

Abstract
Purpose
Platelets are essential for primary hemostasis; however, platelet activation also plays an important proinflammatory role. Inflammation promotes the development of cardiac fibrosis and heart failure induced by hypertension. In this study, we aimed to determine whether inhibiting platelet activation using clopidogrel could inhibit hypertension-induced cardiac inflammation and fibrosis.
Methods
Using a mouse model of angiotensin II (Ang II) infusion (1,500 ng/[kg·min] for 7 days), we determined the role of platelet activation in Ang II infusion-induced cardiac inflammation and fibrosis using a P2Y12 receptor inhibitor, clopidogrel (50 mg/[kg·day]).
Results
CD41 staining showed that platelets accumulated in Ang II-infused hearts. Clopidogrel treatment inhibited Ang II infusion-induced accumulation of α-SMA+ myofibroblasts and cardiac fibrosis (4.17 ± 1.26 vs. 1.46 ± 0.81, p < 0.05). Infiltration of inflammatory cells, including Mac-2+ macrophages and CD45+Ly6G+ neutrophils (30.38 ± 4.12 vs. 18.7 ± 2.38, p < 0.05), into Ang II-infused hearts was also suppressed by platelet inhibition. Real-time PCR and immunohistochemical staining showed that platelet inhibition significantly decreased the expression of interleukin-1β and transforming growth factor-β. Acute injection of Ang II or PE stimulated platelet activation and platelet-leukocyte conjugation, which were abolished by clopidogrel treatment.
Conclusion
Thus, inhibition of platelet activation by clopidogrel prevents cardiac inflammation and fibrosis in response to Ang II. Taken together, our results indicate Ang II infusion-induced hypertension stimulated platelet activation and platelet-leukocyte conjugation, which initiated inflammatory responses that contributed to cardiac fibrosis.
Keywords: Clopidogrel, Platelet activation, Inflammation, Hypertension, Cardiac fibrosis
Introduction
Cardiac fibrosis is an important pathological feature of cardiac remodeling in hypertension [1]. Increased fibrosis and subsequent cardiac dysfunction can cause heart failure, arrhythmia, and even sudden death [2]. Among the numerous risk factors for cardiac fibrosis, the renin-angiotensin-aldosterone system (RAAS), particularly the key effector molecule angiotensin II (Ang II), is of primary importance. Elevated Ang II levels are a well-established risk factor for the development of hypertension, and Ang II is also an important proinflammatory and profibrotic factor in cardiac remodeling [3–5]. Increasing evidence has indicated that inflammation plays a key role in the process of cardiac remodeling [6]. Multiple inflammatory cells and cytokines were reported to be involved in the process of cardiac fibrosis. Elevated blood pressure is important for initiating inflammatory responses; hydralazine, a direct-acting smooth muscle relaxant, decreased collagen expression and production of several profibrotic cytokines in a rat model of DOCA-salt induced inflammation and fibrosis or Ang II infusion-induced cardiac fibrosis through lowering of the elevated blood pressure [7, 8]. However, the early events that initiate inflammation in response to hypertension remain unknown.
Platelets are unnucleated fragments of bone marrow megakaryocytes. In addition to hemostasis and thrombosis, platelets are involved in inflammatory and immune processes. As a “monitor” of blood flow, platelets are sensitive to any circulation anomaly. Endothelial cell damage or sheer stress conditions [9, 10] activate platelets and stimulate the expression of specific adhesion molecules (e.g., P-selectin, CD40, CD154, and GP IIb/IIIa) and secretion of cytokines and chemokines [11, 12]. Activated platelets interact with neutrophils, monocytes/macrophages, and T cells via surface molecule binding to specific ligands or receptors, such as P-selectin/P-selectin glycoprotein ligand-1 (PSGL-1), CD40/CD154, and Mac-1/αIIbβ3 [13, 14]. Platelet activation is involved in the process of atherosclerosis, myocardial infarction, and other cardiovascular diseases [13]. We hypothesized that Ang II-induced hypertension might activate platelets, which initiate inflammation and contribute to cardiac fibrosis development.
In the present study, we used clopidogrel, a P2Y12 subtype ADP receptor inhibitor, to block platelet activation [15]. We found that platelet activation was an early event in response to Ang II infusion-induced hypertension, which led to cardiac inflammation and fibrosis.
Materials and Methods
Reagents
Ang II and phenylephrine (PE) were purchased from sigma chemicals (Sigma,St Louis, Missouri, USA). Clopidogrel (Plavix, Sanofi-Aventis) was purchased from local pharmacy. A 75 mg/tablet clopidogrel was dissolved in 15 ml 0.9 % saline, and the clopidogrel solution was freshly prepared every day. Clopidogrel or vehicle was administered intragastrically to mice for 7 days during the Ang II infusion (at a concentration of 50 mg/kg, once daily) as previously described. [16, 17]
Animal Model
Two-month old male C57/BL6 mice (22 g to 25 g) were purchased from Academy of Military Sciences (Beijing, China). All animal care and experimental protocols complied with the Animal Management Rule of the Ministry of Health, People’s Republic of China (Documentation no. 55, 2001) and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996) and were approved by the Institutional Animal Care and Use and Committee of the Capital University of Medical Science.
Mice were randomly divided into 4 groups (8 in each group). The mice were anesthetized with sodium pentobarbital (50 mg/kg IP). Ang II was dissolved in 0.01 N acetic acid saline solution. With a sterile technique, osmotic minipumps (Alzet MODEL 1007D, DURECT, Cupertino, CA, USA) with Ang II or vehicle (untreated) were placed subcutaneously in the intrascapular area to deliver Ang II at an infusion rate of 1,500 ng/kg per minute for 7 days. Drug or vehicle administration was well tolerated by all animals and no animal died or displayed signs of discomfort in any group.
Blood Pressure Measurement
Mice were trained daily for 3 days to have systolic blood pressure (SBP) determined with a computerized mouse tail-cuff system (BP-98A softron, Tokyo, Japan). Ten to twenty repeated values were averaged at each determination point. SBP was determined before and at the 4th-7th days of the Ang II infusion.
