

Abstract
Two strategies for the synthesis of the icetexane diterpenoids icetexone and epi-icetexone that rely on Ga(III)-catalyzed cycloisomerization of alkynyl indene substrates to yield fused [6-7-6] tricycles have been explored. In the first approach, access to a tricycle bearing a gem-dimethyl group paved the way for explorations of C–H functionalization of one of the methyl groups in close proximity to a hydroxyl-directing group. This approach was ultimately unsuccessful and led only to ring cleaved products. In the second approach, an alkynyl indene substrate bearing a cyano substituent was utilized, which was effective in providing a functional handle to access the icetexone subclass of diterpenoids. A key epoxide opening/diazene rearrangement sequence was utilized to complete a formal synthesis of icetexone and epi-icetexone, which is discussed in detail. Furthermore, the cyano-containing substrate has been prepared in enantioenriched form using a Rh-catalyzed conjugate addition reaction, which now provides a route to the enantioselective synthesis of these natural products.
Keywords: Icetexanes, Total synthesis, Ga(III) cycloisomerization, Cycloheptadienes, Diterpenes, Enantioselective conjugate addition
1. Introduction
The icetexane family of diterpenoids is a diverse group of natural products that exhibit a unique structural skeleton and a wide range of biological activity (see Fig. 1). Members of this diterpenoid family possess a [6-7-6] tricyclic motif, which has the systematic name 9(10→20)-abeo-abietane. Congeners vary by the level of oxidation in each ring and the degree of oxygenation of the arene portion. In a recent review, a classification scheme was proposed that divided this family into five groups on the basis of the oxygenation at the C3, C11, C12, and C14 positions (see Fig. 1 for numbering).1 Of these groups, the icetexones are the most complex, exhibiting oxygenation at the C11, C12, and C14 positions, as well as possessing a lactone or ether linkage between C19 and either the C10 or C6 position. Members of the highly oxygenated icetexone subclass have been isolated from different plants of the Salvia genus. Icetexone (1) was the first compound in this family to be isolated and characterized from the aerial parts of Salvia ballotaeflora Benth in 1976 along with the ortho-quinone tautomer, romulogarzone (3).2 X-ray crystallographic analysis enabled a determination of the absolute configuration of 1, the name of which has been used to represent this family of natural products.3 Other members of the icetexone subclass, such as 7,20-dihydroanastomosine (4), 19-deoxyisoicetexone (5), and 19-deoxyicetexone (6) have subsequently been isolated from the same source.4
Fig. 1.
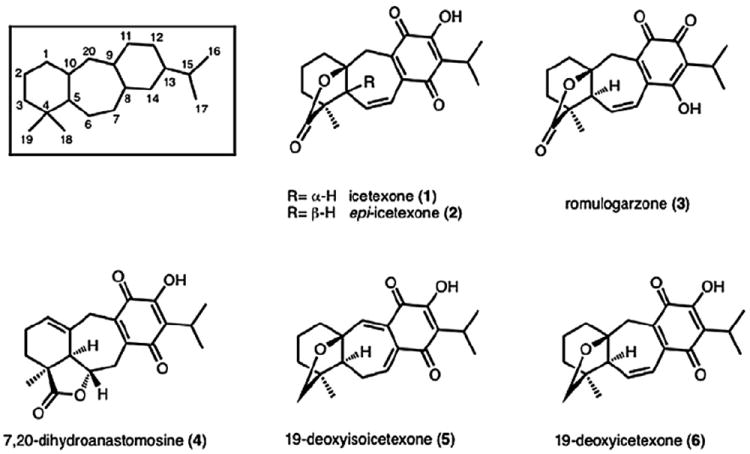
Selected icetexone natural products.
In 2000, Nieto et al. reported the isolation of epi-icetexone (2) from the aerial parts of Salvia gillessi Benth and later, Sanchez et al. reported activity of this compound against Trypanosoma cruzi (IC50 of 6.5±0.75 μM).5 The trypanocidal activity of these diterpenoids has been postulated to arise from a redox cycling process of the quinone moiety, a structural motif that is shared with other trypanocidal agents.6 Of note, in the account pertaining to the biological activity of 2, Sanchez et al. reported the spectral data for isolated epi-icetexone, which matches the data for synthetic icetexone (and not synthetic epi-icetexone) reported by Majetich.7 As such, the Nieto et al. report on the isolation of these compounds may have switched the assignment of the two structures and therefore the reported biological activity for epi-icetexone may in fact be that of icetexone.
The biological activity and interesting structures of the icetexone subclass have garnered interest from the synthetic community. Majetich and Grove completed the first enantioselective syntheses of icetexone, epi-icetexone, and 19-deoxyicetexone in 2009 using a ‘cyclialkylation’ strategy to form the [6-7-6] tricyclic core.7 As noted above, the Majetich synthesis led to reassignment of the spectral data reported in the literature for both icetexone and epi-icetexone. In 2010, we reported formal syntheses of racemic icetexone and epi-icetexone.8 In this full account, we present the details of our studies that led to formal syntheses of icetexone and epi-icetexone as well as the development of a route that accomplishes the formal enantioselective syntheses of these natural products.
Our foray into the synthesis of the icetexone family began with the development of a Ga(III)-catalyzed cycloisomerization reaction of alkynyl indenes (e.g., 9, Scheme 1B) to form benzannulated cycloheptadienes (e.g., 10). This transformation was inspired by a report from Chatani and Murai on the Ga(III)-catalyzed skeletal reorganization of enyne substrates that appeared in 2002 (Scheme 1A).9 By using an appropriate tether for our substrates, a [6-7-6] tricyclic motif could be synthesized selectively and efficiently. Other transition metal salts and complexes commonly utilized for the cycloisomerization of enynes (e.g., PtCl2, PtCl4, [Ru(CO)3Cl]2, [Rh(CO)2Cl]2)10 led to the competing formation of a [7-5-6] tricyclic isomer (e.g., 11). These isomeric byproducts, as well as the starting material, are difficult to separate from the desired products and so it was important that the cycloisomerization reaction was highly selective and proceeded to completion. This demanding criteria was met by the Ga(III) salts that we employed as catalysts. Our initial reports in this area demonstrated that various arene substitution patterns and a gem-dimethyl group on the alkyl tether were well tolerated. The selectivity observed in the formation of the benzannulated cycloheptadiene products utilizing substrates with varying substitution on the benzenoid portion of the indene substrate demonstrated that transposition of the indene double bond (which would ultimately lead to isomeric products) was not a significant competing process. This methodology was subsequently applied to the total synthesis of several members of the icetexane diterpenoid family.11
Scheme 1.
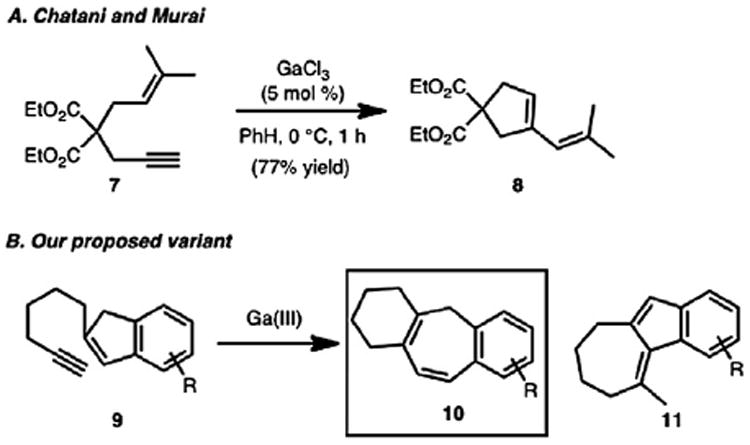
Ga(III)-catalyzed cycloisomerization reaction.
As outlined in Scheme 2, with icetexone as the target, we envisioned a similar strategy for the synthesis of the highly oxygenated icetexone subclass of the icetexane diterpenoids. This reasoning led to our first approach to icetexone and epi-icetexone, which would capitalize on substrates closely related to others that we had prepared previously.
Scheme 2.
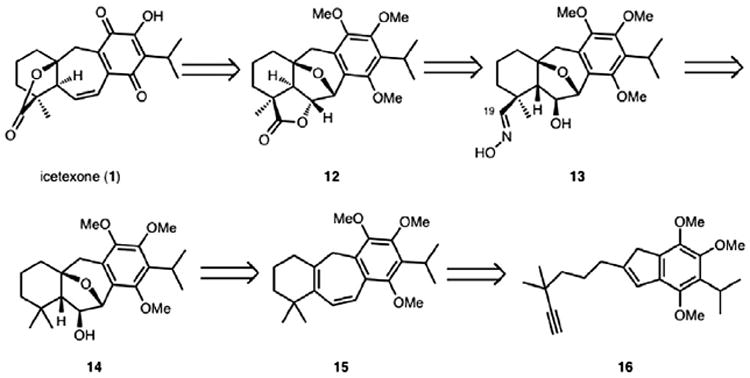
Retrosynthetic analysis of icetexone (1).
2. Results and discussion
2.1. A first approach to icetexone and epi-icetexone
Retrosynthetically (Scheme 2), icetexone (1) could arise from lactone 12 by a concluding oxidation of the hydroquinone moiety to the corresponding quinone as well as cleavage of the lactone ring in 12 and a translactonization. Lactone 12 would be formed from oxime 13 or the corresponding aldehyde, which could be oxidized to the carboxylic acid. Specifically, lactone 12 would arise from the acid derived from oxime 13 using a Mitsunobu displacement to forge the lactone. The oxime (13) was envisioned to be accessible from alcohol 14 using a Barton nitrite ester reaction.12a This classical and powerful C–H functionalization method has been used previously for the functionalization of a range of remote C–H bonds in complex molecule synthesis.12b The hydroxy group of alcohol 14 could be installed through an epoxidation of the tetrasubstituted double bond of diene 15, isomerization to the corresponding dihydrofuran, and hydroboration of the resulting alkene from the β-face. Cycloheptadiene 15 could be obtained from 16 using the Ga(III)-catalyzed cycloisomerization method.
To arrive at alkynyl indene 16, indanone 21 (Scheme 3) was first prepared. This commenced with known alcohol 17,8,13 which was converted to aldehyde 18 using Parikh–Doering oxidation conditions. Aldehyde 18 was subsequently subjected to Horner–Wadsworth–Emmons conditions to provide α,β-unsaturated ester 19. Hydrogenation of 19 (H2, Pd/C) followed by saponification led to the formation of carboxylic acid 20 in 89% yield over the two steps. Indanone 21 arose from an intramolecular Friedel–Crafts acylation of the acid chloride derived from acid 20. The overall sequence for the preparation of indanone 21 is robust and can be scaled to prepare 5 g of 21 in one pass.
Scheme 3.
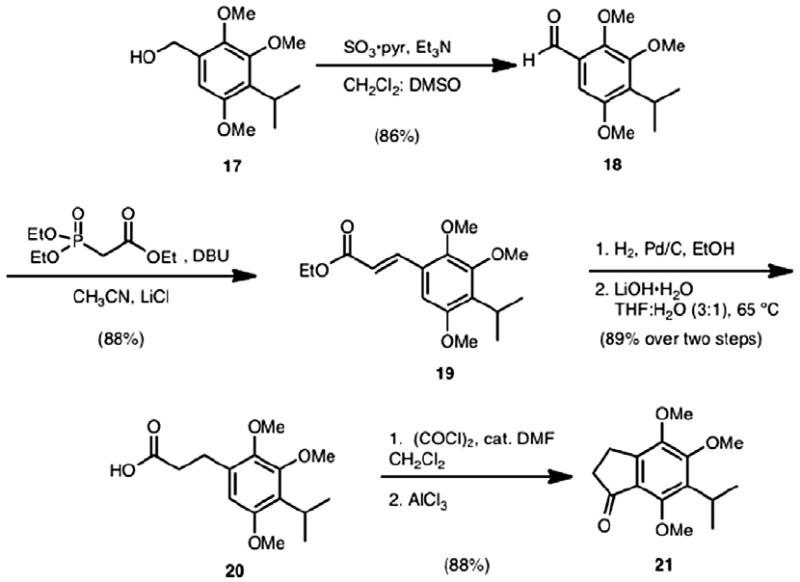
Synthesis of indanone 21.
Formation of β-ketoester 22 (Scheme 4) was accomplished using NaHMDS and Mander’s reagent to favor the desired C-alkylation of indanone 21. Subsequent alkylation of β-ketoester 22 with alkyl iodide 2311a at 65 °C led to adduct 24 in 66% yield over the two steps.
Scheme 4.
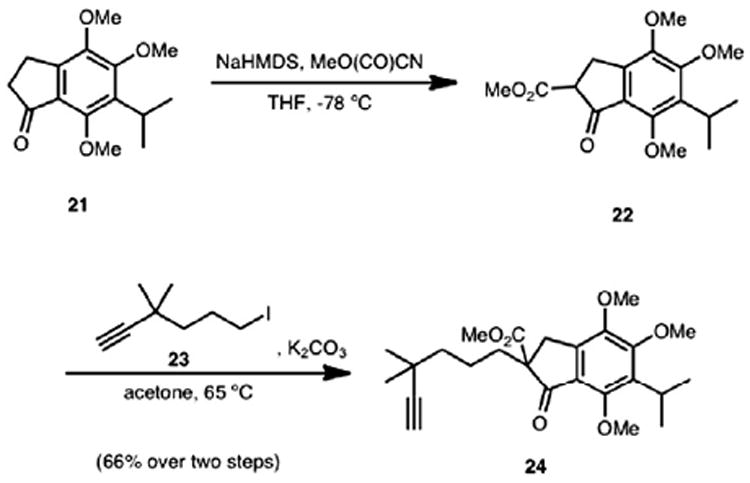
Synthesis of adduct 24.
