

Abstract
The flavoprotein monooxygenase (FPMO) from Stenotrophomonas maltophilia (SMFMO, Uniprot: B2FLR2) catalyses the asymmetric oxidation of thioethers and is unusual amongst FPMOs in its ability to use the non-phosphorylated cofactor NADH, as well as NADPH, for the reduction of the FAD coenzyme. In order to explore the basis for cofactor promiscuity, structure-guided mutation of two residues in the cofactor binding site, Gln193 and His194, in SMFMO were performed in an attempt to imitate the cofactor binding site of the NADPH-dependent FMO from Methylophaga aminisulfidivorans sp. SK1 (mFMO), in which structurally homologous residues Arg234 and Thr235 bind the NADPH 2′-ribose phosphate. Mutation of His194 to threonine proved most significant, with a switch in specificity from NADH to NADPH [(kcat/Km NADH)/kcat/Km NADPH) from 1.5:1 to 1:3.5, mostly as a result of a reduced Km for NADPH of approximately sevenfold in the His194Thr mutant. The structure of the Gln193Arg/His194Thr mutant revealed no substantial changes in the backbone of the enzyme or orientation of side chains resulting from mutation. Mutation of Phe52, in the vicinity of FAD, and which in mFMO is an asparagine thought to be responsible for flavin hydroperoxide stabilisation, is, in SMFMO, a determinant of enantioselectivity in sulfoxidation. Mutation of Phe52 to valine resulted in a mutant that transformed para-tolyl methyl sulfide into the (S)-sulfoxide with 32% e.e., compared to 25% (R)- for the wild type. These results shed further light both on the cofactor specificity of FPMOs, and their determinants of enantioselectivity, with a view to informing engineering studies of FPMOs in the future.
Keywords: Flavoprotein monoxygenase, Baeyer–Villiger monoxygenase, Sulfoxide, Biocatalyst, NADPH
Abbreviations: SMFMO, Stenotrophomonas maltophilia flavin monoxygenase; mFMO, flavin monooxygenase from Methylophaga aminisulfidivorans; PAMO, phenylacetone monooxygenase; CHMO, cyclohexanone monooxygenase
Highlights
-
•
SMFMO was mutated to investigate cofactor specificity and enantioselectivity.
-
•
The Gln193Arg/His194Thr mutant displayed a preference for NADPH, rather than NADH.
-
•
The structure of the Gln193Arg/His194Thr mutant was determined.
-
•
Active site mutants were assessed for enantioselectivity in sulfoxidation reactions.
-
•
The Phe52Val mutant displayed inverted enantioselectivity.
1. Introduction
Flavoprotein monooxygenases [1] are a class of oxidative enzymes which, in addition to having important roles in metabolism in eukaryotes [2] and in the production of secondary metabolites in lower organisms [3,4], have potential for applications in asymmetric organic synthesis. This is due to the ability of different subgroups of these enzymes (‘A’–‘F’ [1]) to catalyse useful oxygenation reactions such as hydroxylation and Baeyer–Villiger (BV) oxidation. FPMOs of Class ‘B’, which include Baeyer–Villiger monooxygenases (BVMOs), N-hydroxylating monooxygenases and flavin monooxygenases (FMOs), also catalyse the asymmetric oxidation of heteroatom-containing compounds (Fig. 1) such as prochiral thioethers [5]. Class B FPMOs most often exist in the form of a single polypeptide that employs the nicotinamide cofactor NADPH to reduce bound flavin (FAD), forming FADH2, which reacts with molecular oxygen to form the oxidising species, a (hydro)peroxy flavin intermediate [1], which then undergoes a nucleophilic attack by the sulphur atom of the thioether. The majority of these enzymes have been shown to be dependent on the phosphorylated nicotinamide cofactor NADPH as the hydride donor in the flavin reduction step. There are also NADH-dependent flavoprotein monooxygenases, but in these cases, the reduction of flavin (in that case FMN) and the oxygenation of substrate are accomplished by different enzymes, an NADH oxidase and an oxygenating subunit respectively. (Class ‘C’ flavoprotein monooxygenases [1].) Because of the lower cost of the non-phosphorylated cofactor NADH, there have been several attempts to re-engineer nicotinamide cofactor specificity in, for example, the Baeyer–Villiger monooxygenase group of FPMOs from NADPH to NADH [6,7], as such a change may provide an advantage in the commercial application of FPMOs.
Fig. 1.
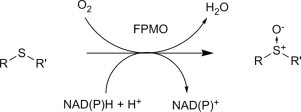
Asymmetric sulfoxidation reactions catalysed by flavoprotein monooxygenases (FPMOs). NAD(P)H is used to reduce the flavin prior to reaction with molecular oxygen, yielding (hydroperoxy)flavin intermediate, which is the oxidant in the reaction.