Cardiac Echocardiography
Mice were anesthetized with isoflurane and analyzed for structural and function by use of the Vevo 770 high-resolution microimaging system with a 30 MHz transduser (Visualsonic, Toronto, Canada). The heart was imaged in the 2D mode in the parasternal short-axis view.
Echocardiographic measurements were taken on M-mode in triplicate from all 8 mice per group as previously described with minor modification [18, 19].
Bleeding Time Measurement
Bleeding time was measured through a tail transection method as previously described with minor modification [20] . Briefly, the maximum bleeding time for recording was 900 s, and the terminal point was the end of bleeding; Moreover, bleeding time recording continued until a new arrest lasting for more than 30 s had occurred if the bleeding restarted within 30 s.
Preparation of Tissue Sections and Histopathology
Hearts were embedded in paraffin serial sections (5 μm) and cut and placed on polysinecoated glass slides as described [21–23]. To measure the size of cardiomyocytes, heart sections were deparaffinized and incubated with 100 μg/ml FITC-labeled WGA (Sigma) for 90 min. At least 50 cells were measured to calculate the area per slide. For morphological staining, heart sections were also stained with hematoxylin and eosin or Masson’s trichrome reagent. Cardiac fibrosis was quantitated in a blinded fashion by measuring the total blue area (mm2) with a NIS- ELEMENTS quantitative automatic program (Nikon, Japan) in the Masson’s trichrome-stained heart sections. For immunostaining, deparaffinized heart sections (5 μm) were incubated with avidin, biotin (Maixin, Fuzhou, China) for 15 min. After the inactivation of endogenous peroxides, they were incubated with anti-mouse monoclonal antibodies against Mac-2(1:400 dilution), interleukin-1β (IL-1β) (1:200 dilution), transforming growth factor-β (TGF-β) (1:300 dilution) and α-smooth muscle actin (α-SMA) (1:200 dilution, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C overnight. The sections were then incubated with biotinylated secondary goat anti rabbit antibody for 30 min at 37 °C. After being incubated with diaminobenzidine tetrahydrochloride, sections were counter stained using hematoxylin, dehydrated through gradient alcohols, and mounted with cover slips. Images were obtained using a CCD camera under a microscope (ECLIPSE80i/90i, Nikon, Japan) with a ×200 lens, and 10–20fields/section were chosen randomly from 8 mice per group. For CD41 staining, frozen heart sections were labeled with anti mouse monoclonal antibody against CD41 (1:100 dilution, Abcam, Cambridge, MA), then incubated with FITC-conjugated secondary antibody (JacksonImmunoResearch Laboratories, West Grove, PA). Images were captured by use of a Nikon Eclipse TE2000-S microscope (Nikon, Japan).
Flow Cytometric Analysis
The inflammatory cells infiltrated were quantified by flow cytometry as described previously by our laboratory [18, 24]. Briefly, heart tissues were quickly minced into multiple small tubes and digested in an enzyme mixture, which contains collagenase type I (0.05 mg/ml) and type IV (0.05 mg/ml), hyaluronidase (0.025 mg/ml), DNase I (0.01 mg/ml) and soybean trypsin inhibitor (0.01 mg/ml) dissolved in DMEM for 45 min at 37 °C. The cell suspension was centrifuged and preincubated with Fc-γ block antibody (anti-mouse CD16/32; Pharmingen, San Diego, CA) to prevent nonspecific binding, further stained with antibodies PE-conjugated antimouse CD45 and PerCP/Cy5.5-conjugated antimouse Ly6G for 30 min at 4 °C in the dark. Data were collected by use of an EPICS XL flow cytometer (Beckman Coulter, Miami, FL) and analyzed by use of Cellquest (Beckman). Total cell population was recorded by cell size (forward scatter) and internal complexity (side scatter) and showed even distribution in a representative cell suspension prepared from heart tissues. Leukocytes were stained with monoclonal anti-CD45 and gated with CD45 fluorescence versus side scatter, while Ly6G+ neutrophils were further gated on CD45+ cells. Appropriate isotype controls of irrelevant specificity were performed.
To quantify the platelet activation and consequently platelet-leukocyte conjugation, peripheral blood was obtained from mice via lateral caudal vein, then underwent erythrocyte lysis using FACS lysing solution according to the manufacture’s instruction (BD Biosciences). Leukocytes were labeled with PerCP/Cy5.5-conjugated anti-mouse CD45.2 (Biolegend, San Diego, CA), while platelets were labeled with PE-conjugated anti-mouse CD41 and FITC-conjugated anti-mouse CD62P (both BD Bioscience) for 30 min at 4 °C in the dark. Data were collected by flow cytometry (Beckman Coulter).
RNA Analysis
Total RNA was extracted by the Trizol reagent method (Invitrogen, Carslbad, CA, USA), and cDNA was synthesized according to the manufacturers’ instruction (Promega, Southhampton, UK). The housekeeping gene GAPDH was used as control. Primers used in this study were for collagen I, forward 5′-GAGCGGAGAGTACTGGATCG-3′ and reverse 5′-TACTCGAACGGGAATCCATC-3′; IL-1β, forward 5′-CTTCAGGCAGGCAGTATCACTCAT-3′ and reverse 5′-TCTAATGGGAACGTCACACACCAG-3′; TGF-β, forward 5′-CAACAATTCCTGGCGTTACCTTGG-3′ and reverse 5′-GAAAGCCCTGTATTCCGTCTCCTT-3′ and GAPDH, forward 5′-CCTGGAGAAACCTGCCAAGTATGA-3′ and reverse 5′-TTGAAGTCACAGGAGACAACCTGG-3′. Real-time PCR was carried out with the use of Bio-Rad iQ5 (Hercules, CA, USA).