With β-ketoester 24 in hand, a saponification/decarboxylation protocol was effected to afford indanone 25 (Scheme 5), which underwent reduction using diisobutylaluminum hydride and elimination of the resulting hydroxyl using methanesulfonic anhydride to generate the key alkylnyl indene (16) in 79% yield over three steps. Utilizing the previously optimized conditions for cycloisomerization established by our group (20 mol % GaCl3 and 4 Å molecular sieves at 40 °C), cycloheptadiene 15 was formed in 87% yield. With a route to 15 in place, we initiated a survey of conditions to achieve the remote functionalization of one of the geminal methyl groups (C19).
Scheme 5.
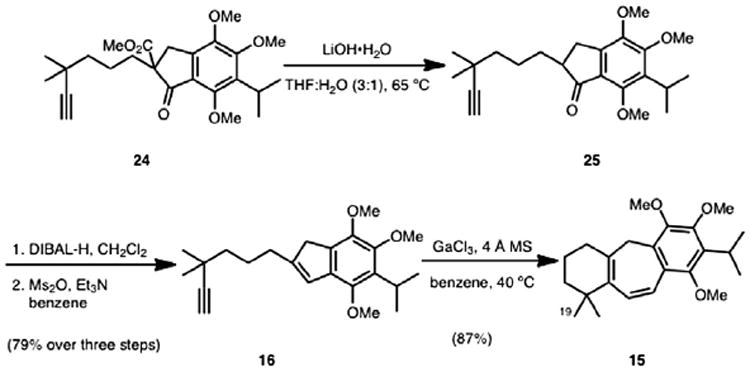
Synthesis of cycloheptadiene 15.
Formation of the substrate for methyl group functionalization (alcohol 26, Scheme 6) was initiated through selective epoxidation of the tetrasubstituted double bond of diene 15 using m-chloroperbenzoic acid (m-CPBA), followed by isomerization of the vinyl epoxide to the dihydrofuran. Hydroboration of the resulting dihydrofuran double bond from the β-face afforded hydroxy tetrahydrofuran 26 in 73% yield over the two steps. Although we were able to synthesize the nitrite ester of 26, an attempted Barton nitrite ester functionalization14 upon irradiation led only to decomposition products. A second attempt at remote functionalization using a Norrish type II protocol was then undertaken. There was cause for optimism with this procedure because of the existing precedent of Jeger, who has demonstrated the formation of a cyclobutanol product by functionalization of an angular methyl group using a Norrish type II procedure.15 Synthesis of Norrish substrate 27 was accomplished by oxidation of alcohol 26 to the corresponding ketone followed by epimerization of the C5 stereocenter. However, upon irradiation of ketone 27, the only identifiable products appeared to arise from the fragmentation of the seven-membered ring to form trace amounts of aldehyde 28.
Scheme 6.
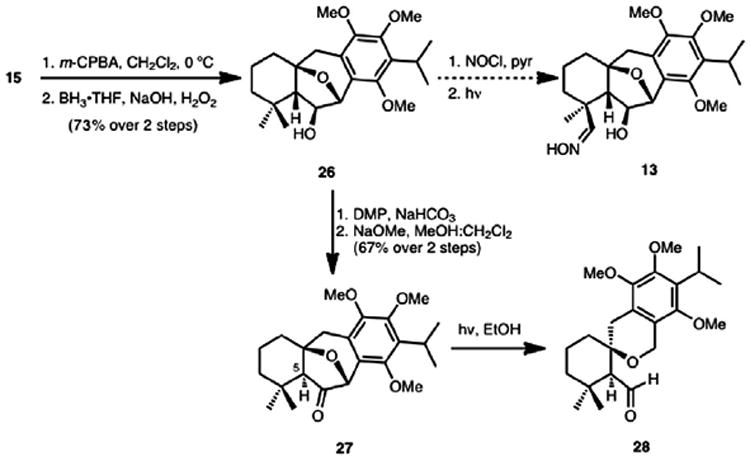
Remote functionalization attempts.
2.2. A second, successful approach to icetexone
The myriad challenges encountered with the remote functionalization strategy led us to devise a new plan to access the icetexone core. We elected to pursue a pre-functionalization of the alkynyl indene cycloisomerization substrate, which would provide a benzannulated cycloheptadiene (e.g., 30, Scheme 7) already possessing a functional handle at C19. Preliminary studies using a range of alcohol derivatives proved futile, but eventually led to the identification of a cyano group as a masked carboxylic acid group that was tolerated by our cycloisomerization conditions as well as subsequent steps.
Scheme 7.
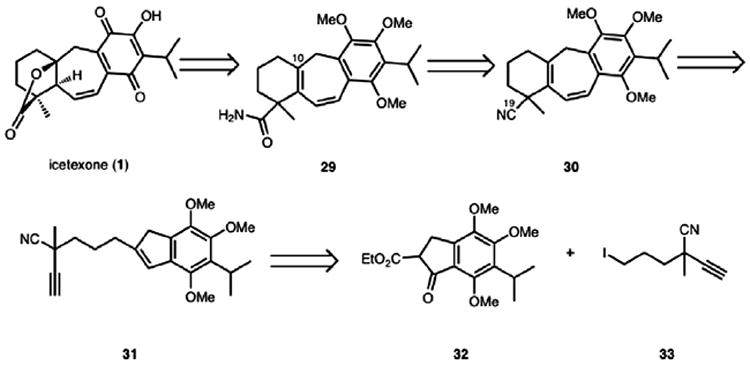
Second generation retrosynthesis.
In accord with this plan (Scheme 7), icetexone would arise from primary amide 29 by oxygenation of the C10 position and subsequent lactone formation. The amide group could form from hydrolysis of nitrile 30, which in turn could come from the Ga(III)-cycloisomerization of alkynyl indene 31. This key substrate could be synthesized by the alkylation of 32 with alkyl iodide 33. Importantly, the use of a nitrile substrate would extend the scope of the Ga(III)-catalyzed cycloisomerization chemistry to prepare [6-7-6] ring systems. Finally, because 33 possesses a stereocenter that would direct the installation of the two additional stereocenters in 1, the preparation of alkyl iodide 33 in enantioenriched form would set the stage for an enantioselective synthesis of icetexone and epi-icetexone.
The synthesis of alkyl iodide 33 (Scheme 8) commenced with commercially available 3-bromo-1-propanol (34). Protection of the hydroxy group with triisopropylsilyl chloride and displacement of the bromide with methyl cyanoacetate in the presence of K2CO3 gave 35 in 90% yield over the two steps. Formation of the quaternary center was accomplished using a standard methylation reaction with methyl iodide and a subsequent chemoselective reduction of the ester group in the presence of the nitrile group was achieved with an excess of sodium borohydride to provide alcohol 36. Oxidation of the alcohol group using Swern conditions followed by an Ohira–Bestmann homologation16 gave rise to alkyne 37 in 85% yield over the two steps. Finally, deprotection of the primary alcohol followed by mesylation and displacement of the mesylate with sodium iodide completed the synthesis of alkyl iodide 33. Using this scalable sequence, 25 g of iodide 33 can be routinely prepared in one pass.
Scheme 8.
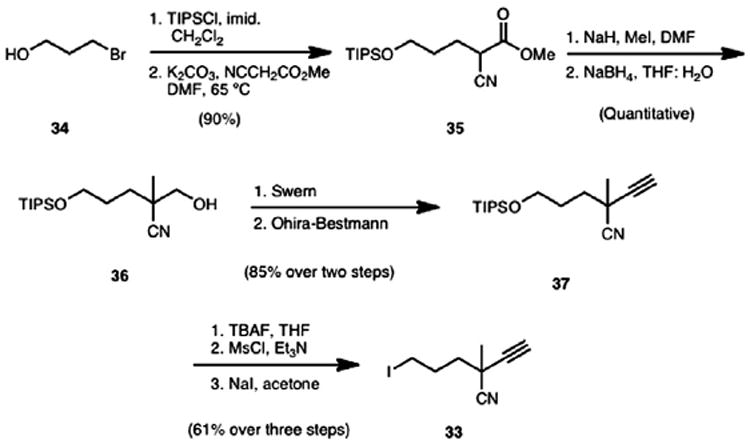
Synthesis of alkyl iodide 33.
Synthesis of the desired cycloisomerization substrate (31, Scheme 9) commenced with a Claisen condensation of indanone 21 with diethyl carbonate to give ketoester 32. β-Ketoester 32 underwent alkylation with alkyl iodide 33 to give adduct 38 in 86% yield over the two steps. A saponification/decarboxylation sequence gave rise to indanone 39, which was reduced with sodium borohydride to afford the indanol as an inconsequential mixture of alcohol diastereomers. Dehydration using KHSO4 completed the synthesis of alkynyl indene 31 in 63% yield over the latter two steps.
Scheme 9.
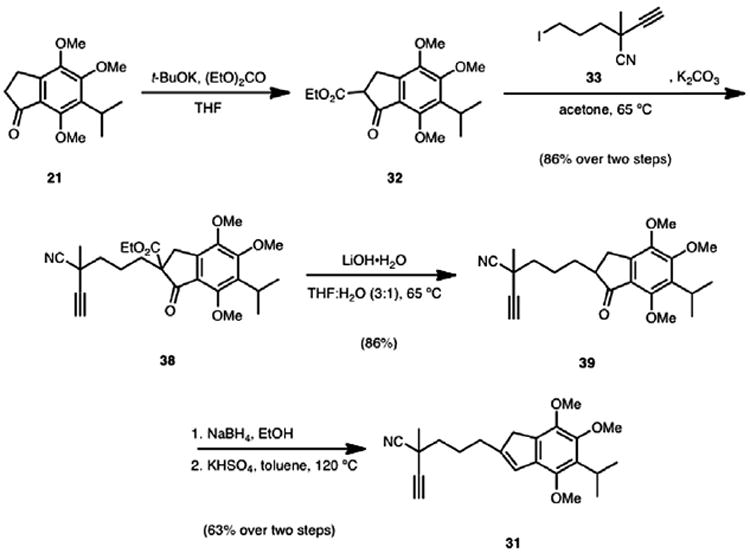
Synthesis of cycloisomerization substrate 31.
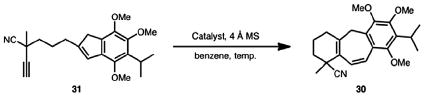
With a route to the key alkynyl indene substrate in hand, several cycloisomerization conditions were surveyed (Table 1). The standard conditions employed in our earlier cycloisomerization studies (entry 1) yielded a mixture of starting material and the desired product in a ratio of 1.36:1.0. It was gratifying to observe only trace decomposition or isomeric byproducts. A study of other Ga(III) salts under the same conditions yielded similar conversion ratios, with GaI3 faring slightly better (compare entries 1–3). In one experiment, complete conversion of alkynyl indene 31 to cycloheptadiene 30 was achieved using 50 mol % GaI3 with heating at 65 °C over 4 days (entry 4). The temperature was then increased in an attempt to decrease the catalyst loading and reaction time. At 80 °C, the catalyst loading could be decreased to 25 mol %, and complete conversion was observed after 4 days (entry 5). Ultimately, it was determined that the reaction time could be decreased to 2 days by heating at 100 °C in toluene (entry 6).
Table 1.
Cycloisomerization optimization
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Equiv | Temp (°C) | Time (d) | Result (SM/Product)a |
| 1 | GaCl3 | 0.2 | 40 | 2 | 1.36:1.0 |
| 2 | GaBr3 | 0.2 | 40 | 2 | 1.29:1.0 |
| 3 | Gal3 | 0.2 | 40 | 2 | 0.63:1.0 |
| 4 | Gal3 | 0.5 | 65 | 4 | Complete |
| 5 | Gal3 | 0.25 | 80 | 4 | Complete |
| 6 | GaCl3 | 0.25 | 100b | 2 | Complete |
Conversion of substrate based on 1H NMR of crude product after aqueous workup.
Reaction run in toluene.
Using the optimized sequence, cycloisomerization of alkynyl indene 31 proceeded in 91% isolated yield to afford cycloheptadiene 30 as the sole product (Scheme 9). Hydrolysis of the tertiary nitrile group was initially attempted using standard acid and base-mediated protocols. However, these proved to be ineffective and either led to no reaction or the non-specific decomposition of the starting material. Perusal of the literature brought to our attention a platinum-based catalyst (see 40) developed by Ghaffar and Parkins,17 which accomplishes the conversion of nitrile groups to primary carboxamides in excellent yields under mild conditions. Gratifyingly, hydrolysis of nitrile 30 using Ghaffar and Parkins’ platinum complex (40) as a catalyst afforded amide 29 in 87% yield.