In previous work [8], we reported the cloning and expression of a gene from the marine bacterium Stenotrophomonas maltophilia that encoded a Class B FPMO, a flavin monooxygenase named SMFMO, which was unusual in its ability to preferentially employ NADH as the nicotinamide cofactor for flavin reduction. SMFMO therefore catalysed the NADH-dependent asymmetric oxidation of prochiral thioethers and also the regioselective Baeyer–Villiger oxidation of the model Baeyer–Villiger monooxygenase substrate bicyclo[3.2.0]hept-2-en-6-one, the latter albeit with poor enantioselectivity. Recently, a more extended group of FMOs related to SMFMO, which also accepts either NADH or NADPH as cofactor, has been described encoded in the genome of Rhodococcus jostii RHA1 [9]. In the interests of investigating the molecular basis of cofactor dependence, the structure of SMFMO was also determined [8] and compared with the structure of a known NADPH-dependent FMO, called mFMO, from the methylotrophic bacterium Methylophaga aminisulfidivorans strain SK1 [10]. mFMO has recently been employed for the asymmetric oxidation of a series of prochiral sulfides to give predominantly (S)-sulfoxide products with moderate to good enantiomeric excess [11]. In structural studies by two groups [12,13], the molecular determinants governing the binding of the 2′ ribose phosphate of NADPH in mFMO, and which discriminates between NADPH and NADH, have been revealed (PDB: 2XLP, PDB: 2XVJ and related structures). The comparison of the structures of SMFMO and mFMO [6] provided leads for further investigation of the cofactor preference observed in SMFMO, and suggested avenues for the potential alteration of cofactor specificity in FPMOs as a whole.
In this paper we report the results of investigations designed to probe the determinants of cofactor promiscuity in SMFMO, and also mutagenesis of the active site immediately surrounding the flavin coenzyme, which has revealed a residue, Phe52, having influence over the enantioselectivity of sulfoxidation by this enzyme.
2. Results and discussion
2.1. Mutations affecting cofactor specificity in SMFMO
The structure of SMFMO was determined to be a dimer, with a monomer fold similar to that of mFMO, and featured one molecule the coenzyme FAD in each subunit [8]. Although NAD(P)H was not observed in the active site, the growth of the crystals in high concentrations of lithium sulfate resulted in sulfate ions being bound to NAD(P)H phosphate binding sites of SMFMO, as identified by superimposition of the structure with the mFMO–NADPH complex [8]. This superimposition revealed that the relaxation in nicotinamide cofactor specificity in this enzyme might be attributed to the substitution of two residues found in NADPH-dependent mFMO: Arg234 and Thr235 (observed to bind the 2′ phosphate on the NADPH ribose in that enzyme) for a glutamine (Gln193) and histidine (His194) in SMFMO (Fig. 2). In order to explore the determinants of cofactor promiscuity in SMFMO, we therefore constructed mutants Gln193Arg, His194Thr and the double mutant Gln193Arg/His194Thr, each of which was expressed in the soluble fraction of cells of Escherichia coli to approximately the same extent as the wild-type enzyme. The oxidation of NADPH and NADH by each variant was measured and compared to the wild-type (Table 1).
Fig. 2.
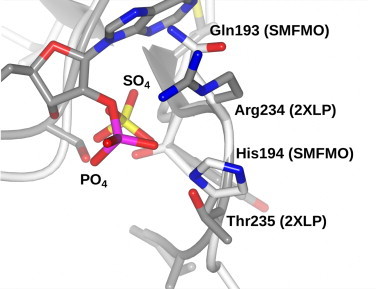
Superimposition of nicotinamide cofactor binding site in flavoprotein monooxygenase mFMO from Methylophaga aminisulfidivorans (2XLP, grey) and SMFMO (white), showing residues Gln193 and His194 in SMFMO targeted for mutation. The SMFMO structure was determined in the presence of sulfate ions, which bind in the phosphate site(s).
Table 1.
