

Abstract
Androgen excess is one of the most common and disturbing endocrine disorder of reproductive-aged women, affecting approximately 7% of this population Androgen excess results in the development of androgenic features in the women affected, with the development of hirsutism, androgenic alopecia, ovulatory dysfunction, and, if extreme, even virilization and masculinization. Adrenocortical carcinoma (ACC) is a rare malignancy accounting for 0.02% of all annual cancers reported. About 60% are functional tumors secreting hormones, with its consequent clinical manifestations, the Cushing's syndrome due to cortisone, virilization due to androgens, feminization due to estrogens, or hypertension due to aldosterone. Adrenal tumors that secrete androgens exclusively are extremely rare. Here, we present a rare case of androgen-secreting adrenocortical carcinoma with non-classical congenital adrenal hyperplasia.
Keywords: Adrenocortical carcinoma, hirsutism, non classical congenital adrenal hyperplasia
INTRODUCTION
Adrenocortical carcinoma (ACC) is a rare malignancy, accounting for 0.02% of all annual cancer.[1] The majority of these tumors are benign, non-functioning adenomas that are incidentally discovered on abdominal image studies. Others are functional adenomas secreting cortisol, aldosterone, or less commonly androgens or estrogens. Pure androgen-secreting adrenal tumors are very rare,[2] and their diagnosis represents a clinical challenge. Hirsutism and virilization syndrome, characterized by clitorimegaly, male pattern baldness, and deepening of the voice along with menstrual irregularities are the most common findings. We present a case of 42-year-old female who presented with secondary amenorrhea, hirsutism, and virilization associated with an incidental adrenal mass.
CASE REPORT
A 42-year-old multiparous came to the hospital with pain in the right upper abdomen since two days. Pain was sudden in onset and colicky in nature; routine ultrasound was done, which revealed large adrenal mass, and then patient was referred to the endocrine department. Patient gave history of amenorrhea since seven years associated with increased hair growth of chest, lower abdomen, and face. Facial hair growth required shaving 3-4 times per week. She ignored her symptoms, attributing them as menopausal symptoms.
On examination, heart rate was 82 per minute, blood pressure 134/80 mm of Hg. Hirsutism was evaluated based on Ferriman-Gallwey modified score (result = 24). Clitorimegaly, male pattern baldness were also present [Figures 1–3]. During evaluation, CT scan abdomen was done, which showed 10 cm × 9.2 cm left adrenal mass [Figure 4]. Total testosterone was 4.33 ng/ml (0.06 - 0.82), dehydroepiandrosterone sulfate (DHEA-S) 1263 μg/dl (35-430 μg/dl), 17- hydroxyprogesterone 47 ng/ml and 119 ng/ml after ACTH. Basal serum cortisol was 12.6 mcg/dl; serum aldosterone and 24 hours urinary VMA were normal. A diagnosis of androgen-secreting adrenocortical cancer with non-classical CAH was made.
Figure 1.
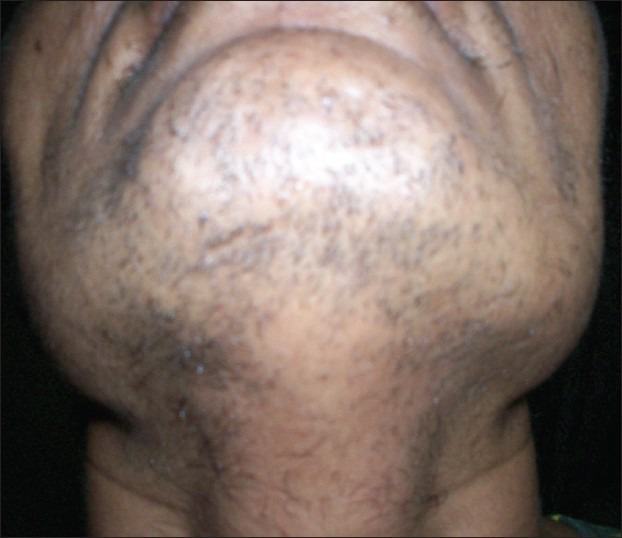
Increased beard and shaving
Figure 3.
