

Abstract
Four different gold nanoparticle (GNP) preparations, including nude GNP and GNP coated either with thiolated undecane (S-C11H23), or with dithiolated diethylenetriaminepentaacetic (DTDTPA) or gadolinium (Gd) DTDTPA chelating agents were synthesized. The average diameters, for each type of nanoparticle are 5 nm, 10 and 13 nm, respectively. Dry films of plasmid DNA pGEM-3Zf(-), DNA with bound GNP and DNA with coated GNP were bombarded with 60 keV electrons. The yields of single and double strand breaks were measured as a function of exposure by electrophoresis. The binding of only one GNP without coating to DNA containing 3197 base pairs increases single and double strand breaks by a factor of 2.3 while for GNP coated with S-C11H23 this factor is reduced to 1.6. GNP coated with the DTDTPA and DTDTPA:Gd in same ratio with DNA, produce essentially no increment in damage. These results could be explained by the attenuation by the coatings of the intensity of low energy photoelectrons emitted from GNP. Thus, coatings of GNP may considerably attenuate short-range low energy electrons emitted from gold, leading to a considerable decrease of radiosensitization. According to our results, the highest radiosensitization should be obtained with GNP having the shortest possible ligand, directed to the DNA of cancer cells.
1. Introduction
Gold nanoparticles (GNP) of well-controlled dimensions are currently being intensively investigated for applications in cancer diagnosis, imaging and therapy [1,2]. Molecular labeling with GNP enables the detection of the biodistribution of various biomolecules by techniques, such as UV-vis spectrophotometry [3], surface plasmon resonance spectroscopy [4 ] and magnetic resonance imaging (MRI) [5]. Moreover, site-specific cellular targeting of GNP could serve as diagnostic probes and radiosensitizers of cancer cells [6,7,8]. In this case, they usually need to be bound to a ligand with a targeting group having a specific biological function [2]. For example, GNP coated with gadolinium chelates have been demonstrated as a good contrast agent in MRI for clinical diagnosis [5]. Hence, the physical and chemical characteristics of GNP associated with specific ligands have become the important factor influencing its application and need to be determined.
Recently, the potential of GNP as a radiosensitizer in cancer radiotherapy has been demonstrated [2, 9 ]. Clinical applications are mainly limited by the amount that can be administered to the patient and the method to preferentially retain GNP in cancer cells. Hainfeld et al. reported that administration of 1.9 nm GNP with X-ray radiation can eradicate EMT-6 mouse tumours [9]. However, the amount of GNP administered to mice in these experiments was 1.35 g/kg, which is much too large for human applications. In order to decrease the amount administered to the patient and maintain a similar radiosensitization, GNP should be targeted to the tumor cells and in addition should be close to nuclear DNA. Zheng et al. showed adding GNP to plasmid DNA with ratio of 2:1 increased damage to the plasmid from exposure to fast electron irradiation by a factor of 2.5 compared to pure DNA [10]. It was suggested that the presence of GNP increases the absorption of ionizing radiation which led to the production of additional low-energy secondary electrons around DNA. Since the low-energy electrons (LEE) are short-range and are produced in large amounts by ionizing radiation, GNP have the potential to focus radiation energy to the DNA of cancer cells. The approach is of obvious interest for applications in radiotherapy, if the nanoparticles can be bound to specific ligands that serve as a vehicle to direct GNP to cancer cells and the nucleus.
In subsequent investigations, Zheng et al. showed that the presence of GNP near DNA also enhanced the DNA damage induced by 1, 10 and 100 eV electrons [11]. Surprisingly, GNP increased single strand breaks (SSB) induced by 1 and 10 eV electrons by factors of 2 and 3.5, respectively. Since 1 eV electrons do not ionize matter and 10 eV electrons do not produce considerable ionization compared to higher energy electrons or 20–100 keV X-rays, the study suggests that DNA is chemically sensitized to LEE damage by GNP. In other words, Zheng et al. [11] suggested that the radiosensitization of DNA by GNP takes place via one or both of the following mechanisms: (1) a local increase in the absorption of ionizing radiation, which increases the production of short range secondary electrons that have a high probability of damaging DNA, and (2) an increase in the sensitivity of DNA to fragmentation induced by secondary LEE.
The most common ligands and stabilizers bound to GNP are thiolated organic molecules [2]. The general synthetic method is the Shiffrin-Brust biphasic synthesis [12]. In this case, HAuCl4, a thiol, and NaBH4 in water-toluene are used to yield thiolated-GNP, in which the thiol ligand is self-assembled on the gold surface through an Au-S covalent bond. Upon subsequent biomolecular substitution, various functional thiolates such as oligonucleotides, peptides etc. can be attached to GNP [2]. Although with appropriate ligands, it is technically possible to introduce GNP into cancer cells, the role of these ligands on the mechanism of GNP radiosensitization is not clear. Xie et al. reported that the chemical linkage of GNP with alkane ligands can enhance the photoexcitation of the nanoparticle and photoemission from them [13]. However, GNP with another similar ligand, gadolinium (Gd) chelating dithiolated diethylentriaminepentaacetic (DTDTPA), which was expected to increase the additional absorption of radiation in cancer cells with a subsequent increase of secondary electrons, showed no radiosensitization in vivo and in vitro [14]. In the present study, we attempt to find the reason for this lack of radiosensitization with DTDTPA:Gd, while obtaining data on a typical thiolated ligand for GNP.
We investigate the role of GNP ligands as well as that of LEE in GNP radiosensitization of DNA. GNP without any ligands or coated either with DTDTPA, DTDTPA:Gd or thiolated C11H23 are mixed with pGEM-3Zf(-) plasmid DNA to form thin films [10]. These films are bombarded by 60 keV electrons. The yields of DNA damage are measured by electrophoresis. Films made of GNP and the two DTDTPA ligands are characterized by X-ray photoelectron spectroscopy (XPS). From the comparison of these yields and XPS spectra from the different type of films, we find that the coating of GNP of about 4 nm considerably attenuates the emitted LEE produced in the core of GNP and makes DNA more resistant to LEE-induced damage. When the GNP are coated with the shorter ligand (C11H23) of 2.5 nm length, the emitted electrons are less attenuated and consequently, DNA damage increases compared to that observed with the DTDTPA ligands. These results can be explained by the short effective range of LEE of the order of 10 nm [15], and the geometrical factor reducing the electron flux as a function of distance from the center of the GNP. Thus, radiosensitization of DNA by GNP may be considerably reduced by LEE attenuation in the ligand.
