

Abstract
Endo-inulinase INU2 from Aspergillus ficuum belongs to glycosidase hydrolase family 32 (GH32) that degrades inulin into fructo oligosaccharides consisting mainly of inulotriose and inulotetraose. The 3D structure of INU2 was recently obtained (Pouyez et al., 2012, Biochimie, 94, 2423–2430). An enlarged cavity compared to exo-inulinase formed by the conserved motif W-M(I)-N-D(E)-P-N-G, the so-called loop 1 and the loop 4, was identified. In the present study we have characterized the importance of 12 residues situated around the enlarged cavity. These residues were mutated by site-directed mutagenesis. Comparative activity analysis was done by plate, spectrophotometric and thin-layer chromatography assay. Most of the mutants were less active than the wild-type enzyme. Most interestingly, mutant N42G differed in the size distribution of the FOS synthesized.
Keywords: Endo-inuinase, Site directed mutagenesis, N42G mutant, Activity modification
Highlights
-
•
Endo-inulinase INU2 degrades inulin into fructo oligosaccharides.
-
•
12 residues around the catalytic pockets of INU2 enzyme were determined.
-
•
These residues were mutated to either a G or A residue.
-
•
The activity has been tested by plate, spectrophotometric and TLC assays.
-
•
One mutation, N42G, which changes the specificity of activity, has been identified.
1. Introduction
Inulin, a storage carbohydrate in plants such as chicory, Jerusalem artichoke and dahlia, is composed of linear chains of ß-(2-1) linked fructose residues terminated by a glucose residue. It can be hydrolysed by endo-inulinases in short-chain fructoinulooligosaccaharides (FOS). Over recent years, different applications of inulin have sparked great interest for the production of ethanol, fructose syrup or inulooligosaccharides (IOS) used as prebiotics in food industries such as those producing baked goods, milk desserts and chocolate [1,2]. The positive effects of IOS on human health have been widely described [3]. Major IOS obtained after inulin hydrolysis with endo-inulinase show a degree of polymerization of 3 or 4.
Inulinases belong to glycoside hydrolase family 32 (GH32) and can be divided into exo- and endo-inulinases. Exo-inulinases (EC 3.2.1.80) cleave the terminal fructose whereas endo-inulinases (EC 3.2.1.7) hydrolyse inulin by endocleavage, producing fructo oligosaccharides consisting mainly of inulotriose and inulotetraose [4,5]. This means that these enzymes hydrolyse inulin between the third and fourth residues. Classically, random attack of inulin by endo-inulinase yielded principally inulotriose (F3) and inulotetraose (F4) in equal amounts.
In the GH32 family, three acidic residues, an aspartate or a glutamate in the WMN(D/E)PN motif, an aspartate in the RDP motif and E in the ECP motif have been identified as the essential residues for enzyme activity based on mutational and/or crystallographic data [6–8]. They are respectively a catalytic nucleophile, a transition state stabilizer and a general acid/base catalyst [7].
The difference in the mechanism of action between exo- and endo-inulinases is thought to be due to a larger pocket in the endo-enzymes [9]. Recently, we described the 3D structure of endo-inulinase INU2 from Aspergillus ficuum and identified an enlarged cavity compared to exo-inulinase formed by the conserved motif W-M(I)-N-D(E)-P-N-G, the so-called loop 1 and loop 4. These two loops among the four identified are conserved among all the endo-inulinases with known amino acid sequence. Docking studies of the substrate-like kestopentaose revealed five subsites and their constitutive residues [10]. Based on these recent results, we investigated the importance of 12 residues, located around the catalytic pocket, on the activity and specificity of INU2 from A. ficuum. The effect of each mutation on the enzyme activity was characterized. Most affected the enzyme activity, while one in particular changed the size distribution of the FOS synthesized.
2. Materials and methods
2.1. Strains, plasmids, and culture conditions
Escherichia coli DH10B (Gibco BRL) was used as the host strain for plasmid amplification. Pichia pastoris X-33 (Invitrogen, Leek, Netherlands) was used for the expression of recombinant endo-inulinases.
The plasmid used in this study was pPICZαA [11] for expression in P. pastoris.
E. coli strains were grown at 37°C in low-salt Luria–Bertani (LB) medium (DIFCO) containing 100 μg/ml ampicillin for selection of recombinant clones.