Acute Studies
To study the role of the platelets in Ang II-induced acute hypertension, male mice were anesthetized with sodium pentobarbital (50 mg/kg IP). Two hours before Ang II or PE injection, mice were treated with clopidogrel or PBS intragastrically at a concentration of 50 mg/kg. Mice were then placed on a heating platform for 10 min. Blood pressure was measured every 30 s for 25 min with a mouse tail-cuff system (BP-98A softron). After 5 min of baseline SBP recording, acute hypertension in anesthetized mice was induced with an Ang II or PE injection as reported (0.2 mg/kg, IP)[25, 26]. About 10 min after Ang II or PE injection, a blood sample was collected from mice via lateral caudal vein using glass pipettes dipped in 3.8 % sodium citrate solution (Sigma-Aldrich) for flow cytometric analysis. Bleeding time was monitored after blood sample collection.
Statistical Analysis
Data are expressed as mean ± SEM. Statistical significance of the results was calculated by Student’s t test using Graphpad software (GraphPad Prism version 5.00 for Windows; GraphPad Software Inc. San Diego, California, USA); A p < 0.05 was considered statistically significant. The Mann Whitney test was applied to the studies of bleeding time.
Results
Clopidogrel Treatment Prolonged the Bleeding Time, but it did not Affect Blood Pressure and Cardiac Function in Ang II-infused Mice
To determine the role of platelets in Ang II infusion-induced cardiac fibrosis, clopidogrel was used to inhibit platelet activation [27]. As shown in Fig. 1a, there was an increase in systolic blood pressure after Ang II infusion in both the vehicle- and clopidogrel-treated groups. As expected, clopidogrel treatment significantly prolonged tail bleeding time (Fig. 1b). WGA staining showed that Ang II infusion increased cardiomyocyte size, which was partly suppressed by clopidogrel treatment (Fig. 1c). Echocardiography revealed that clopidogrel treatment reduced Ang II infusion-induced increased LV mass (Fig. 1d), while the cardiac ejection fraction, LV wall thickness, diameter, and fractional shortening were similar between the groups (data not shown).
Fig. 1.

Clopidogrel treatment does not affect the blood pressure and cardiac function after Ang II infusion. a Blood pressure measurement in clopidogrel or vehicle treated groups before and on the 4th day and 6th day of Ang II infusion (n = 8). b The tail transection method was used to measure the bleeding time on the 7th day of Ang II or vehicle infusion with clopidogrel or vehicle treatment; c Wheat germ agglutinin staining was used to evaluate cardiomyocyte size in heart sections of Ang II or vehicle infused heart with clopidogrel or vehicle treatment (top, Bar = 50 μm); Quantitative analysis of cardiomyocyte cross-sectional area (bottom); d Echocardiography was performed at day 7 after Ang II infusion. *p < 0.05,**p < 0.01 vs. vehicle group, # p < 0.05, ## p < 0.01 vs. Ang II group. Ang II: angiotensin II
Clopidogrel Treatment Inhibited Ang II Infusion-Induced Platelet Deposition and Cardiac Fibrosis
To examine the effect of platelet activation on Ang II infusion-induced cardiac fibrosis, we performed CD41 immunofluorescence staining after 7 days in mice hearts. Ang II infusion stimulated CD41+ platelet deposition in the heart, which was inhibited by clopidogrel treatment (Fig. 2a). As shown in Fig. 2b and c, Ang II infusion significantly increased perivascular and interstitial fibrosis (4.17 ± 1.26 vs. 1.46 ± 0.81, p < 0.05), as well as α-SMA+ myofibroblast accumulation, which were decreased with clopidogrel treatment. Furthermore, Ang II infusion increased collagen I mRNA expression, which was also suppressed by clopidogrel treatment (Fig. 2d).
Fig. 2.
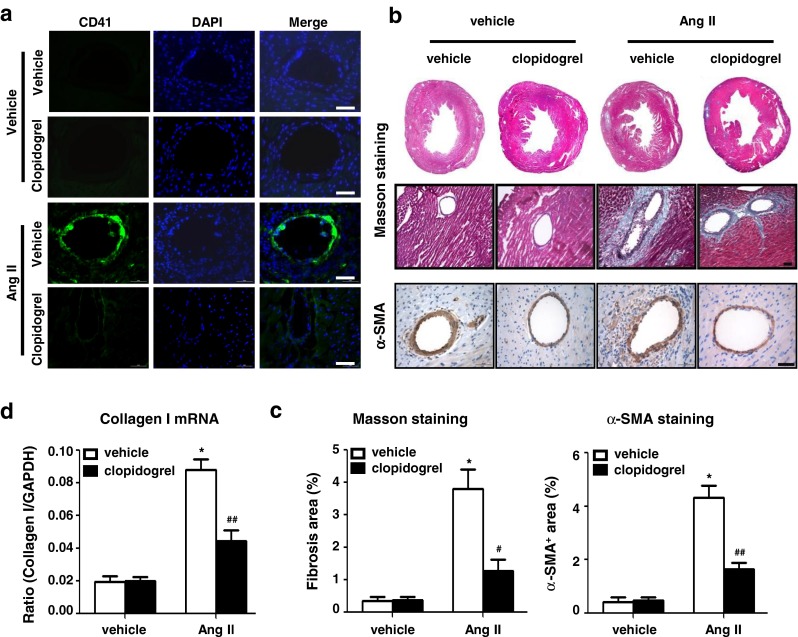
Clopidogrel treatment inhibits platelet deposition and cardiac fibrosis in Ang II infused mice hearts a Immunofluorescence staining for CD41 showed platelet deposition in Ang II or vehicle infused heart (Bar = 50 μm); b Representative photomicrographs from histological sections of hearts stained with Masson’s trichrome staining (top) and immunohistochemical staining for α-SMA (bottom) at day 7 of Ang II or vehicle infusion in each group (Bar = 50 μm). c Histogram shows less fibrosis area and α-SMA+ area in Ang II + Clopidogrel group and d lower mRNA level of collagen I.*p < 0.05 vs. vehicle group, # p < 0.05, ## p < 0.05 vs. Ang II group (n = 8)
Inhibition of Platelet Activation Prevented Ang II Infusion-Induced Inflammatory Cell Infiltration in the Heart
It has been reported that inflammatory cell infiltration into the heart is responsible for cardiac fibrosis [28], and platelets interact with several types of inflammatory cells that are critical for initiation of inflammation [12]. We therefore examined the effect of platelet activation on inflammatory cell infiltration. As shown in Fig. 3a, a large number of inflammatory cells infiltrated the hearts of Ang II-infused mice, which was reduced by clopidogrel treatment. The number of Mac-2+ macrophages was also decreased by clopidogrel treatment in Ang II-infused mouse hearts (Fig. 3a). To further quantitate the inflammatory cell infiltrates in Ang II-infused hearts, flow cytometric analysis was used to measure CD45+ inflammatory cells and CD45+ Ly6G+ neutrophils. As shown in Fig. 3b and c, Ang II infusion increased CD45+ cell infiltration compared to the vehicle-treated group, with CD45 and Ly6G double-positive neutrophils comprising a large proportion (30.38 ± 4.12 vs. 18.7 ± 2.38, p < 0.05), whereas clopidogrel treatment significantly suppressed both CD45+ and CD45+Ly6G+ cell infiltration.