2. Materials and Methods
2.1. Preparation of Plasmid DNA
pGEM-3Zf(-) plasmid DNA (3197 base pairs, Promega) was extracted from E. coli DH5α, purified with QIAfilter Plasmid Giga Kit (Qiagen) [16]. The DNA was diluted in distilled and deionized water (dd H2O). Agarose gel electrophoresis analysis showed that 95% of the purified plasmid DNA was in the supercoiled form and the rest was in the concatemeric (1%) and nicked circular form (4%). The absolute amount of plasmid DNA was determined by measuring its UV absorption at 260 nm [17,18,19]. The relative amount of proteins in the plasmid was obtained from the ratio of the UV absorption of DNA at 260 nm to that of at 280 nm. The ratio is 1.98 in this study which corresponds to less than 15% by weight of proteins remaining with DNA [17,18,19].
2.2. GNP synthesis
GNP without ligands were prepared by the NaBH4 reduction of HAuCl4 [12]. Ten ml of 8 mM NaBH4 was added to 10 ml of 3 mM HAuCl4 (Sigma). The mixture was vigorously stirred for 10 min at room temperature, until no bubble formed in the solution. The final solution was stored in the dark at 5 °C until further manipulation. In the following text gold nanoparticles without ligands was referred as GNP.
The GNP with DTDTPA ligands were synthesized according to the method of Debouttiere et al. [5]. Briefly, 200 mg HAuCl4 in 70 mL methanol was mixed with a solution of 482 mg DTDTPA, 50 ml methanol and 2 mL of acetic acid. Then a solution of 192 mg of NaBH4 was added under vigorous stirring for 1 hr. The color of the mixture instantly changed to dark brown indicating the presence of GNP. The solution was filtered through the nylon membrane (0.2 μm) and subsequently washed by different solvents to obtain the GNP with the thiolated DTDTPA (Au@DTDTPA). By adding 5 mM GdCl3 to a solution of Au@DTDTPA, calibrated with the colormetric titration of xylenol orange, the gadolinum was chelated to the GNP forming Au@DTDTPA:Gd as the second coated GNP in this study.
The GNP with undecanethiol (Au@C11H23) were prepared by the Brust-Schiffrin method [12]. 80 ml of Tetraoctylammonium bromide in toluene was added to 30 ml of hydrogen tetrachloroaurate with vigorously stirring until the water phase turned to colorless, indicating all the tetrachloroaurate was transferred to the organic phase. l70 mg of undecanethiol was added to the subtracted organic phase and mixed with 0.4 M sodium borohydride while vigorously stirring for 3 hrs. After rotary evaporation and washing with 400ml ethanol the mixture was kept at −18 °C for 4 hrs to form the dark brown precipitate. The precipitate was filtered, redissolved in toluene and washed with ethanol twice for purification to obtain Au@C11H23.
2.3. GNP and coating characterization
Ten microliters of freshly made GNP solution was deposited onto a 230-mesh carbon-coated copper grid (Beijing Zhongjingkeyi Tech.). Excess water was blotted off the grids with filter paper, and the sample was dried on a specimen mounting heated plate at 130 °C before observation. The transmission electron microscopy (TEM) (Tecnai G2 F20 S-TWIN, FEI) was operated in the high resolution mode, at an accelerating voltage of 200 KV and with a beam current of 4200 μA. The TEM images were recorded with a digital camera (Gatan DigitalMicrograph). The size distribution of GNP was analyzed by ImageJ (Version 1.24o) using at least 200 particles in the TEM image. The average diameter of GNP was 5 ± 2 nm, as characterized by TEM.
Since the TEM images only display the heavy element Au in GNP with chelating agent, the size of the molecular assembly was deduced from thermogravimetric analysis (TGA). The TGA analysis was performed with a NETZSCH STA449C device on 35 mg of dry, purified samples under an air flow of 40 mL min−1 at a heating rate of 5°C min−1 in the temperature range of 25–700°C. The result of TGA revealed a weight loss around 43% and 49% between 200 and 600°C for Au@DTDTPA and Au@DTDTPA:Gd, respectively (Fig. 2B). According to the density of GNP and DTDTPA:Gd of 19.3 and 1.2 g cm−3, with the inner GNP of radius of 2.5 nm, the length of the binding DTDTPA:Gd linker was estimated to be 4 nm. Since the chelating gadolinium ions are entrapped in the DTDTPA shell (Fig.1A) [20], we assume that the size of GNP without adding of gadolinium is the same as that of Au@DTDTPA:Gd. Thus, the diameter of GNP with linker DTDTPA was deduced to be 13 nm which compares to 5 nm for GNP without the linker. The result of TGA analysis of Au@C11H23 showed a weight loss of 26% for the S- C11H23 ligand. The length of undecane was calculated to be 2.5 nm, from the estimated density of 0.95 g cm−3 for SC11H23.
Fig. 2.
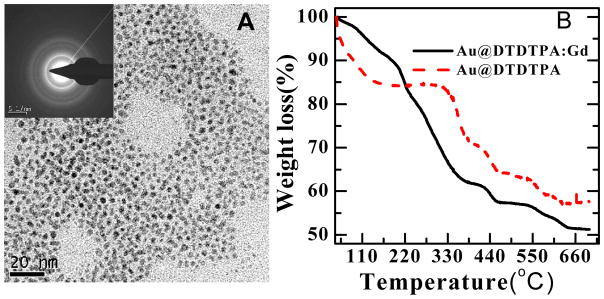
A) The TEM image of Au@DTDTPA with the electron diffraction pattern in the insert. B) The TGA analysis of Au@DTDTPA and Au@DTDTPA:Gd.
Fig. 1.