P. pastoris was grown in flasks shaken at 30°C in buffered YEPS medium containing 1% yeast extract, 2% peptone, and 1% sorbitol. The transformants were selected on the appropriate medium containing 25 μg/ml zeocin. Recombinant cultures of P. pastoris were grown in flasks at 30°C in BMGY and BMMY media containing 1% yeast extract, 2% peptone, 100 mM potassium phosphate at pH 6.0, 1.34% YNB, 4 × 10−5 % biotin and 1% glycerol or 0.5% methanol.
2.2. Recombinant DNA techniques
Standard recombinant DNA techniques (preparation and transformation of competent E. coli cells, DNA cloning, restriction enzymes digestion, ligation) were performed according to published procedures [12].
2.3. Site-directed mutagenesis
All mutations were performed using the “quickchange® site-directed mutagenesis kit” (Stratagene). The mutagenic primers used to produce the desired gene alteration based on the induced amino acid alterations (mutated bases shown in bold) are shown in Table 1. Beckman Coulter Genomics performed the sequencing.
Table 1.
Oligonucleotides employed for mutagenesis. Forward (F) and reverse (R) sequences are shown with the mutations in bold letters.
| Mutant | Orientation | Sequence |
|---|---|---|
| Inu M41A | F | ccggaccaatattgggcgaacgagccaaacggcc |
| R | ggccgtttggctcgttcgcccaatattggtccgg | |
| Inu N42G | F | gaccagtactggatgggagagccaaacggcctg |
| R | caggccgtttggctctcccatccagtactggtc | |
| Inu E43D | F | ccggaccagtattggatgaacgatccaaacggcctg |
| R | caggccgtttggatcgttcatccaatactggtccgg | |
| Inu Q59A | F | cctggcacctgttctttgcgcacaatccgacggcc |
| R | ggccgtcggattgtgcgcaaagaacaggtgccagg | |
| Inu P62G | F | Ttctttcaacacaatggcacggccaatgtatggggcaatatttgctgggg |
| R | Ccccagcaaatattgccccatacattggccgtgccattgtgttgaaagaa | |
| Inu W67A | F | ccgacggccaatgtagcgggcaatatatgctgggggc |
| R | gcccccagcatatattgcccgctacattggccgtcgg | |
| Inu I70A | F | ccaatgtatggggcaacgcatgctgggggcacgctacg |
| R | cgtagcgtgcccccagcatgcgttgccccatacattgg | |
| Inu F99A | F | ggatgagaacggagtcgaagcggctaccggtaccgcc |
| R | ggcggtaccggtagccgcttcgactccgttctcatcc | |
| Inu R175A | F | cgggcggccttgagagtgcggatccaaaggtattcttcc |
| R | ggaagaatacctttggatccgcactctcaaggccgcccg | |
| Inu N265A | F | ggatcccctgccggtggtgccggggtgctagctatcaccgg |
| R | ccggtgatagctagcaccccggcaccaccggcaggggatcc | |
| Inu R295A | F | ggctggacaatggggctgatttcgatggagctctgagc |
| R | gctcagagctccatcgaaatcagccccattgtccagcc | |
| Inu D298A | F | ggacaatgggcgtgatttcgctggagctctgagctggg |
| R | cccagctcagagctccagcgaaatcacgcccattgtcc |
A molecular model of the N42G mutant was built from the X-ray structure of the wild-type enzyme (PDB: 3SC7). The EsyPred3D program was used [13].
2.4. Expression of recombinant and wild-type enzymes
P. pastoris X-33 cells were transformed by electroporation with 10 μg of SacI-linearized plasmid DNA carrying wild-type or mutated endo-inulinase encoding genes. Freshly transformed cells were plated onto solid YEPS medium containing 25 μg/ml zeocin. Positive transformants were checked for the endo-inulinase gene by PCR analysis. Recombinant cultures were grown in flasks (shaking at 150 rpm) at 30°C in 10 ml of BMGY medium for 1 day, reaching A600 = 2.0–6.0 (approximately 16–18 h). The cells were harvested and resuspended to an A600 of 1.0 in 50 ml of BMMY medium. Methanol (100% v/v) was added to the cultures to a final concentration of 0.5% (v/v) every 24 h to maintain induction. Proteins were recovered from the culture supernatants by centrifugation at 4000 rpm for 20 min and then filtered on 0.22 μMillipore filters (Millipore).