Fig. 3.
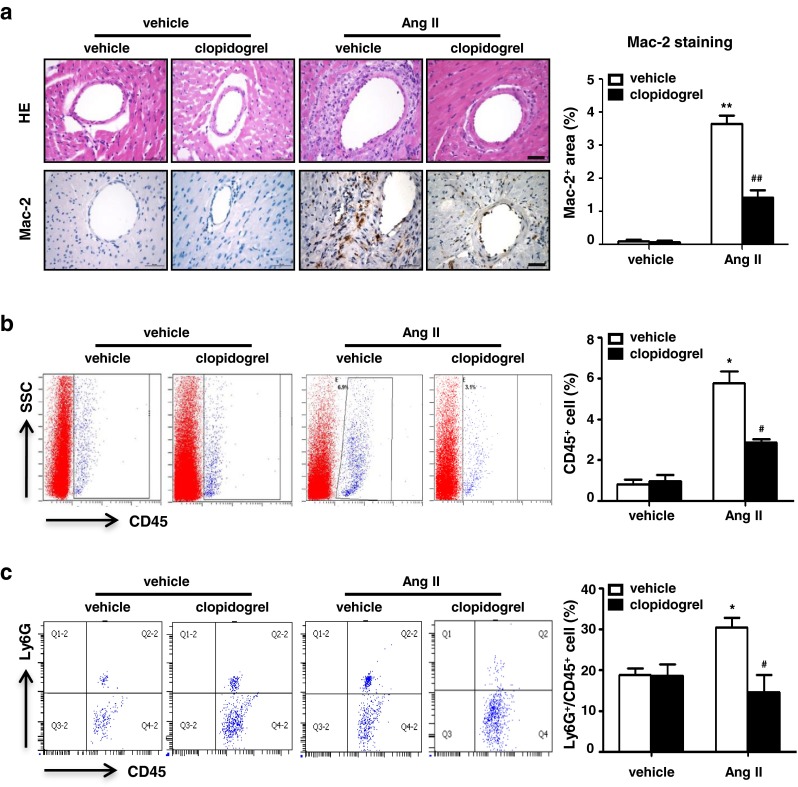
Clopidogrel treatment inhibits inflammatory cell infiltration in Ang II infused mice hearts. a Representative photomicrographs from histological sections of hearts with hematoxylin-eosin staining and immunohistochemical staining for Mac-2 at day 7 of Ang II infusion in each group (left panel, Bar = 50 μm, n = 8). Histogram shows less Mac-2+ area in Ang II+ Clopidogrel group (right panel). b Leukocytes are gated with CD45 fluorescence versus side-angle scatter (SS). Scatter plots are gated on CD45+population cells (left panel); Histogram shows decreased leukocytes infiltrated in Ang II infused heart after clopidogrel treatment (right panel, n = 4). c Infiltrated neutrophils (CD45+Ly6G+ cells) were analyzed in each group after 7 days of Ang II or vehicle infusion. Data are mean ± SEM (n = 4). *p < 0.05 vs. vehicle group, # p < 0.05 vs. Ang II group
Inhibition of Platelet Activation Suppressed Ang II Infusion-Induced Cytokine Expression
TGF-β is a well-established profibrotic cytokine, and IL-1β is an important cytokine that is involved in cardiac fibrosis [29, 30]. As both of these cytokines are secreted by activated platelets, we examined their expression in the mouse hearts. Immunohistochemical staining showed that clopidogrel treatment inhibited both TGF-β (8.09 ± 3.21 vs. 2.34 ± 0.59, p < 0.01) and IL-1β expression (7.14 ± 1.91 vs. 0.99 ± 0.27, p < 0.01), which were significantly upregulated in Ang II-infused mouse hearts (Fig. 4a and b). Moreover, real-time PCR analysis showed that Ang II increased mRNA expression of these two cytokines, which was also suppressed by clopidogrel treatment (Fig. 4c).
Fig. 4.