On the basis of our previous synthetic studies on the icetexane diterpenoid salviasperanol,11a we anticipated that oxygenation at the C10 position of diene 29 could occur through a chemoselective epoxidation of the tetrasubstituted olefin. We were pleased to observe exclusive formation of vinyl oxirane 41 (as a single diastereomer) when diene 29 was treated with m-CPBA. Presumably, epoxidation is directed by the amide functional group to the face bearing the amide group. Initial attempts to open the vinyl epoxide moiety of vinyl oxirane 41 included the use of DIBAL and Red-Al®, as well as the use of Pd(0) catalysts in an attempt to form an intermediate π-allyl species. These efforts simply returned the starting material. Also, on the basis of our previous studies, camphorsulfonic acid was expected to effect an acid-catalyzed isomerization of the vinyl epoxide to the corresponding dihydrofuran, but instead, lactone 42 was isolated. This unexpected, yet fortuitous, outcome could arise from opening of the vinyl epoxide under acidic conditions, followed by trapping of an intermediate allylic carbocation by adventitious water and lactonization with the primary amide.
Although lactone 42 was merely a deoxygenation away from a per-methylated precursor to icetexone and epi-icetexone, all attempts to directly remove the hydroxy group resident at C5 were unsuccessful. For example, we were unable to transform the hydroxy group to a xanthate group in preparation for a Barton deoxygenation, presumably because of the high steric congestion at C5. An attempted ionic reduction of the allylic alcohol with BF3·OEt2 and triethylsilane led only to non-specific decomposition. Because of the potential role of sterics in the lack of reactivity of the hydroxy group of 42, transposition of the hydroxyl using chromium reagents was attempted, but this also led to decomposition products. A stepwise transposition of the hydroxy group of 42 could be achieved by first hydrogenating the double bond followed by elimination using thionyl chloride, which yielded trisubstituted alkene 43 (Scheme 11). Attempted isomerization of the trisubstituted double bond in alkene 43 into conjugation with the arene only returned starting material. Oxidation of the doubly activated benzylic and allylic position of 43 with manganese triacetate gave a stable peroxide intermediate that could be converted to an alcohol (not shown) using an aluminum/mercury amalgam procedure developed by Yu and Corey.18
Scheme 11.
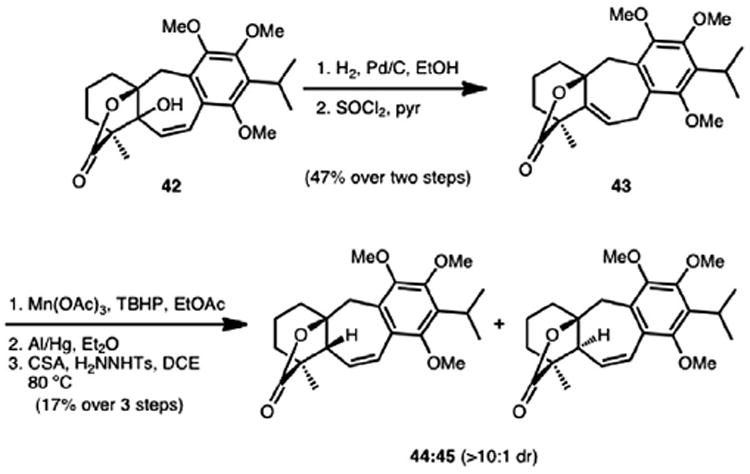
An initial formal synthesis of epi-icetexone.
Subjecting the alcohol generated from Mn(OAc)3/TBHP oxidation of alkene 43 to Myers’ reductive allylic alcohol transposition conditions19 only led to the recovery of the starting material. However, it was reasoned that ionization of the allylic hydroxy group could be possible since a doubly stabilized carbocation would result. Thus, exposure of the alcohol intermediate to acidic conditions in the presence of tosyl hydrazide at elevated temperatures (ca. 80 °C) led to diastereomeric lactones 44 and 45 (44 as the major product, >10:1 dr). Presumably this occurs by the tosyl hydrazide engaging the incipient carbocation followed by loss of sulfinic acid and an ensuing diazene rearrangement. Comparison of the 1H NMR and 13C NMR spectra of 44 to that reported in the literature by Majetich confirmed its structure and thus a formal synthesis of epi-icetexone was achieved.
Although our access to 44 as described in Scheme 11 was serviceable, several attempts were made to improve the synthetic route. It was theorized that the ionization of tertiary alcohol 42 could lead directly to the same reactive carbocation intermediate that was presumed to engage the tosyl hydrazide nucleophile. To test this hypothesis, allylic alcohol 42 was exposed to camphorsulfonic acid in the presence of tosyl hydrazide. Gratifyingly, lactone 44 was obtained (Scheme 12), albeit in low yield. This observation eliminated the need to undergo the lengthy and low yielding transposition sequence previously described (see Scheme 11). Furthermore, on the basis of the formation of 42 from a CSA-catalyzed isomerization of epoxide 41, a one-pot epoxide ring-opening followed by reductive transposition was proposed. Upon subjecting vinyl epoxide 41 to the reaction conditions described previously, a mixture of 44 and 45 (2.5:1 diastereomeric ratio) was obtained, allowing access to both icetexone and epi-icetexone, respectively. A brief optimization study established that this transformation could be accomplished in a 42% combined yield of lactones 44 and 45 from vinyl epoxide 41.
Scheme 12.
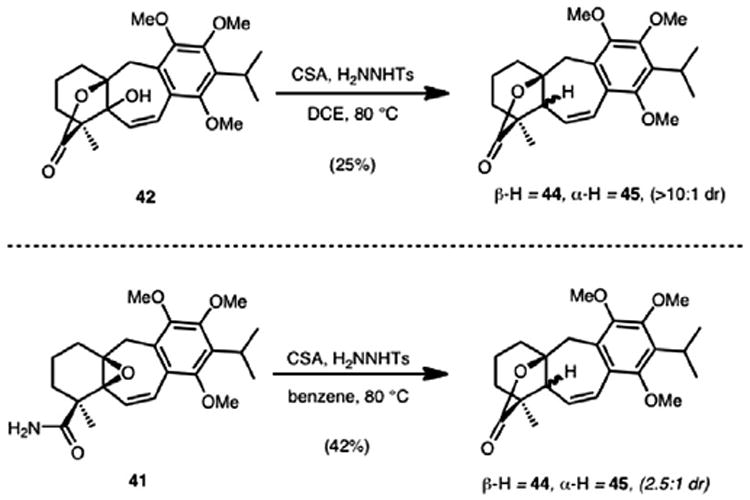
Formal synthesis of icetexone and epi-icetexone.
The variability in diastereomeric ratio of lactones 44 and 45 obtained from vinyl oxirane 41 (Scheme 12) as compared to that obtained from allylic alcohol 42 (or alkene 43, Scheme 11) can be rationalized by the following proposed mechanism (Scheme 13). Acid-catalyzed opening of the vinyl epoxide likely leads to tertiary carbocation 46, which can be stabilized by the styrenyl double bond. Attack by tosyl hydrazide at the less sterically hindered benzylic position from either face can give rise to intermediate 47. From this intermediate, a tandem lactone formation and diazene transposition yields lactones 44 and 45, wherein the stereochemistry at C5 is determined by which face nucleophilic attack occurs from. For allylic alcohol 42 (or alkene 43), because the lactone is formed prior to tosyl hydrazide attack, there is significant facial control in the addition step.
Scheme 13.
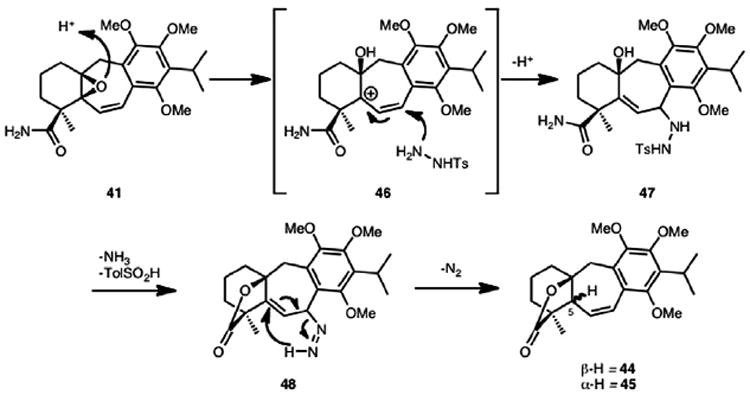
Proposed mechanism.
2.3. Enantioselective formal synthesis of icetexone and epi-icetexone
It is evident from our retrosynthetic analysis of icetexone and epi-icetexone (Scheme 7) that rendering the synthesis of alkyl iodide 33 enantioselective would complete enantioselective formal syntheses of these natural products given that the single stereocenter in nitrile 33 controls the stereoselectivity during the installation of all additional stereocenters. Our planned approach to enantioenriched nitrile 33 sought to capitalize on the remarkable enantioselective conjugate addition reaction of cyanopropionates (e.g., 49, Scheme 14) to acrolein (50) using Rh(I)-2,2′-bis[1-(diarylphosphino)ethyl]-1,1′-biferrocenes (ArylTRAP) complexes (51) as first reported by Sawamura et al.20 In accordance with the protocols developed by Sawamura et al., reacting cyanoester 49 (1 equiv) with acrolein (1.5 equiv) in benzene at 0 °C yielded the conjugate adduct, which was immediately reduced to provide alcohol 52. The enantiomeric excess of quaternary cyanoester 52 (89% ee)21 was determined after its derivatization to the corresponding p-nitro benzoate. To convert enantioenriched 52 to alkyl iodide 33, the hydroxy group was protected with TIPSCl and, the tert-butyl ester group could be reduced selectively to the corresponding alcohol in the presence of the nitrile group using sodium borohydride in EtOH/THF (4:1). Swern oxidation of the hydroxy group and alkynylative homologation of the resulting aldehyde using the Ohira–Bestmann protocol gave enantioenriched 37 (not shown in Scheme 14) in good yield. Unveiling the primary alcohol of 37 (with TBAF) set the stage for its replacement with an iodide group via the intermediacy of the mesylate. Enantioenriched 33 obtained in this way provided identical 1H and 13C NMR spectra as the racemic material that we had prepared previously. Because we began with the (S,S)-(R,R)-2,2″-bis[1-(diphenylphosphino)ethyl]-1,1″-biferrocene complex (51), we wanted to ensure that the absolute stereochemistry was as is depicted for naturally occurring icetexone and epi-icetexone. Thus, using an analogous sequence as described in Schemes 9 and 10, enantioenriched 33 was advanced to tricyclic nitrile 30. Reduction of the nitrile group using LAH proceeded without event to afford a primary amine, which was derivatized with p-bromo benzoyl chloride to give amide derivative 54. X-ray crystallographic analysis of 54 (see ORTEP in Scheme 14) confirmed the C(4) stereocenter as R, consistent with that found in the natural product, thus completing the formal synthesis of icetexone and epi-icetexone.
Scheme 14.
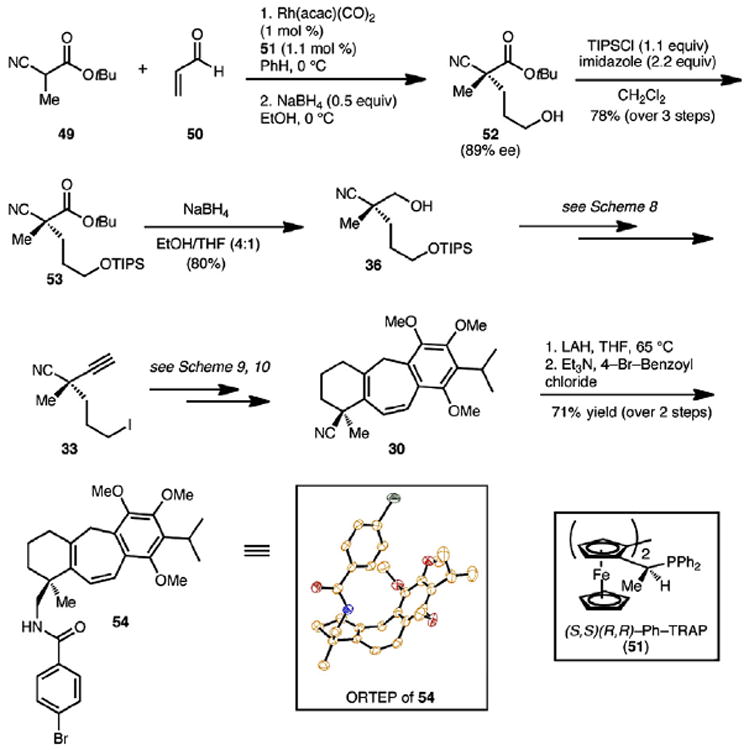
Synthesis of an enantioenriched common intermediate.
Scheme 10.
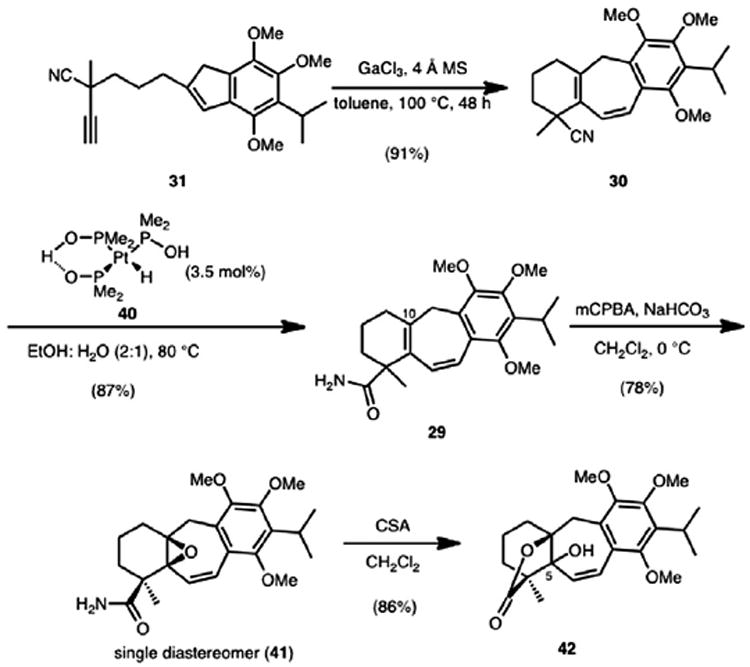
Lactone formation.