2.5. Inulinase purification
After filtration, supernatant was dialyzed at 4°C in 10 mM phosphate buffer at pH 7.5. The resulting enzyme was applied to a DEAE sepharose CL-6B column pre-equilibrated with the same buffer. Endo-inulinase was eluted with a linear gradient of NaCl from 0 to 500 mM in the same buffer. The protein was then dialyzed against a 50 mM sodium acetate buffer (pH 5.0) in order to keep the protein in its optimal conditions. The purity of the protein was checked on SDS–PAGE stained with Coomassie Blue.
2.6. Enzymatic determination
2.6.1. Plate assay
An inulin-agar plate was prepared with 20 mM phosphate buffer (pH6) containing 4% inulin. Equal amounts of proteins from the P. pastoris supernatant were spotted onto the inulin-agar plate and incubated at 50°C for 12 h.
2.6.2. Spectrophotometric assays
Inulinase activity was assayed by measuring the amount of reducing sugars released from inulin using Somogy–Nelson's method [21]. The reaction mixture was composed of 60 μl of suitably diluted protein, 440 μl of 4% inulin from a solution of dahlia tubers (Sigma Chemical Co.) in 50 mM phosphate buffer at pH 6. The reaction was carried out for 10 min at 50°C. Inulinase activity was determined spectrophotometrically by recording the increase in optical density (OD) at 520 nm.
2.6.3. Thin-layer chromatography assay
Thin-layer chromatography (TLC) was carried out on silica gel 60 plate. The plates were developed at room temperature for 2 h with a solvent system of ethyl acetate–acetic acid–water (2:1:1 vol/vol/vol). The sugar spots were visualised by spraying the plates with 5% sulfuric acid in methanol and heating them at 100°C for 3 min.
2.6.4. Kinetic analysis of inulin hydrolysis by N42G mutant and wild-type enzyme
Kinetic assays were performed using wild-type endo-inulinase at a final concentration of 0.6 μg ml−1 and N42G mutant at a final concentration of 9 μg ml−1. The reaction was carried out for 48 h. Samples were taken after 0.1, 0.5, 2, 5, 22, 30 and 48 h. After 48 h of incubation, the mutant sample was separated into three samples. Wild-type enzyme was added to the first, one mutant enzyme to the second, and one inulin to the third. The same proportions as described above were used. After 5 and 12 h, aliquots were taken. The amount of equivalent fructose was determined by Somogy–Nelson's method [21]. The hydrolysis products were analysed in parallel by TLC.
3. Results
3.1. Structural localization of different mutations and site-directed mutagenesis
Several residues located in the neighbourhood of the two catalytic glutamic acids, E43 and E233 [5,14], and /or belonging to the substrate pocket [10] may play a role in the catalytic mechanism of endo-inulinase leading to the cleavage of inulin to generate mainly inulotriose (DP3) during the late stage of the reaction [15,16]. These residues (shown in Fig. 1) are as follows: M41 and N42 in the WMN(D/E)PN conserved motif; P62, W67 and I70 from the so-called loop 1; N265 at the end of the so-called loop 3; R295 and D298 at the beginning of loop 4 and R175 from the conserved motif RDP. Residues Q59 and the F99 that are also found in the substrate cavity were also selected. These 11 residues were substituted in the wild-type enzyme by direct mutagenesis to a G or an A in order to potentially increase the size of the pocket. Finally, although this leads to highly decreased activity [5], the catalytic E 43 in the conserved WMN(D/E)PN motif was replaced by a D that is a smaller residue and is conserved in most hydrolases from GH32, 43, 62 and 68 [14].
Fig. 1.