Schematic diagram of A) Au@DTDTPA:Gd; ● represents the Gd3+ chelating ion. B) the surface geometrical structure of a layer of GNP with and without linkers. The packing of Au@ligand is illustrated in the left panel with orange and gray circles representing the GNP in the first and second layer, respectively.
According to Hostetler et al., gold atoms cluster in the cubic-face configuration to form the GNP [21]. The geometrical arrangements of a surface layer of spherical GNP with and without linkers are shown schematically in this configuration in Fig. 1B. The packing of Au@ligand is illustrated in the left panel with orange and gray circles representing the GNP in the first and second layer, respectively. The model serves in following calculations of GNP density in our films.
2.4. 60 keV Electron irradiation
GNP, Au@DTDTPA and Au@DTDTPA:Gd were dissolved in dd H2O and mixed with DNA forming GNP-DNA complexes. In the case of Au@C11H23, 4.2 mg of the compound was dissolved by ultrasound in 1 ml ethanol before mixing with DNA. Five μl aliquots of DNA and GNP-DNA solutions were deposited on a tantalum foil (99.99%, Goodfellow), 25-μm-thick with a surface area of 4.6 mm2. The samples were dried in a glove box at ambient temperature, at a relative humidity of 10 %. For 7.9 μg of DNA, this procedure produced films of 1 μm thickness estimated from the density of DNA of 1.7 g cm−3[22,23]. In this study, the ratio of GNP to DNA is fixed at a molar ratio of 1:1. The amount of DNA in films of GNP-DNA complexes was reduced to maintain the same film thickness of 1μm. Furthermore, since the yields are expressed as the number of SSB and DSB per DNA molecule in the sample, these yields are independent of the amount of DNA in each film. Hence, in all experiments the irradiated volume remains the same. The thickness of 1 μm was chosen to absorb sufficient energy from the electron beam, while avoiding the effects of secondary electrons emitted from the metal substrate.
Once prepared, the samples were transferred to the TEM (H-7100 Hitachi chamber, where they were irradiated or not by a 60 keV electron beam with a current of 15 μA for periods varying from 5 to 30 s. Data were recorded at five different doses and with no dose under identical experimental conditions. The incident electron fluence of the TEM was measured with a radiochromatic dosimetry film as described previously [10]. Taking into account the area of the electron beam of 4.6 mm2, the incident electron flux was determined to be 5.3 ×1013 electrons s−1 cm−2.
2.5. Analysis of plasmid DNA by Agarose Gel Electrophoresis
After irradiation, the gold foil was removed from the UHV chamber and the sample was immediately dissolved in dd H2O. The recovery of DNA was approximately 90%. The different forms of DNA were separated by 1% neutral agarose gel electrophoresis run in TAE buffer (40 mM Tris acetate, 1 mM EDTA, pH 8.0) at 100 V for 7 min and 75 V for 90 min. Both the gel and the DNA samples were prestained by SYBR Green I (Molecular Probes), 10 000X for gel and 100X for samples, respectively. After electrophoresis, gels were scanned with the STORM860 using the blue fluorescence mode (Molecular Dynamics) at an excitation wavelength of 430 nm. A correction factor of 1.8 was applied because of the weaker binding of SYBR Green I to the supercoiled form of DNA compared to nicked circular (SSB) and linear (i.e., double strand break: DSB) configurations [24]. The percentage of each form was obtained from Image Quant analysis. The gel image of Au@DTDTPA-DNA analysis with increasing irradiation time and one corresponding scanned intensity are shown in Fig. 3. Special care was taken to accurately assess the magnitude of the band associated with the linear form of DNA. This band, when very weak, is convoluted with the much more intense band corresponding to the nicked circular form. In most gels, it was possible to quantify the DSB peak using a well-chosen baseline that correctly exempted the SSB peak. In extreme cases, it was necessary to manually deconvolute the bands with two pseudo-Voigt functions (a linear combination of a Gaussian and a Lorentzian function at the same position and with the same area) using a software such as Origin 8.
Fig. 3.
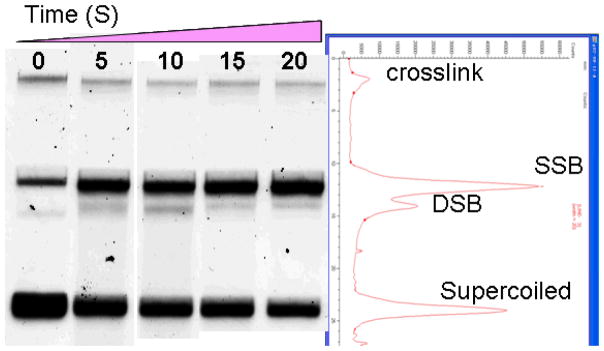
Agarose gel electrophoresis of Au@DTDTPA-DNA samples of 348 nm thickness deposited on the tantalum substrate and irradiated by 60 keV electrons at four different doses. The first band on the left is that of the non-irradiated control sample. Starting from the top, the bands represent the crosslink, circular (SSB), linear (DSB), and supercoiled DNA configurations, respectively. The magnitude of the last 20 s lane is shown by the ImageQuant scan in the right panel with peak corresponding to each DNA conformation.
2.6. XPS characterization
The XPS measurements were conducted using the commercial system (Thermo Scientific ESCALAB 250) at Fuzhou University. The apparatus was operated at an emission current of 6 mA producing a monochromatized Al Kα beam in a chamber at a base pressure of 3.8×10−10 mbar. The neutralizing electron gun was turned on in the low energy Mode with emission current of 100 mA to eliminate the charging of the samples during X-ray irradiation. The hemispherical electron energy analyzer input axis was normal to the sample surface. XPS survey spectra were recorded in the fixed analyzer transmission mode with a pass energy of 100 eV and energy steps of 1 eV. The typical peaks of elements, Au 4f, Gd 3d, S 2p, C 1s, N 1s and O 1s, were recorded separately with a pass energy of 20 eV and energy steps of 0.1 eV. The energy scale of XPS spectra was adjusted according to the standard C 1s binding energy line of 284.6 eV.