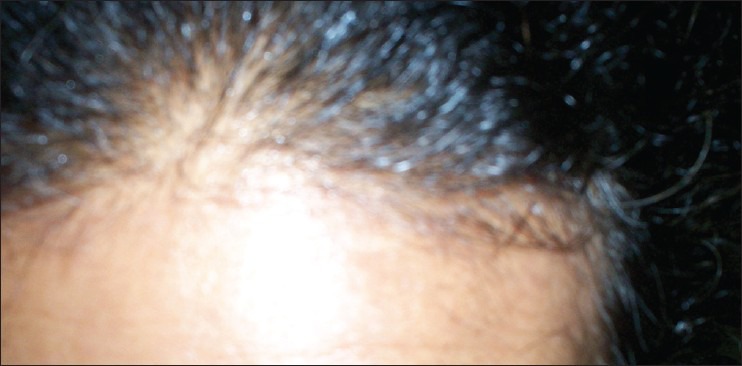
Male pattern baldness
Figure 4.
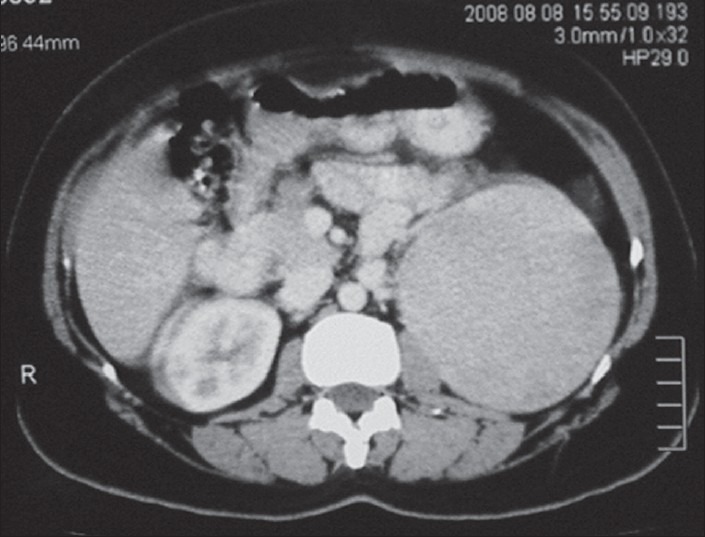
CT scan abdomen showing large left adrenal mass
Figure 2.
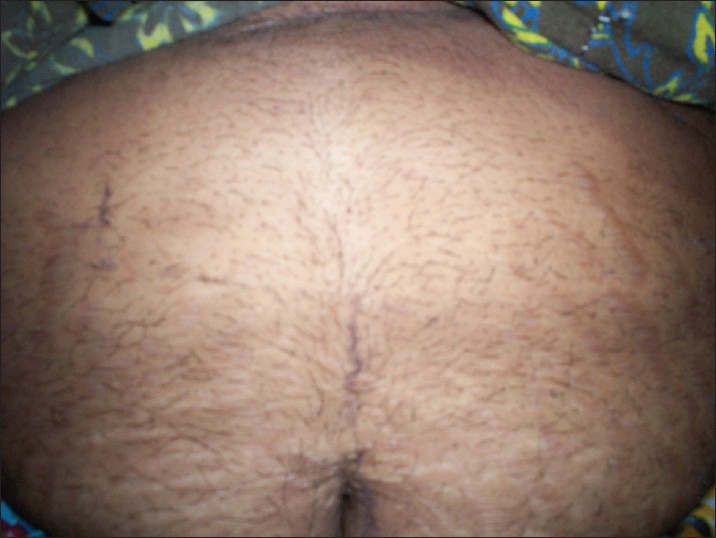
Abdominal hair growth
Patient underwent exploratory laparotomy with excision of large adrenal tumor without any complication. Patient's post-operative stay was uneventful and was discharged on tab Prednisolone 5 mg once-daily. Histopathology report was consistent with adrenocortical carcinoma, but no staining was done for androgen.
On follow-up after three months, patient was comfortable, but she still had amenorrhea. Total testosterone was 0.43 ng/ml (0.06-0.82), dehydroepiandrosterone sulfate (DHEA-S) was 163 μg/dl (35-430 μg/dl).
DISCUSSION
This case illustrates a long delay in the diagnosis of a virilizing syndrome due to a pure androgen-secreting adrenal carcinoma that presented with simultaneous non-classical CAH. Pure androgen-secreting adrenal tumors are extremely rare.[2] ACCs are classified as functional tumors (FT) or non-functional tumors (NFT). Functional tumors usually presents clinically as a result of hormonal secretions like Cushing syndrome, virilization, feminization, or a mixed Cushing-virilizing syndrome. Functional tumors in adults are more frequently a mixed Cushing virilizing syndrome. Pure virilizing ACC are uncommon in adult women,[3] as found in this 42-year-old woman.