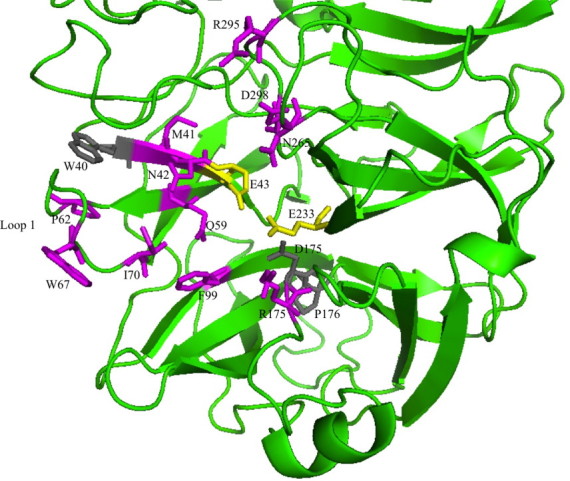
N-terminal part of the crystal structure of endo-inulinase INU2 from A. ficuum with the mutated residues indicated and coloured in purple. The two catalytic residues, E43 and E 233, are shown in yellow. The conserved W40, D175 and P176 are shown in grey. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Comparative activity and specificity assays
Each mutant obtained was overexpressed in P. pastoris and the recombinant protein was efficiently recovered in the supernatant as previously described [17]. After gel filtration on a PD10 column, the supernatants were analysed by SDS gel electrophoresis.
The concentration of proteins was determined and quantities of proteins between 0.5 and 10 μg were used in the spectrophotometric enzymatic assay. The wild-type enzyme was optimally active at pH 5 and 50°C. The 12 single mutants exhibited the same optimal pH and temperature as the wild type.
Activity was compared by plate, spectrophotometric and thin-layer chromatography assay. The experimental results from the plate and spectrophotometric assays led to class the mutants into 3 groups (Fig. 2, Table 2). In the first group, mutants R175A, P62G, Q59A, W67A showed <2.5% of the wild-type activity. The activity of group 2, i.e. mutants E43D, F99A and N42G, was about 6% of the wild-type activity. The other mutants, I70A, M41A, D298 A, N265A and R295A in group 3 retained good activity, albeit lower than that of the wild-type enzyme.
Fig. 2.
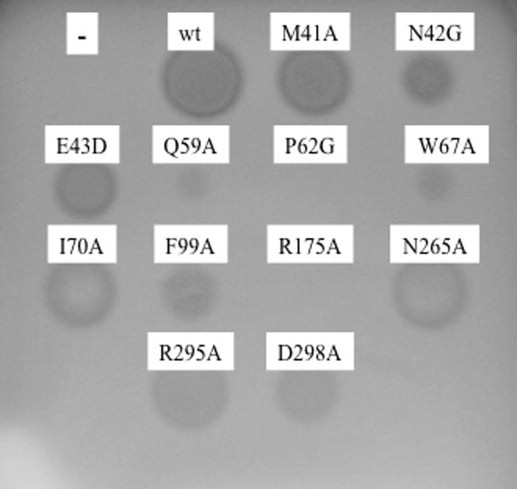
Inulinase activities of wild-type and mutant enzymes. Supernatants of P. pastoris carrying each recombinant plasmid were spotted onto agar plates containing 4% inulin 3D structure and incubated for 12 h at 50 °C.
Table 2.
Enzymatic activity of the wild-type and mutant inulinases. Enzymatic activities were determined by the Somogy method as mentioned in Section 2.6.2. The relative activities were estimated based on the mean of three experiments.
| Mutation | Localisation | Relative activity |
|---|---|---|
| Wild type | 100 | |
| R175A | +2 | 0.1 ± 0,02 |
| P62G | Loop 1 | 0.2 ± 0,05 |
| Q59A | −1 | 2.2 ± 0,4 |
| W67A | −2 | 2.4 ± 0,3 |
| E43D | −1 | 5.4 ± 0,8 |
| F99A | −1 | 6.1 ± 0,75 |
| N42G | −1 | 7.1 ± 0,5 |
| I70A | −2 | 38.9 ± 2,3 |
| M41A | – | 56.2 ± 3,8 |
| D298A | – | 59.7 ± 5,6 |
| N265A | +1 | 72.9 ± 4,1 |
| R295A | – | 84.6 ± 3,9 |
The specificity of the different mutants was then analysed by thin-layer chromatography. The typical reaction mixture was incubated at 50°C for 22 h. Aliquots were taken after 6 and 22 h and analysed by TLC for the hydrolysis products. All the mutants showed a classical wild-type profile of hydrolysis with an equal amount of inulotetraose (F4) and inulotriose (F3), except for the N42G mutant that produced more F4 than F3 (Fig. 3).
Fig. 3.