The secondary electron (SE) emission measurements were performed in the low-energy electron mode with the pass energy of 0.8 eV and energy step of 0.05 eV without the neutralizing gun. The freshly synthesized GNP were deposited onto a carbon-coated copper grid (SPI Supplies), dried in a nitrogen filled glove box and formed a thick GNP film. The thickness of the GNP film was sufficiently large to avoid detection of background signal from the carbon-coated copper grid in our measurements. Au@DTDTPA and Au@DTDTPA:Gd sample powders were first formed into pellets and deposited on metal sample holders. Then the GNP@ samples were circumscribed with a conducting silver gel (SPI Scientific), shown schematically in Fig. 5, so as to remove the charging on the surface. The work function at the surface was 5.19 eV. During the measurements the sample platform of the XPS was connected to ground.
Fig. 5.
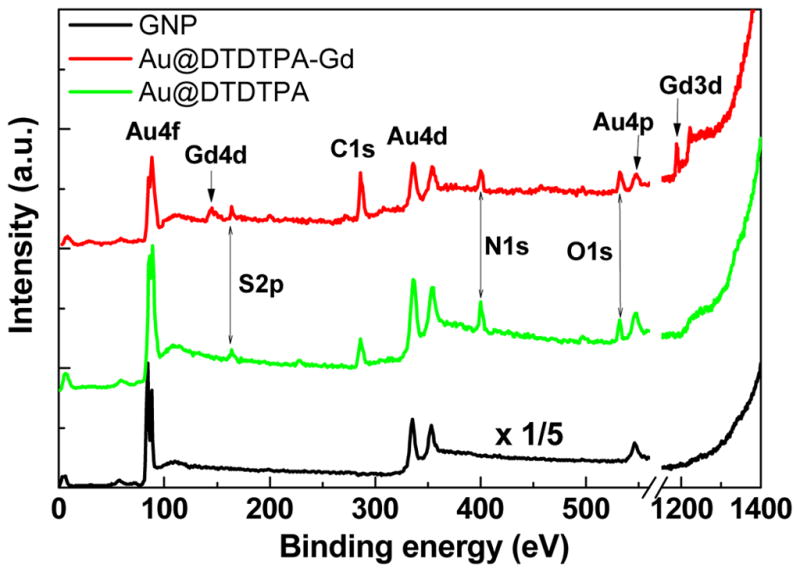
XPS elemental analysis of GNP, Au@DTDTPA and Au@DTDTPA:Gd. The intensity of GNP without DTDTPA is reduced by 1/5 for comparison.
3. Results and discussion
Fig. 4 exhibits the fluence response curve for the induction of SSB and DSB by 60 keV electrons in films of DNA, Au@DTDTPA-DNA and Au@DTDTPA:Gd-DNA held under high vacuum conditions. As expected, the number of breaks increases with incident electron fluence. All curves exhibit a similar rise and almost parallel slopes in the linear region. Previously published fluence-response curves for GNP-DNA films with no linkers recorded under similar experimental conditions exhibited a much steeper slope than those for pure DNA films [10]. The same result was obtained in the present experiments. The initial slope of the corresponding response curve represents the yield per incident 60 keV electron per DNA molecule at low fluences. The yields for the formation of SSB, DSB and loss of supercoiled DNA are listed in Table 1 for pure DNA and different GNP-DNA complexes. Taking into account standard deviations, we find that the yields are the same for naked DNA, Au@DTDTPA-DNA and Au@DTDTPA:Gd-DNA. On the other hand, the difference between the yields from the latter three samples and the yields from the GNP-DNA and GNP@C11H23-DNA samples are large and statistically significant. In all cases, the ratio of SSB to DSB is situated around 20, a value which is typical of high energy radiation with low linear energy transfer [25]. Previous studies [10] have shown that the formation of SSB and DSB is considerably increased by the binding of GNP to DNA. The present results (2nd line in Table I) corroborate this finding. The formation of SSB and DSB is increased by a factor of 2.3 in the presence of GNP. When the GNP is coated with thiolated undecane (S-C11H23), which has a thickness of 2.5 nm, this factor is reduced to 1.6; e.g., as shown in the last line of Table I, production of SSB is reduced from 17.1 to 12.0 × 10-15/electron-mol. However, enhancement of radiation damage by 60 keV electrons is not observed with GNP coated with DTDTPA or DTDTPA:Gd; i.e., DNA is not radiosensitized by GNP coated with these organic linkers of 4 nm average thickness. Thus, the amount of DNA damage appears to be related to the thickness of the linker. The results obtained with the longer thicker coatings are consistent with the study of Hébert et al. [14], which showed no radiosensitization in MC7-L1 tumor cells incubated with Au@DTDTPA:Gd, nor in animals bearing MCT-L1 tumors injected with Au@DTDTPA:Gd. The ensemble of the results raise the question: why does coating GNP with these organic linkers destroy the radiosensitization action of GNP in vivo and at the cellular and DNA levels?
Fig. 4.

Exposure-response curves for SSB and DSB induced by 60 keV electron impact on films of pure DNA (▲), Au@DTDTPA-DNA (●) and Au@DTDTPA:Gd-DNA (■) complexes in ratios of 1:1, respectively. The curves are the corresponding exponential and spline fit for SSB and DSB, respectively, as well as the slope.
Table I.
