

Abstract
Understanding how cells regulate and transport metal ions is an important goal in the field of bioinorganic chemistry, a frontier research area that resides at the interfaces of chemistry and biology. This Current Topics article reviews recent advances from the authors' group in using single-molecule fluorescence imaging techniques to identify the mechanisms of metal homeostatic proteins, including metalloregulators and metallochaperones. It emphasizes the novel mechanistic insights into how dynamic protein–DNA and protein–protein interactions offer efficient pathways for MerR-family metalloregulators and copper chaperones to fulfill their functions. The article also summarizes other related single-molecule studies of bioinorganic systems, and gives an outlook toward single-molecule imaging of metalloprotein functions in living cells.
Transition metals, such as iron, copper, and zinc, play a variety of important roles in biological processes, including catalyzing reactions, providing structural supports, mediating charge transfer, and transducing signals (1, 2). Many transition metals are thus essential in organisms ranging from bacteria to mammals. Yet some transition metals are highly toxic, such as mercury and lead, threatening organisms living in environments that contain high levels of such metals. Even essential metals can turn harmful if their concentrations and availabilities go awry inside cells. Therefore, it is crucial to understand how cells harness the power of essential metals for function, while preventing toxicity, and how they defend against toxic metals. This understanding is one of the major research goals in the field of bioinorganic chemistry (also known as inorganic biochemistry or metallobiochemistry), an active research field at the interface of chemistry and biology.
Most metal-related biological processes are done by proteins, i.e., metalloproteins. These proteins work either individually or with one another to carry out their biological functions. For the latter, the interactions among the proteins are often key determinants of their functionality. These interactions are often difficult to study in ensemble-averaged measurements due to their dynamic nature, which makes it necessary to synchronize molecular actions (as done in stopped-flow measurements) for probing interaction intermediates.
Single-molecule techniques emerged in the past two decades as powerful methods to study dynamic protein interactions (for example, see reviews (3–8)). Their applications thrived in the field of biophysics — a quick look at the technical programs in the recent biophysics society national meetings can spot many lectures on single-molecule studies; examples include nucleic-acid-processing enzymes, molecular motors, cytoskeleton structures, protein synthesis and folding, to name a few. A search of the keywords “single molecule” and “biophysics” in PubMed generates >1500 publications. On the other hand, much less single-molecule studies have been reported on bioinorganic systems (see Section 3), even though bioinorganic chemistry is extensively intertwined with biology (1, 2). Yet, many compelling problems in bioinorganic chemistry can be solved using the advances in single-molecule techniques, as shown by the examples in this article and others.
About eight years ago our group started an effort to develop and apply single-molecule fluorescence microscopy methods to bioinorganic problems, partly to target the shortage of this type of research as well as to push the frontiers of both bioinorganic chemistry and single-molecule research. We chose metal homeostasis as the topic of interest, which comprises many processes that involve dynamic protein–protein and protein–DNA interactions. Focusing on metalloregulators and metallochaperones, we have developed engineered DNA Holiday junctions as reporters in single-molecule FRET (smFRET) measurements of protein–DNA interactions, as well as adapted a lipid vesicle trapping approach to enable single-molecule studies of weak, dynamic protein interactions (see our previous review (9)). In this Current Topics article we highlight the mechanistic insights gained from our latest smFRET studies of metalloregulators and metallochaperones.
1. Metalloregulators: novel pathways for transcription deactivation
Metalloregulators are metal-sensing transcription factors; they regulate the transcription of genes that protect the cell from metal excess or enable the cell to acquire essential metals (see recent reviews (10–26)). MerR-family metalloregulators are a distinct family among these metalloregulators; they enable cells to sense and defend against many metal ions such as Hg2+, Cu+, and Zn2+, with high selectivity and sensitivity; MerR, the archetype in the family, responds to Hg2+ and regulates the mercury resistance genes (11, 13, 14, 25, 27–29).
How MerR-family metalloregulators activate transcription in response to metal ions has been well studied — they operate via a DNA distortion mechanism (Figure 1) (14, 27, 28, 30, 31). These homodimeric regulators recognize specific dyad symmetric DNA sequences within a promoter, and both their apo and holo forms bind DNA tightly. In the absence of metal, the metalloregulator bends the DNA; in this configuration, RNA polymerase (RNAp) cannot interact with both −10 and −35 sequences properly, and transcription is repressed. Upon binding metal, the metalloregulator changes its conformation and further unwinds the DNA slightly to allow proper RNAp interactions with the −10 and −35 sequences; transcription is then activated.
Figure 1.
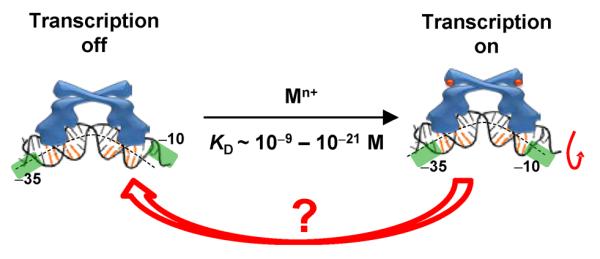
The DNA distortion mechanism for transcription activation by MerR-family metalloregulators. The orange-colored base pairs denote the dyad-symmetric sequence that the metalloregulator recognizes. The green shades denote the −10 and −35 elements of the promoter. The mechanism for transcription deactivation is unclear (red arrow).
Until recently, little had been known about the mechanism by which MerR-family metalloregulators deactivate transcription (Figure 1). It is important, however, to deactivate transcription promptly, as it wastes energy for the cell to continue expressing metal resistance genes after metal stress is relieved. Metal dissociation to convert a holo-metalloregulator to its apo form would be the simplest way for deactivation, but is unlikely, as the metal is bound tightly (often by cysteine ligands) and metal–cysteine bond dissociation is slow (32). For example, CueR, the Cu+-responsive MerR-family metalloregulator in E. coli, has a Cu+ binding affinity of ~10−21 M (33). Although thiol ligand exchange can possibly facilitate Cu+ removal from the binding site as observed for copper chaperones (34), no evidence exists that CueR can undergo similarly facile ligand exchange reactions. Then, in order to deactivate transcription, a holo-metalloregulator has to be either replaced somehow by its apo-protein or removed completely to result in a vacant promoter which is also a weakly repressed state. Here, a simple scenario would be for the holo-protein to unbind from DNA, followed by the binding of an apo-protein, which will not only repress transcription but also prevent the rebinding of the holo-protein. But, are there any alternative, and more efficient, pathways to deactivate transcription?
Using smFRET measurements we have discovered novel pathways for transcription deactivation by MerR-family metalloregulators (35). We focused on CueR, which regulates the transcriptions of CopA, a membrane transporter that pumps Cu+ out of the cytoplasm, and CueO, a periplasmic multicopper oxidase that is also involved in maintaining copper homeostasis (36–39). In our experiment (Figure 2A), we immobilized on a surface an oligomeric DNA (25 or 121 base pairs), which encoded the copA promoter sequence and was labeled at one end with a FRET donor Cy3 whose fluorescence was excited by a laser directly. We then flowed in the protein molecule, which was labeled with a single FRET acceptor Cy5. Upon protein binding to the DNA, FRET occurs from the donor to the acceptor, and the corresponding changes in the FRET efficiency (EFRET ≈ IA/(IA+ID), where ID and IA are the donor and acceptor fluorescence intensities) report the protein–DNA interactions. As CueR is a homodimer, labeling it with a single FRET acceptor breaks its symmetry. Consequently, its two binding orientations onto DNA are differentiated. The EFRET versus time trajectory from a single immobilized DNA interacting with proteins in solution shows transitions between three different EFRET states: the E0 state corresponds to the free DNA, and E1 and E2 correspond to the two different binding orientations of labeled CueR on DNA (Figure 2B and C).
Figure 2.
