared with the control group at the same time point; # P<0.05 compared with the KA-Veh group by one-way ANOVA. Cont: control group injected with vehicle and saline; KA-Veh: vehicle-treated KA group; KA-LF: LF-treated KA group
MDA content reflects the status of lipid peroxidation. Increased MDA content was first found 6 h after SE in the acute seizure period. Similar to the change in SOD activity, the content of MDA returned to baseline 1 d after SE. It increased 3 d after SE and remained high through to 28 d (Fig. 2b). These data suggest increased oxidant stress induced by seizure.
3.2. LF treatment reversed the changes in SOD activity and MDA content
Following the pretreatment, LF-treated mice showed normal SOD activity 1 h after KA-induced SE. Although LF-treated mice exhibited increased SOD activity at 6 h, 3 d, and 7 d compared to the control mice, SOD activity was markedly decreased compared to the vehicle-treated KA group (Fig. 2a). Similarly, MDA content was significantly decreased compared to the vehicle-treated KA group at 6 h, 3 d, 7 d, and 28 d. No difference in MDA content was observed between the control group and the LF-treated group at 7 and 28 d (Fig. 2b), indicating a robust antioxidant effect of LF on KA-induced SE.
Following post-treatment, we analyzed SOD activity and MDA content at 7 and 28 d. While SOD activity increased in the LF-treated KA group compared to control mice, it markedly decreased compared to the vehicle-treated KA group at 7 d. At 28 d after SE, no significant difference was observed between the control and LF-treated KA groups, whereas the vehicle-treated KA group exhibited higher SOD activity (Fig. 3a). Similar results were observed for MDA content (Fig. 3b).
Fig. 3.

Effect of LF post-treatment on KA-induced changes in SOD activity (a) and MDA content (b)
Mice were injected with saline or 25 mg/kg KA to induce seizure. Mice were treated with saline or LF 24 h after KA injection and once daily thereafter for 7 d and then sacrificed on Day 7 or 28. SOD activity (a) and MDA content (b) were analyzed in brain homogenate. Data are expressed as mean±SEM (n=12‒15 mice/group). * P<0.05 compared with the control group at the same time point; # P<0.05 compared with the KA-Veh group by one-way ANOVA. Cont: control group injected with vehicle and saline; KA-Veh: vehicle-treated KA group; KA-LF: LF-treated KA group
3.3. LF treatment protected against KA-induced cell death
KA seizure-induced neuronal cell death was detected by FJB staining. No obvious FJB positive cells were observed in control mice (Fig. 4a). However, FJB-positive cells were abundant in vehicle-treated seizure mice in hippocampal CA1, CA3, and hilus (Fig. 4b, CA1 is shown). Mice pre- (Fig. 4c) or post-treated (Fig. 4d) with LFs had significantly fewer FJB positive cells, indicating a neuronal protective effect of LFs.
Fig. 4.

Ameliorative effect of LF on KA seizure-induced neuronal death in hippocampal CA1
Mice were treated with vehicle (Veh) or LF before (LF-pre) or after (LF-post) KA injection for 3 d and sacrificed on Day 7 for FJB staining. (a‒d) Representative sections of FJB staining in hippocampus from mice of different groups; (e) Quantitative analysis of different groups. Data are expressed as mean±SEM (n=8‒10 mice/group). * P<0.05 compared to the control (Cont) group; # P<0.05 compared to the vehicle (Veh) group by one-way ANOVA
3.4. LF treatment protected against cognitive impairment
Cognitive impairment is a common consequence of KA-induced SE. Since LFs protected against seizure-induced cell death, we then assessed whether LFs had any protective effect against seizure-induced cognitive impairment. The vehicle-treated KA group had an obvious prolongation of escape latency (Fig. 5a) and a reduction in the number of crossings of the target quadrant (Fig. 5b) compared to control mice, indicating cognitive impairment after KA-induced seizure. However, mice pre-or post-treated with LFs showed the same escape latency and number of crossings of the target quadrant as the control mice, indicating that LF treatment reverses seizure-induced cognitive impairment (Figs. 5a and 5b). No change in swimming speed was observed among different groups (data not shown). LFs also showed a protective effect against seizure-induced cognitive impairment during training (Figs. 5c and 5d) on Day 3 and/or Day 4.