The yields (Y in 10−15 electron-molecule−1) for the formation of SSB, DSB and loss of supercoiled DNA induced by 60 keV electrons in different DNA films deposited on a tantalum substrate. The error is the standard deviation obtained from the slope of the exposure curve. The ratio of SSB to DSB is displayed in the last column.
| Samples | Y
|
Ratio (SSB/DSB) | ||
|---|---|---|---|---|
| SSB | DSB | Supercoiled | ||
| DNA | 7.4 ± 1.6 | 0.4 ± 0.1 | −9.9 ± 1.9 | 19 |
| GNP:DNA=1:1 | 17.1 ± 1.3 | 0.9 ± 0.2 | −18.1 ± 1.3 | 19 |
| Au@DTDTPA:DNA=1:1, | 8.8 ± 0.5 | 0.4 ± 0.1 | −10.0 ± 1.0 | 22 |
| Au@DTDTPA-Gd:DNA=1:1 | 8.8 ± 0.9 | 0.4 ± 0.1 | −9.9 ± 0.6 | 22 |
| Au@C11H23= 1:1 | 12.0 ± 2.0 | 0.6 ± 0.1 | −13.2 ± 2.2 | 20 |
Since the decrease of radiosensitization by GNP depends on the thickness of the coating, it is possible that this reduction be due to considerable absortion of the emitted low energy photoelectrons by the linker or any biological material lying between the GNP and DNA. Our DNA samples contain approximately 15% proteins by weight; so, we can not rule out the possibility that coated GNP could preferentially bind to proteins. However, the aliphatic hydrocarbon linker is expected to be quite inert with respect to binding to either DNA or proteins. Thus, most of the GNP-S-C11H23 nanoparticles should lie near DNA. In such a case, electrons escaping the undecane coating could still damage DNA as seen from the results of the last line in Table I. However, it is not known if DTDTPA has a stronger affinity for protein or DNA. We therefore conclude that in this case most of the emitted photoelectrons are absorbed by either the linker alone or by a combination of linker and proteins. In the following text, we show from XPS data that attenuation of LEE by the DTDTPA and DTDTPA:Gd coatings alone is sufficient to suppress most LEE and keep them from reaching DNA; i.e., LEE absorption by these ligands alone can explain the lost of radiosensitivity.
Typical elemental compositions of Au, S, N and Gd from the XPS spectra of films of GNP, Au@DTDTPA and Au@DTDTPA:Gd are shown in Fig. 5. The spectra characterize the GNP synthesized with and without linkers. The Au 4f peaks for each film appear in more detail in Fig. 5. The Au 4f7/2 binding energy in the GNP film is 84.1 eV with a full width at half-maximum (FWHM) of 0.8 eV. This is identical to that measured for the pure Au reference film [2,13]. The Au 4f7/2 peaks of Au@DTDTPA and Au@DTDTPA:Gd are broader than that of the reference with a FWHM of ~1.6 eV. Inelastic electron scattering within the linker probably contributes to the enlargement of the peak. An additional contribution could come from chemical bonding of the linker to gold. This is illustrated in Fig. 6 by deconvoluting the broad peaks into two components C1 and C2. Component C1 is located at the same energy as the peak of pure GNP, which is attributed to unbound inner Au atoms in the GNP. Component C2 is located at a higher binding energy of ~85.0 eV which is associated with the formation of Au-S chemical bonding in the complexes [13]. For a diameter of 5 nm, the number of gold atoms at the surface and the total number of gold atoms in a GNP are 1563 and 3725, respectively [21]. Thus, 42% of the gold atoms are on the surface of the GNP. According to the XPS spectra in Fig. 6, of the total area under the Au@DTDTPA and Au@DTDTPA:Gd peaks, 52% and 46% of the signal arises from the C2 component, respectively. This is in fairly good agreement with the Au atom density if one considers that some electrons from the core will be absorbed by the surface layer. These results support the hypothesis [13] that formation of the Au-S bond contributes to the broadening of the Au peak.
Fig. 6.
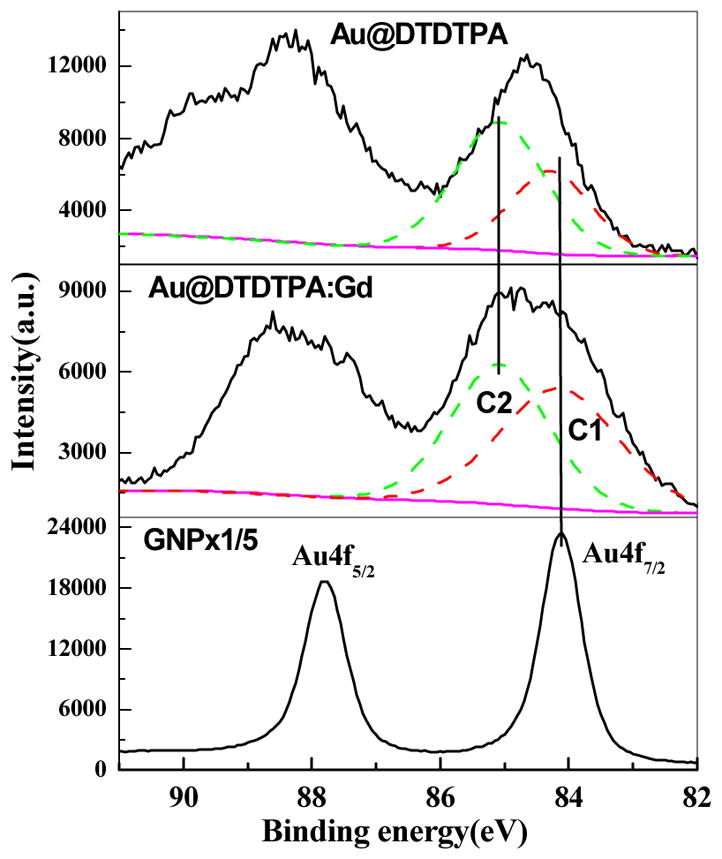
The Au 4f spectra of GNP, Au@DTDTPA and Au@DTDTPA:Gd, respectively. The Au 4f7/2 is deconvoluted into two components C1 and C2 for the latter two.
Fig. 7 shows the 0–14.8 eV distribution of electrons emitted from different films, and a gold surface, by the 1.5 keV photons. Under the same measurement conditions, the intensity of secondary electrons (SE) from different GNP films can be quantitatively compared. The SE spectrum of GNP without the linker was almost the same as that of the SE spectrum from the pure gold substrate, while the SE spectra of the two films of GNP with linkers nearly overlapped. The intensity of SE spectra from the GNP with linkers were about one order of magnitude smaller than that of pure GNP. As shown from the bottom curve in Fig. 7, the lowest intensity was measured from films made of only DTDTPA.
Fig. 7.