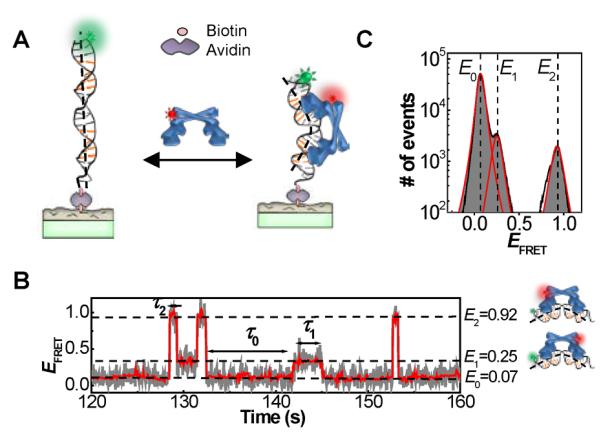
(A) Experimental scheme of surface immobilization of DNA; CueR is supplied in a continuously flowing solution. Upon CueR binding to DNA, FRET occurs from the donor Cy3 (green sphere) to the acceptor Cy5 (red sphere). (B) Single-molecule EFRET trajectory of an immobilized Cy3-DNA interacting with holo-CueRCy5-C129 (2 nM), where Cy5-C129 designates the labeling position on one monomer. τ0, τ1 and τ2 are the microscopic dwell times on the E0, E1 and E2 states, respectively. The cartoons on the right show CueRCy5-C129 in two binding orientations. (C) Histogram of EFRET trajectories as in B of holo-CueRCy5-C129–DNA interactions, showing the three EFRET states.
Kinetic mechanism of CueR–DNA interactions and its functional implications
Figure 3 gives the kinetic mechanism and the associated rate constants of apo- and holo-CueR interacting with a specific DNA that contains the dyad symmetric sequence recognized by CueR. The protein (P) binds to DNA (D) reversibly (k1 and k−1 processes) to form a complex (I) in which CueR recognizes the specific sequence and distorts the DNA structure. The reversible binding processes are manifested experimentally by the reversible transitions between E0 and E1 states and those between E0 and E2 states in the EFRET trajectories (Figure 2B). The structural distortion of DNA in the CueR–DNA complex was well known from the structural studies on other MerR-family regulators in complex with DNA (14, 27, 30, 31).
Figure 3.
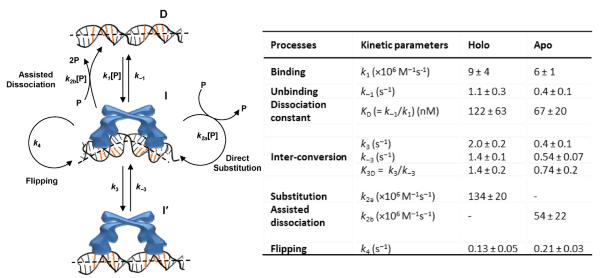
Left: kinetic mechanism of CueR interactions with a specific DNA, which includes the protein (P), DNA (D), two protein–DNA complexes that differ in protein binding modes (I and I′ ), and the rate constants for the kinetic processes. [P]: protein concentration. Between k2a and k2b, the direct substitution process k2a is dominant for holo-CueR–DNA interactions, whereas the assisted dissociation process k2b is dominant for apo-CueR–DNA interactions. Right: the kinetic parameters. The rate constants for CueR (holo) binding and unbinding with nonspecific DNA are k1 = 0.016 ± 0.001 nM−1 s−1 and k−1 = 5.9 ± 0.1 s−1; other kinetic processes do not occur to nonspecific DNA (35).
Interestingly, we observed that both apo- and holo-CueR could spontaneously flip their binding orientations on DNA without completely detaching from DNA (k4, Figure 3). This spontaneous flip is experimentally manifested by the direct E1 ↔ E2 transitions that are observable down to protein concentrations as low as 0.5 nM (e.g., at ~130 s in Figure 2B). Furthermore, this flipping occurs only when CueR binds to the specific DNA sequence, where CueR distorts DNA structure (complex I in Figure 3), as it is not observed in CueR interactions with a nonspecific DNA (35). Similar flipping behaviors were also observed for HIV reverse transcriptase on DNA/RNA duplexes (40). This spontaneous flipping indicates that CueR is highly dynamic when bound at its recognition site on DNA. Being dynamic, especially for holo-CueR, may facilitate transcription initiation, which involves large structural rearrangements of associated proteins and DNA (28).
We further discovered that for both apo- and holo-CueR, within each protein-binding orientation on DNA, there was another binding mode besides the one where CueR recognized the targeting sequence and distorted the DNA structure — i.e., CueR has two different binding modes on DNA (I and I′ in Figure 3). These two different binding modes are experimentally manifested in the distribution of τ2 (and the equivalent τ1), the microscopic dwell time on the protein-bound E2 state (Figure 2B). The distribution of τ2 follows a double-exponential decay (Figure 4A). Furthermore, the two different binding modes of CueR on DNA are present only when CueR recognizes the specific DNA sequence, because, when CueR interacts with a nonspecific DNA, the distribution of the dwell time on the protein-bound state follows a single-exponential decay (35). We attributed the second CueR-binding mode to a CueR–DNA complex in which the CueR binds DNA in a way as if the DNA is nonspecific; this attribution is reasonable because CueR does bind nonspecific DNA and the binding mode here must be different from that of the specific DNA binding mode. No structural information is yet available about CueR, or any MerR-family regulators, in complex with a nonspecific DNA. Just for illustrative purposes, we drew the cartoon of this complex having the DNA structure undistorted (I′ in Figure 3). The two different binding modes, in which CueR interacts with DNA specifically or nonspecifically, are beneficial for CueR in its searching for the recognition sequence in the large bacterial chromosome (~4.6 million base pairs for E. coli (41)) — being able to interact with DNA nonspecifically would help CueR slide along the chromosome, and upon locating the recognition sequence, the CueR–DNA complex can interconvert to the specific binding mode, thereby distorting the DNA structure to regulate transcription (Figure 4B). This sliding along DNA via nonspecific interactions has long been realized to be advantageous for reducing the dimensionality of site search for DNA-binding proteins (42) and has been directly visualized via single-molecule tracking, for example, for an adenovirus proteinase (43).
Figure 4.
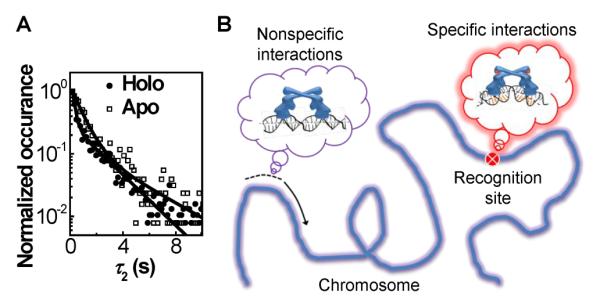
Two different binding modes of CueR on specific DNA. (A) Double-exponential distributions of τ2 for holo- and apo-CueRCy5-C129 interactions with specific DNA, both at 2 nM protein concentration. (B) Schematic of CueR sliding on chromosome via nonspecific interactions to help search for the recognition site where specific interactions apply.
Most strikingly, we discovered that CueR could undergo two novel processes at the specific DNA site, both of which interrupt an existing CueR–DNA complex. In one, a direct substitution process, which is dominant for holo-CueR–DNA interactions, a protein coming from the surrounding solution directly replaces the incumbent protein on DNA (k2a, Figure 3); in the other, an assisted dissociation process, which is dominant for apo-CueR–DNA interactions, a protein from solution helps carry away the incumbent one on DNA (k2b, Figure 3). Both of these processes lead to a shortening of the average single-molecule dwell time (e.g., 〈τ2〉) of each protein-binding orientation when the holo- or apo-protein concentration increases (Figure 5A). (Note that overall, the protein still spends more time on DNA at higher protein concentrations because of the higher binding rates.)
Figure 5.
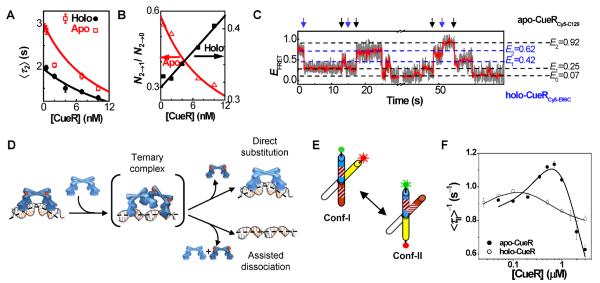
Direct substitution and assisted dissociation pathways of CueR–DNA interactions. (A) [CueR] dependence of 〈τ2〉 for CueRCy5-C129–DNA interactions. (B) Dependence of N2→1/N2→0 on [holo-CueRCy5-C129] and [apo-CueRCy5-C129]. N2→1/N2→0 is the ratio between the observed numbers of E2 → E1 and E2 → E1 transitions in such EFRET trajectories as in Figure 2B. (C) Single-molecule EFRET trajectory of an immobilized Cy3-DNA interacting with a mixture of apo-CueRCy5-C129 and holo-CueRCy5-E96C of 5 nM each. The blue arrows denote the transitions from the holo-protein bound states to the apo-protein bound states, and the black arrows denote the reverse transitions; these transitions report the direct substitution of a DNA-bound holo-protein by an apo-protein or the reverse. (D) Schematic of the proposed mechanism involving a ternary CueR2–DNA complex as a common intermediate (or transition state) for the direct substitution and assisted dissociation to a CueR that is bound at the specific DNA site. Note here CueR = homodimer. (E) Structural dynamics of an engineered HJ between its two conformers, conf-I and conf-II, with FRET donor (green) and acceptor (red) labels. The stripes on two arms indicate the encoded dyad symmetric sequence recognized by CueR. (F) CueR concentration dependences of 〈τII〉−1, where τII is the single-molecule dwell time on conf-II in EFRET trajectories of HJ structural dynamics; reprinted from reference (45), Copyright 2009, with permission from Elsevier.