3. Conclusion
In this article, formal syntheses of icetexone (1) and epi-icetexone (2) that rely on a Ga(III)-catalyzed cycloisomerization to form 6-7-6 fused tricyclic carbocycles was presented. Two distinct approaches were pursued. The first, which entailed attempts to oxygenate the carbocyclic skeleton, involved a remote functionalization strategy that ultimately led to fragmentation of the seven-membered ring. A pre-functionalization approach was then undertaken, and the synthesis of a cyano group-containing tether allowed for a successful Ga(III)-catalyzed alkynyl indene cycloisomerization. Of note, this work served as the first demonstration that a Ga(III)-catalyzed cycloisomerization is possible in the presence of a nitrile group. The presence of this functional handle was exploited in achieving a formal synthesis of icetexone and epi-icetexone through a tandem vinyl epoxide opening/diazene rearrangement sequence. Finally, using a Rh-catalyzed enantioselective conjugate addition, the formal enantioselective syntheses of these natural products was achieved.
4. Experimental section
4.1. General experimental details
All reagents were obtained from commercial chemical suppliers and used without further purification unless otherwise noted. All reactions were performed in round-bottomed flasks sealed with rubber septa, under an atmosphere of N2, and stirred with a Teflon™-coated magnetic stir bar unless otherwise noted. Temperatures above 23 °C were controlled by an IKA® temperature modulator. Pre-dried tetrahydrofuran (THF), benzene, toluene, acetonitrile (MeCN), methanol (MeOH), and triethylamine (Et3N), were degassed with argon for 60 min and passed through activated alumina columns. Dichloromethane (CH2Cl2) was distilled over calcium hydride before use. Reactions were monitored by thin layer chromatography (TLC) using Silicycle Siliaplate™ glass backed TLC plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) and p-anisaldehyde stain or KMnO4 stain. Volatile solvents were removed using a rotary evaporator under reduced pressure. Silica gel chromatography was performed using Sorbent Technologies 60 Å, 230×400 mesh silica gel (40–63 μm). 1H NMR and 13C NMR were obtained in CDCl3 on Bruker 400, 500, or 600 MHz spectrometers with 13C operating frequencies of 100, 126, or 151 MHz, respectively. Chemical shifts are reported in parts per million (δ) relative to residual chloroform (7.26 ppm for 1H and 77.16 ppm for 13C). Data for 1H NMR spectra are reported as follows: chemical shift (multiplicity, coupling constants, number of hydrogens). Multiplicity is designated as s (singlet), d (doublet), t (triplet), q (quartet), or m (multiplet). IR spectra were obtained using a Nicolet MAGNA-IR 850 spectrometer as thin films on NaCl plates and reported in frequency of absorption (cm−1). High resolution mass spectral (HRMS) data was obtained from the Mass Spectral facility at the University of California, Berkeley.
4.1.1. 4-Isopropyl-2,3,5-trimethoxybenzaldehyde (18)
To a solution of 4-isopropyl-2,3,5-trimethoxybenzyl alcohol8,13 (17, 4.09 g, 17.0 mmol) in 4:1 CH2Cl2/DMSO (45 mL) at 0 °C were added SO3·pyr (4.73 g, 29.8 mmol) and Et3N (11.8 mL, 85 mmol). The resulting mixture was stirred at 0 °C and allowed to warm to rt over 18 h. The reaction mixture was poured into a mixture of saturated aq NaHCO3 (25 mL) and water (25 mL), then extracted with Et2O (3×100 mL). The combined organic layers were washed with saturated aq NaHCO3 (100 mL), and brine (100 mL), dried over MgSO4, and concentrated. The crude product was passed through a plug of silica gel to give 3.49 g (14.6 mmol, 86%) of 18 as an orange oil. Rf 0.66 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 10.28 (s, 1H), 6.99 (s, 1H), 3.89 (s, 3H), 3.81 (s, 3H), 3.77 (s, 3H), 3.52 (m, 1H), 1.26 (d, J=7.2 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 189.2, 155.0, 152.0, 151.5, 138.8, 127.3, 103.1, 62.3, 60.9, 55.6, 25.7, 20.7; IR (film) νmax 2959, 1688, 1595, 1465, 1414, 1384, 1348, 1273, 1235, 1208, 1136, 1063, 1024 cm−1; HRMS (EI) calcd for C13H18O4 ([M]+): m/z 238.1205, found 238.1205.
4.1.2. Ethyl(4-isopropyl-2,3,5-trimethoxy)cinnamate (19)
Triethyl phosphonoacetate (3.19 mL, 16.1 mmol) was added to a solution of 18 (3.49 g, 14.6 mmol) and LiCl (643 mg, 15.2 mmol) in CH3CN (52 mL) under N2 at 0 °C. After the addition of DBU (2.40 mL, 16.1 mmol), the resulting mixture was allowed to warm to rt and was stirred for 13 h. The mixture was poured into water (100 mL) and extracted with CHCl3 (3×50 mL). The combined organic layers were washed with brine (100 mL), dried over MgSO4, and concentrated. Flash chromatography (9:1 hexanes/EtOAc) gave 3.95 g (12.8 mmol, 88%) of 19 as a thick yellow oil. Rf 0.48 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J=16.1 Hz, 1H), 6.73 (s, 1H), 6.43 (d, J=16.1 Hz, 1H), 4.24 (q, J=7.1 Hz, 2H), 3.80 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 3.53–3.46 (m, 1H), 1.31 (t, J=7.1 Hz, 3H), 1.28 (d, J=7.1 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 167.6, 155.1, 152.6, 147.5, 139.7, 134.1, 126.1, 118.7, 104.3, 61.6, 61.2, 60.7, 55.9, 25.8, 21.4, 14.7; IR (film) νmax 2958, 1713, 1633, 1471, 1410, 1346, 1284, 1230, 1175, 1134, 1055, 1026 cm−1; HRMS (ESI) calcd for C12H25O5 ([M+H]+): m/z 309.1697, found 309.1701.
4.1.3. 4-Isopropyl-2,3,5-trimethoxydihydrocinnamic acid (20)
A mixture of 19 (2.83 g, 9.18 mmol) and 10% Pd/C (283 mg) in EtOH (95%, 30 mL) was placed under an atmosphere of H2 (balloon) and stirred rapidly at rt for 12 h. The mixture was then filtered through Celite and concentrated to give 2.53 g (8.15 mmol, 89%) of a light yellow oil, which was used without further purification. To a solution of ethyl(4-isopropyl-2,3,5-trimethoxy)dihydrocinnamate (3.96 g, 12.8 mmol) in 3:1 THF/water (60 mL) was added LiOH·H2O (1.34 g, 31.9 mmol). The resulting mixture was stirred at rt for 18 h. The mixture was acidified with 1 N HCl (50 mL) and extracted with EtOAc (2×100 mL). The combined organic layers were washed with brine (100 mL), dried over MgSO4, and concentrated to give 3.6 g (12.8 mmol, 100%) of 20 as a thick yellow oil, which was used without further purification. Rf 0.45 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.45 (s, 1H), 3.81 (s, 3H), 3.80 (s, 3H), 3.76 (s, 3H), 3.47 (m, 1H), 2.92 (t, J=7.8 Hz, 2H), 2.68 (t, J=7.8 Hz, 2H), 1.29 (d, J=7.1 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 179.3, 154.4, 151.7, 145.3, 130.8, 128.9, 107.5, 60.6, 60.4, 55.7, 34.8, 25.5, 25.0, 21.2; IR (film) νmax 2956, 2835, 1710, 1603, 1578, 1452, 1409, 1340, 1286, 1231, 1130, 1072, 1028 cm−1; HRMS (EI) calcd for C15H22O5 ([M]+): m/z 282.1467, found 282.1463.
4.1.4. 6-Isopropyl-4,5,7-trimethoxy-1-indanone (21)
To a flask containing 20 (1.43 g, 5.06 mmol) in CH2Cl2 (16 mL) under N2 at 0 °C was added oxalyl chloride (0.44 mL, 5.06 mmol), followed by DMF (3 drops) after 20 min. After stirring at 0 °C for 2 h, the resulting solution was warmed to rt. The resulting solution was then added to a suspension of AlCl3 (1.35 g) in CH2Cl2 (8 mL) at 0 °C slowly via syringe. After stirring for 1 h, the mixture was poured into ice-water in a separatory funnel and diluted with 1 N HCl (25 mL) and CH2Cl2 (25 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (2×25 mL). The combined organic layers were washed with 1 N NaOH (25 mL), dried over MgSO4, and concentrated to give 1.18 g (4.46 mmol, 88%) of 21 as a thick light yellow syrup, which was used without further purification. Rf 0.51 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.94 (s, 3H), 3.90 (s, 3H), 3.86 (s, 3H), 3.52 (m, 1H), 3.06–2.99 (m, 2H), 2.68–2.60 (m, 2H), 1.31 (d, J=7.1 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 203.5, 158.4, 153.7, 147.9, 146.1, 134.9, 125.5, 62.8, 61.1, 60.4, 37.3, 25.7, 22.5, 22.1; IR (film) νmax 2936, 1708, 1583, 1470, 1418, 1320, 1274, 1136, 1060, 1035 cm−1; HRMS (EI) calcd for C15H21O4 ([M]+): m/z 264.1366, found 264.1362.
4.1.5. 2-(Carbomethoxy)-2-(4′,4′-dimethyl-5′-hexynyl)-6-isopropyl-4,5,7-trimethoxy-1-indanone (24)
To a flask containing sodium bis(trimethylsilyl)amide (1.0 M in THF, 6.06 mL, 6.06 mmol) in THF (12 mL) under N2 at −78 °C was added a solution of 21 (641 mg, 2.43 mmol) in THF (8 mL) over 2 min. After 45 min at −78 °C, the resulting bright orange solution was treated with methyl cyanoformate (0.21 mL, 2.67 mmol). The mixture was stirred at −78 °C for 15 min, then poured into water (30 mL) and extracted with EtOAc (3×30 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, and concentrated to give a dark yellow oil, which was used immediately without further purification. To a solution of the crude β-ketoester (22, 783 mg, 2.43 mmol) in acetone (10 mL) in a Schlenk flask was added K2CO3 (1.01 g, 7.28 mmol), and the resulting suspension was stirred at rt for 15 min. A solution of 2311a (573 mg, 2.43 mmol) in acetone (6 mL) was added, and the flask was evacuated and backfilled with N2 (3×). The flask was sealed and heated to 65 °C for 40 h. After cooling to rt, the mixture was diluted with water (30 mL) and extracted with Et2O (3×30 mL). The combined organic layers were washed with water (30 mL) and brine (30 mL), dried over MgSO4, and concentrated. Flash chromatography (9:1 to 4:1 hexanes/EtOAc) gave 689 mg (1.60 mmol, 66% over two steps) of 24 as a viscous pale yellow syrup. Rf 0.59 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.93 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H), 3.67 (s, 3H), 3.60 (d, J=17.5 Hz, 1H), 3.48 (septet, J=7.1 Hz, 1H), 2.95 (d, J=17.5 Hz, 1H), 2.15–2.06 (m, 1H), 2.00 (s, 1H), 1.83–1.73 (m, 1H), 1.48–1.32 (m, 4H), 1.28 (d, J=7.1 Hz, 6H), 1.14 (s, 3H), 1.12 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 198.3, 171.6, 158.6, 154.0, 145.3, 145.0, 134.8, 122.9, 91.4, 67.8, 62.3, 61.1, 60.6, 60.1, 52.6, 43.1, 35.2, 32.7, 30.9, 29.1, 29.0, 25.3, 21.6, 20.6; IR (film) νmax 3290, 2963, 2872, 1746, 1709, 1582, 1471, 1420, 1323, 1275, 1200, 1172, 1136, 1039 cm−1; HRMS (EI) calcd for C25H34O5 ([M]+): m/z 430.2355, found 430.2351.