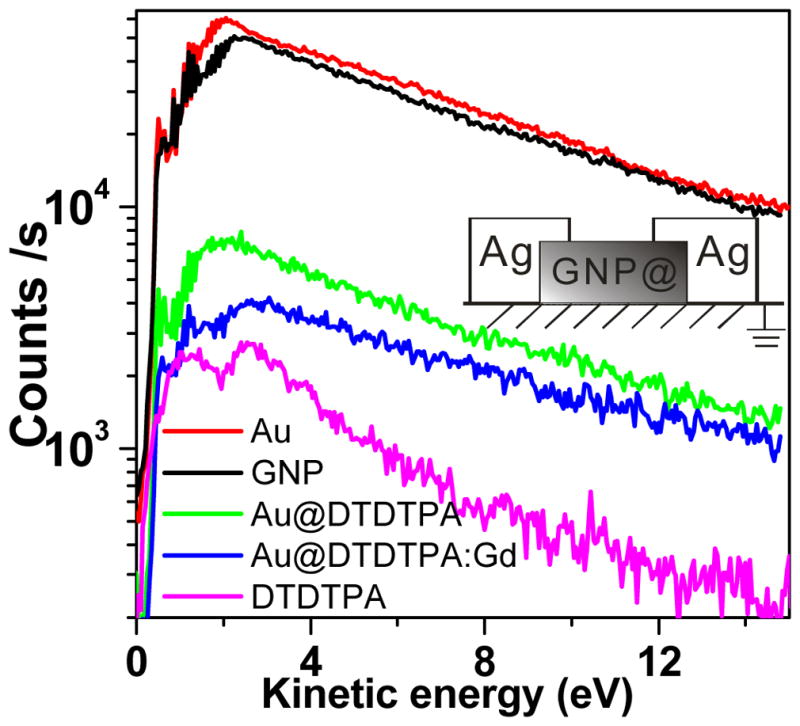
Low energy portion of secondary electron spectrum resulting from irradiation of GNP, Au@DTDTPA, Au@DTDTPA:Gd, pure gold and DTDTPA samples by 1.5 keV X-rays. The GNP with linker (GNP@) and DTDTPA samples are circumscribed with Ag gels to remove surface charging (shown in the insert).
The energy-absorption coefficients of 1.5 keV X-ray by gold and water are 409 ×103 and 25 ×103 cm2/mol, respectively [26]. According to this difference of a factor of 16, around 94 % of the X-rays are absorbed by GNP, whereas 6% are absorbed by the linkers which have a coefficient similar to water [26]. Thus, high-energy photoelectrons induced by 1.5 keV X-rays are mainly produced from GNP rather than from the linkers.
Fig. 1B shows a schematic model of ordered layers of GNP spheres in the cubic face-centered configuration with and without linkers. In this configuration, the inverse density ratio of gold at the first surface layer of each film depends on (r/R)2, where r and R are the radii of a sphere composed of a GNP and a Au@DTDTPA complex, respectively. This ratio of 6.6 is listed in the first column of Table II. When we integrate the peak areas of Au 4f7/2 in Fig. 6, a decrease by a factor of 4.6 and 5.4 is found for the Au@DTDTPA:Gd and Au@DTDTPA, with respect to the GNP film. These factors are listed in the second column of Table II. They can be compared to the inverse density ratio of gold (1st column). Considering that (1) the GNPs in the second layer could also contribute to the signal of the Au4f peak, and (2) the geometry and the stacking conditions of our film are probably different from the ideal model in Fig. 1B, the numbers in the first two column of Table II show reasonable agreement. Thus, within the limits of the model, we can conclude that the decrease of the magnitude of the 4f7/2 signal observed experimentally can be explained by the smaller density of the GNP at the surface of the Au@DTDTPA:Gd or Au@DTDTPA film. In other words, 1384 eV photoelectrons are not significantly absorbed by the coating and these 4.6 and 5.4 factors can be used to evaluate the stronger absorption of LEE in the same films.
Table II.
Comparison of parameters for different GNP preparations.
| Samples | Inverse density ratio of gold (r/R)2 | Decrease in magnitude of Au peak in XPS | Decrease in number of LEE | % of LEE absorbed by coating | Estimated decrease in DNA damage | Measured decrease in DNA damage |
|---|---|---|---|---|---|---|
| GNP | 1 | 1 | 1 | 0 | 0 | |
| Au@DTDTPA=1:1, | 6.6 | 5.4 | 7.3 | 46% | 82% | 86% |
| Au@DTDTPA-Gd=1:1 | 6.6 | 4.6 | 11.3 | 76% | 92% | 86% |
The total number of LEE between 0 and 14.8 eV produced from each film was calculated by integrating the SE curves in the Fig. 7. The LEE produced by Au@DTDTPA and Au@DTDTPA:Gd films decrease by factors of about 7.3 and 11.3, respectively, compared to the pure GNP film. These values are listed in the third column of Table II. Considering the factors of 4.6 or 5.4 as due to the decrease of the density of GNP, the remaining LEE attenuation translates into a further decrease of 26% and 59% of LEE produced by the GNP with DTDTPA and DTDTPA:Gd coatings, respectively. These percentages however do not take into consideration LEE produced by the linkers, whose distribution is shown at the bottom of Fig. 5. If we subtract the LEE yield of a pure film of DTDTPA from the SE curves with linkers in the calculation, more realistic estimates are obtained. In this case, we find that 46% of the LEEs produced by the GNP are absorbed in the DTDTPA coating, whereas 76% of the LEEs produced by the GNP are absorbed in the DTDTPA:Gd coating (4th column in Table II). The overall results in Table II indicate that LEE produced from the GNP are significantly reduced by linkers only 4 nm long and that the addition of a heavy element (e.g., Gd) within the linker further increases the absorption of LEE. These values are consistent with the thermalization distances of the order of 10 nm for 1–20 eV electrons in water [15] and the measurements of Cai et al. [24]. These authors measured the attenuation length for 8–18 eV electrons in DNA to be 1.6–2.5 nm [27].