Kinetics of nicotinamide cofactor oxidation by SMFMO and mutants of Gln193 and His194.
| SMFMO variant | Km (NADH M x 10−6) | kcat (NADH s−1) | kcat/Km (NADH s−1 M−1) | Km (NADPH M x 10−6) | kcat (NADPH s−1) | kcat/Km (NADPH s−1 M−1) |
|---|---|---|---|---|---|---|
| Wild-type | 23.7 ± 9.1 | 0.029 ± 0.003 | 1223 | 27.3 ± 5.3 | 0.022 ± 0.002 | 805 |
| Gln193Arg | 21.2 ± 4.3 | 0.031 ± 0.003 | 1476 | 8.3 ± 1.6 | 0.007 ± 0.0005 | 875 |
| His194Thr | 14.1 ± 2.9 | 0.012 ± 0.0008 | 857 | 4.2 ± 0.7 | 0.012 ± 0.0004 | 3000 |
| Gln193Arg/His194Thr | 12.8 ± 6.9 | 0.010 ± 0.002 | 769 | 4.1 ± 2.8 | 0.003 ± 0.0003 | 743 |
Mutation of Gln193 to arginine appears to have resulted in a mutant with a fourfold reduced Km for NADPH, although turnover was compromised, resulting in a mutant of comparable catalytic efficiency (kcat/Km) to the wild-type with NADPH. Binding and turnover of NADH was not greatly affected in this mutant, compared to wild-type activity. Mutation of His194 to threonine on the other hand, resulted in an enzyme of approximately threefold increased catalytic efficiency with NADPH owing largely to a sevenfold decrease in the Km for NADPH. Binding of NADH was also slightly improved, although with a drop in turnover rates. A combination of these effects was observed with the double mutant Gln193Arg/His194Thr,in which the lower Km and lower kcat of the Gln193Arg and His194Thr mutants respectively combined to give a mutant of lower catalytic efficiency overall. Thus, the His194Thr mutation resulted in reduced catalytic efficiency with NADH and increased efficiency with NADPH, mostly through a significantly reduced Km, and an overall change in ratio of (kcat/Km NADH)/kcat/Km NADPH) from 1.5:1 to 1:3.5. The greater contribution of the threonine residue to NADPH binding is perhaps reflected in the bond distances from the threonine 235 side chain to the ribose 2′ phosphate oxygen in mFMO structures such as 2XLP: this is 2.6 Å compared to 3.2–3.5 Å for the arginine 234 side-chain nitrogen atoms with relevant phosphate oxygens.
In order to exclude the possibility that substitution of Gln193 and His194 for the residues arginine and threonine had caused major structural changes in the cofactor binding site, we determined the crystal structure of the Gln193Arg/His194Thr mutant of SMFMO at 2.60 Å resolution. In contrast to the wild-type structure, which featured two molecules in the asymmetric unit arranged as a dimer, the double mutant structure solution revealed eight monomers, with four dimers. Omit maps after refinement in the absence of flavin clearly revealed residual density for the FAD at the active site of each subunit. In common with the wild-type structure, there was no density corresponding to a stretch of residues in the region of Glu210 to Asp234, and these areas could not be modeled. Each subunit was otherwise largely complete, although subunit ‘A’ featured the poorest density for side-chains and had average B-factors of 71 versus an average value of 50 for the assembly overall. Subunits ‘A’ from the wild-type structure and subunit ‘H’ (which had the lowest ‘B’-factors of 40) from the Gln193Arg/His194Thr mutant structure were superposed in order to examine whether the introduction of the new residues was likely to have distorted the cofactor 2′ phosphate binding region in the enzyme. An overlap of the cofactor binding region of these subunits from the wild-type and double-mutant structures (Fig. 3) actually revealed very little movement of the backbone and the position of the sulfate ion (which overlays with the NADPH 2′ phosphate from NADPH complex structures of mFMO), is also conserved from wild-type to double mutant enzyme.
Fig. 3.
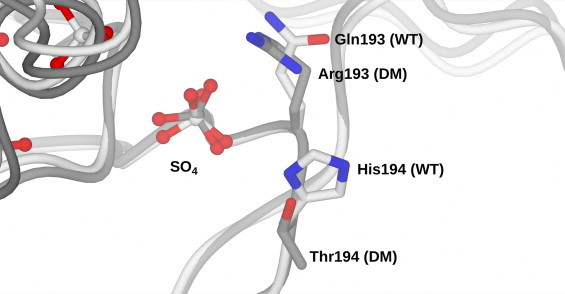
Superimposition of nicotinamide cofactor binding site in wild-type SMFMO (WT, white) and Gln193Arg/His194Thr double mutant (DM, grey). The r.m.s.d. between the wild-type structure and double mutant structure over 330 C-alpha atoms was 0.42 Å.