But clear differences between holo- and apo-CueR behaviors differentiate these two processes. For holo-CueR, at higher protein concentrations, more frequent transitions are observed between the two protein-binding orientations (Figure 5B), because direct substitution has a 50% probability that the replacing protein ends up at an opposite binding orientation to the incumbent one. In contrast, for apo-CueR, at higher protein concentrations, more frequent transitions are observed from a bound state to the free DNA state because of the assisted dissociation process (Figure 5B). Using two differently labeled CueR variants, each prepared in the apo or holo form, we further observed the direct substitution processes on a specific DNA between apo- and holo-CueR, reflected by the direct transitions from the bound states of one variant to those of the other (Figure 5C). Moreover, both k2a and k2b, the rate constants for direct substitution and assisted dissociation respectively, are significantly larger than k1, the protein binding rate constant; these larger rate constants indicate that the presence of a CueR on DNA facilitates another CueR molecule in finding the recognition sequence, leading to either direct protein substitution or assisted protein dissociation.
Relatedly, Marko et al. has observed that some sequence-neutral DNA-binding proteins can readily exchange with themselves or with each other on DNA (44); we hypothesize that the direct substitution or the assisted dissociation pathway could be the underlying mechanism for these facile protein exchanges on DNA. Besides being novel mechanistic pathways for protein–DNA interactions, these two pathways are also functionally significant for MerR-family metalloregulators, as will be discussed later.
Ternary protein2–DNA complex as a possible intermediate (or transition state) for direct substitution and assisted dissociation pathways
The direct substitution and assisted dissociation processes that occur to a CueR molecule bound at the specific DNA site pose an immediate question: how do they occur at the molecular level? Here we propose a mechanism that involves a ternary protein2–DNA complex as a common intermediate (or transition state**) for both pathways (here each protein is a functional dimer of CueR; Figure 5D). Start with a CueR–DNA complex, where each of the two DNA-binding domains of the homodimeric CueR attaches to one half of the dyad symmetric sequence. Under thermal fluctuation, one of the DNA-binding domains could detach momentarily, allowing another CueR molecule to bind to one half of the dyad with one of its DNA-binding domains and leading to a ternary CueR2–DNA complex (note CueR = homodimer dimer). Owing to the low stability of the ternary complex, it could proceed in either of the two pathways. In one, the incumbent CueR falls off DNA, resulting in a direct substitution (the incoming one could fall off too, leading to no observable change). In the other, both proteins fall off, resulting in an assisted dissociation.
A ternary CueR2–DNA complex was indeed observed in our previous study (45), where we used an engineered DNA Holliday junction (HJ) to probe CueR–DNA interactions. A DNA HJ is a four-way junction of DNA. In the presence of Na+ and Mg2+, it folds into two X-shaped stacked conformers (conf-I and conf-II, Figure 5E), where each conformer could be viewed approximately as two B-form helices forming a cross structurally (46, 47). The two conformers interconvert dynamically at room temperature (Figure 5E), and the structural dynamics of a single HJ molecule can be followed in real time by smFRET measurement where two of the HJ's four arms are labeled with a FRET pair (45, 48–50). We engineered a HJ and encoded in its arms the dyad symmetric sequence recognized by CueR (Figure 5E). Because the part of HJ that contains the encoded sequence has distinct spatial orientations in the two conformers, CueR binds to the two conformers differentially and causes changes in the interconversion kinetics between the two conformers. These changes are readily measurable by smFRET and thus report the associated protein–DNA interactions (45, 49). We found that both apo- and holo-CueR interacted with conf-II in a two-step manner — they initially bind to conf-II to form a binary complex that can facilitate its structural transition to conf-I; this binary complex can then bind a second protein molecule to form a ternary complex that stabilizes conf-II. The two-step interactions of CueR with conf-II are manifested by the biphasic protein concentration dependence of 〈τII〉−1, the time-averaged single-molecule rate of conf-II → conf-I transition (Figure 5F) — with increasing CueR concentration, 〈τII〉−1 initially increases, reflecting the formation of the binary complex that helps structural transition to conf-I, and then it decays at higher protein concentrations after reaching a maximum, reflecting the subsequent formation of the ternary complex.
The observation of a ternary complex here likely results from that in the HJ conf-II, the spatial orientation of the two halves of the dyad symmetric sequence is significantly distorted from that in a double-strand DNA helix (Figure 5E), allowing two CueR molecules to bind, each of which accesses one half of the dyad sequence. In contrast, in a normal double-strand DNA helix, its structural distortion upon CueR binding is small (14, 27, 30, 31); the small magnitude of this structural distortion likely renders the ternary complex unstable and merely a short-lived intermediate (or transition state), which nevertheless could provide a possible molecular mechanism for the direct substitution and assisted dissociation pathways observed experimentally.
Efficient pathways for transcription deactivation and their broader relevance
Besides being novel pathways in protein–DNA interactions, the direct substitution and assisted dissociation could both be functionally significant for deactivating transcription in CueR's regulatory function. For transcription deactivation, the holo-CueR bound promoter needs to return to the apo-CueR bound state or the free DNA form (Figure 1). The direct substitution of holo-protein by an apo-protein can reach the apo-bound state in a single step: its kinetics depends on the intracellular concentration of CueR. Depending on growth conditions, an E. coli cell has about 60 to 400 copies of monomeric CueR (51), corresponding to [P] ≈ 30 to 220 nM with a cell volume of ~1.5 fL (33) (here P is the functional homodimer). Using the rate constant k2a (Figure 3), the direct substitution takes about 0.03-to-0.2 s to reach the apo-protein bound, transcription-repressed state (Figure 6, step iii). On the other hand, the assisted dissociation takes about 0.08-to-0.6 s to reach the free DNA form (Figure 6, step iv), using the rate constant k2b. Compared with the generic pathway of protein unbinding-and-binding (Figure 6, step i and ii, total about 1.7-to-5.9 s) or that of just unbinding (step i, ~0.9 s), the direct substitution and assisted dissociation are both tens of times faster. Therefore, both the direct substitution of holo-CueR by apo-CueR and the assisted dissociation of holo-CueR at the promoter site could be the more efficient pathways for transcription deactivation.
Figure 6.
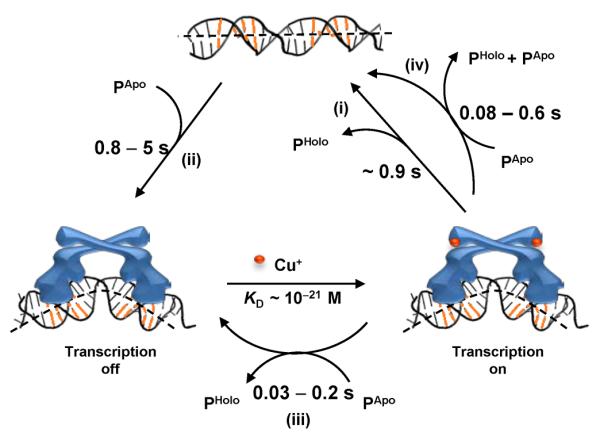
Pathways for transcription deactivation by CueR. The timescales are denoted for relevant kinetic steps, including (i) unbinding, (ii) binding, (iii) direct substitution, and (iv) assisted dissociation. PApo: apo-CueR; PHolo: holo-CueR.