Finally, these calculations do not consider that the electron flux (i.e., number of electrons per surface area) emanating from a point source varies as the inverse of the square of the distance. The center of a GNP lies at about 5 nm from DNA whereas that of a coated GNP resides 13 nm from DNA. Within a point source approximation these different radii would result in another decrease by a factor of 6.8 in the number of LEE reaching DNA in the case of GNP with linkers. This factor is probably lower since the source is a sphere rather than a point and LEE which traverse the coating and/or DNA can multiply scatter from other complexes within the film. Taking a conservative factor of 3 for the reduction of LEE current density we find that damage to DNA induced by LEE from GNP should be reduced by at least 82% and 92% with Au@DTDTPA and Au@DTDTPA:Gd, respectively These values, given in the fifth column of Table II, can be compared with those obtained from direct measurements of DNA damage, appearing in the last column of this table. Such a comparison shows that the percentage absorption of LEE by DTDTPA linkers corresponds to the percentage decrease of the radiosensitization of DNA induced by naked GNP.
The role of the secondary LEE produced in large number by ionizing radiation (~3×104/MeV) is well established in radiation physics and chemistry. They induce various DNA damages such as strand breaks, base fragmentation and base release [28]. Below 15 eV, these electrons have a high cross section for forming transient negative ions, which damage DNA by decaying into dissociative electron attachment or autoionization [28]. The preceding arguments show from attenuation length measurements, geometrical considerations and electron absorption by the linker that, the flux of LEE at the DNA surface is considerably reduced by Au@DTDTPA or Au@DTDTPA:Gd. It is therefore not surprising to find from Fig. 4 and Table I that radiosensitization of DNA is considerably decreased with these linkers. Indeed, according to Table II, the linkers DTDTPA and DTDTPA:Gd reduce damage to DNA from radiosensitization by 86% which compares with our XPS estimates of 82% and 92%, respectively. Thus, within experimental error, attenuation of the secondary LEE emanating from the GNP could account for the entire reduction of radiosensitization. As pointed out in the introduction, contact of GNP with DNA may also radiosensitize the molecule to secondary electrons [11,29]. This added chemical sensitivity may disappear when a spacer (i.e., the linker) is placed between DNA and the GNP. However, since with the shorter and relatively inert linker (i.e., S-C11H23) radiosensitization is still observed, we can at least conclude that chemical sensitization is not the only factor involved.
4. Conclusions
The present study provides further evidence that secondary LEE are involved in the radiosensitization of DNA by GNP. As shown previously [10], GNP bound to DNA increases absorption of ionizing radiation energy and causes the subsequent production of additional secondary LEE. However, binding linkers to the GNP decreases the number of LEE emitted from their surface that can reach DNA. Owing to their short effective range, the secondary LEE intensity is almost completely attenuated by the binding of relatively long DTDTPA linkers to GNP. This is reflected by the absence of a change in the yield of damage (i.e., by the absence of additional damages with the DTDTPA coated GNP). However, with a shorter linker (S-C11H23), reduced but finite radiosensitization of DNA occurs, indicating that LEE emitted from the GNP may be only partially absorbed. Taken together with the results of the in vitro and vivo studies on Au@DTDTPA:Gd radiosensitization [14], the present study is consistent with the recent theoretical calculations of McMahon et al [30]. These authors related the energy deposition of electrons emitted by GNP to biological outcomes and showed good agreement with cell killing by a combination of X-rays and GNP. Both model calculations [30] and our results indicate that the close proximity of GNP to the DNA molecule appears as an important factor to be considered in the application of GNP in cancer radiotherapy. Considering DNA as the most sensitive target in radiotherapy, the highest radiosensitization should be obtained by directing to the DNA of cancer cells GNP coated with the shortest possible linker.
Acknowledgments
Financial support for this work was provided by the Canadian Institutes of Health Research, the Marie Curie incoming fellowship program of the European Commission, the China Award Program of Minjiang Scholar Professorship, National Basic Research Program of China (973 Program: 2007CB613306), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT0818), and the NNSF of China (20973039). The technical assistance of Mrs Sonia Girouard and Dr. Etienne Hebert are gratefully acknowledged. We would also like to thank Dr. Andrew Bass for helpful comments and suggestions.
References
- 1.Daniel MC, Astruc D. Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications towards biology, catalysis, and nanotechnology. Chem Rev. 2004;104:293. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 2.Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chem Soc Rev. 2009;38:1759. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 3.Jin R, Wu G, Li Z, Mirkin CA, Schatz GC. What conrols the melting properties of DNA-linked gold nanoparticle assemblies? J Am Chem Soc. 2003;125:1643. doi: 10.1021/ja021096v. [DOI] [PubMed] [Google Scholar]
- 4.El-Sayed IH, Huang X, El-Sayed MA. Surface Plasmon resonance scattering and absorption of anti-EGFR antibody conjugated gold nanoparticles in cancer diagnostics: applications in oral cancer. Nano Lett. 2005;5:829. doi: 10.1021/nl050074e. [DOI] [PubMed] [Google Scholar]