Analysis of SMFMO variants with phosphate binding site mutations demonstrates that, while the strength of NADPH binding can, in part, be determined by the identity of the residue at position 194 and also 193, a straight swap of residue(s) for those found in NADPH-dependent FMOs does not engineer absolute NADPH specificity in the cofactor-promiscuous enzyme. This may simply be a steric effect, as residual space is still available in the cofactor binding site to accommodate the smaller NADH ribose 2′ hydroxyl. In NADPH-dependent mFMO, this residual space is also available so it remains difficult to conclude how NADH binding might be disfavoured in that enzyme. The activity of mFMO with NADH has not, to our knowledge, been reported however, so it is possible that some activity with NADH exists. However, the Arg/Thr couple in the cofactor phosphate binding pocket of mFMO is conserved amongst other NADPH-specific flavoprotein monooxygenases such as the BVMOs phenylacetone monooxygenase [14] and cyclohexanone monooxygenase from Rhodococcus [15]. In a study of nicotinamide cofactor binding in PAMO, Fraaije and co-workers determined that the relevant arginine 217 was essential for NADPH binding, but that the mutation of the adjacent threonine (218) did not have a significant effect in that instance [7]. Interestingly, substitution of a histidine residue at position 220 in PAMO for a glutamine, increased the catalytic efficiency of the enzyme with NADH threefold, however, the new glutamine is not structurally homologous with Gln193 in SMFMO. Although mutation of Gln193 in our study did not radically affect cofactor specificity in SMFMO, there is extensive precedent for glutamine as a facilitating determinant of cofactor promiscuity in, for example, formate dehydrogenases (FDHs). Most FDHs are NAD+ dependent, yet both mutational studies [16] and genomic screens [17] have revealed enzymes of either promiscuous cofactor recognition ability or of strict NADPH dependence. The structural basis for this change is partly the exchange of a glutamate or aspartate residue in NADH-dependent enzymes, which repel negatively charged phosphate, for a glutamine, which is neutral. In an effort, therefore, to engineer strict NADH-specificity in SMFMO, Gln193 was mutated to Glu, but unfortunately this resulted in a mutant that was not expressed in the soluble fraction of the E. coli cells used for the study. Further structural and mechanistic studies of SMFMO and the larger group of cofactor-promiscuous FMOs identified by Riebel and co-workers [9] may help to shed further light on cofactor specificity in this group of FPMOs.
2.2. Mutations affecting enantioselectivity of sulfoxidation in SMFMO
Mutations in and around the active site of Baeyer–Villiger monooxygenases and flavin-dependent monooxygenases such as mFMO have revealed amino acid residues that have effects on both the activity and selectivity of the enzymes [18,19]. The structure of SMFMO revealed the environment surrounding the flavin, and allowed for a structure-informed approach to the analysis of the active site, with a view to either improving or altering the enantioselectivity of the enzyme. In order to identify residues with possible direct effects on ligand binding, the active site was superimposed with the structure of mFMO that had been determined in complex with the substrate indole ([13]; PDB: 2XVJ; Fig. 4). On this basis, Phe52, Asn173 and Ser174 were selected as the first residues for mutation, as being close to the indole ligand and also the C4 position of the flavin which is the site of formation of the (hydro)peroxy flavin intermediate. The mutants were tested for their ability to oxidise the substrates methyl-p-tolyl sulfide and ethyl phenyl sulfide and also for their enantioselectivity towards those substrates (Table 2).
Fig. 4.
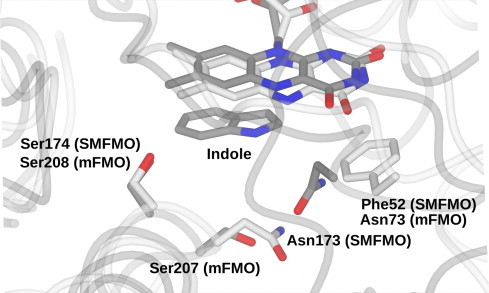
Superimposition of flavin-binding site of SMFMO (carbon atoms in white) with flavin-binding site of mFMO (carbon atoms in grey) in complex with the substrate indole (taken from PDB: 2XVJ; [13]) and illustrating sites chosen for mutation in SMFMO in order to investigate enantioselectivity in SMFMO.
Table 2.
Enantioselectivity of selected SMFMO mutants towards the substrates methyl-p-tolyl sulfide and ethyl phenyl sulphide.
| Variant | Absolute configuration and enantiomeric excess of sulfoxide product |
|
|---|---|---|
| Methyl-p-tolyl sulfoxide product of biotransformation of methyl-p-tolyl sulfide | Ethyl phenyl sulfoxide product of biotransformation of ethyl phenyl sulfide | |
| Wild-type | (R)-, 25% e.e. | (R)-, 71% e.e. |
| Asn173Ser | (S)-, 12% e.e. | n.d. |
| Ser174Cys | (R)-, 37% e.e. | (R)-, 48% e.e. |
| Phe52Val | (S)-, 32% e.e. | Racemic |
| Phe52Arg | (R)-, 38% e.e. | n.d. |
| Phe52Leu | (R)-, 24% e.e. | (R)-, 67% e.e. |
n.d. = not determined.