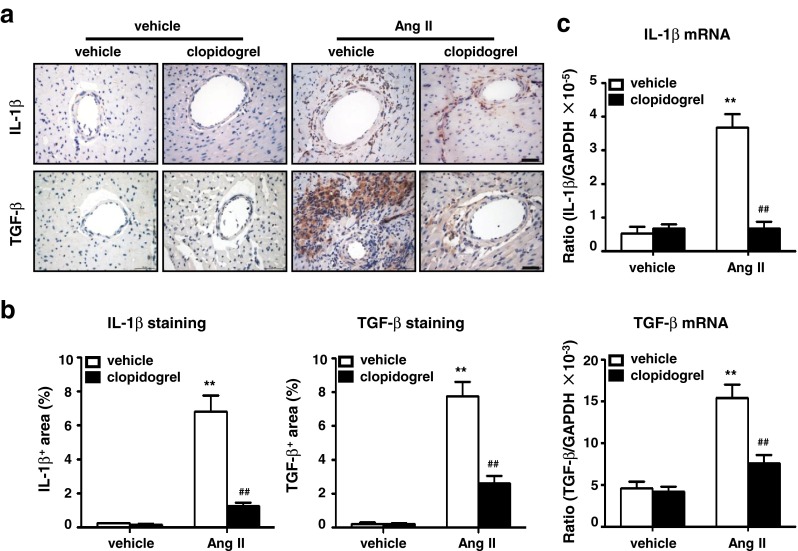
Clopidogrel treatment decreases inflammatory cytokine production in Ang II infused mice hearts. a Immunohistochemical analysis of cytokine expression was detected by anti-IL-1β and anti-TGF-β in Ang II or vehicle infused heart sections with clopidogrel or vehicle treatment (Bar = 50 μm, n = 8). b Histogram shows less IL-1β+ and TGF-β+ area in clopidogrel treated group and c a lower mRNA level of IL-1β and TGF-β. Data are mean ± SEM for n = 8 mice with 10 fields per animal. **p < 0.01 vs. vehicle group, ## p < 0.01 vs. Ang II group. IL-1β: interleukin-1β, TGF-β: transforming growth factor-β
Platelet Activation is an Early Event in Ang II Injection-Induced Hypertension
We previously reported that Ang II infusion-induced cardiac inflammation and fibrosis is pressure dependent [8]. Therefore, we tested whether platelet activation is the early event associated with this elevation in blood pressure. Acute hypertension was induced via intraperitoneal injection of Ang II. As expected, Ang II caused a rapid increase in blood pressure (Fig. 5a). Ang II injection stimulated platelet-leukocyte conjugation (shown as CD45+CD41+) in peripheral blood, which was abolished by clopidogrel treatment (Fig. 5b and c). Furthermore, Ang II stimulated CD62P (P-selectin), which is expressed on activated platelets; this was also inhibited by clopidogrel treatment. As studies have demonstrated that Ang II can activate platelets in an AT1 dependent fashion in response to Ang II infusion [31], we therefore used acute PE injection as reported to increase blood pressure in an Ang II/AT1 independent fashion [25, 26]. As shown in Fig. 5a, similar to Ang II injection, PE injection also increased blood pressure. Flow cytometric analysis also showed that PE injection caused platelet-leukocyte conjugation (shown as CD45+CD41+) and platelet activation (shown as CD41+CD62P+) in the peripheral blood, which was reduced by clopidogrel treatment (Fig. 5d). These results demonstrated that in addition to a direct effect of Ang II on its receptor AT1, an acute increase in blood pressure could activate platelet-leucocyte complexes.
Fig. 5.
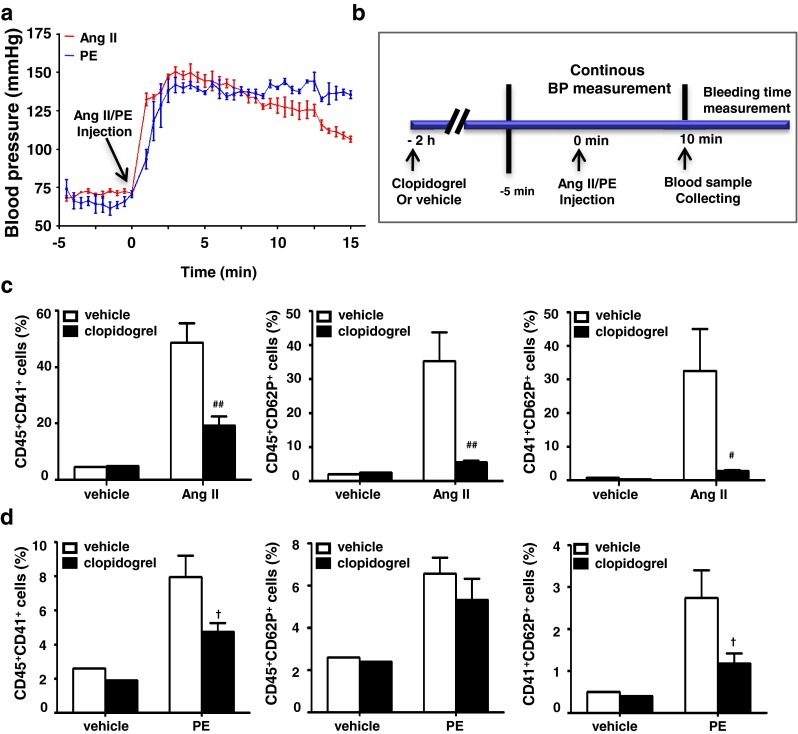
Clopidogrel treatment suppresses Ang II injection induced platelet activation and platelet-leukocyte conjugation. a Continuous blood pressure measurement shows a rapid blood pressure increase in response to Ang II or PE injection. b Clopidogrel was administrated 2 h before Ang II injection to test its effect on platelet activation in response to elevated blood pressure. Leukocytes were gated with CD45 fluorescence versus side-angle scatter (SS). Platelet-leukocyte conjugation (CD45+CD41+, CD45+CD62P+) and platelet activation (CD41+CD62P+) were analyzed in the blood samples after c Ang II injection d PE injection with clopidogrel or vehicle treatment. Data are mean ± SEM for n = 4. # p < 0.05, ## p < 0.01 vs. Ang II group; † p < 0.05 vs. PE group. PE: phenylephrine
Discussion
We previously reported that Ang II infusion-induced cardiac inflammation is pressure-dependent [8]; however, the early event that occurs in response to elevated blood pressure and triggers this inflammation remains unknown. Our present study revealed several novel findings. (1) Ang II infusion stimulates platelet deposition on microvessels; (2) the P2Y12 antagonist clopidogrel used to inhibit the platelet activation decreases Ang II infusion-induced cardiac inflammation and fibrosis; and (3) acute elevation of blood pressure stimulates platelet activation and platelet-leukocyte conjugation, which was abrogated by clopidogrel treatment. Thus, inhibition of platelet activation by clopidogrel prevents cardiac inflammation and fibrosis in response to Ang II. Our results suggest that platelet activation in response to elevated blood pressure may be an early event that initiates inflammation in Ang II-infused mice.