One expects that cellular conditions are possibly not the same as in our experiments and kinetic constants may thus differ. The percentage of cellular CueR being holo or apo is not known under different copper exposures and growth conditions. The turnover rates of apo and holo CueR in cells could also provide another layer of control in affecting CueR interaction kinetics with DNA. With these complications, the two novel pathways we discovered here do not prove that they must operate in vivo, but they do can occur, making them possible mechanistic pathways for transcription deactivation.
Moreover, past studies have shown that for the archetype Hg2+-responsive metalloregulator MerR, a protein called MerD might mediate the unbinding of holo-MerR from DNA for transcription deactivation (25), a scenario analogous to assisted dissociation. No evidence has yet been found, however, for a MerD homologue for CueR or other MerR-family metalloregulators. As all known MerR-family metalloregulators share the DNA distortion mechanism for transcription activation (14, 27, 30), it is thus reasonable to think that most of them share a common mechanism for transcription deactivation. Therefore, the direct substitution and assisted dissociation pathways, if operating, might be common mechanisms for MerR-family metalloregulators to deactivate transcription efficiently after transcription activation.
2. Metallochaperones: versatile pathways for copper trafficking
Metallochaperones are transport proteins that deliver metal ions to their destinations or intermediate locations inside cells, while protecting the metals from adventitious binding sites or harmful reactions (52–56). In human cells, the copper chaperone Hah1 (also named Atox1), delivers Cu+, an essential but potentially harmful metal ion, to two homologous PIB-type ATPases: the Wilson's disease protein (WDP) and Menkes disease protein (MNK), which use ATP hydrolysis to further drive Cu+ translocation through membrane for either subsequent incorporation into copper enzymes or export (52, 57–59).
Both WDP and MNK have six N-terminal metal-binding domains (MBDs), connected by flexible peptides of various lengths (Figure 7). All these MBDs, as well as Hah1, are homologous, each with a βαββαβ protein fold and a CXXC motif that binds Cu+ with an affinity of ~1018 M–1 (60–65). Under a shallow thermodynamic gradient, Hah1 can transfer Cu+ to each MBD (61, 66), and the transfer is mediated by weak and dynamic protein interactions and involves metal-bridging of the CXXC motifs of the two proteins (52, 57, 59, 67–70).
Figure 7.
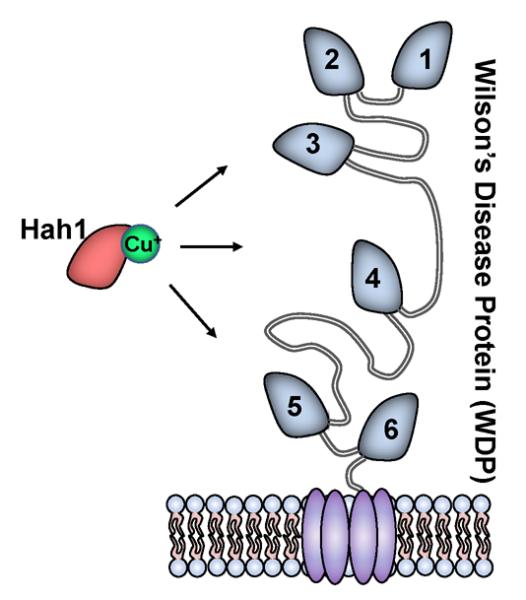
Schematic of Cu+ trafficking from Hah1 to the six MBDs of WDP (or MNK) anchored on a membrane.
Both intermolecular Hah1–MBD interactions and intramolecular MBD–MBD interactions are vital to the copper transport from Hah1 to WDP/MNK. The multiplicity of the WDP/MNK MBDs also seems important because the number of MBDs, which varies between one and six in homologous proteins (71, 72), tends to be larger for higher organisms (for example, the yeast homologue Ccc2 has merely two MBDs (73)). Characterizing and understanding these interactions is thus an important task, but it is challenging. Surface plasmon resonance has been used to study the kinetics of these interactions (74, 75), but the non-specific protein–surface interactions therein may perturb the kinetics. NMR (66, 76–83), x-ray crystallography (67, 84–86), protein docking (68), and molecular dynamic simulations (70, 87–89) have provided detailed structural information on the interaction interfaces, but they only provided estimates on the interaction thermodynamics and kinetics.
To complement these studies while overcoming some of their limitations, we have used smFRET in combination with lipid vesicle trapping (90–93) to quantify weak and dynamic interactions between Hah1 and WDP MBDs (93–96). In this approach, we label the two interacting partners with a FRET donor-acceptor pair and co-trap them within a surface-immobilized ~100-nm diameter unilamellar lipid vesicle. The FRET pair allows us to detect protein interactions at the single-molecule level, similar to the studies of metalloregulator–DNA interactions discussed earlier. The lipid vesicle trapping gives a confined volume, resulting in an effective protein concentration of ~μM for a single molecule inside, which is needed for studying weak interactions (KD ~ μM). The vesicle trapping also eliminates nonspecific protein interactions with glass surfaces while being immobilized, and allows us to selectively remove homodimeric interactions between two proteins of the same type during data analysis; these homodimeric interactions inevitably convolute ensemble-averaged measurements and are particularly relevant in studies of Hah1–WDP interactions, as Hah1 (and likely WDP MBDs) can form homodimers in solution (53, 67).
By placing the FRET pair on Hah1, MBD3, and/or MBD4 site-specifically, we systematically studied interactions between Hah1 and the isolated WDP MBD4 (denoted MBD4SD; SD = single domain), between Hah1 and each of the two domains of the double-domain WDP construct MBD34, and between the two MBDs of MBD34 (Figure 8A, C, E, G) (93–96). The labeling positions in Hah1 and each WDP MBD were all at their C-termini of these homologous protein domains. The interactions between Hah1 and MBD4SD provided the foundation for understanding Hah1–WDP interactions, which involve many MBDs. The double-domain construct MBD34 represented a simplest multi-domain system, which allowed us to gain insights into the multi-domain effect on Hah1–WDP interactions.
Figure 8.

(A, C, E, G) Protein labeling schemes to probe pair-wise interactions. The lipid vesicles were immobilized on a surface for smFRET measurements. For nomenclature, for example, in MBD34L4, the “L4” superscript denotes that the label is on MBD4 within MBD34. (B, D, F, H) Single-molecule EFRET trajectories corresponding to the labeling schemes in A, C, E, and G, respectively. (I–M) Compiled EFRET distributions for Cy5-Hah1 + Cy3-MBD4SD, Cy5-Hah1 + Cy3-MBD34L4, Cy5-Hah1 + Cy3-MBD34L3, Cy3Cy5-MBD34L34, and Cy3Cy5-MBD34L34 with excess Hah1. The histograms are Gaussian resolved; the relative peak areas in each histogram reflect the relative stabilities of corresponding states. Reprinted with permission from references (94, 96). Copyright 2008 and 2012 American Chemical Society.
Persistent dynamic interactions with two major interconverting complexes regardless of metallation states
For all Hah1–MBD and MBD–MBD interactions, we have consistently observed two major interaction complexes that interconvert dynamically, regardless of the protein metallation states. This is best illustrated by Hah1–MBD4SD interactions: the single-molecule EFRET trajectory shows dynamic transitions between the dissociated state (EDisso) and two major complexes (EMid and EHigh) (Figure 8B). For Hah1 interactions with MBD3 or MBD4 within the double-domain construct MBD34, the two higher EFRET states, EMid and EHigh, are preserved (Figure 8D, F); they are also present in MBD3–MBD4 interactions (Figure 8H) (note we use the terms “MBD3” and “MBD4” to refer to the respective MBDs within the double-domain MBD34 construct). The lowest EFRET state (i.e., ELow) in the EFRET trajectories of Hah1–MBD3 or Hah1–MBD4 interactions contains the dissociated state (EDisso) and the state (ELow′) in which Hah1 interacts with the respective unlabeled MBD of MBD34; these two states overlap significantly but can be resolved in global fitting of the EFRET histograms (Figure 8I–M); ELow′ is approximated by the lowest EFRET state in MBD3–MBD4 interactions (Figure 8H and L), where MBD3 and MBD4 are separated with their peptide linker in an extended conformation.
The conservation of the two major interaction geometries among Hah1–MBD and MBD–MBD interactions is reflected by the similarity of the EFRET values of EMid and EHigh (Figure 8I–L). This conservation is attributable to the sequence and structural homology across Hah1 and WDP MBDs. One should note that the geometric information from FRET measurements here is limited to the 1-D coordinate between the FRET donor and acceptor; 3-D geometric information needs multiple labeling schemes that cover multiple directions, which we are currently pursuing.