Mutation of asparagine 173 resulted largely in variants of low activity, although soluble expression was comparable to that obtained with the wild-type SMFMO. Mutation of Serine 174 resulted again in inactive mutants except where the mutation was conservative, to cysteine. Although neither interacts directly with flavin, superimposition of the mFMO structure in complex with NADPH suggests that residues 173 and 174 may be in proximity to the nicotinamide ring of the cofactor; indeed position 173 is occupied by tyrosine 212 in mFMO, in which it is thought to act as a ‘backdoor’ residue, protecting the reactive site from bulk solvent [20].
Phe52 mutants proved to be more active, with mutation to hydrophobic leucine giving a mutant which was also moderately enantioselective with the chosen substrates. However, the Phe52 mutant with a smaller hydrophobic group, valine, gave an oxidised product of inverted absolute configuration compared to the wild-type enzyme. In an effort to augment this effect, Phe52 was also mutated to smaller alanine, but in this case, the mutant was not expressed well, and cell extracts that contained it did not appear yellow, suggesting that a side-chain of minimum size is required in this position to maintain flavin binding. Using the Phe52Val mutant therefore, the inversion of enantioselectivity was explored with other thioether substrates (Table 3). Whilst enantioselectivity was poor in most cases, the absolute configuration of products was opposite to that obtained with the wild-type enzyme save for the p-chloro thioanisole, in which the (R)-selectivity was maintained, although substantially reduced.
Table 3.
Biotransformation of sulfides by Phe52Val mutant of SMFMO compared to biotransformations of equivalent substrates using the wild-type enzyme.
| Substrate | Absolute configuration and enantiomeric excess of sulfoxide product |
|
|---|---|---|
| WT SMFMO | Phe52Val mutant | |
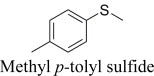 |
(R)-, 25% e.e. | (S)-, 32% e.e. |
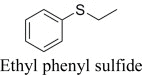 |
(R)-, 71% e.e. | (S)-, 11% e.e. |
 |
(R)-, 24% e.e. | (S)-, 7% e.e. |
 |
(R)-, 21% e.e. | (S)-, 8% e.e. |
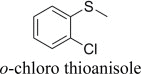 |
(S)-, 19% e.e. | (S)-, 19% e.e. |
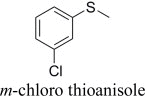 |
(R)-, 30% e.e. | (S)-, 41% e.e. |
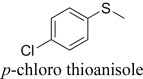 |
(R)-, 80% e.e. | (R)-, 28% e.e. |
The role of Phe52 in SMFMO appears to be different to that served by structurally homologous residues in other FPMOs. In mFMO, the position is occupied by asparagine [11]. Mutation of this residue, even conservatively to serine, resulted in an inactive enzyme [20]. It has been suggested that Asn73 is involved in stabilisation of the flavin hydroperoxide in mFMO, as its mutation results in an enzyme that is unable to oxygenate exogenous substrates [20]. However, in other FPMOs, the mutation of residues in this position has had little or no effect on enzyme activity [21]. In SMFMO, the residue appears to have a role in substrate binding, as the substitution of a smaller residue, valine, has resulted in mutants of altered enantioselectivity. The enantioselectivity of FPMOs with respect to sulfoxidation has successfully been altered previously through mutation at, for example, Met446 in the active site in PAMO, for which structure-guided mutation resulted in a variant, Met446Gly, which catalysed the oxidation of methyl p-tolyl sulfide to its (R)-sulfoxide enantiomer with 92% e.e. compared to 6% for the wild-type [19]. In the absence of a structure, Reetz and co-workers used directed evolution to create mutants of cyclohexanone monooxygenase (CHMO) from Acinetobacter calcoaceticus, which were enantiocomplementary in respect of the oxidation of methyl p-methyl benzyl thioether, with mutant Phe432Ser giving the (R)-sulfoxide enantiomer with 98.7% e.e. and the Phe16Leu/Phe277Ser variant giving the (S)-product with 95.2% e.e. [18]. At this point it is interesting to note that a number of different sites in FPMOs have proved to be valuable in respect of mutation for altered or improved enantioselectivity, including sites both proximal and remote from the flavin binding site.