- 5.Debouttiere P, Roux S, Vocanson F, Billotey C, Beuf O, Favre-Reguillon A, Lin Y, Pellet-Rostaring S, Lamartine R, Perriat P, Tillement O. Design of gold nanoparticles for magnetic resonance imaging. Adv Funct Mater. 2006;16:2330. [Google Scholar]
- 6.Huang X, El-Sayed IH, Qian W, El-Sayed MA. Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. J Am Chem Soc. 2006;128:2115. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 7.Chithrani D, Dunne M, Stewart J, Allen C, Jaffray D. Cellular uptake and transport of gold nanoparticles incorporated in a liposomal carrier. Nanomedicine:NBM. 2010:6161. doi: 10.1016/j.nano.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U, Schmid G, Brandau W, Jahnen-Dechent W. Size-dependent cytotoxicity of gold nanoparticles. Small. 2007;3:1941. doi: 10.1002/smll.200700378. [DOI] [PubMed] [Google Scholar]
- 9.Hainfeld JF, Slatkin D, Smilowitz H. Gold nanoparticles greatly enhance x-ray therapy in mice. Phys Med Biol. 2004;49:N309. doi: 10.1088/0031-9155/49/18/n03. [DOI] [PubMed] [Google Scholar]; Hainfeld JF, Dilmanian F, Zhong Z, Slatkin D, Kalef-Ezra J, Smilowitz H. Gold nanoparticles enhance the radiation therapy of amurine squamous cell carcinoma. Phys Med Biol. 2010;55:3045. doi: 10.1088/0031-9155/55/11/004. [DOI] [PubMed] [Google Scholar]
- 10.Zheng Y, Hunting DJ, Ayotte P, Sanche L. Radiosensitization of DNA by gold nanoparticles irradiated by high energy electrons. Radiat Res. 2008;169:19. doi: 10.1667/RR1080.1. Errata, 2008 Radiat Res 169, 481. [DOI] [PubMed] [Google Scholar]
- 11.Zheng Y, Cloutier P, Hunting DJ, Sanche L. Radiosensitization by gold nanoparticles: Comparison of DNA damage induced by low and high-energy electrons. J Biomed Nanotech. 2008;4(4):469. [Google Scholar]
- 12.Brust M, Walker D, Bethell DJ, Schiffrin R, Whywan J. Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid-liquid system. Chem Soc Chem Commu. 1994:801. [Google Scholar]
- 13.Xie X, Gao X, Qi D, Xie Y, Shen L, Yang S, Sow C, Wee A. Chemically linked AuNP-alkane network for enhanced photoemission and field emission. ACS Nano. 2009;3:2722. doi: 10.1021/nn9005335. [DOI] [PubMed] [Google Scholar]
- 14.Hebert E, Debouttiere P, Lepage M, Sanche L, Hunting D. Preferential tumour accumulation of gold nanoparticles, visualised by magnetic resonance imaging: radiosensitisation studies in vivo and in vitro. Int J Radiat Biol. 2010;86:692. doi: 10.3109/09553001003746067. [DOI] [PubMed] [Google Scholar]
- 15.Meesungnoen J, Jay-Gerin J-P, Filali-Mouhim A, Mankhetkorn S. Low-energy electron penetration range in liquid water. Radiat Res. 2002;158:657. doi: 10.1667/0033-7587(2002)158[0657:leepri]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 16.QIAGEN Inc. Available from: www.qiagen.com/goto/plasmidinfo.
- 17.Glasel JA. Validity of nucleic-acid purities monitored by 260 nm 280 nm absorbency ratios. BioTechniques. 1995;18:62. [PubMed] [Google Scholar]
- 18.Manchester KL. Use of UV methods for measurement of protein and nucleic acid concentrations. BioTechniques. 1996;20:968. doi: 10.2144/96206bm05. [DOI] [PubMed] [Google Scholar]
- 19.Manchester KL. Value of A(260)/A(280) ratios for measurement of purity of nucleic-acids. BioTechniques. 1995;19:208. [PubMed] [Google Scholar]
- 20.Alric C, Taleb J, Le Duc G, Mandon C, Billotey C, Meur-Herland A, Brochard T, Vocanson F, Janier M, Perriat P, Roux S, Tillement O. Gadolinium chelate coated gold nanoparticles as contrast agents for both X-ray computed tomography and magnetic resonance imaging. J Am Chem Soc. 2008;130:5908. doi: 10.1021/ja078176p. [DOI] [PubMed] [Google Scholar]
- 21.Hostetler MJ, et al. Alkanethiolate Gold Cluster Molecules with Core Diameters from 1.5 to 5.2 nm: Core and Monolayer Properties as a Function of Core Size. Langmuir. 1998;14:17. [Google Scholar]
- 22.Fashman GD. Handbook of Biochemistry and Molecular Biology. 3. Boca Raton (FL): CRC Press; 1995. [Google Scholar]
- 23.Kago K, Matsuoka H, Yoshitome R, Yamaoka H, Ijiro H, Shimonura M Direct in situ observation of a lipid monolayer-DNA complex at the air-water interface by X-ray reflectometry. Langmuir. 1999;15:5193. [Google Scholar]
- 24.Cai Z, Cloutier P, Hunting D, Sanche L. Comparison between x-ray photon and secondary electron damage to DNA in vacuum. J Phys Chem B. 2005;109:4796. doi: 10.1021/jp0459458. [DOI] [PubMed] [Google Scholar]
- 25.Ito T, Baker SC, Stickly CD, Peak JG, Peak MJ. Dependence of the yield of strand breaks induced by gamma-rays in DNA on the physical conditions of exposure: water content and temperature. Int J Radiat Biol. 1993;63:289. doi: 10.1080/09553009314550391. [DOI] [PubMed] [Google Scholar]
- 26.Hubbell JH, Seltzer SM. NISTIR 5632. National Institute of Standards and Technology; Gaithersburg, MD: 1995. [Google Scholar]
- 27.Cai Z, Dextraze M, Cloutier P, Hunting D, Sanche L. Induction of strand breaks by low-energy electrons (8–68 eV) in a self-assembled monolayer of oligonucleotides : effective cross sections and attenuation lengths. J Chem Phys. 2006;124:024705. doi: 10.1063/1.2141505. [DOI] [PubMed] [Google Scholar]
- 28.Sanche L. Low-Energy Electron Interaction with DNA: Bond Dissociation and Formation of Transient Anions, Radicals and Radical Anions. In: Greenberg M, editor. Wiley Series on Reactive Intermediates in Chemistry and Biology entitled Radicals in Nucleic Acids. John Wiley & Sons; New Jersey: 2009. pp. 239–293. [Google Scholar]
- 29.Zheng Y, Sanche L. Gold nanoparticles enhance DNA damage induced by anti-cancer drugs and radiation. Radiat Res. 2009;172:114. doi: 10.1667/RR1689.1. [DOI] [PubMed] [Google Scholar]
- 30.McMahon SJ, Hyland WB, Muir MF, Coulter JA, Jain S, Butterworth KT, Schettino G, Dickson GR, Hounsell AR, O’Sullivan M, Prise KM, Hirst DG, Currell FJ. Biological consequences of nanoscale energy deposition near irradiated heavy atom nanoparticles. Scientific Reports. 2011;1:18, 1. doi: 10.1038/srep00018. [DOI] [PMC free article] [PubMed] [Google Scholar]