4.1.6. 2-(4′,4′-Dimethyl-5′-hexynyl)-6-isopropyl-4,5,7-trimethoxyin dene (16)
To a solution of 24 (1.88 g, 4.37 mmol) in 3:1 THF/water (20 mL) was added LiOH·H2O (916 mg, 21.8 mmol). The resulting mixture was heated at 65 °C for 16 h in a sealed tube under N2. After cooling to rt, the reaction mixture was acidified with 1 N HCl (40 mL) and extracted with Et2O (3×40 mL). The combined organic layers were washed with brine (40 mL), dried over MgSO4, and concentrated to give 25 as a light yellow oil, which was used without further purification, Rf 0.64 (2:1 hexanes/EtOAc). To a solution of the 25 (1.63 g, 4.37 mmol) in CH2Cl2 (35 mL) under N2 at 0 °C was added diisobutylaluminum hydride (1.0 M in toluene, 5.28 mL, 5.28 mmol) dropwise over 5 min. The resulting solution was stirred at 0 °C for 30 min and then quenched by the addition of an aq solution of Rochelle’s salt (4.47 g in 40 mL water). The resulting biphasic mixture was allowed to warm to rt and stirred vigorously for 1 h before it was diluted with water (40 mL) and extracted with CH2Cl2 (3×40 mL). The combined organic layers were washed with brine (40 mL), dried over MgSO4, and concentrated to give a pale yellow oil, which was used immediately without further purification, Rf 0.40 (4:1 hexanes/EtOAc). To a solution of the crude indanol (1.63 g, 4.37 mmol) and Et3N (1.84 mL, 13.2 mmol) in benzene (30 mL) was added methanesulfonic anhydride (1.53 g, 8.81 mmol). The resulting mixture was stirred at rt for 35 min before it was poured onto water (40 mL) and extracted with Et2O (3×40 mL). The combined organic layers were washed with saturated aq NaHCO3 (40 mL), and brine (40 mL), dried over MgSO4, and concentrated. Flash chromatography (20:1 hexanes/EtOAc) to give 1.23 g (3.45 mmol, 79% over three steps) of 16 as a viscous pale yellow oil. Rf 0.57 (4:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 6.61 (s, 1H), 3.92 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.50 (septet, J=7.1 Hz, 1H), 3.35 (s, 2H), 2.51 (t, J=7.3 Hz, 2H), 2.11 (s, 1H), 1.86–1.73 (m, 2H), 1.51–1.43 (m, 2H), 1.37 (d, J=7.1 Hz, 6H), 1.24 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 148.5, 148.4, 146.9, 145.5, 133.9, 133.1, 133.0, 122.8, 91.7, 67.9, 61.8, 60.9, 59.6, 42.6, 38.7, 31.3, 30.9, 29.1, 25.7, 24.6, 22.1; IR (film) νmax 3288, 2963, 2937, 2870, 2831, 2107, 1467, 1419, 1338, 1278, 1213, 1121, 1058, 1042, 999, 980, 918, 834 cm−1; HRMS (EI) calcd for C23H32O3 ([M]+): m/z 356.2351, found 356.2350.
4.1.7. 8-Isopropyl-6,7,9-trimethoxy-1,1-dimethyl-2,3,4,5-tetrahydro-1H-dibenzo[a,d][7]annulene (15)
In an N2 dry box, a Schlenk flask was charged with GaCl3 (122 mg, 0.69 mmol) and powdered 4 Å molecular sieves (244 mg). The flask was removed from the dry box and a solution of 16 (1.23 g, 3.45 mmol) in freshly distilled benzene (85 mL) was added via syringe under N2 to give a dark red solution. The flask was sealed and placed in an oil bath that was preheated to 40 °C. After being stirred at 40 °C for 24 h, the mixture was allowed to cool to rt and the sieves were removed by filtration through a plug of glass wool. The filtrate was poured onto a solution of saturated aq NaHCO3 (50 mL) and extracted with Et2O (3×50 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, and concentrated to give a viscous dark yellow syrup. Flash chromatography (30:1 hexanes/EtOAc) gave 1.07 g (3.00 mmol, 87%) of 15 as a viscous pale yellow oil, which crystallized to a white solid on standing at rt. Rf 0.66 (4:1 hexanes/EtOAc); mp 101.1–102.0 °C; 1H NMR (500 MHz, CDCl3) δ 7.12 (d, J=12.0 Hz, 1H), 6.66 (d, J=12.0 Hz, 1H), 3.92 (s, 3H), 3.84 (s, 3H), 3.69 (s, 3H), 3.49 (septet, J=7.0 Hz, 1H), 2.97 (br s, 2H), 2.36 (t, J=5.9 Hz, 2H), 1.69–1.59 (m, 2H), 1.51–1.42 (m, 2H), 1.37 (d, J=7.1 Hz, 6H), 1.08 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 152.4, 151.2, 145.4, 136.5, 132.5, 132.3, 131.5, 129.6, 126.6, 125.4, 61.6, 60.7, 60.3, 38.9, 33.4, 32.27, 32.26, 29.1, 25.6, 22.2, 19.6; IR (film) νmax 2933, 2868, 2827, 1580, 1454, 1400, 1341, 1315, 1120, 1101, 1061, 1047, 996, 969, 740 cm−1; HRMS (EI) calcd for C23H32O3 ([M]+): m/z 356.2351, found 356.2351.
4.1.8. 8-Isopropyl-6,7,9-trimethoxy-1,1-dimethyl-1,2,3,4,5,10,11,11aoctahydro-4a,10-epoxydibenzo[a,d][7]annulen-11-ol (26)
A mixture of cycloheptadiene 15 (229 mg, 0.64 mmol) and NaHCO3 (108 mg, 1.28 mmol) in CH2Cl2 (6.4 mL) at 0 °C was treated with m-chloroperbenzoic acid (77%, 173 mg, 0.77 mmol). The resulting mixture was stirred at 0 °C for 1 h before it was quenched by the addition of saturated aq. NaHSO3 (7 mL). The mixture was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with saturated aq. NaHCO3 (2 × 15 mL), and brine (15 mL), dried over MgSO4, and concentrated. Flash chromatography (20:1 hexanes/EtOAc) gave 174 mg (0.47 mmol, 73%) of the dihydrofuran as a viscous colorless oil. Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 6.06 (d, J = 1.5 Hz, 1H), 5.47 (d, J = 2.0 Hz, 1H), 3.81 (s, 3H), 3.73 (s, 3H), 3.70 (s, 3H), 3.33 (septet, J = 7.1 Hz, 1H), 2.94 (d, J = 17.3 Hz, 1H), 2.66 (d, J = 17.3 Hz, 1H), 2.16-2.07 (m, 1H), 1.83-1.69 (m, 3H), 1.55-1.49 (m, 1H), 1.40-1.34 (m, 1H), 1.32 (d, J = 7.1 Hz, 3H), 1.30 (d, J = 7.1 Hz, 3H), 1.13 (s, 3H), 1.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 151.3, 150.3, 148.6, 148.5, 131.5, 127.82, 127.78, 125.8, 83.3, 74.8, 62.6, 60.2, 59.4, 40.0, 38.6, 33.8, 30.5, 29.8, 27.5, 25.6, 22.2, 22.0, 19.0; IR (film) nmax 2936, 2868, 1454, 1411, 1341, 1285, 1244, 1166, 1121, 1092, 1058, 1040, 1018, 1001, 965, 738 cm-1; HRMS (EI) calcd for C23H32O4 ([M]+): m/z 372.2301, found 372.2299. To a solution of dihydrofuran (72 mg, 0.19 mmol) in THF (1.5 mL) under N2 was added a solution of BH3•THF (1.0 M in THF, 1.54 mL, 1.54 mmol). The resulting solution was stirred at rt for 11 h. After cooling the solution to 0 °C, the excess borane was carefully quenched by slow, dropwise addition of EtOH (0.54 mL). The resulting mixture was then treated with a solution of NaOH (125 mg, 3.12 mmol) in water (2.19 mL), followed by a solution of H2O2 (30% in water, 1.15 mL, 10.2 mmol). After addition was complete, the mixture was allowed to warm to rt and was stirred for 4 h. The reaction mixture was poured onto a 1:1 mixture of NH4Cl:water (15 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (15 mL), dried over MgSO4, and concentrated to give 77 mg (0.19 mmol, 100%) of crude alcohol 26 as a colorless oil, which was used without further purification. Rf 0.16 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 5.04 (s, 1H), 3.99 (d, J=7.3 Hz, 1H), 3.82 (s, 3H), 3.75 (s, 3H), 3.74 (s, 3H), 3.37 (dd, J=14.2, 7.1 Hz, 1H), 3.20 (d, J=18.3 Hz, 1H), 2.66 (d, J=18.3 Hz, 1H), 2.03 (m, 1H), 1.81–1.74 (m, 2H), 1.74–1.70 (m, 2H), 1.70–1.65 (m, 2H), 1.56 (m, 1H), 1.33 (d, J=7.1 Hz, 3H), 1.31 (d, J=7.1 Hz, 3H), 1.07 (s, 3H), 0.94 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 151.2, 148.2, 147.3, 132.5, 128.2, 124.7, 82.1, 80.5, 79.3, 68.8, 62.7, 60.2, 59.5, 41.1, 40.8, 33.3, 33.1, 31.1, 25.7, 22.2, 22.0, 21.1, 20.2; IR (film) νmax 3422, 2938, 2870, 1646, 1456, 1415, 1360, 1341, 1284, 1173, 1123, 1071, 1042, 1013 cm−1; HRMS (EI) calcd for C23H34O5 ([M]+): m/z 390.2406, found 390.2411.
4.1.9. 8-Isopropyl-6,7,9-trimethoxy-1,1-dimethyl-1,3,4,5,10,11a-hexahydro-4a,10-epoxydibenzo[a,d][7]annulen-11(2H)-one (27)
To a solution of the alcohol 26 (82 mg, 0.21 mmol) in CH2Cl2 (4.2 mL) were added NaHCO3 (53 mg, 0.63 mmol) and Dess–Martin periodinane (106 mg, 0.25 mmol) at rt. After 1 h, the mixture was poured onto saturated aq Na2SO3 (8 mL) and extracted with EtOAc (3×16 mL). The combined organic layers were washed with saturated aq NaHCO3 (2×16 mL) and brine (16 mL), dried over MgSO4, and concentrated to give 62 mg (0.16 mmol, 76%) of a yellow oil. To a solution of the crude cis-fused ketone (62 mg, 0.16 mmol) in 3:1 MeOH/CH2Cl2 (2.0 mL) was added sodium methoxide (9.6 mg, 0.18 mmol). The resulting pale yellow solution was stirred at rt for 21 h before it was poured onto a 1:1 mixture of water/saturated aq NH4Cl (20 mL) and extracted with EtOAc (3×15 mL). The combined organic layers were washed with brine (15 mL), dried over MgSO4, and concentrated to give a colorless oil. Flash chromatography (20:1 hexanes/EtOAc) gave 53 mg (0.14 mmol, 88%) of ketone 27 as a colorless oil. Rf 0.64 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.16 (s, 1H), 3.83 (s, 3H), 3.81 (s, 3H), 3.74 (s, 3H), 3.36 (m, 1H), 2.90 (d, J=18.1 Hz, 1H), 2.43 (d, J=18.0 Hz, 1H), 2.13–2.06 (m, 1H), 1.91–1.81 (m, 1H), 1.88 (s, 1H), 1.69–1.60 (m, 1H), 1.59–1.48 (m, 2H), 1.32 (d, J=7.1 Hz, 3H), 1.29 (d, J=7.1 Hz, 3H), 1.29–1.23 (m, 1H), 1.16 (s, 3H), 1.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 213.2, 152.5, 149.9, 147.5, 133.7, 125.5, 123.2, 79.7, 75.2, 63.5, 60.1, 59.4, 59.0, 40.0, 37.5, 35.1, 32.8, 31.8, 25.8, 22.8, 22.0, 21.9, 17.4; IR (film) νmax 2952, 2872, 2838, 1749, 1459, 1414, 1345, 1271, 1250, 1174, 1121, 1067, 1040, 961, 836, 795, 737 cm−1; HRMS (EI) calcd for C23H32O5 ([M]+): m/z 388.2250, found 388.2246.
4.1.10. Methyl 2-cyano-5-(triisopropylsilyloxy)pentanoate (35)
To a solution of 3-bromo-1-propanol (34, 20.0 mL, 230 mmol) in CH2Cl2 (575 mL) were added imidazole (39.0 g, 575 mmol) and triisopropylsilyl chloride (59.0 mL, 276 mmol). The cloudy reaction mixture was stirred at rt for 12 h. The solution was poured into water (500 mL) and extracted with CH2Cl2 (2×500 mL). The combined organic layers were washed with brine (300 mL), dried over MgSO4, and concentrated to give of a colorless oil that was used without purification. To a solution of (3-bromopropoxy)triisopropylsilane (67.5 g, 230 mmol) in DMF (460 mL) were added K2CO3 (95.0 g, 690 mmol) and methyl cyanoacetate (40 mL, 3460 mmol) at rt. The flask was fitted with a reflux condenser, then heated to 65 °C for 1 h. After cooling to rt, the mixture was diluted with water (500 mL) and extracted with Et2O (3×100 mL). The combined organic layers were washed with brine (1500 mL), dried over MgSO4, and concentrated to give a thick yellow oil. Flash chromatography (9:1 hexanes/EtOAc) gave 64.8 g (207 mmol, 90% over two steps) of 35 as a clear oil. Rf 0.68 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.85 (s, 3H), 3.79 (m, 2H), 3.69 (dd, J=8.4, 5.9 Hz, 1H), 2.12 (m, 2H), 1.81–1.72 (m, 2H), 1.08 (m, 21H); 13C NMR (126 MHz, CDCl3) δ 167.1, 116.9, 62.6, 37.6, 30.1, 18.4, 18.1, 12.7, 12.3; IR (film) νmax 2943, 2867, 1754, 1463, 1250, 1110, 1013 cm−1; HRMS (ESI) calcd for C16H32NO3Si ([M+H]+): m/z 314.2146, found 314.2143.