FPMOs continue to be an interesting group of enzymes in respect of their potential application in industrial biotechnology. In order to realise this potential, applications of the enzymes will benefit from both structure-guided and random mutagenesis studies that may help to expand their substrate repertoire, or provide catalysts of altered or improved selectivity. Amongst the FPMOs, SMFMO is, despite its poor enantioselectivity, interesting as it is one of the only enzymes, along with the BVMO mekA [22] and the recently identified group of enzymes from Rhodococcus sp. RHA1 [17] to demonstrate acceptance of the cheaper nicotinamide cofactor NADH. The studies reported herein identify His194 in particular as a determinant of cofactor promiscuity in SMFMO and suggest avenues for engineering of other FPMOs for NADH acceptance. In addition, the mutation of Phe52 in SMFMO has revealed not an absolute requirement for this residue but rather an interesting effect on the active site recognition of sulfide substrates, such that enantioselectivity is altered by its substitution with an amino acid side-chain of smaller size. The nature and role of this residue in SMFMO and structurally homologous residues in other FPMOs is therefore very much still under scrutiny.
3. Materials and methods
3.1. Generation of mutant SMFMO genes and purification of SMFMO variants
The gene encoding wild-type SMFMO (B2FLR2) was the same as that described previously [8]. Point mutations in the gene were generated using site-directed mutagenesis employing either mutation specific primers or an NDT codon-based approach for the generation of restricted libraries at certain positions, as described by Reetz and co-workers [23]. A full list of primers for mutagenic PCR can be found in Table 4. Following PCR, reaction mixtures were subjected to digest with the enzyme DpnI, after which the mixtures were used to transform cells of E. coli Novablue singles™ (Merck-Millipore, USA). Plasmids recovered using standard miniprep techniques from colony transformants were subjected to DNA sequencing to confirm the incorporation of the desired mutations. Using the mutant plasmids, the SMFMO muteins were expressed using techniques established for WT-SMFMO [8] (Table 4).
Table 5.
Data collection and refinement statistics for Q193R/H194T double mutant of SMFMO (figures in brackets correspond to data corresponding to the highest resolution shell).
| Q193R/H194T double mutant of SMFMO | |
|---|---|
| Beamline | Diamond I03 |
| Wavelength (Å) | 0.97630 |
| Resolution (Å) | 59.78–2.60 (2.65–2.60) |
| Space group | P32 |
| Unit cell (Å) | a = 170.54; b = 170.54; c = 101.80 |
| α = β = 90.00; γ = 120 | |
| No. of molecules in the asymmetric unit | 8 |
| Unique reflections | 102,301 (6014) |
| Completeness ( %) | 97.8 (99.4) |
| Rmerge ( %) | 0.15 (0.56) |
| Rp.i.m. | 0.12 (0.44) |
| Multiplicity | 4.8 (4.6) |
| 〈I/σ(I)〉 | 4.6 (2.1) |
| CC1/2 | 0.99 (0.71) |
| Overall B factor from Wilson plot (Å2) | 43 |
| Rcryst/ Rfree ( %) | 25.5/29.5 |
| r.m.s.d 1–2 bonds (Å) | 0.011 |
| r.m.s.d 1–3 angles (o) | 1.675 |
| Avge main chain B (Å2) | 50 |
| Avge side chain B (Å2) | 51 |
| Avge water B (Å2) | 34 |
Table 4.
PCR oligonucleotide primers used in this study.
| Mutant | Forward primer | Reverse primer |
|---|---|---|
| Q193R | GAAACGACTTGGATCACACGTCACGAGCCGGCCTTTCTGGC | GCCAGAAAGGCCGGCTCGTGACGTGTGATCCAAGTCGTTTC |
| H194T | GACTTGGATCACACAGACCGAGCCGGCCTTTCTGG | CCAGAAAGGCCGGCTCGGTCTGTGTGATCCAAGTC |
| N173X | GCAATTATCGGTGGCGGTNDTTCTGGCGCACAG | n/a |
| S174X | CGGTGGCGGTAATNDTGGCGCACAGATCCTGGC | n/a |
| F52X | GGCATTCTCTGCATCTGNDTAGCCCAGCGGGCTGG | n/a |
3.2. Enzyme assays and biotransformations
Steady-state kinetic constants for the NADH and NADPH oxidation by SMFMO and its variants were determined using the method employed for hydroxyacetophenone monooxygenase (HAPMO, [6]) and employed previously for SMFMO [8]. Biotransformations using cell lysates with cofactor recycling were performed using the method of Faber and co-workers [24] and as used for WT-SMFMO previously [8]. Standard GC and chiral GC analysis of the biotransformation of thioether substrates used in this study was performed as for the WT-SMFMO [8].
3.3. Crystallisation of SMFMO Gln193Arg/His194Thr double mutant
The SMFMO Gln193Arg/His194Thr double mutant was crystallised in conditions similar to that used for the wild-type structure [8]. The crystals used for diffraction experiments were obtained from hanging crystal drops set up in 24-well plate crystallisation dishes. The reservoir contained 0.9 M lithium sulphate in 100 mM bis–tris propane buffer (pH 5.6). The protein concentration used was 3 mg mL−1. Crystals were tested for diffraction using a Rigaku Micromax-007HF fitted with Osmic multilayer optics and a MARRESEARCH MAR345 imaging plate detector. The crystals that diffracted to a resolution of greater than 3 Å were flash-cooled in liquid nitrogen in a cryogenic solution containing the mother liquor that also contained 10% (v/v) glycerol.