Fig. 5.

LF protection against KA seizure-induced cognitive impairment in Morris water maze test
Mice were treated with vehicle (Veh) or LF before (LF-pre) or after (LF-post) KA injection for 7 d and then trained for four consecutive days after SE. (a) Escape latency on Day 5 after training in all groups; (b) Number of crossings of the target quadrant on Day 5; (c) Mean time to reach the platform during four consecutive days; (d) Mean swimming length during four consecutive days. Data are expressed as mean±SEM (n=13–20 mice/group). * P<0.05 compared to the control (Cont) group; # P<0.05 compared to the vehicle (Veh) group by one-way ANOVA
4. Discussion
The main findings of the present study are: (1) KA-induced SE results in increased oxidative stress, acutely and chronically; (2) LF treatment partially reverses SE-induced oxidative stress; and (3) LF treatment ameliorates KA seizure-induced neuronal cell death and cognitive impairment.
4.1. KA-induced SE and oxidative stress
The KA-induced seizure model is widely used as a model of human temporal lobe epilepsy (Ben-Ari and Cossart, 2000). A single systemic administration of KA at a relatively high dose results in SE followed by spontaneous recurrent seizures in both rats (Zeng et al., 2009) and mice (Royle et al., 1999). As a structural analog of glutamate, KA activates excitatory amino acid receptors and triggers neuronal membrane depolarization, which results in the release of calcium ions and subsequently induces the formation of ROS. In turn, the increased ROS lead to impairment of mitochondrial respiratory chain function and damage to the cell structure, subsequently resulting in brain damage (Bruce and Baudry, 1995; Shin et al., 2011; Kovac et al., 2012). The brain has an array of antioxidant defense enzymes, such as SOD, catalase (CAT), and glutathione (GSH), to prevent it from over-oxidative damage. However, controversy remains about the change in endogenous antioxidant reagents after SE. It has been reported that the activities of SOD and CAT are increased 48 h and 5 d after KA-induced SE (Bruce and Baudry, 1995), respectively. In a pilocarpine model, SOD and CAT activities were increased at 2 h and 60 d after SE, respectively, indicating that endogenous antioxidant reagents are up-regulated due to increased oxidant radicals (Freitas, 2009). However, other reports have demonstrated no change or even a decrease in SOD activity (Bellissimo et al., 2001; Freitas et al., 2005). This discrepancy may result from differences in the animal models, severity of injury, selected time points and/or methods of detection. To reveal the details of the change in SOD activity after SE, we assessed the activity at multiple time points in the present study. SOD activity was obviously decreased immediately after SE (1 h), indicating increased oxidative stress and deprivation of SOD shortly after SE onset. SOD activity then dramatically increased both in the acute seizure (6 h) and chronic periods (3 to 28 d), which may represent over-compensation due to continuous oxidative stress. Correspondingly, MDA content was elevated at 3, 7, and 28 d after KA seizure. These results are slightly different from those reported previously (Bruce and Baudry, 1995).
4.2. LFs and antioxidant effects
Flavonoids are compounds found in numerous plants, fruits, and vegetables (Jäger and Saaby, 2011). They have been reported to have diverse bioactivities including anti-inflammatory, antioxidant, anti-tumor, and anti-viral effects. Licorice is a widely-used herb in Chinese medicine and contains several kinds of bioactive ingredient, such as glucose, glycyrrhizin, oleane triterpenoid, and flavonoids. Among them, LFs, such as liquiritigenin, isoliquiritigenin, and liquiritin, have recently been studied widely. Liquiritin has a potential effect against focal cerebral ischemia/reperfusion through its antioxidant and antiapoptotic properties (Sun et al., 2010). Zhan and Yang (2006) also demonstrated that isoliquiritigenin has protective potential against cerebral ischemia injury through the amelioration of cerebral energy metabolism and its antioxidant property. Several reports have indicated that flavonoids, such as naringin, baicalin, and vitexin, have neuroprotective effects on animal seizure models induced by KA, pilocarpine, or pentylenetetrazole (Golechha et al., 2011; Abbasi et al., 2012; Liu et al., 2012). In the present study, we found that pretreatment with LFs for 7 d significantly, albeit not completely, reversed the KA seizure-induced changes of SOD and MDA, indicating that LFs have a potential anti-oxidative effect. Similar results were also found in LF post-treated mice. These results are identical to those of our previous report (Xie et al., 2009), which demonstrated that LFs reduce neutrophil-mediated oxidative injury in lipopolysaccharide-induced acute pulmonary inflammation.