Increasing evidence has shown that platelets function as an immune modulator in several inflammation-associated processes [32]. Clinical studies have reported that low-grade inflammation and persistent platelet activation are present in patients with hypertension, particularly those with complications (e.g. vascular lesions and microalbuminuria) [33–36]. Consistent with these studies, our study provides direct evidence that platelet activation is involved in hypertension-induced cardiac inflammation and fibrosis (Fig. 2). Our results show that Ang II infusion induces platelet activation and platelet-leukocyte conjugation. These results are consistent with studies showing that Ang II injection induces platelet activation in humans [37]. It has also been reported that Ang II promotes platelet aggregation at lower concentrations in human platelets via the AT1 receptor, which can be inhibited by AT1 receptor antagonists [31]. Moreover, AT1 receptor antagonists inhibit platelet adhesion (collagen-stimulated) and aggregation (U46619-stimulated) in a dose-dependent manner [38]. Taken together with our results, we conclude that elevated blood pressure and the Ang II receptor stimulate platelet activation, platelet-leukocyte conjugation, and cardiac inflammation in response to Ang II infusion.
Activated platelets have been shown to stimulate platelet-leukocyte interactions, which are involved in the pathogenesis of several inflammatory diseases [39]. Liu et al. found that myocardial infarction evokes platelet accumulation and platelet-leukocyte conjugation in infarcted hearts in as early as 6 h, which is responsible for local and systemic inflammation, leading to cardiac remodeling and cardiac rupture in a P-selectin/PSGL-1-dependent manner [40]. Importantly, antiplatelet interventions including clopidogrel or anti-CD41 antibody-mediated platelet deletion suppressed these inflammatory responses. In a murine model of cutaneous arthus reaction in which immune complex challenge-induced neutrophil and mast cell accumulation is necessary for edema and hemorrhage, platelets were found to regulate leukocyte recruitment through the interaction of P-selectin/PSGL-1 [41]. Several studies have demonstrated that circulating activated platelets and platelet-leukocyte conjugation are causal factors of inflammation in rescue experiments with P-selectin-deficient or wild-type platelets. Huo et al. [42] reported that activated platelet-monocyte complex formation promotes leukocyte binding to VCAM-1 and increases their adhesiveness to inflamed or atherosclerotic endothelium. Importantly, injection of activated wild-type platelets, but not P-selectin-deficient platelets, increased monocyte arrest on the surface of atherosclerotic lesions in Apo E−/−mice. Furthermore, Pitchford et al. [43] found that platelet-leukocyte aggregates form in the circulating blood of patients with asthma after allergen exposure. They demonstrated that platelets are necessary for lung leukocyte recruitment in a murine model of asthma; pulmonary leukocyte recruitment in platelet-depleted mice transfused with unstimulated platelets was abolished, while transfusion with activated platelets from wild-type but not from P-selectin-deficient mice, or wild-type platelets in the presence of an anti-P-selectin antibody, were able to restore pulmonary leukocyte recruitment. In addition, using antibody targeting of integrin Mac-1 reduced platelet-dependent leukocyte adhesion and recruitment following vascular injury [44]. Along with these studies, our finding that Ang II-infusion induced platelet-leukocyte conjugation could be a causal event for initiation of cardiac inflammation.
In this study, platelet-leukocyte conjugation was also involved in Ang II infusion-induced cardiac fibrosis, whereas clopidogrel treatment attenuated its formation and consequently inhibited the inflammatory response and fibrosis in the heart (Figs. 2 and 3). Activated platelets aggregate with circulating leukocytes and adhere to the subendothelial matrix and intact endothelium to mediate and recruit leukocytes into the vascular wall [11, 45, 46]. In our study, inhibition of platelet activation using clopidogrel decreased the circulating platelet-leukocyte conjugates, reducing inflammatory cell infiltration into the heart (Figs. 3 and 5). Formation of platelet-leukocyte conjugates may be physiologically important, as this process not only mediates leukocyte infiltration but also releases a set of potent inflammatory substances during the adhesion process, including growth factors (e.g. TGF-β, platelet-derived growth factor, and fibroblast growth factor), cytokine-like factors (e.g. IL-1β, CD40 ligand, and β-thromboglobulin), and chemokines (e.g. RANTES, platelet factor 4 [CXC chemokine ligand 4] [11]. TGF-β, the primary activator of myofibroblasts, is a central mediator of fibrosis in various conditions [3, 47, 48]. Platelets contain the largest amount of TGF-β in the body [49]; indeed, our results showed that inhibiting platelet activation decreased both mRNA and protein levels of TGF-β in Ang II-infused hearts (Fig. 4). IL-1 plays a central role in the regulation of inflammatory and fibrotic responses by inducing synthesis of proinflammatory mediators, promoting leukocyte infiltration and activation, and modulating fibroblast function [30]. Bujak et al. examined the effects of disrupted IL-1β signaling on myocardial ischemia/reperfusion-induced cardiac remodeling using IL-1 receptor knockout mice, and noted that the absence of IL-1 signaling suppressed inflammation, followed by an attenuated fibrotic response [30]. In the present study, we found that Ang II infusion-induced IL-1β expression was suppressed by clopidogrel treatment (Fig. 4), suggesting that platelet activation might be responsible for the release of this cytokine.