4.1.11. 2-(Hydroxymethyl)-2-methyl-5-(triisopropylsilyloxy)pentanenitrile (36)
To a solution of 35 (64.8 g, 207 mmol) in DMF (400 mL) was added NaH (60% in mineral oil, 9.89 g, 248 mmol) portion-wise, followed by methyl iodide (25.7 mL, 414 mmol) at rt. The reaction mixture was stirred for 1 h, after which the reaction was quenched with water (500 mL) then extracted with Et2O (3×500 mL). The combined organic layers were washed with water (500 mL) and brine (300 mL), dried over MgSO4, and concentrated to give 67.8 g (207 mmol, 100%) of a clear oil, which was used without further purification. Rf 0.54 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.81 (s, 3H), 3.71 (t, J=5.9 Hz, 2H), 2.08–2.00 (m, 1H), 1.93–1.85 (m, 1H), 1.79–1.69 (m, 1H), 1.60 (s, 3H), 1.63–1.55 (m, 1H), 1.04 (m, 21H). To a solution of the ester (68.7 g, 207 mmol) in 14:1 THF/water (690 mL) was added NaBH4 (39.0 g, 1.03 mol) at rt. After stirring for 3 h, saturated aq NH4Cl (300 mL) was added slowly at 0 °C. The mixture was stirred until gas evolution ceased. The reaction mixture was extracted with Et2O (3×500 mL) and the combined organic layers were washed with saturated aq NaHCO3 (300 mL), and brine (300 mL), dried over MgSO4, and concentrated to give 62 g (207 mmol, 100%) of 36 as a colorless oil, which was used immediately without purification. Rf 0.47 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.77 (t, J=5.8 Hz, 2H), 3.67 (m, 2H), 2.04 (t, J=6.7 Hz, 1H), 1.90–1.76 (m, 1H), 1.67–1.59 (m, 3H), 1.38 (s, 3H), 1.09 (m, 21H); 13C NMR (151 MHz, CDCl3) δ 123.2, 68.0, 62.8, 39.4, 32.0, 28.0, 21.0, 18.0, 11.9; IR (film) νmax 3454, 2943, 2866, 1463, 1383, 1106, 1069 cm−1; HRMS (ES) calcd for C13H26NO2Si ([M−C3H7]+): m/z 256.1733, found 256.1737.
4.1.12. 2-Ethynyl-2-methyl-5-(triisopropylsilyloxy)pentanenitrile (37)
To a solution of anhydrous DMSO (31.0 mL, 436 mmol) in CH2Cl2 (500 mL) was added oxalyl chloride (18.3 mL, 218 mmol) under N2 at −78 °C. After stirring for 120 min, a solution of 36 (32.5 g, 109 mmol) in CH2Cl2 (100 mL) was added at −78 °C via cannula. After an additional 4 h, Et3N (121 mL, 872 mmol) was added at −78 °C. The resulting mixture was allowed to warm to rt and stirred for 12 h and then poured into a mixture of saturated aq NH4Cl (300 mL) and water (300 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (3×500 mL). The combined organic layers were washed with brine (300 mL), dried over MgSO4, and concentrated to give a yellow oil, which was taken on immediately without further purification. To a solution of the crude aldehyde (32.4 g, 109 mmol) in MeOH (600 mL) were added K2CO3 (30.1 g, 218 mmol) and the Ohira–Bestmann reagent (23.0 g, 120 mmol) at 0 °C. After stirring for 2 h, the reaction mixture was poured into a mixture of saturated aq NaHCO3 (150 mL) and water (150 mL), and extracted with Et2O (3×500 mL). The combined organic layers were washed with brine (300 mL), dried over MgSO4, and concentrated to give a yellow oil. Flash chromatography (9:1 hexanes/EtOAc) gave 27.4 g (93 mmol, 85% over two steps) of 37 as a light yellow oil. Rf 0.67 (EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.79 (t, J=5.5 Hz, 2H), 2.40 (s, 1H), 1.99–1.81 (m, 4H), 1.68 (s, 3H),1.08 (m, 21H); 13C NMR (126 MHz, CDCl3) δ 120.8, 81.4, 72.4, 62.8, 38.2, 31.5, 29.4, 27.6, 18.4, 12.3; IR (film) νmax 3312, 2943, 2866, 1463, 1382, 1367, 1240, 1107, 1070, 1014 cm−1; HRMS (ESI) calcd for C17H31NOSi ([M+H]+): m/z 294.2248, found 294.2259.
4.1.13. 2-Ethynyl-5-iodo-2-methylpentanenitrile (33)
To a solution of 37 (27.4 g, 93 mmol) in THF (370 mL) was added tert-butylammonium fluoride (1.0 M in THF, 112 mL) at rt. After stirring for 12 h, the reaction mixture was poured into saturated aq NaHCO3 (300 mL) and extracted with Et2O (3×300 mL), the combined organic layers were washed with brine (200 mL), dried over MgSO4, and concentrated to give a crude colorless oil. Flash chromatography (2:1 hexanes/EtOAc) gave 9.79 g (71 mmol, 76%) of the alcohol as a colorless oil. To a solution of resulting alcohol (9.79 g, 71 mmol) in CH2Cl2 (280 mL) was added methanesulfonyl chloride (6.0 mL, 78 mmol) and Et3N (14.8 mL, 107 mmol) under N2 at 0 °C. The resulting mixture was allowed to warm to rt. After stirring for 2 h, the reaction mixture was washed with 1 N HCl (150 mL), saturated aq NaHCO3 (150 mL), and brine (150 mL), dried over MgSO4, and concentrated to give a light yellow oil that was immediately used without further purification. The resulting mesylate (15.2 g, 71 mmol) was added to a Schlenk flask with acetone (280 mL). NaI (16.0 g, 107 mmol) was added and the flask was sealed and placed in an oil bath preheated to 65 °C. After stirring at 65 °C for 15 h, the mixture was allowed to cool to rt and quenched with water (280 mL). The reaction mixture was extracted with pentane (3×300 mL) and the combined organic layers were washed with brine (200 mL), dried over MgSO4, and concentrated to give 14.0 g (57 mmol, 80% over two steps) of 27 as a light yellow oil. Rf 0.62 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 3.26 (m, 2H), 2.45 (s, 1H), 2.17 (m, 2H), 1.99–1.86 (m, 2H), 1.69 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 120.4, 80.8, 73.1, 42.1, 31.0, 29.7, 27.6, 5.0; IR (film) νmax 3290, 2939, 1451, 1382, 1294, 1253, 1185, 1148 cm−1; HRMS (EI) calcd for C8H18NI ([M]+): m/z 246.9858, found 246.9858.
4.1.14. Ethyl 2-(4-cyano-4-methylhex-5-yn-1-yl)-6-isopropyl-4,5,7-trimethoxy-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (38)
To a flask containing KOt-Bu (850 mg, 7.57 mmol) under N2 were added 21 (1.00 g, 3.78 mmol) and diethyl carbonate (0.97 mL, 7.57 mmol) in THF (19 mL) at rt. The resulting mixture was heated to 70 °C in a flask fitted with a reflux condenser for 30 min. After cooling to rt over 1 h, the mixture was diluted with EtOAc (40 mL) and saturated aq NaHCO3 (40 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2×40 mL). The combined organic layers were washed with brine (40 mL), dried over MgSO4, and concentrated to give 32 as a thick orange oil that was used without further purification. Rf 0.43 (2:1 hexanes/EtOAc). To a solution of the 32 (1.23 g, 3.78 mmol) in acetone (20 mL) were added K2CO3 (1.56 g, 11.3 mmol) and 33 (933 mg, 3.78 mmol) in a Schlenk flask at rt. The flask was evacuated and backfilled with N2 (3×), then sealed and heated at 65 °C for 48 h. After cooling to rt, the mixture was diluted with water (40 mL) and extracted with Et2O (3×40 mL). The combined organic layers were washed with brine (40 mL), dried over MgSO4, and concentrated. Flash chromatography (9:1 to 4:1 hexanes/EtOAc) gave 1.48 g (3.25 mmol, 86% over two steps) of 38 as a viscous dark orange syrup. Rf 0.48 (2:1 hexanes/EtOAc); Mixture of diastereomers (1:1) 1H NMR (600 MHz, CDCl3) δ 4.20–4.07 (m, 4H), 3.93 (s, 6H), 3.84 (s, 6H), 3.84 (s, 6H), 3.60 (d, J=17.4 Hz, 1H), 3.58 (d, J=17.4 Hz, 1H), 3.52–3.45 (m, 2H), 2.93 (d, J=17.4 Hz, 2H), 2.36 (s, 1H), 2.32 (s, 1H), 2.21–2.10 (m, 2H), 1.87–1.67 (m, 6H), 1.55 (m, 4H), 1.59 (s, 6H), 1.28 (d, J=3.8 Hz, 12H), 1.20 (t, J=7.1 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 198.1, 198.0, 170.9, 170.8, 158.8, 154.1, 145.36, 145.35, 144.9, 135.0, 134.9, 122.9, 122.7, 120.1, 120.0, 80.7, 80.6, 72.24, 72.19, 62.3, 62.2, 61.61, 61.57, 60.9, 60.8, 60.6, 60.14, 60.12, 41.0, 34.0, 33.9, 32.7, 31.14, 31.13, 27.13, 27.09, 25.3, 21.6, 21.1, 21.0, 14.0; IR (film) νmax 3262, 2939, 2873, 1740, 1705, 1582, 1472, 1420, 1322, 1275, 1198, 1136, 1037 cm−1; HRMS (ESI) calcd for C26H33NO6 ([M+H]+): m/z 456.2381, found 456.2396.
4.1.15. 2-Ethynyl-5-(6-isopropyl-4,5,7-trimethoxy-1-oxo-2,3-dihydro-1H-inden-2-yl)-2-methylpentanenitrile (39)
To a solution of 38 (1.04 g, 2.35 mmol) in 3:1 THF/water (12 mL) was added LiOH·H2O (246 mg, 5.88 mmol). The resulting mixture was heated at 65 °C for 12 h in a sealed flask. After cooling to rt, the reaction mixture was acidified with 1 N HCl (12 mL) and extracted with EtOAc (3×12 mL). The combined organic layers were washed with brine (12 mL), dried over MgSO4, and concentrated to give 774 mg (2.01 mmol, 86%) of crude indanone 39 as a yellow oil, which was used without further purification. Rf 0.48 (2:1 hexanes/EtOAc); Mixture of diastereomers (1:1) 1H NMR (600 MHz, CDCl3) δ 3.95 (s, 6H), 3.92 (s, 6H), 3.88 (s, 6H), 3.57–3.49 (m, 2H), 3.31 (d, J=8.0 Hz, 1H), 3.28 (d, J=8.0 Hz, 1H), 2.73 (m, 1H), 2.70 (m, 1H), 2.68–2.58 (m, 2H), 2.43 (s, 1H), 2.41 (s, 1H), 2.08–1.97 (m, 2H), 1.91–1.78 (m, 8H), 1.68 (s, 6H), 1.58–1.45 (m, 2H), 1.34 (d, J=7.1 Hz, 12H); 13C NMR (151 MHz, CDCl3) δ 204.3, 158.2, 153.4, 145.7, 134.7, 124.6, 124.5, 120.3, 80.9, 72.2, 62.41, 62.39, 60.7, 60.1, 47.7, 47.5, 41.0, 40.9, 31.29, 31.26, 31.1, 31.0, 29.1, 29.0, 27.22, 27.18, 25.3, 23.8, 23.5, 21.6; IR (film) νmax 3260, 2939, 2871, 1704, 1583, 1470, 1419, 1322, 1273, 1201, 1172, 1138, 1058, 1037 cm−1; HRMS (ESI) calcd for C23H30NO4 ([M+H]+): m/z 384.2169, found 384.2188.
4.1.16. 2-Ethynyl-5-(5-isopropyl-4,6,7-trimethoxy-1H-inden-2-yl)-2-methylpentanenitrile (31)
To a solution of indanone 39 (748 mg, 1.95 mmol) in EtOH (10 mL) was added NaBH4 (74.0 mg, 1.95 mmol) at rt in a water bath to maintain temperature. After 3 h, saturated aq NH4Cl (30 mL) was added slowly at 0 °C, along with Et2O (30 mL). The reaction mixture was stirred until bubbling was no longer observed. The layers were separated and the aqueous layer was extracted with Et2O (2×30 mL). The combined organic layers were washed with saturated aq NaHCO3 (30 mL) and brine (30 mL), dried over MgSO4, and concentrated to give 563 mg (1.45 mmol) of a clear oil, which was used immediately without purification. To a solution of the crude indanol (543 mg, 1.45 mmol) in toluene (7 mL) was added KHSO4 (191 mg, 1.45 mmol) in a sealed vial. The reaction mixture was heated to 120 °C and stirred for 4 h. After cooling to rt, the reaction mixture was passed through a silica plug with 2:1 hexanes/EtOAc to give 456 mg (1.24 mmol, 63% over two steps) of 31 as a light yellow oil. Rf 0.63 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 6.64 (s, 1H), 3.93 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.51 (m, 1H), 3.37 (s, 2H), 2.59 (t, J=7.2 Hz, 2H), 2.43 (s, 1H), 2.05–1.93 (m, 2H), 1.91–1.79 (m, 2H), 1.68 (s, 3H), 1.37 (d, J=7.1 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 148.8, 147.1, 147.0, 145.6, 133.6, 133.2, 133.0, 123.6, 120.3, 80.9, 72.2, 62.0, 61.0, 59.8, 40.7, 38.7, 31.3, 30.5, 27.2, 25.7, 24.9, 22.2; IR (film) νmax 3275, 2937, 2832, 1460, 1418, 1337, 1278, 1218, 1121, 1057 cm−1; HRMS (ESI) calcd for C23H29NO3 ([M+H]+): m/z 368.2220, found 368.2229.