4.3. LFs and neuronal death
As one consequence of seizure, neuronal death is commonly seen in KA-induced SE. Although the precise molecular mechanisms of seizure-induced neuronal death remain unknown, oxidative stress and excessive glutamate receptor activation may exert sequential, as well as interacting effects on neuronal vulnerability. FJB staining has been used as a fluorescence marker for neuronal degeneration. We have detected a large number of FJB-positive neurons in the hippocampus 7 d after SE induced by KA in rats (Zeng et al., 2009). In the present study, we also found obvious neuronal death in the hippocampus after KA-induced SE in ICR mice. Moreover, treatment with LFs before or after KA-induced SE markedly inhibited the increase in FJB-positive neurons in the hippocampus. Since LFs had an antioxidant effect in this study, it seems that the neuroprotective effect of LFs is related to their antioxidative effect, at least to some extent.
4.4. LFs and cognitive impairment
In the present study, we observed that KA-induced SE was associated with cognitive impairment in mice as shown by a prolonged escape latency and decreased number of crossings of the right quadrant in Morris water maze tests. These results are identical to previous findings which demonstrated cognitive impairment after administration of chemoconvulsants (Royle et al., 1999). The cognitive impairment was partially reversed by LF treatment both before and after KA-induced SE, which may result from a protective effect of LFs on neuronal cell death.
5. Conclusions
In summary, our data suggest that LFs have a protective effect on KA-induced SE and may have potential for use in the prevention and treatment of seizure-induced brain injury. The protective action of LFs might at least partly be due to their antioxidant activity. Nevertheless, further studies are needed at the molecular level to gain a full understanding of the mechanisms underlying the effects of LFs during seizure.
Footnotes
Project supported by the Scientific Research Foundation for Returned Scholars of the Ministry of Education of China (2011), the Project of Experiment Animal Platform of Department of Science and Technology of Zhejiang Province (No. 2013C37026), and the Hangzhou Science and Technology Development Plan (No. 20100333T24), China
Compliance with ethics guidelines: Ling-hui ZENG, Hua-dan ZHANG, Cai-ju XU, Yu-jia BIAN, Xue-jiao XU, Qiang-min XIE, and Rong-hua ZHANG declare that they have no conflict of interest.
All institutional and national guidelines for the care and use of laboratory animals were followed.
References
- 1.Abbasi E, Nassiri-Asl M, Shafeei M, Sheikhi M. Neuroprotective effects of vitexin, a flavonoid, on pentylenetetrazole-induced seizure in rats. Chem Biol Drug Des. 2012;80(2):274–278. doi: 10.1111/j.1747-0285.2012.01400.x. [DOI] [PubMed] [Google Scholar]
- 2.Aguiar CC, Almeida AB, Araújo PV, de Abreu RN, Chaves EM, do Vale OC, Macêdo DS, Woods DJ, Fonteles MM, Vasconcelos SM. Oxidative stress and epilepsy: literature review. Oxid Med Cell Longev. 2012;2012:795259. doi: 10.1155/2012/795259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asha MK, Debraj D, Prashanth D, Edwin JR, Srikanth HS, Muruganantham N, Dethe SM, Anirban B, Jaya B, Deepak M, et al. In vitro anti-Helicobacter pylori activity of a flavonoid rich extract of Glycyrrhiza glabra and its probable mechanisms of action. J Ethnopharmacol. 2013;145(2):581–586. doi: 10.1016/j.jep.2012.11.033. [DOI] [PubMed] [Google Scholar]