Thin-layer chromatogram of hydrolysed inulin. (a) Inulin hydrolysed 22 h with endo-inulinase (A). 1, 2, 3, 4 lines are standard oses like fructose (F), kestose (GF2), 1,1-kestotetraose (GF3) and 1,1,1-kestotetraose (GF4). (b) Inulin hydrolysed after treatment for 6 and 22 h with the wild-type (A) and mutant (N/G) endo-inulinase (B). Inulotriose (F3) and inulotetraose (F4) were produced.
3.3. Hydrolysis kinetics of the N42G mutant
To study the basic reaction kinetics, N42G and the wild-type proteins were purified. The amount of the N42G mutant used was 15 times higher than that of the wild type in order to remain within a similar range of OD after reaction with 4% inulin. Aliquots of the reaction mixture were withdrawn periodically and analysed for the hydrolysis products by spectrophotometric methods and TLC. After 48 h, the hydrolysed products from the N42G mutant were enhanced in DP4 compared to those of the wild type enzyme. At that point, the sample was divided into three parts. In the first batch, the N42G enzyme was added but the OD did not change. In the second, wild-type enzyme was added. OD increased and the TLC revealed appearance of DP3 in conjunction with disappearance of DP4. When inulin was added, OD increased and DP4 was still the main product observed (Fig. 4). These results show that the N42G enzyme is still active after 48 h, meaning that it is unable to hydrolyse more of its target. On the contrary, the hydrolysed product from N42G is still a substrate for the wild-type enzyme.
Fig. 4.
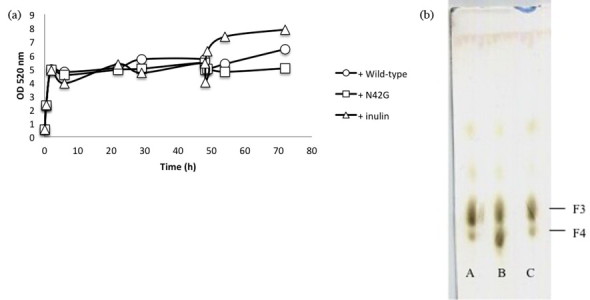
Analysis of N42G activity specificity (a) by following the appearance of reducing sugars at 590 nm. After 48 h of digestion of inulin with the N42G mutant, wild-type enzyme (circle) or the N42G mutant (square) or inulin (triangle) were added and the change of reducing sugar amounts was observed. (b) Thin-layer chromatography. The digestion products were produced by a first hydrolysis of inulin with the wild-type enzyme (A) or the N42G mutant (B, C). After 48 h, a second digestion was performed by adding wild-type enzyme (A, C) or the N42G mutant (B). Inulotriose (F3) and inulotetraose (F4) were produced.
In order to understand the particular behaviour of the N42G mutant, a molecular model was built from the crystal structure of the wild-type (WT) enzyme. This substitution does not change the global folding of the protein. Since the WT N42 side chain points to the centre of the catalytic site, its removal clearly induces the enlargement of the substrate cavity, which is then less obstructed to stabilize F4, the major product of inulin hydrolysis of the mutant.
4. Discussion
Multiple analyses of ligand-bound structures and site-directed mutagenesis experiments were performed to identify key residues in substrate binding and recognition in the GH32 family and GH68 and particularly in invertases and exo-inulinases.
Until now, in endo-inulinase enzymes, except for catalytic residues E43, D176 and E 233 that have been well characterized, only residue W40 has been described as essential [5]. Specific cleavage of inulin by endo-inulinase has been shown to form inulotriose and inulotetraose in equal amounts. The role of conserved residues around or in the catalytic site on the specificity of the cleavage was examined here. These residues were chosen based on the recently determined crystal structure of INU2 from A. ficuum [10].
Those that were most affected in their total activity were the group 1 mutants, i.e. R175A, P62G, Q59A and W67A. Residue D176 is conserved and present in the active sites of fructan metabolizing enzymes and in invertases. The mutation of R175 from the RDP motif into A resulted in a complete loss of activity as already described by Chen et al. for invertase [18] and by Kuzuwa et al. for Lactobacillus casei exo-inulinase [19]. In Arabidopsis thaliana cell wall invertase (AtcwINV1), this same residue, R148, together with N149, W82 and D239, was reported to play a role in stabilization of the fructose ring [20]. A similar role of substrate binding orientation is proposed for endo-inulinase. R175 was actually observed to form an H bond in subsite +2 with the substrate analogue [10].