It is worth noting that earlier NMR studies on interactions between Hah1 and various WDP/MNK constructs did not detect interaction complexes at their apo states (66, 76–82), even though earlier SPR studies did observe apo protein interactions (74, 75). Our smFRET results represented the first evidence that multiple complexes exist for metallochaperone–target protein interactions regardless of the protein metallation states, and further corroborated that Hah1 can form complexes with WDP without Cu+. Moreover, more recent NMR studies by Fatemi et al. on the interactions between Hah1 and WDP MBD4–6 resolved interactions at their apo states (83), consistent with our results.
We proposed two structural models for the two observed interaction complexes between Hah1 and WDP MBDs on the basis of our smFRET results and past structural studies of these proteins and other homologs, all of which have the βαββαβ fold and contain the CXXC motif. The two α-helices are on one side of the protein (i.e., the “face” side), and the four β-strands form a β-sheet on the other side (i.e., the “back” side). We proposed a face-to-face interaction geometry that gave rise to the EMid state observed in our smFRET measurements, and a face-to-back geometry for the EHigh state (Figure 9A, B). The face-to-face geometry has been observed by NMR between Hah1 and MNK1, the first N-terminal MBD of MNK (82), and in homodimeric complexes of Hah1 (67). The CXXC motifs of the two proteins face each other in this geometry, where Cu+ can coordinate to cysteines from both proteins, thus offering a facile pathway for copper transfer via ligand exchange (52, 57, 59, 67–69). The face-to-back geometry was based on the crystal structure of an asymmetric dimer of the MBD of Hma7, a Cu+- transporting ATPase in Arabidopsis thaliana; this MBD is homologous to Hah1 and WDP/MNK MBDs (85). The FRET donor–acceptor distances in the face-to-face models are longer than those in the face-to-back models, consistent with EMid < EHigh. The validity of these two models was further supported by detailed interface thermodynamic analysis and molecular dynamics simulations (96).
Figure 9.
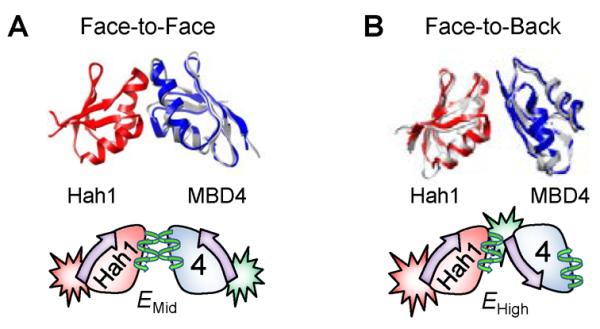
Structural models (top) of face-to-face (A) and face-to-back (B) Hah1–MBD4 interaction complexes with corresponding cartoon representations (bottom). In the cartoons, the “face” side of a protein is represented by a helix and the “back” side by an arrow. The FRET label positions are also indicated. Reprinted with permission from reference (96). Copyright 2012 American Chemical Society.
Hah1 interactions with MBD3 and MBD4 within MBD34 have similar stabilities, reflected by their similar peak areas of the associated states (EMid and EHigh) relative to the area of the dissociated state (EDisso) in the EFRET histograms (Figure 8J vs. K). But the Hah1–MBD4 interactions are significantly more stable than the Hah1–MBD4SD interactions whose peak areas of the EMid and EHigh states are much smaller relative to that of the dissociated state (Figure 8I). This enhanced stability in Hah1's interactions with MBD4 within the double-domain construct MBD34 is related to an increase in the rate of protein association rate, not a decrease in the rate of protein dissociation, because the interaction complexes have similar lifetimes regardless if Hah1 is interacting with MBD4 in MBD34 or with MBD4SD (96).
In the presence of one equivalent of Cu+ (i.e., one protein is at the apo state and the other is at the holo state), the face-to-face interaction geometry gets stabilized significantly for Hah1–MBD4SD interactions, reflected by the increased peak area of the EMid state relative to that of the EDisso state in EFRET histograms (Figure 10B vs. A). This stabilization of the face-to-face geometry at the apo-holo interaction state is attributable to possible Cu+ bridging via the CXXC motifs at the protein interaction interface (ref. Figure 9A). Consistently, this stabilization vanishes when both proteins are metallated in the presence of excess Cu+ (Figure 10C vs. A). In contrast, this Cu+-bridging induced stabilization of the apo-holo interactions in the face-to-face geometry is insignificant for Hah1 interactions with MBD4 in the double-domain construct MBD34, as no stabilization of the EMid state was observed in the presence of 1 equivalent of Cu+ (Figure 10E vs. D). Moreover, under excess Cu+, the Hah1–MBD4 interactions in both geometries are destabilized relative to those in the absence of Cu+ (Figure 10F vs. D), possibly due to a disruption of concerted interactions within the double-domain MBD34, as this destabilization was not observed in Hah1–MBD4SD interactions (Figure 10C vs. A).
Figure 10.
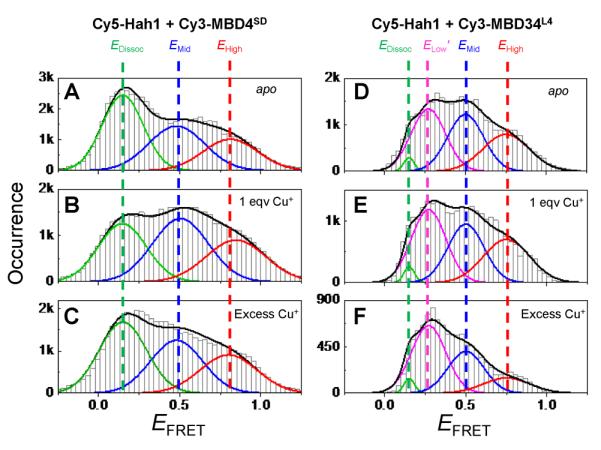
Compiled EFRET distributions for Cy5-Hah1 + Cy3-MBD4SD (A–C) and for Cy5-Hah1 + Cy3-MBD34L4 (D–F) in the absence and presence of 1 equivalent (eqv) and excess Cu+ per protein pair. Reprinted with permission from reference (96). Copyright 2012 American Chemical Society. And reproduced from reference (95) by permission of The Royal Society of Chemistry.
Three-body interactions
Quantitative population analysis of different interaction states in different labeling schemes for Hah1–MBD34 interactions also suggested the presence of 3-body interactions among Hah1, MBD4, and MBD3. First, an overlap population was observed between Hah1–MBD4 complexes and Hah1–MBD3 complexes, attributed to Hah1 interacting with MBD4 and MBD3 simultaneously. Second, Hah1 can interact with the intramolecular-interdomain MBD3–MBD4 complexes, reflected in part by the population changes in EMid and EHigh states in the Cy3Cy5-MBD34L34 labeling scheme when an excess of Hah1 was introduced (Figure 8M vs. L).
In our two proposed interaction geometries, the face and back interfaces are spatially distinct (i.e., non-overlapping), making it possible for 3-body interactions between Hah1, MBD3, and MBD4. We thus made models for 3-body interactions using combinations of face-to-face and face-to-back interactions. Figure 11 illustrates two possible 3-body interactions: in one Hah1 is sandwiched between MBD3 and MBD4, and in the other Hah1 interacts with an intramolecular MBD3–MBD4 complex. We should emphasize that the interaction geometries here are only models that are supported by data and deduced from known structures of protein complexes. Within either EMid or EHigh states, additional subpopulations could exist that are unresolved in our measurements. The dynamic peptide linker between MBD4 and MBD3 may also play a role in the complex formation (89).
Figure 11.
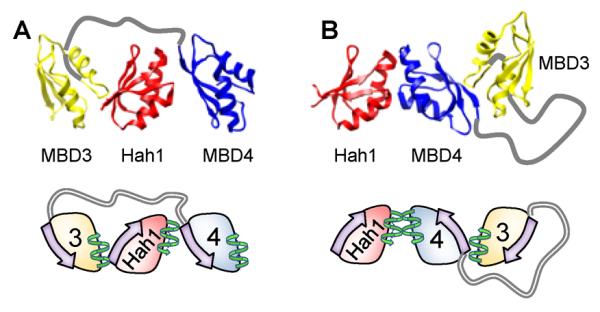
Structural models of 3-body interactions where Hah1 is sandwiched between MBD3 and MBD4 (A) and where Hah1 is interacting with an MBD34 intramolecular-interdomain adduct (B). Reprinted with permission from reference (96). Copyright 2012 American Chemical Society.