- 4.Ashrafi MR, Shams S, Nouri M, Mohseni M, Shabanian R, Yekaninejad MS, Chegini N, Khodadad A, Safaralizadeh R. A probable causative factor for an old problem: selenium and glutathione peroxidase appear to play important roles in epilepsy pathogenesis. Epilepsia. 2007;48(9):1750–1755. doi: 10.1111/j.1528-1167.2007.01143.x. [DOI] [PubMed] [Google Scholar]
- 5.Bellissimo MI, Amado D, Abdalla DS, Ferreira EC, Cavalheiro EA, Naffah-Mazzacoratti MG. Superoxide dismutase, glutathione peroxidase activities and the hydroperoxide concentration are modified in the hippocampus of epileptic rats. Epilepsy Res. 2001;46(2):121–128. doi: 10.1016/S0920-1211(01)00269-8. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23(11):580–587. doi: 10.1016/S0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Menachem E, Kyllerman M, Marklund S. Superoxide dismutase and glutathione peroxidase function in progressive myoclonus epilepsies. Epilepsy Res. 2000;40(1):33–39. doi: 10.1016/S0920-1211(00)00096-6. [DOI] [PubMed] [Google Scholar]
- 8.Bruce AJ, Baudry M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med. 1995;18(6):993–1002. doi: 10.1016/0891-5849(94)00218-9. [DOI] [PubMed] [Google Scholar]
- 9.Chen LL, Feng HF, Mao XX, Ye Q, Zeng LH. One hour of pilocarpine-induced status epilepticus is sufficient to develop chronic epilepsy in mice, and is associated with mossy fiber sprouting but not neuronal death. Neurosci Bull. 2013;29(3):295–302. doi: 10.1007/s12264-013-1310-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elger CE, Schmidt D. Modern management of epilepsy: a practical approach. Epilepsy Behav. 2008;12(4):501–539. doi: 10.1016/j.yebeh.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Floyd R, Carney J. Free radical damage to protein and DNA: mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann Neurol. 1992;32(S1):S22–S27. doi: 10.1002/ana.410320706. [DOI] [PubMed] [Google Scholar]
- 12.Frantseva MV, Perez Velazquez JL, Tsoraklidis G, Mendonca AJ, Adamchik Y, Mills LR, Carlen PL, Burnham MW. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience. 2000;97(3):431–435. doi: 10.1016/S0306-4522(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 13.Freitas RM. Investigation of oxidative stress involvement in hippocampus in epilepsy model induced by pilocarpine. Neurosci Lett. 2009;462(3):225–229. doi: 10.1016/j.neulet.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 14.Freitas RM, Vasconcelos SM, Souza FC, Viana GS, Fonteles MM. Oxidative stress in the hippocampus after pilocarpine induced status epilepticus in Wistar rats. FEBS J. 2005;272(6):1307–1312. doi: 10.1111/j.1742-4658.2004.04537.x. [DOI] [PubMed] [Google Scholar]
- 15.Golechha M, Chaudhry U, Bhatia J, Saluja D, Arya DS. Naringin protects against kainic acid-induced status epilepticus in rats: evidence for an antioxidant, anti-inflammatory and neuroprotective intervention. Biol Pharm Bull. 2011;34(3):360–365. doi: 10.1248/bpb.34.360. [DOI] [PubMed] [Google Scholar]
- 16.Jäger AK, Saaby L. Flavonoids and the CNS. Molecules. 2011;16(12):1471–1485. doi: 10.3390/molecules16021471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovac S, Domijan AM, Walker MC, Abramov AY. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J Cell Sci. 2012;125(7):1796–1806. doi: 10.1242/jcs.099176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehtinen MK, Tegelberg S, Schipper H, Su H, Zukor H, Manninen O, Kopra O, Joensuu T, Hakala P, Bonni A, et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J Neurosci. 2009;29(18):5910–5915. doi: 10.1523/JNEUROSCI.0682-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu YF, Gao F, Li XW, Jia RH, Meng XD, Zhao R, Jing YY, Wang Y, Jiang W. The anticonvulsant and neuroprotective effects of baicalin on pilocarpine-induced epileptic model in rats. Neurochem Res. 2012;37(8):1670–1680. doi: 10.1007/s11064-012-0771-8. [DOI] [PubMed] [Google Scholar]