4.1.17. 8-Isopropyl-6,7,9-trimethoxy-1-methyl-2,3,4,5-tetrahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (30)
In a N2 dry box, a Schlenk flask was charged with GaCl3 (9 mg, 0.05 mmol) and powdered 4 Å molecular sieves (18 mg). The flask was removed from the dry box and a solution of 31 (78 mg, 0.21 mmol) in freshly distilled toluene (2.1 mL) was added via syringe under N2 to give a dark brown solution. The flask was sealed and placed in an oil bath that was preheated to 100 °C. After stirring at 100 °C for 48 h, the mixture was allowed to cool to rt and the reaction mixture was passed through a silica column with 4:1 hexanes/EtOAc to give 71 mg (0.19 mmol, 91%) of 30 as a viscous yellow oil. Rf 0.48 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.27 (d, J=11.9 Hz, 1H), 6.66 (d, J=11.9 Hz, 1H), 3.92 (s, 3H), 3.84 (s, 3H), 3.70 (s, 3H), 3.53–3.41 (m, 1H), 3.10 (s, 1H), 3.00 (s, 1H), 2.50–2.37 (m, 2H), 2.16–2.10 (m, 1H),1.85–1.78 (m,1H), 1.74 (m, 2H),1.49 (s, 3H), δ 1.39 (d, J=3.4 Hz, 3H), 1.38 (d, J=3.4 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 153.3, 151.5, 145.7, 136.6, 132.4, 131.2, 129.0, 127.8, 127.3, 125.1, 124.6, 61.9, 60.8, 60.4, 35.6, 35.3, 32.1, 31.1, 26.3, 25.7, 22.09, 22.08, 19.1; IR (film) νmax 2937, 2870, 1453, 1417, 1400, 1342, 1316, 1260, 1121, 1060, 1043 cm−1; HRMS (EI) calcd for C23H29NO3 ([M]+): m/z 367.2147, found 367.2149.
4.1.18. 8-Isopropyl-6,7,9-trimethoxy-1-methyl-2,3,4,5-tetrahydro-1H-dibenzo[a,d][7]annulene-1-carboxamide (29)
To a solution of cycloheptadiene 30 (285 mg, 0.780 mmol) in 2:1 EtOH/water (4 mL) was added Pt catalyst 4017 (11 mg, 0.03 mmol) in a vial at rt. The vial was sealed and the reaction mixture was heated to 80 °C. After stirring for 4 days, the solvent was removed and CH2Cl2 (1 mL) was added. The solution was filtered through a pad of Celite and concentrated to give 260 mg (0.67 mmol, 87%) of 29 as a light yellow oil that was used without further purification. Rf 0.25 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.18 (d, J=11.9 Hz, 1H), 6.54 (d, J=11.9 Hz, 1H), 5.36 (s, 2H), 3.90 (s, 3H), 3.82 (s, 3H), 3.64 (s, 3H), 3.45 (m, 1H), 3.13 (s, 1H), 2.94 (s, 1H), 2.44 (t, J=5.9 Hz, 2H), 2.07–1.99 (m, 1H), 1.66 (m, 1H), 1.58–1.52 (m, 2H), 1.35 (s, 3H), 1.33 (d, J=7.1 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 179.9, 153.2, 151.5, 145.6, 137.1, 132.3, 131.6, 131.1, 129.0, 128.1, 124.8, 61.9, 60.8, 60.3, 46.2, 36.3, 32.2, 31.8, 25.7, 24.2, 22.1, 22.0, 19.6; IR (film) νmax 3468, 3179, 2935, 2870, 1676, 1581, 1453, 1400, 1378, 1341, 1316, 1260, 1120, 1059, 1042 cm−1; HRMS (ESI) calcd for C23H32NO4 ([M+H]+): m/z 386.2326, found 386.2336.
4.1.19. 8-Isopropyl-6,7,9-trimethoxy-1-methyl-2,3,4,5-tetrahydro-1H-4a,11a-epoxydibenzo[a,d][7]annulene-1-carboxamide (41)
A mixture of 29 (260 mg, 0.670 mmol) and NaHCO3 (113 mg, 0.81 mmol) in CH2Cl2 (7 mL) at 0 °C was treated with m-chloroperbenzoic acid (70%, 185 mg, 0.810 mmol). The resulting mixture was stirred at 0 °C for 15 min, and then warmed to rt. After stirring for 3 h, the reaction mixture was quenched with saturated aq NaHSO3 (5 mL). The solution was stirred for 1 h and then extracted with Et2O (2×10 mL). The combined organic layers were washed with saturated aq NaHCO3, (3×10 mL), and brine (10 mL), dried over MgSO4, and concentrated to give 208 mg (0.52 mmol, 78%) of 41 as a clear oil. Rf 0.25 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 6.82 (d, J=11.6 Hz, 1H), 6.71 (s, 1H), 6.19 (d, J=11.6 Hz, 1H), 5.38 (s, 1H), 3.94 (s, 3H), 3.85 (s, 3H), 3.73 (s, 3H), 3.56 (d, J=13.2 Hz,1H), 3.47 (m,1H), 2.32 (d, J=13.0 Hz,1H), 2.15 (m, 2H), 2.06–1.98 (m, 2H), 1.84 (m, 2H), δ 1.40 (d, J=7.1 Hz, 3H), 1.35 (d, J=7.1 Hz, 3H),1.09 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 178.7,153.2, 152.2, 147.1, 133.6, 129.1, 128.5, 127.0, 125.5, 64.8, 62.4, 60.5, 60.4, 53.5, 45.2, 35.7, 32.5, 26.4, 25.7, 22.0, 19.9, 16.2; IR (film) νmax 3461, 2936, 1680, 1454, 1416, 1395, 1340, 1257, 1121, 1044 cm−1; HRMS (ESI) calcd for C23H32O5 ([M+H]+): m/z 402.2275, found 402.2281.
4.1.20. 11a-Hydroxy-8-isopropyl-6,7,9-trimethoxy-1-methyl-1,2,3,4, 5,11a-hexahydro-4a,1-(epoxymethano)dibenzo[a,d][7]annulen-13-one (42)
To a solution of 41 (190 mg, 0.470 mmol) in CH2Cl2 (18 mL) was added camphorsulfonic acid (109 mg, 0.470 mmol) at rt. After 2 h, the reaction mixture was quenched with saturated aq NaHCO3 (20 mL) and extracted with Et2O (2×30 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, and concentrated to give 163 mg (0.40 mmol, 86%) of 42 as a white foam that was used without further purification. Rf 0.33 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.15 (d, J=12.6 Hz, 1H), 5.88 (d, J=12.6 Hz, 1H), 3.91 (s, 3H), 3.76 (s, 3H), 3.69 (d, J=17.0 Hz, 1H), 3.65 (s, 3H), 3.45 (d, J=17.0 Hz, 1H), 3.43 (m, 1H), 1.83–1.76 (m, 2H), 1.71–1.65 (m, 3H), 1.65–1.58 (m, 2H), 1.34 (d, J=6.7 Hz, 3H),1.32 (d, J=6.7 Hz, 3H),1.30 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 179.0, 154.1, 154.0, 148.7, 133.6, 128.1, 127.7, 123.03, 122.96, 85.6, 80.1, 62.8, 60.4, 59.6, 51.9, 31.5, 30.8, 30.6, 26.1, 21.9, 21.8, 18.2, 14.0; IR (film) νmax 3466, 2932, 2874, 1781, 1653, 1570, 1558, 1507, 1455, 1416, 1397, 1341, 1256, 1123, 1092, 1035 cm−1; HRMS (ESI) calcd for C23H31O6 ([M+H]+): m/z 403.2115, found 403.2118.
4.1.21. 8-Isopropyl-6,7,9-trimethoxy-1-methyl-1,2,3,4,5,10-hexahydro-4a,1-(epoxymethano)dibenzo[a,d][7]annulen-13-one (43)
A mixture of 42 (100 mg, 0.25 mmol) and 10% Pd/C (40 mg) in EtOH (1.2 mL) was placed under an atmosphere of H2 (balloon) and stirred rapidly at rt for 12 h. The mixture was then filtered through Celite and concentrated to give 77 mg (0.19 mmol) of a yellow foam, which was used without further purification. To a solution of the resulting alcohol (77 mg, 0.19 mmol) in dry pyridine (1.6 mL) was added thionyl chloride (82 μL, 1.14 mmol) at rt. The solution instantly turned yellow. After 90 min, the reaction mixture was quenched with 1 N HCl (3 mL) and extracted with Et2O (3×3 mL). The combined organic layers were washed with brine (3 mL), dried MgSO4, and concentrated to give 45 mg (0.12 mmol, 47% over two steps) of 43 as a yellow oil that was used without further purification. Rf 0.53 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 5.50 (m, 1H), 3.84 (s, 3H), 3.80 (s, 3H), 3.62 (s, 3H), 3.41 (m, 2H), 3.35 (m, 1H), 3.26 (m, 1H), 1.94 (m, 1H), 1.81–1.74 (m, 3H), 1.48–1.39 (m, 3H), δ 1.34 (d, J=4.4 Hz, 3H), 1.33 (d, J=4.4 Hz, 3H),1.19 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 177.8,151.3,150.8,148.5, 146.7, 133.5, 127.5, 113.3, 84.5, 62.3, 60.3, 60.2, 49.0, 37.0, 30.3, 32.5, 26.0, 24.1, 24.2, 22.2, 22.1, 19.3, 16.3; IR (film) νmax 2936, 2873, 1779, 1454, 1414, 1381, 1341, 1253, 1122, 1094, 1043, 1033; HRMS (ESI) calcd for C23H30O5 ([M+H]+): m/z 387.2166, found 387.2167.
4.1.22. 8-Isopropyl-6,7,9-trimethoxy-1-methyl-1,2,3,4,5,11a-hexahydro-4a,1-(epoxymethano)dibenzo[a,d][7]annulen-13-one (44+45, from 43)
To a solution of 43 (60 mg, 0.15 mmol) in EtOAc (1.5 mL) were added tert-butyl hydrogen peroxide (0.15 mL) and 3 Å molecular sieves (150 mg) at rt. After 30 min, Mn(OAc)3·H2O (5 mg, 0.02 mmol) was added at rt and kept in the dark for 18 h. The reaction mixture was filtered through Celite to give 73 mg (0.15 mmol) as a yellow oil that was used without further purification. To a solution of the resulting peroxide (73 mg, 0.15 mmol) in Et2O (1.5 mL) was added a freshly prepared amalgam from aluminum foil (150 mg) in 2% HgCl2 solution that was washed with water (1 mL), ethanol (1 mL), and ether (1 mL). The reaction mixture was heated to 40 °C. After 2 h, the reaction mixture was filtered through Celite with Et2O and concentrated to give 34 mg (0.085 mmol) of a clear oil that was used without further purification. To a solution of the crude alcohol (34.0 mg, 0.085 mmol) in 1,2 dichloroethane (8.5 mL) were added tosyl hydrazide (79 mg) and camphorsulfonic acid (9.8 mg, 0.085 mmol) at rt. The reaction mixture was sealed and heated to 80 °C for 12 h. The reaction mixture was cooled to rt and passed through a pad of Celite. Flash chromatography (4:1 hexanes/EtOAc) gave 10 mg (0.025 mmol, 17% over three steps) of a colorless oil. 1H NMR analysis showed >10:1 dr with the intermediate leading to epi-icetexone (44) as the major diastereomer. Rf 0.55 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 6.97 (dd, J=12.2, 2.8 Hz,1H), 5.68 (dd, J=12.2, 3.1 Hz,1H), 3.88 (s, 3H), 3.73 (s, 3H), 3.64 (s, 3H), 3.58 (d, J=16.4 Hz,1H), 3.46–3.37 (m,1H), 3.13 (d, J=16.4 Hz,1H), 2.87 (s,1H), 1.84–1.76 (m, 1H), 1.70–1.60 (m, 3H), 1.56–1.46 (m, 2H), δ 1.34 (d, J=2.6 Hz, 3H), 1.32 (d, J=2.6 Hz, 3H), 1.23 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 180.1, 153.0, 148.6, 133.6, 127.0, 126.5, 124.7, 122.1, 85.3, 62.3, 60.4, 59.9, 55.4, 47.0, 37.3, 27.4, 27.0, 26.0, 22.0, 21.9, 19.3, 18.9; IR (film) νmax 2935, 1778, 1451, 1396, 1340, 1254, 1121, 1034; HRMS (ESI) calcd for C23H31O5 ([M+H]+): m/z 387.2166, found 387.2176.