3.4. Data collection, structure solution, model building and refinement of SMFMO Gln193Arg/His194Thr double mutant
A complete dataset was collected on beamline I03 at the Diamond Light Source, Oxfordshire, UK. The data were processed and integrated using XDS [25] and scaled using SCALA [26] as included within the Xia2 processing system [27]. The data collection statistics are given in Table 5. The crystals were in space group P32. The structure was solved using the program MOLREP [28] using a monomer of the WT SMFMO structure (PDB: 4a9w) as the model. The solution on this occasion contained eight monomers (A–H) in the asymmetric unit, which constituted four dimers in the final model. The structure was improved using iterative rounds of building in Coot [29] and refinement using REFMAC [30]. In refinement, non-crystallographic symmetry (NCS) restraints were applied for chains B–H against chain A. Following building of the protein and the water molecules, the flavin coenzyme FAD was modelled into residual density in the omit maps in each of the eight subunits. The final structure had Rcryst and Rfree values of 25.5% and 29.5% respectively and was finally validated using PROCHECK [31]. The Ramachandran plot showed 96% of residues to be situated in the most favoured regions, 3.7% in additional allowed and only tyrosine residue Tyr139 in each subunit exhibiting unusual backbone conformations. The coordinates and structure factors have been deposited in the Protein Databank with the accession code PBD: 4c5o.
Acknowledgements
We are grateful to the Biotechnology and Bioscience Research Council and PML Applications Ltd for funding a CASE studentship to C.N.J.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.van Berkel W.J.H., Kamerbeek N.M., Fraaije M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 2.Cashman J.R., Zhang J. Human flavin-containing monooxygenases. Annu. Rev. Pharmacol. Toxicol. 2006;46:65–100. doi: 10.1146/annurev.pharmtox.46.120604.141043. [DOI] [PubMed] [Google Scholar]
- 3.Beam M., Bosserman M.A., Noinaj N., Wehenkel M., Rohr J. Crystal structure of Baeyer–Villiger monooxygenase MtmOIV, the key enzyme of the mithramycin biosynthetic pathway. Biochemistry. 2009;48:4476–4487. doi: 10.1021/bi8023509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seo M.-J., Zhu D., Endo S., Ikeda H., Cane D.E. Genome mining in Streptomyces. Elucidation of the role of Baeyer–Villiger monooxygenases and non-heme iron-dependent dehydrogenase/oxygenases in the final steps of the biosynthesis of pentalenolactone and neopentalenolactone. Biochemistry. 2011;50:1739–1754. doi: 10.1021/bi1019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grogan G. Asymmetric enzymatic sulfoxidation. In: Carreira E.M., Yamamoto H., editors. Comprehensive Chirality. Elsevier; 2012. pp. 295–328. [Google Scholar]
- 6.Kamerbeek N.M., Fraaije M.W., Janssen D.B. Identifying determinants of NADPH specificity in Baeyer–Villiger monooxygenases. Eur. J. Biochem. 2004;271:2107–2116. doi: 10.1111/j.1432-1033.2004.04126.x. [DOI] [PubMed] [Google Scholar]
- 7.Dudek H., Torres Pazmiño D., Rodríguez C., de Gonzalo G., Gotor V., Fraaije M.W. Investigating the coenzyme specificity of phenylacetone monooxygenase from Thermobifida fusca. Appl. Microbiol. Biotechnol. 2010;88:1135–1143. doi: 10.1007/s00253-010-2769-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen C.N., Cartwright J., Ward J., Hart S., Turkenburg J.P., Ali S.T., Allen M.J., Grogan G. A flavoprotein monooxygenase that catalyses a Baeyer–Villiger reaction and thioether oxidation using NADH as the nicotinamide cofactor. Chembiochem. 2012;13:872–878. doi: 10.1002/cbic.201200006. [DOI] [PubMed] [Google Scholar]
- 9.Riebel A., de Gonzalo G., Fraaije M.W. Expanding the biocatalytic toolbox of flavoprotein monooxygenases from Rhodococcus jostii RHA1. J. Mol. Catal. B Enzym. 2013;88:20–25. [Google Scholar]
- 10.Choi H.S., Kim J.K., Cho E.H., Kim Y.K., Kim J.I., Kim S.W. A novel flavin-containing monooxygenase from Methylophaga sp. strain SK1 and its indigo synthesis in Escherichia coli. Biochem. Biophys. Res. Commun. 2003;306:930–936. doi: 10.1016/s0006-291x(03)01087-8. [DOI] [PubMed] [Google Scholar]