- 20.Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs: innovative strategies. Epilepsy Res. 2006;69(3):183–272. doi: 10.1016/j.eplepsyres.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinc B, Grabnar I, Vovk T. The role of reactive species in epileptogenesis and influence of antiepileptic drug therapy on oxidative stress. Curr Neuropharmacol. 2012;10(4):328–343. doi: 10.2174/157015912804143504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nazıroğlu M, Akay MB, Çelik Ö, Yıldırım Mİ, Balcı E, Yürekli VA. Capparis ovata modulates brain oxidative toxicity and epileptic seizures in pentylentetrazol-induced epileptic rats. Neurochem Res. 2013;38(4):780–788. doi: 10.1007/s11064-013-0978-3. [DOI] [PubMed] [Google Scholar]
- 23.Royle SJ, Collins FC, Rupniak HT, Barnes JC, Anderson R. Behavioural analysis and susceptibility to CNS injury of four inbred strains of mice. Brain Res. 1999;816(2):337–349. doi: 10.1016/S0006-8993(98)01122-6. [DOI] [PubMed] [Google Scholar]
- 24.Ryan K, Backos DS, Reigan P, Patel M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J Neurosci. 2012;32(33):11250–11258. doi: 10.1523/JNEUROSCI.0907-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiihara T, Kato M, Ichiyama T, Takahashi Y, Tanuma N, Miyata R, Hayasaka K. Acute encephalopathy with refractory status epilepticus: bilateral mesial temporal and claustral, associated with a peripheral marker of oxidative DNA damage. J Neurol Sci. 2006;250(1):159–161. doi: 10.1016/j.jns.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Shin EJ, Ko KH, Kim WK, Chae JS, Yen TP, Kim HJ, Wie MB, Kim HC. Role of glutathione peroxidase in the ontogeny of hippocampal oxidative stress and kainate seizure sensitivity in the genetically epilepsy-prone rats. Neurochem Int. 2008;52(6):1134–1147. doi: 10.1016/j.neuint.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Shin EJ, Jeong JH, Chung YH, Kim WK, Ko KH, Bach JH, Hong JS, Yoneda Y, Kim HC. Role of oxidative stress in epileptic seizures. Neurochem Int. 2011;59(2):122–137. doi: 10.1016/j.neuint.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun YX, Tang Y, Wu AL, Liu T, Dai XL, Zheng QS, Wang ZB. Neuroprotective effect of liquiritin against focal cerebral ischemia/reperfusion in mice via its antioxidant and antiapoptosis properties. J Asian Nat Prod Res. 2010;12(12):1051–1060. doi: 10.1080/10286020.2010.535520. [DOI] [PubMed] [Google Scholar]
- 29.Tejada S, Sureda A, Roca C, Gamundí A, Esteban S. Antioxidant response and oxidative damage in brain cortex after high dose of pilocarpine. Brain Res Bull. 2007;71(4):372–375. doi: 10.1016/j.brainresbull.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42(4):515–524. doi: 10.1046/j.1528-1157.2001.28900.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang KL, Hsia SM, Chan CJ, Chang FY, Huang CY, Bau DT, Wang PS. Inhibitory effects of isoliquiritigenin on the migration and invasion of human breast cancer cells. Expert Opin Ther Targets. 2013;17(4):337–349. doi: 10.1517/14728222.2013.756869. [DOI] [PubMed] [Google Scholar]
- 32.Xie YC, Dong XW, Wu XM, Yan XF, Xie QM. Inhibitory effects of flavonoids extracted from licorice on lipopolysaccharide-induced acute pulmonary inflammation in mice. Int Immunopharmacol. 2009;9(2):194–200. doi: 10.1016/j.intimp.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29(21):6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhan C, Yang J. Protective effects of isoliquiritigenin in transient middle cerebral artery occlusion-induced focal cerebral ischemia in rats. Pharmacol Res. 2006;53(3):303–309. doi: 10.1016/j.phrs.2005.12.008. [DOI] [PubMed] [Google Scholar]