Versatile pathways for receiving, redistributing, and exporting copper ions via multi-body protein interactions
The ways that Hah1 and the double-domain construct MBD34 can interact suggest versatile pathways for copper trafficking from Hah1 to WDP (or MNK) inside cells. This versatility is better illustrated by an analogy to the modern cargo transportation industry. In this analogy Hah1 is a delivery truck with Cu+ as its cargo. The N-terminal region of WDP, with multiple MBDs, is a warehouse distribution center, and the MBDs are the loading docks. The distribution center must operate with both efficiency and versatility to receive, re-route, and export shipments from many trucks.
The operation versatility of the WDP distribution center is accomplished by providing multiple MBD docking sites for the Hah1 truck to deliver its cargo. The truck can park frontward or backward at the dock (i.e., with two major Hah1–MBD interaction geometries, Figure 12A). Even better, the truck can interconvert between its docking geometries dynamically, thus allowing either of the two interfaces to be exposed for interaction with an additional MBD. The 3-body interactions where Hah1 is sandwiched between MBDs allow for the re-routing of the delivery truck, i.e., a Hah1 molecule can be handed over directly from one MBD to another (Figure 12D). This re-routing of Hah1 would especially be useful when the initially targeted MBD is already loaded with Cu+. WDP's intramolecular MBD–MBD interactions provide a way for internal redistribution of the Cu+ cargo, either to vacate space for the next Hah1 delivery or to traffic Cu+ downstream. This redistribution also occurs in a versatile manner, as two major binding geometries were observed between MBD3 and MBD4 (Figure 12B). This internal cargo redistribution among MBDs can be directly coupled to the cargo delivery or export, through Hah1 interactions with the intramolecular MBD–MBD complexes (Figure 12C). All of the above interactions occur on a time scale of ~1 s (96), including the protein associations at ~μM concentrations (note the intracellular concentration of the yeast Hah1 homologue Atx1 is also ~μM (97)). Therefore, all these processes should occur comparably inside cells for function.
Figure 12.

Versatile pathways for copper trafficking from Hah1 to WDP MBDs illustrated by the major features of Hah1–MBD34 interaction dynamics. (A, B) Intermolecular and intramolecular Hah1–MBD4, Hah1–MBD3, and MBD3–MBD4 interactions can occur in two major geometries, providing versatile docking with interconversion for Cu+ transfer. (C) Hah1 can interact with intramolecular-interdomain MBD34 complexes linking MBD–MBD and Hah1–MBD interactions. (D) The 3-body interaction where Hah1 is sandwiched provides a mechanism to re-route Hah1 between MBDs. Reprinted with permission from reference (96). Copyright 2012 American Chemical Society.
The ways that Hah1 and MBD34 interact also shed light on the possible regulatory function of the MBDs, in which Hah1-MBD or MBD-MBD interactions modulate the ATPase activity associated with Cu+-translocation (71, 98–101) or the kinase-mediated phosphorylation associated with the relocalization of WDP/MNK for Cu+-efflux (71, 98–101). It has been proposed that large-scale conformational changes within the N-terminal tail of WDP/MNK can act as a regulatory switch (80, 89, 102–105): these changes would disrupt MBD interactions with the catalytic core affecting Cu+-translocation or expose/hide phosphorylation sites in the linker regions. The 3-body interactions where Hah1 is sandwiched between MBDs (Figure 12D) could induce large-scale conformational changes in the cytoplasmic tail of WDP, and hence may play a role in this regulatory switching mechanism.
Although our study was limited to Hah1–WDP interactions for Cu+ trafficking, we suspect that their versatile metal trafficking mechanism may also operate in other PIB-ATPases: many of these ATPases contain multiple MBDs and could have associated metallochaperones, which regulate other metals such as Cd2+, Zn2+, Pb2+, and Ag+ (72, 106).
3. Related single-molecule bioinorganic work
In the past few years, more single-molecule studies of bioinorganic systems have emerged. A few earlier ones were reviewed by us in 2008 (107). Below we briefly summarize these studies according to the techniques employed, as well as refer to relevant reviews for in-depth reviews on these topics. These single-molecule studies provide a context for the single-molecule fluorescence studies our group has pursued. The strengths and limitations of the techniques are also discussed.
Single-molecule fluorescence microscopy studies
Several groups have used the single-molecule fluorescence quenching strategy to study metalloproteins; this strategy is a variant of FRET in which the acceptor, here a metal-based active site in protein, is a strong chromophore at a certain oxidation state but nonfluorescent, and acts as a quencher to an introduced fluorescent label that acts as the donor. Erker, Basché, and coworkers used this strategy to study oxygen binding by binuclear copper protein hemocyanin (108, 109). Aartsman, Canters, Schmidt and coworkers used this strategy to probe the redox states of blue copper protein azurin (110, 111). Takahashi and coworkers used it to study the folding dynamics of cytochrome c (112). Herten and coworkers used it to monitor the formation and dissociation of metal complexes (113). Canters, Moerner, and coworkers used it to study the enzymatic reactions by the copper enzyme nitrite reductase (114, 115). Spies and coworkers used it to study the interactions of an iron-sulfur-cluster-containing helicase with DNA (116).
Moreover, Ha, Lu, and coworkers have used smFRET to study the metal-ion dependent folding of DNAzymes. Rigler and coworkers developed the fluorogenic reaction approach to study catalysis by the heme-enzyme horseradish peroxidase (117, 118), in which the fluorescence signal of a reaction product was detected at the single-molecule level to monitor catalytic reactions. The intrinsic fluorescence of Mg2+-containing chlorophyll has also been utilized widely to study light-harvesting complexes at the single-molecule level and have been reviewed elsewhere (119–126).
Single-molecule fluorescence imaging, including single-molecule FRET and fluorescence quenching discussed here, is broadly applicable for studying protein function and dynamics. The use of external fluorescent probes is general. Site-specific labeling of proteins is readily achievable with many accessible labeling schemes including site-directed mutagenesis, GFP fusion, and unnatural amino acids (127, 128). Many fluorescent probes suitable for singe-molecule detections are also available covering a wide spectral range (128). The single-molecule fluorescence quenching strategy has particular potential for studying metalloproteins — in principle, any metalloprotein that shows intense absorption properties can be targeted using this approach. Moreover, transition-metal-based chemistry often involves species that have intense ligand-to-metal charge transfer absorptions. These strong chromophoric species can be exploited as quenching centers for single-molecule fluorescence detection.
Limitations also exist to fluorescence-based single-molecule methods. For example, photobleaching of the probe limits the observation time window. With a good oxygen scavenging system, a single fluorescent probe molecule can last for up to a few minutes before being photobleached (129). The introduction of fluorescent probes always bring concerns of perturbation to protein structure and function, for which careful controls need to be carried out to ensure that the perturbation is not significant.
Single-molecule scanning probe microscopy studies
Scanning probe microscopies have also been applied to study metalloproteins at the single-molecule level. These studies are approximately in two categories: scanning tunneling microscopy (STM) and atomic force microscopy (AFM). STM was used to study the electron transfer properties of single metalloproteins via electrical current measurements. The STM studies by Ulstrup (130, 131), Facci (132) and coworkers on electron transfer by metalloproteins, and by Cannistraro (133) and coworkers on electron conduction and recognition by metalloproteins have been reviewed by these researchers. Wingginton (134), Jones (135), and coworkers have used STM to measure tunneling currents through single cytochrome molecules. AFM was used to manipulate metalloproteins mechanically (i.e., by force). Cannistraro and coworkers used single-molecule force measurements to study the recognition between cytochrome c551 and azurin (136, 137) and the interaction between p53 and azurin (138, 139). Yersin (140), Ikuta (141), and coworkers studied transferrin-receptor interactions. Li and coworkers studied the metal-thiolate bond ruptures in rubredoxin (142, 143).
The electric-current-based detection of scanning probe microscopy (e.g., STM) is powerful for interrogating the electron transfer properties of redox-active proteins, many of which contain a redox-active metal center. The measurements can also be performed for an extended time on a single protein molecule, allowing the study of time-dependent behaviors. The mechanics-based approach (e.g., AFM) measures force directly, which readily connects to thermodynamic properties, such as the interaction affinity between proteins. The scanning probe can also be used to manipulate protein molecules, offering a way to control and change protein structure or function.