4.1.23. 8-Isopropyl-6,7,9-trimethoxy-1-methyl-1,2,3,4,5,11a-hexahy-dro-4a,1-(epoxymethano)dibenzo[a,d][7]annulen-13-one (44+45, from 41)
To a solution of 41 (30 mg, 0.08 mmol) in benzene (1.5 mL) were added tosyl hydrazide (74.5 mg, 0.400 mmol) and camphorsulfonic acid (8.70 mg, 0.038 mmol) at rt. The reaction vessel was sealed and brought to 80 °C for 12 h. The reaction mixture was cooled to rt and passed through a pad of Celite. Basic alumina chromatography (4:1 hexanes/EtOAc) gave 12 mg (0.031 mmol, 42%) of 44 and 45 as a colorless oil. 1H NMR analysis showed 2.5:1 dr with the intermediate leading to epi-icetexone (44) as the major diastereomer. Rf 0.53 (2:1 hexanes/EtOAc); Major diastereomer: 1H NMR (500 MHz, CDCl3) δ 6.97 (dd, J=12.2, 3.0 Hz, 1H), 5.68 (m, 1H), 3.89 (s, 3H), 3.73 (s, 3H), 3.64 (s, 3H), 3.59 (d, J=16.4 Hz, 1H), 3.46–3.37 (m, 1H), 3.13 (d, J=16.4 Hz, 1H), 2.87 (s, 1H), 1.88–1.74 (m, 2H), 1.64 (m, 4H), δ 1.34 (d, J=2.9 Hz, 3H), 1.33 (d, J=2.8 Hz, 3H), 1.24 (s, 3H); Minor diastereomer: 1H NMR (500 MHz, CDCl3) 6.79 (dd, J=11.0, 2.4 Hz, 1H), 6.09 (dd, J=11.0, 2.4 Hz, 1H), 3.87 (s, 3H), 3.79 (s, 3H), 3.66 (s, 3H), 3.42 (m, 1H), 2.72 (m, 1H), 2.44–2.37 (m, 1H), 2.27 (s, 1H), 1.96–1.36 (m, 6H), δ 1.34 (d, J=2.9 Hz, 3H), 1.33 (d, J=2.8 Hz, 3H), 1.27 (s, 3H); IR (film) νmax 2935, 1778, 1451, 1396, 1340, 1254, 1121, 1034 cm−1; HRMS (ESI) calcd for C23H31O5 ([M+H]+): m/z 387.2166, found 387.2176.
4.1.24. (R)-tert-Butyl 2-cyano-2-methyl-5-(triisopropylsilyloxy)pentanoate (53)
A solution of tert-butyl 2-cyanopropanoate20d (49, 2.33 g, 15.0 mmol) in benzene (47 mL) was evacuated and backfilled with N2 (3×). Acrolein (50, 90%, 0.5 mL, 7.5 mmol) was added to the solution via syringe and the mixture was cooled to 0 °C. In a separate flask, Rh(acac)(CO)2 (128 mg, 0.15 mmol) and (S,S)-(R,R)-PhTRAP20b (51, 128 mg, 0.16 mmol) were dissolved in 3 mL of benzene, when the bubbling ceased the catalyst solution was added slowly to the reaction mixture via syringe. The resulting mixture was stirred at 0 °C for a 2 h and monitored by TLC. Once the reaction was completed, ethanol (99%, 10 mL) was added to the reaction mixture at 0 °C, NaBH4 (283 mg, 7.5 mmol) was then slowly added. The reaction was warmed up to rt and stirred for ~3 h. The reaction was quenched with a saturated solution of NH4Cl (50 mL) and extracted with EtOAc (4 × 30 mL). The combined organic layers were dried over MgSO4, and concentrated. The crude product was used without further purification. Rf 0.40 (6:4 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 3.68 (t, J=5.6 Hz, 2H), 2.07–1.96 (m, 1H), 1.87–1.73 (m, 2H), 1.73–1.61 (m, 1H),1.56 (s, 3H),1.50 (s, 9H),1.22–1.13 (m,1H). The crude alcohol (52) was dissolved in CH2Cl2 (50 mL). Imidazole (2.25 g, 33 mmol) and triisopropylsilyl chloride (3.5 mL,16.5 mmol) were added to the flask and the reaction mixture was stir at rt. When the reaction was complete by TLC (~4 h), saturated aq NH4Cl (30 mL) was added and the aqueous layer was extracted with CH2Cl2 (3×30 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, and concentrated. The crude material was purified via flash chromatography (9:1 hexanes/EtOAc) to give 4.32 g (11.7 mmol, 78%) of 53 as a clear oil. The desired product was often isolated along with triisopropyl silanol and it was carried on as is in the following reaction. Rf 0.66 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3): δ 3.71 (t, J=5.9 Hz, 2H), 1.99 (td, J=12.9, 4.4 Hz, 1H), 1.82 (td, J=13.0, 4.3 Hz, 1H), 1.78–1.69 (m, 1H), 1.64–1.57 (m, 1H), 1.54 (s, 3H), 1.48 (s, 9H), 1.09–1.02 (m, 21H); 13C NMR (126 MHz, CDCl3): δ 168.4, 120.5, 83.7, 62.5, 44.6, 35.2, 28.9, 27.8, 23.3, 18.1, 12.0; IR (film) νmax 2943, 2867, 1739 cm−1; HRMS (ESI) calcd for C20H39O3 NNaSi ([M+Na]+): m/z 392.2591, found 392.2594; −33.32 (c 1.17, CHCl3).
4.1.25. (R)-2-(Hydroxymethyl)-2-methyl-5-(triisopropylsilyloxy)pentanenitrile (36)
To a solution of 53 (4.32 g, 11.7 mmol) in a mixture of ethanol(99%)/THF (4:1, 100 mL, 0.15 M) was added NaBH4 (2.84 g, 75 mmol). The resulting mixture was stirred at ambient temperature for 18 h. The reaction mixture was cooled to 0 °C and then quenched slowly by addition of a saturated aq NH4Cl (100 mL). The aqueous layer was extracted with EtOAc (4×30 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, and concentrated. The crude product was purified via flash chromatography (9:1 to 3:2 hexanes/EtOAc) to give 2.82 g (9.41 mmol, 80%) alcohol 36 as a clear oil. The alcohol 36 showed identical NMR data to the previously reported data for the racemic alcohol. Rf 0.27 (8:2 hexanes/EtOAc).
4.1.26. (S)-N-((8-Isopropyl-6,7,9-trimethoxy-1-methyl-2,3,4,5-tetra hydro-1H-dibenzo[a,d][7]annulen-1-yl)methyl)-4-bromobenzamide (54)
To a flame dried Schlenk flask equipped with a stir bar and under N2 atmosphere was added lithium aluminum hydride (38 mg, 1.0 mmol). Nitrile 30 (79 mg, 0.20 mmol) was added as a solution in anhydrous THF (4 mL). The Schlenk flask was sealed and the reaction was heated to 65 °C for 2 h. The reaction mixture was cooled to rt and slowly quenched by the addition of powdered Na2SO4·10H2O (~200 mg). The solution was stirred overnight open to air, filter through Celite, wash with EtOAc (50 mL), and concentrated to give 73 mg (0.20 mmol, 100%) of the amine that was used without further purification. The amine (25 mg, 0.07 mmol) was dissolved in CH2Cl2 (1.4 mL) and to the solution was added triethylamine (20 μL, 0.14 mmol) followed by the 4-bromobenzoyl chloride (23 mg, 0.11 mmol). The reaction was stirred at ambient temperature and monitored by TLC. When the reaction was completed, the solution was concentrated and loaded directly onto a silica column. The product was purified by flash chromatography (4:1 hexanes/EtOAc) to yield 27 mg (0.05 mmol, 71%) of 54 as a white solid. Slow diffusion crystallization of hexanes into EtOAc provided X-ray quality crystals. Rf 0.60 (2:1 hexanes/EtOAc); mp 148–157 °C; 1H NMR (600 MHz, CDCl3) δ 7.34 (d, J=8.0 Hz, 2H), 7.22 (d, J=11.9 Hz, 1H), 6.98 (s, 2H), 6.67 (d, J=11.9 Hz, 1H), 5.45 (s, 1H), 3.97 (s, 3H), 3.83 (s, 3H), 3.60 (dd, J=13.2, 8.7 Hz,1H), 3.52–3.31 (m, 5H), 3.08 (d, J=12.7 Hz,1H), 2.69 (s, 1H), 2.38 (t, J=6.1 Hz, 2H), 1.70–1.60 (m, 2H), 1.47 (t, J=5.2 Hz, 2H), 1.29 (d, J=7.1 Hz, 3H), 1.28–1.24 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 165.8, 153.4, 151.6, 145.8, 133.1, 132.7, 132.5, 132.0, 131.9, 128.9, 128.1, 128.0,126.0,124.8, 61.7, 61.1, 60.6, 49.4, 38.2, 34.1, 32.7, 32.5, 25.8, 24.1, 22.2, 22.2, 19.3; IR (film) νmax 3338, 3425, 2931, 2869, 1656, 1590, 1525 cm−1; HRMS (ESI) calcd for C30H37O4N79Br ([M+H]+): m/z 554.1900, found 554.1900; +72.0 (c 0.65, CHCl3).
Supplementary Material
Acknowledgments
The authors are grateful to the NIH (NIGMS RO1 086374 to R.S. and F31GM089139-01 to F.C.) and Amgen for generous, unrestricted financial support. D.L. thanks the FQRNT–Canada for a postdoctoral fellowship. The authors thank Ms. Ariel Yeh (UC Berkeley) and Mr. Christopher Marth (UC Berkeley) for their assistance in the preparation of starting material 31.
Footnotes
Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.tet.2013.04.049
References and notes
- 1.Simmons EM, Sarpong R. Nat Prod Rep. 2009;26:1195–1217. doi: 10.1039/b908984e. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dominguez XA, Gonzalez FH, Aragon R, Guiterrez M, Marroquin JS, Watson W. Planta Med. 1976;30:237–241. doi: 10.1055/s-0028-1097724. [DOI] [PubMed] [Google Scholar]; (b) Watson WH, Taira Z, Dominguez XA, Gonzales H, Guiterrez M, Aragon R. Tetrahedron Lett. 1976;17:2501–2502. [Google Scholar]
- 3.Taira Z, Watson WH, Dominguez XA. J Chem Soc Perkin Trans 2. 1976:1728–1730. [Google Scholar]
- 4.Esquivel B, Calderon J, Flores E, Sanchez A, Rivera R. Phytochemistry. 1997;46:531–534. [Google Scholar]
- 5.(a) Nieto M, Garcia EE, Giordano OS, Tonn CE. Phytochemistry. 2000;53:911–915. doi: 10.1016/s0031-9422(99)00480-x. [DOI] [PubMed] [Google Scholar]; (b) Sanchez AM, Jimenez-Ortiz V, Sartor T, Tonn CE, Garcia EE, Nieto M, Burgos MH, Sosa MA. Acta Trop. 2006;98:118–124. doi: 10.1016/j.actatropica.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Uchiyama N, Kabututu Z, Kubata BK, Kiuchi F, Ito M, Nakajima-Shimada J, Aoki T, Ohkubo K, Fukuzumi S, Martin SK, Honda G, Urade Y. Antimicrob Agents Chemother. 2005;49:5123–5126. doi: 10.1128/AAC.49.12.5123-5126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Majetich G, Grove JL. Org Lett. 2009;11:2904–2907. doi: 10.1021/ol9009128. [DOI] [PubMed] [Google Scholar]
- 8.Cortez F, de J, Sarpong R. Org Lett. 2010;12:1428–1431. doi: 10.1021/ol902959v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Chatani N, Inoue H, Kotsuma T, Murai S. J Am Chem Soc. 2002;124:10294–10295. doi: 10.1021/ja0274554. [DOI] [PubMed] [Google Scholar]; (b) Chatani N, Morimoto T, Muto T, Murai S. J Am Chem Soc. 1994;116:6049–6050. [Google Scholar]
- 10.For a leading discussion on the use of cycloisomerization catalysts in complex molecule synthesis, see: Fürstner A, Davies PW. Angew Chem Int Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335.
- 11.(a) Simmons EM, Sarpong R. Org Lett. 2006;8:2883–2886. doi: 10.1021/ol061037c. [DOI] [PubMed] [Google Scholar]; (b) Simmons EM, Yen JR, Sarpong R. Org Lett. 2007;9:2705–2708. doi: 10.1021/ol0712428. [DOI] [PubMed] [Google Scholar]
- 12.(a) Barton D, Beaton J, Geller L, Pechet M. J Am Chem Soc. 1960;82:2640–2641. [Google Scholar]; (b) Majetich G, Wheless K. Tetrahedron. 1995;51:7095–7129. [Google Scholar]
- 13.Majetich G, Zhang Y. J Am Chem Soc. 1994;116:4979–4980. [Google Scholar]
- 14.See Ref. 12 for conditions for Barton nitrite ester reaction.
- 15.Wehrli H, Cereghetti M, Schaffner K, Jeger O. Helv Chem Acta. 1960;47:367–371. [Google Scholar]
- 16.Müller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521–522. [Google Scholar]
- 17.Ghaffar T, Parkins AW. Tetrahedron Lett. 1995;36:8657–8660. [Google Scholar]
- 18.Yu JQ, Corey EJ. Org Lett. 2002;4:2727–2730. doi: 10.1021/ol0262340. [DOI] [PubMed] [Google Scholar]
- 19.Myers A, Zheng B. Tetrahedron Lett. 1996;37:4841–4844. [Google Scholar]
- 20.(a) Sawamura M, Hamashima H, Ito Y. Tetrahedron: Asymmetry. 1991;2:593–596. [Google Scholar]; (b) Sawamura M, Hamashima H, Sugawara M, Kuwano R, Ito Y. Organometallics. 1995;14:4549–4558. [Google Scholar]; (c) Sawamura M, Hamashima H, Ito Y. J Am Chem Soc. 1992;114:8295–8296. [Google Scholar]; (d) Sawamura M, Hamashima H, Ito Y. Tetrahedron. 1994;50:4439–4454. [Google Scholar]
- 21.Determined using a ChiralPak IC column. For details, see the Supplementary data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.