The present study also showed that platelet activation is an early event in Ang II infusion-induced cardiac inflammation and fibrosis. Ang II injection immediately stimulated blood pressure elevation, which reached peak levels in 5 min. Under this condition we observed that Ang II injection stimulated platelet activation (CD41+CD62P+) and platelet-leukocyte conjugation (CD45+CD41+, CD45+CD62P+) within 10 min (Fig. 5). Along with our previous study showing that Ang II infusion-induced cardiac inflammation and fibrosis is pressure dependent [8], our present study indicates that platelet activation is an early event that initiates cardiac inflammation and fibrosis in response to Ang II stimulation.
In summary, our study demonstrates that platelet activation is an early event in response to Ang II infusion-induced hypertension that stimulates platelet-leukocyte conjugation and recruits inflammatory cells into the heart, which secrete inflammatory cytokines (IL-1β and TGF-β) and lead to cardiac fibrosis. Inhibition of platelet activation by clopidogrel prevents cardiac inflammation and fibrosis in response to Angiotensin II -induced hypertension.
Acknowledgment
None.
Source of Fundings
This study was supported by grants from the Chinese Ministry of Science and Technology (2012CB945104, 2012CB517802, 2009CB522205), the National Natural Science Foundation of China (81230006, 81100094, 31090363).
Conflict of Interest
None.
References
- 1.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83(6):1849–1865. doi: 10.1161/01.CIR.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 2.Brilla CG, Janicki JS, Weber KT. Impaired diastolic function and coronary reserve in genetic hypertension. Role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ Res. 1991;69(1):107–115. doi: 10.1161/01.RES.69.1.107. [DOI] [PubMed] [Google Scholar]
- 3.Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63(3):423–432. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 4.Huang XR, Chung AC, Yang F, Yue W, Deng C, Lau CP, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010;55(5):1165–1171. doi: 10.1161/HYPERTENSIONAHA.109.147611. [DOI] [PubMed] [Google Scholar]
- 5.McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sato K, et al. Modulation of angiotensin II-mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res. 2010;106(5):952–960. doi: 10.1161/CIRCRESAHA.109.209007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117(3):524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klanke B, Cordasic N, Hartner A, Schmieder RE, Veelken R, Hilgers KF. Blood pressure versus direct mineralocorticoid effects on kidney inflammation and fibrosis in DOCA-salt hypertension. Nephrol Dial Transplant. 2008;23(11):3456–3463. doi: 10.1093/ndt/gfn301. [DOI] [PubMed] [Google Scholar]
- 8.Qi G, Jia L, Li Y, Bian Y, Cheng J, Li H, et al. Angiotensin II infusion-induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc Toxicol. 2011;11(2):157–167. doi: 10.1007/s12012-011-9109-z. [DOI] [PubMed] [Google Scholar]
- 9.Massberg S, Gruner S, Konrad I, Garcia Arguinzonis MI, Eigenthaler M, Hemler K, et al. Enhanced in vivo platelet adhesion in vasodilator-stimulated phosphoprotein (VASP)-deficient mice. Blood. 2004;103(1):136–142. doi: 10.1182/blood-2002-11-3417. [DOI] [PubMed] [Google Scholar]
- 10.Massberg S, Enders G, Matos FC, Tomic LI, Leiderer R, Eisenmenger S, et al. Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood. 1999;94(11):3829–3838. [PubMed] [Google Scholar]
- 11.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115(12):3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 13.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100(1):27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 14.Danese S, de la Motte C, Reyes BM, Sans M, Levine AD, Fiocchi C. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: a novel pathway for immune response amplification. J Immunol. 2004;172(4):2011–2015. doi: 10.4049/jimmunol.172.4.2011. [DOI] [PubMed] [Google Scholar]
- 15.Savi P, Zachayus JL, Delesque-Touchard N, Labouret C, Herve C, Uzabiaga MF, et al. The active metabolite of Clopidogrel disrupts P2Y12 receptor oligomers and partitions them out of lipid rafts. Proc Natl Acad Sci U S A. 2006;103(29):11069–11074. doi: 10.1073/pnas.0510446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evangelista V, Manarini S, Dell’Elba G, Martelli N, Napoleone E, Di Santo A, et al. Clopidogrel inhibits platelet-leukocyte adhesion and platelet-dependent leukocyte activation. Thromb Haemost. 2005;94(3):568–577. [PubMed] [Google Scholar]
- 17.Hashimoto M, Sugidachi A, Isobe T, Niitsu Y, Ogawa T, Jakubowski JA, et al. The influence of P2Y12 receptor deficiency on the platelet inhibitory activities of prasugrel in a mouse model: evidence for specific inhibition of P2Y12 receptors by prasugrel. Biochem Pharmacol. 2007;74(7):1003–1009. doi: 10.1016/j.bcp.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 18.Yang M, Zheng J, Miao Y, Wang Y, Cui W, Guo J, et al. Serum-glucocorticoid regulated kinase 1 regulates alternatively activated macrophage polarization contributing to angiotensin II-induced inflammation and cardiac fibrosis. Arterioscler Thromb Vasc Biol. 2012;32(7):1675–1686. doi: 10.1161/ATVBAHA.112.248732. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Xu N, Feng X, Hou N, Zhang J, Cheng X, et al. Targeted disruption of Smad4 in cardiomyocytes results in cardiac hypertrophy and heart failure. Circ Res. 2005;97(8):821–828. doi: 10.1161/01.RES.0000185833.42544.06. [DOI] [PubMed] [Google Scholar]