Limitations also exist to these approaches as well. The current-based detection is limited to redox-active proteins. Many redox-inactive metalloproteins, such as ZnII-containing ones, are thus not accessible. The mechanics-based detection is based on measuring force or distance changes; processes that do not cause force or distance changes, for example most of enzymatic reactions, would be challenging to study. The scanning probe approach is a serial measurement, where only one molecule is studied at one time, leading to low data throughput.
4. Concluding remarks
The reach of single-molecule studies has been expanding rapidly in recent years, not only in biological sciences but also in physical sciences (e.g., in heterogeneous catalysis (144)). Single-molecule research in the bioinorganic field is yet still under-populated. Vast opportunities exist for new research endeavors. Of particular interest to us, the advances in the cell biology of metals continue to unravel new scientific problems. We are currently continuing our studies on metalloregulators and metallochaperones, with more focus on living cell studies at the single-molecule level. Studying how these metalloproteins operate in vivo is important, as a cell presents a much more complicated environment than a test tube, such as compartmentation, localization, crowdedness, and nonspecific interactions, which in vitro experimental conditions may not capture. The mechanism of a metal homeostatic protein, or any protein, can be significantly affected by its cellular spatiotemporal state, i.e., where it is in the cell and when. To obtain such information, one needs to interrogate protein functions in a living cell in a spatiotemporally resolved manner. For bacteria, which are merely a few microns in size, this interrogation requires nanometer spatial resolution, beyond the reach of conventional optical microscopy (at best ~250 nm resolution), while electron microscopy or scanning probe microscopy, with their nanometer resolution, cannot probe into living cells. For eukaryotic cells, many of their internal compartments, such as organelles, are also small in size and require nanometer spatial resolution to resolve. High sensitivity is often needed, too, especially for low copy number proteins (e.g., metalloregulators, which are transcription factors). Single-molecule fluorescence imaging techniques can meet many of these requirements, for example nanometer resolution (via a super-resolution imaging approach based on single-molecule detection (144–147)), single-molecule sensitivity, millisecond temporal resolution, and high specificity in imaging cellular processes (e.g., through genetically tagging with fluorescent proteins). It is the authors' belief that the application of single-molecule imaging could create a new subarea in bioinorganic research, breaking new grounds and establishing new directions. The opportunities are only limited by one's imagination.
ACKNOWLEDGMENT
We thank the National Institute of Health (GM082939, GM106420, and EB009202), the National Science Foundation (CHE-0645392), and Optofluidics Inc. for financial support. We also thank Debashis Panda, Derek Klarin, and Matthew Goldfogel for their contributions, and David Erickson, Peng R. Chen/Chuan He, Ahmed Gaballa/John Helmann, David Huffman, Liliya Yatsunyk/Amy Rosenzweig, and Linghao Zhong for collaborations on the research reviewed here.
Funding Sources: NIH (GM082939, GM106420, EB009202), NSF (CHE-0645392), and Optofluidics, Inc.
ABBREVIATIONS
- CueR
copper efflux regulator
- MerR
mercury resistance regulator
- CopA
copper-exporting ATPase
- CueO
copper efflux oxidase
- RNAp
RNA polymerase
- smFRET
single-molecule fluorescence resonance energy transfer
- HJ
Holliday junction
- MerD
mercury resistance coregulator
- Hah1
human Atx1 homologue
- Atox1
antioxidant protein 1
- WDP
Wilson's disease protein
- MNK
Menkes disease protein
- MBD
metal-binding domain
Footnotes
On the potential energy surface going from the reactant to the product along the reaction coordinate, a transition state is a first-order saddle point, where there is a minimum in all dimensions but one. On the other hand, an intermediate is a local minimum in all dimensions.
REFERENCES
- 1.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley: 1994. [Google Scholar]
- 2.Holm RH, Solomon EI, editors. Thematic Issue: Bioinorganic Enzymology. Chem. Rev. 1996;96(Issue 7) [Google Scholar]
- 3.Michalet X, Weiss S, Jaeger M. Single-molecule fluorescence studies of protein folding and conformational dynamics. Chem. Rev. 2006;106:1785–1813. doi: 10.1021/cr0404343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuang X. Single-Molecule RNA Science. Annu. Rev. Biophys. Biomol. Struct. 2005;34:399–414. doi: 10.1146/annurev.biophys.34.040204.144641. [DOI] [PubMed] [Google Scholar]
- 5.Selvin PR, Ha T, editors. Single Molecule Techniques: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- 6.Smiley RD, Hammes GG. Single Molecule Studies of Enzyme Mechanisms. Chem. Rev. 2006;106:3080–3094. doi: 10.1021/cr0502955. [DOI] [PubMed] [Google Scholar]
- 7.Xie XS, Choi PJ, Li G-W, Lee NK, Lia G. Single-Molecule Approach to Molecular Biology in Living Bacterial Cells. Annu. Rev. Biophys. 2008;37:417–444. doi: 10.1146/annurev.biophys.37.092607.174640. [DOI] [PubMed] [Google Scholar]
- 8.Bustamante C, Macosko JC, Wuite GJL. Grabbing the cat by the tail: Manipulating molecules one by one. Nat. Rev. Mol. Cell Biol. 2000;1:130–136. doi: 10.1038/35040072. [DOI] [PubMed] [Google Scholar]
- 9.Chen P, Andoy NM, Benitez JJ, Keller AM, Panda D, Gao F. Tackling Metal Regulation and Transport at the Single-Molecule Level. Nat. Prod. Rep. 2010;27:757–767. doi: 10.1039/b906691h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Halloran TV. Transition Metals in Control of Gene Expression. Science. 1993;261:715–725. doi: 10.1126/science.8342038. [DOI] [PubMed] [Google Scholar]
- 11.Giedroc DP, Arunkumar AI. Metal Sensor Proteins: Nature's Metalloregulated Allosteric Switch. Dalton Trans. 2007:3107–3120. doi: 10.1039/b706769k. [DOI] [PubMed] [Google Scholar]
- 12.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Metalloproteins and Metal Sensing. Nature. 2009;460:823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 13.Barkey T, Miler SM, Summers AO. Bacterial Mercury Resistance from Atoms to Ecosystems. FEMS Microbiol. Rev. 2003;27:355–384. doi: 10.1016/S0168-6445(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 14.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. The MerR Family of Transcripitional Regulators. FEMS Microbiol. Rev. 2003;27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 15.Busenlehner L, Pennella MA, Giedroc DP. The SmtB/ArsR Family of Metalloregulatory Transcriptional Repressors: Structural Insights into Prokaryotic Metal Resistance. FEMS Microbiol. Rev. 2003;27:131–143. doi: 10.1016/S0168-6445(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 16.Andrews SC, Robinson AK, Rodriguez-Quinones F. Bacterial Iron Homeostasis. FEMS Microbiol. Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 17.Cavet JS, Borrelly GPM, Robinson NJ. Zn, Cu and Co in Cyanobacteria: Selective Control of Metal Availability. FEMS Microbiol. Rev. 2003;27:165–181. doi: 10.1016/S0168-6445(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 18.Mergeay M, Monchy S, Vallaeys T, Auquier V, Benotmane A, Bertin P, Taghavi S, Dunn J, van der Lelie D, Wattiez R. Ralstonia metallidurans, a Bacterium Specifically Adapted to Toxic Metals: Towards a Catalogue of Metal-Reponsive Genes. FEMS Microbiol. Rev. 2003;27:385–410. doi: 10.1016/S0168-6445(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 19.Rensing C, Grass G. Escherichia coli Mechanisms of Copper Homeostasis in a Changing Environment. FEMS Microbiol. Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 20.Solioz M, Stoyanov JV. Copper Homeostasis in Enterococcus hirae. FEMS Microbiol. Rev. 2003;27:183–195. doi: 10.1016/S0168-6445(03)00053-6. [DOI] [PubMed] [Google Scholar]