- 11.Rioz-Martinez A., Kopacz M., de Gonzalo G., Torres Pazmiño D.E., Gotor V., Fraaije M.W. Exploring the biocatalytic scope of a bacterial flavin-containing monooxygenase. Org. Biomol. Chem. 2011;9:1337–1341. doi: 10.1039/c0ob00988a. [DOI] [PubMed] [Google Scholar]
- 12.Alfieri A., Malito E., Orru R., Fraaije M.W., Mattevi A. Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. USA. 2008;105:6572–6577. doi: 10.1073/pnas.0800859105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho H.J., Cho H.Y., Kim K.J., Kim M.H., Kim S.W., Kang B.S. Structural and functional analysis of bacterial flavin-containing monooxygenase reveals its ping-pong-type reaction mechanism. J. Struct. Biol. 2011;175:39–48. doi: 10.1016/j.jsb.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Malito E., Alfieri A., Fraaije M.W., Mattevi A. Crystal structure of a Baeyer–Villiger monooxygenase. Proc. Natl. Acad. Sci. USA. 2004;101:13157–13162. doi: 10.1073/pnas.0404538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirza I.A., Yachnin B.J., Wang S., Grosse S., Bergeron H., Imura A., Iwaki H., Hasegawa Y., Lau P.C.K., Berghuis A.M. Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J. Am. Chem. Soc. 2009;131:8848–8854. doi: 10.1021/ja9010578. [DOI] [PubMed] [Google Scholar]
- 16.Andreadeli A., Platis D., Tishkov V., Popov V., Labrou N.E. Structure-guided alteration of coenzyme specificity of formate dehydrogenase by saturation mutagenesis to enable efficient utilization of NADP+ FEBS J. 2008;275:859–3869. doi: 10.1111/j.1742-4658.2008.06533.x. [DOI] [PubMed] [Google Scholar]
- 17.Hatrongjit R., Packdibamrung K. A novel NADP+-dependent formate dehydrogenase from Burkholderia stabilis 15516: screening, purification and characterization. Enzyme Microb. Technol. 2010;46:557–561. [Google Scholar]
- 18.Reetz M.T., Daligault F., Brunner B., Hinrichs H., Deege A. Directed evolution of cyclohexanone monooxygenases: enantioselective biocatalysts for the oxidation of prochiral thioethers. Angew. Chem. Int. Ed. 2004;43:4078–4081. doi: 10.1002/anie.200460311. [DOI] [PubMed] [Google Scholar]
- 19.Torres Pazmiño D.E., Snajdrova R., Rial D.V., Mihovilovic M.D., Fraaije M.W. Altering the substrate specificity and enantioselectivity of phenylacetone monooxygenase by structure-inspired enzyme redesign. Adv. Synth. Catal. 2007;349:1361–1368. [Google Scholar]
- 20.Orru R., Pazmiño D.E.T., Fraaije M.W., Mattevi A. Joint functions of protein residues and NADP(H) in oxygen activation by flavin-containing monooxygenase. J. Biol. Chem. 2010;285:35021–35028. doi: 10.1074/jbc.M110.161372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald C.A., Fagan R.L., Collard F., Monnier V.M., Palfey B.A. Oxygen reactivity in flavoenzymes: context matters. J. Am. Chem. Soc. 2011;133:16809–16811. doi: 10.1021/ja2081873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Völker A., Kirschner A., Bornscheuer U.T., Altenbuchner J. Functional expression, purification and characterization of the recombinant Baeyer–Villiger monooxygenase MekA from Pseudomonas veronii MEK700. Appl. Microbiol. Biotechnol. 2008;77:1251–1260. doi: 10.1007/s00253-007-1264-6. [DOI] [PubMed] [Google Scholar]
- 23.Reetz M.T., Kahakeaw D., Lohmer R. Addressing the numbers problem in directed evolution. Chembiochem. 2008;9:1797–1804. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- 24.Hall M., Stueckler C., Kroutil W., Macheroux P., Faber K. Asymmetric bioreduction of activated alkenes using cloned 12-oxophytodienoate reductase isoenzymes OPR-1 and OPR-3 fromLycopersicon esculentum (Tomato): a striking change of stereoselectivity. Angew. Chem. Int. Ed. 2007;46:3934–3937. doi: 10.1002/anie.200605168. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 27.Winter G. Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010;3:186–190. [Google Scholar]
- 28.Vagin A., Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 29.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski R.A., Macarthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]