Proline residues can act as structural disruptors of helices, and as turning points in sheets. P62 lies at the beginning of loop 1, which has been hypothesized to be responsible for enlargement of the endo-inulinase catalytic site. P62G is therefore thought to modify the conformation of loop 1, therefore changing the position of W67 thought to stabilize the substrate in the enlarged cavity. W67A also showed less than 2.5% of wild-type activity. This highlights the importance of the conformation of loop 1 for the specific activity of endo-inulinase. The corresponding residue in Thermotoga maritima invertase, F74 (T. maritima numbering), was reported to border on the fructose-binding pocket in T. maritima invertase. In addition, the same residue in Bambusa oldhamii, W159, replaced by an L or F residue and increasing the Km values for sucrose, confirms its role in fructose binding. Furthermore, W67, together with W40 and F99, forms an aromatic zone conserved at the rim of the active site among GH32 and GH68 families [21]. It seems to be important for optimal and stable binding of sucrose in invertases, as described by Le Roy et al. [22]. The known importance of W40 [5] together with the experimental results on W67 and F99 therefore revealed the importance of this aromatic environment for the binding of inulin. In the endo-inulinase of A. ficuumor or the invertase of A. thaliana, W67 or 47 respectively, affects the activity more than mutations of F99 or W82 [22].
The striking reduction in activity of the Q59A mutant confirms its importance in substrate binding at subsite −1 as is suggested not only by the structural data from INU2 [10] but also from Aspergillus awamori's exo-inulinase [23] and Thermotoga maritima's invertase [24].
N42, E43, and F99, from group 2, were also previously shown to surround a substrate analogue in subsite −1. As a consequence, their respective mutants induced a large decrease in the relative activity. F99, which belongs to the aromatic zone discussed above, was less affected than W40 and W67. Mutant N42G results in a drastic drop in activity, but interestingly, this is accompanied by the ability to generate more F4 than F3. In all the other mutations, the F3/F4 ratio was unchanged compared to the wild type. This residue is therefore important for activity as well as product specificity. Based on the modelled structure of this mutant, we can hypothesize that the removal of the N42 side-chain leads to the enlargement of the catalytic site and/or may stabilize the substrate in a new orientation so that the enzyme can produce inulotetraose. In addition, replacement of the corresponding W271 residue preceding the nucleophile D272 in Lactobacillus reuteri, 121 fructansucrase, from the GH68 family also results in a change of specificity of this enzyme. Indeed, in the W271N mutant, larger fructans were produced compared to the wild-type enzyme, probably due to the difference in size between W and N [25]. Moreover, in fructosyltransferase of Allium cepa of the GH32 family, substitution of the corresponding N84 by a G results in an inactive enzyme and the N84S mutant leads to a modification in its specificity. The mutant adds fructose residues preferably to a terminal fructose rather than to glucose although the wild type does both. Finally, N42G may also influence the positioning of E43, partly explaining the 15-fold decrease in activity.
E43, the nucleophile catalytic residue, was substituted by a D which contains a shorter side chain and is present in other glycosylhydrolases from families 32, 43, 62 and 68. The resulting specific activity is about 5% of the wild type, in agreement with the 1.1% residual activity reported previously [5]. This underscores the importance of the orientation and distance requirement for nucleophilic attack. Although the D residue is smaller than E, no change in the size of the products was observed.
The activity of the third group composed of I70A, M41A, D298A, N265A and R295A was slightly affected. I70A activity was the most affected, which could be explained by its position in subsite −2 and loop 1, and its close contact with the second fructose molecule of the substrate analogue, as shown by the wild-type structure [10].
N265A that is predicted to belong to subsite +1 does not affect the total activity. R295 is not located in the catalytic site and was shown to interact with or be close to a mannose molecule in an adjacent pocket. Therefore, its mutation does not significantly affect the enzyme activity.
In the crystal structure, M41A and D298A point outwards from the catalytic site. Their effect on the activity is yet not fully understood.
In conclusion, we have identified residues here that play a critical role in overall activity, including P62, W67, Q59 and R175. We have also found that N42 plays an important role in the specificity of hydrolysis. The mutation of N42 to G leads to the production of F4 as the major product of inulin hydrolysis. These results could lead to interesting applications in the context of the use of IOS as prebiotics in food industry applications such as baked goods, milk desserts, chocolate, and their positive effect on human health.