Hyperandrogenemia in women may be from an ovarian or adrenal source. DHEA-S, DHEA, androstenedione, testosterone, and dihydrotestosterone are the major circulating androgens in women. DHEA-S is produced solely in the adrenal gland. DHEA is produced 50% in the adrenal gland, 30% from conversion of DHEA-S, and 20% in the ovary. Both glands equally produce androstenedione, and testosterone is synthetized in the adrenal gland (25%), in the ovaries (25%), and 50% from androstenedione conversion.[4] Dihydrotestosterone is classically an intracellular androgen. In this case, DHEA-S along with testosterone was elevated. Androgen-secreting tumors can produce hirsutism and virilization in 90-100% of the patients and amenorrhea in 40-60%.[2] Our patient had all three of them.
The onset, progression, and severity of hyperandrogenic signs must be established. Manifestations of functional causes of hyperandrogenism such as polycystic ovary syndrome or non-classic congenital adrenal hyperplasia appear around puberty and progress slowly. A detailed interrogation about the presence of hyperandrogenic symptoms, menstrual disturbances, or infertility during the reproductive years is crucial. More severe causes of hyperandrogenism, including tumors, rarely present around puberty, may progress rapidly, and usually associate signs of virilization or defeminization. Our patient has non-classical CAH, but she on repeated questioning denied any hyperandrogenic symptoms around puberty.
Adrenocortical carcinoma (ACC) is a rare malignancy accounting for 0.02% of all annual cancers reported. Adrenal tumors that secrete androgens exclusively are extremely rare. In one of the largest series, Moreno et al., over a period of 33 years, described 21 cases with pure androgen-secreting adrenal tumors. In their paper, they reported 801 adrenalectomies. Only 2.4% were due to pure androgen-secreting adrenal tumors and 4.4% to primary adrenocortical tumors. Hirsutism was found in all patients, and virilization was only found in 23% of the cases. Tumor size had a mean of 9 cm in the adenomas and 14 cm in the carcinomas. Testosterone and DHEA-S were elevated in virtually every patient.[5]
Non-classical CAH usually presents during childhood or puberty with menstrual disorders, infertility, and hyperandrogenism. Adrenal tumors have rarely been identified among individuals with non-classical CAH. Following discovery of an adrenal incidentaloma, an 88-year-old woman was diagnosed with non-classical CAH. A 57-year-old man ascertained by finding an adrenal incidentaloma was diagnosed with non-classical CAH; he had elevated serum 17-OHP concentrations and urinary 17-ketosteroid excretion. Adrenal myelolipomas have been reported among untreated adults with non-classical CAH.[6] Our patient has androgen-secreting ACC with non-classical CAH. After extensive review of literature, we were not able to find any reported case of androgen-secreting ACC with non-classical CAH.
To conclude, pure androgen-secreting adrenal tumors are extremely rare. Hirsutism, virilization, and menstrual irregularities are the usual clinical findings. Malignancy is difficult to predict, but it is usually related with tumor size, type of hormones secreted, and the tumor progression. Association with non-classical CAH is extremely rare. Detailed history and clinical examination, hormonal workup, and radiological evaluation are the cornerstone for diagnosing hyperandrogenism with hirsutism and virilization in a female.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Roman S. Adrenocortical carcinoma. Curr Opin Oncol. 2006;18:36–42. doi: 10.1097/01.cco.0000198976.43992.14. [DOI] [PubMed] [Google Scholar]
- 2.Cordera F, Grant C, Van Heerden J, Thompson G, Young W. Androgen-secreting adrenal tumors. Surgery. 2003;134:874–80. doi: 10.1016/s0039-6060(03)00410-0. [DOI] [PubMed] [Google Scholar]
- 3.Shamim M, Usmani F, Qureshi IM, Iqbal AS. Virilizing adrenocortical carcinoma in a multiparous woman. J Surg Pak. 2010;15:112–4. [Google Scholar]
- 4.Burger HG. Androgen production in women. Fertil Steril. 2002;77:S3–5. doi: 10.1016/s0015-0282(02)02985-0. [DOI] [PubMed] [Google Scholar]
- 5.Moreno S, Montoya G, Armstrong J, Leteurtre E, Aubert S, Vantyghem MC, et al. Profile and outcome of pure androgen-secreting adrenal tumors in women: Experience of 21 cases. Surgery. 2004;136:1192–8. doi: 10.1016/j.surg.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 6.Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol 2010. 2010 doi: 10.1155/2010/625105. 625105. [DOI] [PMC free article] [PubMed] [Google Scholar]