- 20.Gresele P, Momi S, Berrettini M, Nenci GG, Schwarz HP, Semeraro N, et al. Activated human protein C prevents thrombin-induced thromboembolism in mice. Evidence that activated protein c reduces intravascular fibrin accumulation through the inhibition of additional thrombin generation. J Clin Invest. 1998;101(3):667–676. doi: 10.1172/JCI575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan L, Li Y, Jia L, Qin Y, Qi G, Cheng J, et al. Cathepsin S deficiency results in abnormal accumulation of autophagosomes in macrophages and enhances Ang II-induced cardiac inflammation. PLoS One. 2012;7(4):e35315. doi: 10.1371/journal.pone.0035315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One. 2012;7(5):e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han YL, Li YL, Jia LX, Cheng JZ, Qi YF, Zhang HJ, et al. Reciprocal interaction between macrophages and T cells stimulates IFN-gamma and MCP-1 production in Ang II-induced cardiac inflammation and fibrosis. PLoS One. 2012;7(5):e35506. doi: 10.1371/journal.pone.0035506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Zhang C, Wu Y, Han Y, Cui W, Jia L, et al. Interleukin-12p35 deletion promotes CD4 T-cell-dependent macrophage differentiation and enhances angiotensin II-Induced cardiac fibrosis. Arterioscler Thromb Vasc Biol. 2012;32(7):1662–1674. doi: 10.1161/ATVBAHA.112.249706. [DOI] [PubMed] [Google Scholar]
- 25.Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55(1):90–98. doi: 10.1161/HYPERTENSIONAHA.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavalli A, Lattion AL, Hummler E, Nenniger M, Pedrazzini T, Aubert JF, et al. Decreased blood pressure response in mice deficient of the alpha1b-adrenergic receptor. Proc Natl Acad Sci U S A. 1997;94(21):11589–11594. doi: 10.1073/pnas.94.21.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akan M, Cakir B, Misirlioglu A, Yildirim S, Taylan G, Akoz T. Effects of clopidogrel and high dose aspirin on survival of skin flaps in rats. Scand J Plast Reconstr Surg Hand Surg. 2005;39(1):7–10. doi: 10.1080/02844310410017951. [DOI] [PubMed] [Google Scholar]
- 28.Jia L, Li Y, Xiao C, Du J. Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci. 2012;17:221–231. doi: 10.2741/3923. [DOI] [PubMed] [Google Scholar]
- 29.Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, et al. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res. 2009;105(11):1149–1158. doi: 10.1161/CIRCRESAHA.109.208199. [DOI] [PubMed] [Google Scholar]
- 30.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173(1):57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Utsugisawa K, Kizawa H, Nagane Y, Kondoh R, Iwa Y, Akutsu H, et al. Biphasic effects of angiotensin II and receptor antagonism on aggregability and protein kinase C phosphorylation in human platelets. Thromb Haemost. 2005;94(5):1012–1018. doi: 10.1160/TH05-02-0125. [DOI] [PubMed] [Google Scholar]
- 32.Boilard E, Blanco P, Nigrovic PA. Platelets: active players in the pathogenesis of arthritis and SLE. Nat Rev Rheumatol. 2012;8(9):534–542. doi: 10.1038/nrrheum.2012.118. [DOI] [PubMed] [Google Scholar]
- 33.Ferroni P, Guagnano MT, Falco A, Paoletti V, Manigrasso MR, Michetti N, et al. Association of low-grade inflammation and platelet activation in patients with hypertension with microalbuminuria. Clin Sci (Lond) 2008;114(6):449–455. doi: 10.1042/CS20070307. [DOI] [PubMed] [Google Scholar]
- 34.Davi G, Gresele P, Violi F, Basili S, Catalano M, Giammarresi C, et al. Diabetes mellitus, hypercholesterolemia, and hypertension but not vascular disease per se are associated with persistent platelet activation in vivo. Evidence derived from the study of peripheral arterial disease. Circulation. 1997;96(1):69–75. doi: 10.1161/01.CIR.96.1.69. [DOI] [PubMed] [Google Scholar]
- 35.Spencer CG, Gurney D, Blann AD, Beevers DG, Lip GY. Von Willebrand factor, soluble P-selectin, and target organ damage in hypertension: a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) Hypertension. 2002;40(1):61–66. doi: 10.1161/01.HYP.0000022061.12297.2E. [DOI] [PubMed] [Google Scholar]
- 36.Nadar SK, Blann AD, Kamath S, Beevers DG, Lip GY. Platelet indexes in relation to target organ damage in high-risk hypertensive patients: a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) J Am Coll Cardiol. 2004;44(2):415–422. doi: 10.1016/j.jacc.2004.03.067. [DOI] [PubMed] [Google Scholar]
- 37.Larsson PT, Schwieler JH, Wallen NH. Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coagul Fibrinolysis. 2000;11(1):61–69. [PubMed] [Google Scholar]
- 38.Kalinowski L, Matys T, Chabielska E, Buczko W, Malinski T. Angiotensin II AT1 receptor antagonists inhibit platelet adhesion and aggregation by nitric oxide release. Hypertension. 2002;40(4):521–527. doi: 10.1161/01.HYP.0000034745.98129.EC. [DOI] [PubMed] [Google Scholar]
- 39.Totani L, Evangelista V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol. 2010;30(12):2357–2361. doi: 10.1161/ATVBAHA.110.207480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Gao XM, Fang L, Jennings NL, Su Y, Xu Q, et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler Thromb Vasc Biol. 2011;31(4):834–841. doi: 10.1161/ATVBAHA.110.220467. [DOI] [PubMed] [Google Scholar]
- 41.Hara T, Shimizu K, Ogawa F, Yanaba K, Iwata Y, Muroi E, et al. Platelets control leukocyte recruitment in a murine model of cutaneous arthus reaction. Am J Pathol. 2010;176(1):259–269. doi: 10.2353/ajpath.2010.081117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9(1):61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 43.Pitchford SC, Momi S, Giannini S, Casali L, Spina D, Page CP, et al. Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood. 2005;105(5):2074–2081. doi: 10.1182/blood-2004-06-2282. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, et al. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112(19):2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]
- 45.McEver RP. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb Haemost. 2001;86(3):746–756. [PubMed] [Google Scholar]
- 46.Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, et al. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med. 1999;190(6):803–814. doi: 10.1084/jem.190.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wick G, Backovic A, Rabensteiner E, Plank N, Schwentner C, Sgonc R. The immunology of fibrosis: innate and adaptive responses. Trends Immunol. 2010;31(3):110–119. doi: 10.1016/j.it.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258(11):7155–7160. [PubMed] [Google Scholar]