Acknowledgments
The authors also thank Prof. Johan Wouters (UNamur), Prof. Jean Vandenhaute (UNamur) and Prof. Jean-Marie Frère (Ulg) for helpful advice.
Catherine Michaux thanks FRS-FNRS for her research associate position. The Walloon Region financially supported part of this study through project conventions D31-1167 and D31-1215. The authors thank the local contact people for beamline ProximaI (SOLEIL Synchrotron Radiation Facility, Gif-sur-Yvette, France) and the FRS-FNRS for financial support and access to the synchrotron.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Chi Z., Chi Z., Zhang T., Liu G., Yue L. Inulinase-expressing microorganisms and applications of inulinases. Appl. Microbiol. Biotechnol. 2009;82:211–220. doi: 10.1007/s00253-008-1827-1. [DOI] [PubMed] [Google Scholar]
- 2.Chi Z.M., Zhang T., Cao T.S., Liu X.Y., Cui W., Zhao C.H. Biotechnological potential of inulin for bioprocesses. Bioresour. Technol. 2011;102:4295–4303. doi: 10.1016/j.biortech.2010.12.086. [DOI] [PubMed] [Google Scholar]
- 3.Roberfroid M. Prebiotics: the concept revisited. J. Nutr. 2007;137:830S–837S. doi: 10.1093/jn/137.3.830S. [DOI] [PubMed] [Google Scholar]
- 4.Uhm T.B., Chung M.S., Lee S.H., Gourronc F., Housen I., Kim J.H., Van Beeumen J., Haye B., Vandenhaute J. Purification and characterization of i endoinulinase. Biosci. Biotechnol. Biochem. 1999;63:46–51. doi: 10.1271/bbb.63.146. [DOI] [PubMed] [Google Scholar]
- 5.Park S., Han Y., Kim H., Song S., Uhm T.B., Chae K.S. Trp17 and Glu20 residues in conserved WMN(D/E)PN motif are essential for Aspergillus ficuum endoinulinase (EC 3.2.1.7) activity. Biochemistry (Mosc). 2003;68:658–661. doi: 10.1023/a:1024669810540. [DOI] [PubMed] [Google Scholar]
- 6.Reddy V.A., Maley F. Identification of an active-site residue in yeast invertase by affinity labeling and site-directed mutagenesis. J. Biol. Chem. 1990;265:10817–10820. [PubMed] [Google Scholar]
- 7.Reddy A., Maley F. Studies on identifying the catalytic role of Glu-204 in the active site of yeast invertase. J. Biol. Chem. 1996;271:13953–13957. doi: 10.1074/jbc.271.24.13953. [DOI] [PubMed] [Google Scholar]
- 8.Meng G., Futterer K. Structural framework of fructosyl transfer in Bacillus subtilis levansucrase. Nat. Struct. Biol. 2003;10:935–941. doi: 10.1038/nsb974. [DOI] [PubMed] [Google Scholar]
- 9.Basso A., Spizzo P., Ferrario V., Knapic L., Savko N., Braiuca P., Ebert C., Ricca E., Calabro V., Gardossi L. Endo- and exo-inulinases: enzyme–substrate interaction and rational immobilization. Biotechnol. Prog. 2010;26:397–405. doi: 10.1002/btpr.334. [DOI] [PubMed] [Google Scholar]
- 10.Pouyez J., Mayard A., Vandamme A.M., Roussel G., Perpete E.A., Wouters J., Housen I., Michaux C. First crystal structure of an endo-inulinase, INU2, from Aspergillus ficuum: Discovery of an extra-pocket in the catalytic domain responsible for its endo-activity. Biochimie. 2012;94:2423–2430. doi: 10.1016/j.biochi.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 11.Invitrogen (2001) EasySelect Pichia Expression Kit. A manual of methods for expression of recombinant proteins using pPICZ and pPICZalpha in Picha pastoris. in EasySelect Pichia Expression Kit Version G 122701 (Technologies, I. l., ed).
- 12.Sambrook J., Fritsch E.F., Maniatis T. (second ed.) Cold Spring Harbor Laboratory Press; 1989. Molecular Cloning, A Laboratory Manual. [Google Scholar]
- 13.Lambert C., Leonard N., De Bolle X., Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 14.Pons T., Naumoff D.G., Martinez-Fleites C., Hernandez L. Three acidic residues are at the active site of a beta-propeller architecture in glycoside hydrolase families 32, 43, 62, and 68. Proteins. 2004;54:424–432. doi: 10.1002/prot.10604. [DOI] [PubMed] [Google Scholar]
- 15.Uhm T., C. S., Lee D., Kim S., Cassart J.-P., Vandenhaute J. Cloning and nucleotide sequence of the endoinulinase-encoding gene, inu2, from Aspergillus ficuum. Biotechnol. Lett. 1998;20:809–812. [Google Scholar]
- 16.Uhm T.B., Chung M.S., Lee S.H., Gourronc F., Housen I., Kim J.H., Van Beeumen J., Haye B., Vandenhaute J. Purification and characterization of Aspergillus ficuum endoinulinase. Biosci. Biotechnol. Biochem. 1999;63:146–151. doi: 10.1271/bbb.63.146. [DOI] [PubMed] [Google Scholar]
- 17.Al Balaa B., Brijs K., Gebruers K., Vandenhaute J., Wouters J., Housen I. Xylanase XYL1p from Scytalidium acidophilum: site-directed mutagenesis and acidophilic adaptation. Bioresour. Technol. 2009;100:6465–6471. doi: 10.1016/j.biortech.2009.06.111. [DOI] [PubMed] [Google Scholar]
- 18.Chen T.H., Huang Y.C., Yang C.S., Yang C.C., Wang A.Y., Sung H.Y. Insights into the catalytic properties of bamboo vacuolar invertase through mutational analysis of active site residues. Phytochemistry. 2009;70:25–31. doi: 10.1016/j.phytochem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Kuzuwa S., Yokoi K.J., Kondo M., Kimoto H., Yamakawa A., Taketo A., Kodaira K. Properties of the inulinase gene levH1 of Lactobacillus casei IAM 1045; cloning, mutational and biochemical characterization. Gene. 2012;495:154–162. doi: 10.1016/j.gene.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Lammens W., Le Roy K., Van Laere A., Rabijns A., Van den Ende W. Crystal structures of Arabidopsis thaliana cell-wall invertase mutants in complex with sucrose. J. Mol. Biol. 2008;377:378–385. doi: 10.1016/j.jmb.2007.12.074. [DOI] [PubMed] [Google Scholar]
- 21.Lammens W., Le Roy K., Schroeven L., Van Laere A., Rabijns A., Van den Ende W. Structural insights into glycoside hydrolase family 32 and 68 enzymes: functional implications. J. Exp. Bot. 2009;60:727–740. doi: 10.1093/jxb/ern333. [DOI] [PubMed] [Google Scholar]
- 22.Le Roy K., Lammens W., Verhaest M., De Coninck B., Rabijns A., Van Laere A., Van den Ende W. Unraveling the difference between invertases and fructan exohydrolases: a single amino acid (Asp-239) substitution transforms Arabidopsis cell wall invertase1 into a fructan 1-exohydrolase. Plant Physiol. 2007;145:616–625. doi: 10.1104/pp.107.105049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagem R.A., Rojas A.L., Golubev A.M., Korneeva O.S., Eneyskaya E.V., Kulminskaya A.A., Neustroev K.N., Polikarpov I. Crystal structure of exo-inulinase from Aspergillus awamori: the enzyme fold and structural determinants of substrate recognition. J. Mol. Biol. 2004;344:471–480. doi: 10.1016/j.jmb.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 24.Alberto F., Jordi E., Henrissat B., Czjzek M. Crystal structure of inactivated Thermotoga maritima invertase in complex with the trisaccharide substrate raffinose. Biochem. J. 2006;395:457–462. doi: 10.1042/BJ20051936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozimek L.K., Kralj S., Kaper T., van der Maarel M.J., Dijkhuizen L. Single amino acid residue changes in subsite −1 of inulosucrase from Lactobacillus reuteri 121 strongly influence the size of products synthesized. FEBS J. 2006;273:4104–4113. doi: 10.1111/j.1742-4658.2006.05411.x. [DOI] [PubMed] [Google Scholar]