- 21.Kehres DG, Maguire ME. Emerging Themes in Manganese Transport, Biochemistry and pathogenesis in Bacteria. FEMS Microbiol. Rev. 2003;27:263–290. doi: 10.1016/S0168-6445(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 22.Lloyd JR. Microbial Reduction of Metals and Radionuclides. FEMS Microbiol. Rev. 2003;27:411–425. doi: 10.1016/S0168-6445(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 23.Mulrooney SB, Hausinger RP. Nickel Uptake and Utilization by Microorganism. FEMS Microbiol. Rev. 2003;27:239–261. doi: 10.1016/S0168-6445(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 24.Nies DH. Efflux-Mediated Heavy Meal Resistance in Prokaryotes. FEMS Microbiol. Rev. 2003;27:313–339. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 25.Hobman JL, Wilkie J, Brown NL. A Design for Life: Prokaryotic Metal-Bindng MerR Family Regulators. BioMetals. 2005;18:429–436. doi: 10.1007/s10534-005-3717-7. [DOI] [PubMed] [Google Scholar]
- 26.Laity JH, Andrews GK. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1) Arch. Biochem. Biophys. 2007;463:201–210. doi: 10.1016/j.abb.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 27.O'Halloran TV, Frantz B, Shin MK, Ralston DM, Wright JG. The MerR Heavy Metal Receptor Mediates Positive Activation in A Topologically Novel Transcription Complex. Cell. 1989;56:119–129. doi: 10.1016/0092-8674(89)90990-2. [DOI] [PubMed] [Google Scholar]
- 28.Frantz B, O'Halloran TV. DNA Distortion Accompanies Transcriptional Activation by the Metal-Responsive Gene-Regulatory Protein MerR. Biochemistry. 1990;29:4747–4751. doi: 10.1021/bi00472a001. [DOI] [PubMed] [Google Scholar]
- 29.Chen PR, He C. Selective recognition of metal ions by metalloregulatory proteins. Curr. Opin. Chem. Biol. 2008;12:214–221. doi: 10.1016/j.cbpa.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 30.Newberry KJ, Brennan RG. The Structural Mechanism for Transcription Activation by MerR Family Member Multidrug Transporter Activation, N-Terminus. J. Biol. Chem. 2004;279:20356–20362. doi: 10.1074/jbc.M400960200. [DOI] [PubMed] [Google Scholar]
- 31.Outten CE, Outten FW, O'Halloran TV. DNA Distortion Mechanism for Transcriptional Activation by ZntR, A Zn(II)-responsive MerR Homologue in Escherichia coli. J. Biol. Chem. 1999;274:37517–37524. doi: 10.1074/jbc.274.53.37517. [DOI] [PubMed] [Google Scholar]
- 32.Holm RH, Kennepohl P, Solomon EI. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 33.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O'Halloran TV, Mondragon A. Molecular Basis of Metal-Ion Selectivity and Zeptomolar Sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 34.Rosenzweig AC, O'Halloran TV. Structure and Chemistry of the Copper Chaperone Proteins. Curr. Opin. Chem. Biol. 2000;4:140–147. doi: 10.1016/s1367-5931(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 35.Joshi CP, Panda D, Martell DJ, Andoy NM, Chen T-Y, Gaballa A, Helmann JD, Chen P. Single-Molecule Analysis Suggests Novel Pathways for Turning Off Transcription by A MerR-family Metalloregulator. Proc. Nat. Acad. Sci. USA. 2012;109:15121–15126. doi: 10.1073/pnas.1208508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Outten FW, Outten CE, Hale J, O'Halloran TV. Transcriptional Activation of An Escherichia coli Copper Efflux Regulation by the Chromosomal MerR Homologue, CueR. J. Biol. Chem. 2000;275:31024–31029. doi: 10.1074/jbc.M006508200. [DOI] [PubMed] [Google Scholar]
- 37.Stoyanov JV, Hobman JL, Brown NL. CueR (YbbI) of Escherichia coli Is A MerR Family Regulator Controlling Expression of the Copper Exporter CopA. Mol. Microbiol. 2001;39:502–511. doi: 10.1046/j.1365-2958.2001.02264.x. [DOI] [PubMed] [Google Scholar]
- 38.Outten FW, Huffman DL, Hale JA, O'Halloran TV. The Independent cue and cus Systems Confer Copper Tolerance during Aerobic and Anaerobic Growth in Escherichia coli. J. Biol. Chem. 2001;2001:30670–30677. doi: 10.1074/jbc.M104122200. [DOI] [PubMed] [Google Scholar]
- 39.Grass G, Rensing C. Genes Involved in Copper Homeostasis in Escherichia coli. J. Bacteriol. 2001;133:2145–2147. doi: 10.1128/JB.183.6.2145-2147.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abbondanzieri EA, Bokinsky G, Rausch JW, Zhang JX, Le Grice SFJ, Zhuang X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The Complete Genome Sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 42.von Hippel PH, Berg OG. Facilitated target location in biological systems. J. Biol. Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 43.Blainey PC, Graziano V, Pérez-Berná AJ, McGrath WJ, Flint SJ, Martín CS, Xie XS, Mange WF. Regulation of a Viral Proteinase by a Peptide and DNA in One-dimensional Space: IV. VIRAL PROTEINASE SLIDES ALONG DNA TO LOCATE AND PROCESS ITS SUBSTRATES. J. Biol. Chem. 2013;288:2092–2102. doi: 10.1074/jbc.M112.407460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graham JS, Johnson RC, Marko JF. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucl. Acids Res. 2011;39:2249–2259. doi: 10.1093/nar/gkq1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andoy NM, Sarkar SK, Wang Q, Panda D, Benitez JJ, Kalininskiy A, Chen P. Single-Molecule Study of Metalloregulator CueR-DNA Interactions Using Engineered Holliday Junctions. Biophys. J. 2009;97:844–852. doi: 10.1016/j.bpj.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eichman BF, Vargason JM, Mooers BHM, Ho PS. The Holliday junction in an inverted repeat DNA sequence: sequence effects on the structure of four-way junctions. Proc. Natl. Acad. Sci. USA. 2000;97:3971–3976. doi: 10.1073/pnas.97.8.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ortiz-Lombardía M, González A, Eritja R, Aymamí J, Azorín F, Coll M. Crystal structure of a DNA Holliday junction. Nat. Struct. Biol. 1999;6:913–917. doi: 10.1038/13277. [DOI] [PubMed] [Google Scholar]
- 48.McKinney SA, Declais AC, Lilley DMJ, Ha T. Structural Dynamics of Individual Holliday Junctions. Nat. Struct. Biol. 2003;10:93–97. doi: 10.1038/nsb883. [DOI] [PubMed] [Google Scholar]
- 49.Sarkar SK, Andoy NM, Benitez JJ, Chen PR, Kong JS, He C, Chen P. Engineered Holliday Junctions as Single-Molecule Reporters for Protein-DNA Interactions with Application to a MerR-Family Regulator. J. Am. Chem. Soc. 2007;129:12461–12467. doi: 10.1021/ja072485y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karymov MA, Chinnaraj M, Bogdanov A, Srinivasan AR, Zheng G, Olson WK, Lyubchenko YL. Structure, dynamics, and branch migration of a DNA Holliday junction: a single-molecule fluorescence and modeling study. Biophys J. 2008;95:4372–4383. doi: 10.1529/biophysj.108.135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamamoto K, Ishihama A. Transcriptional Response of Escherichia coli to External Copper. Mol Microbiol. 2004;56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 52.O'Halloran TV, Culotta VC. Metallochaperones, An Intracellular Shuttle Service for Metal Ions. J. Biol. Chem. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- 53.Rosenzweig AC. Copper Delivery by Metallochaperone Proteins. Acc. Chem. Res. 2001;34:119–128. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 54.Donnelly PS, Xiao Z, Wedd AG. Copper and Alzheimer's disease. Curr. Opin. Chem. Biol. 2007;11:128–133. doi: 10.1016/j.cbpa.2007.01.678. [DOI] [PubMed] [Google Scholar]
- 55.Cobine PA, Pierrel F, Winge DR. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim. Biophys. Acta. 2006;1763:759–772. doi: 10.1016/j.bbamcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Subramanian P, Rodrigues AV, Ghimire-Rijal S, Stemmler TL. Iron chaperones for mitochondrial Fe-S cluster biosynthesis and ferritin iron storage. Curr. Opin. Chem. Biol. 2011;15:312–318. doi: 10.1016/j.cbpa.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pufahl RA, Singer CP, Peariso KL, Lin S-J, Schmidt PJ, Fahrni CJ, Culotta VC, Penner-Hahn JE, O'Halloran TV. Metal Ion Chaperone Function of the Soluble Cu(I) Receptor Atx1. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 58.Hamza I, Schaefer M, Klomp LWJ, Gitlin JD. Interaction of the Copper Chaperone Hah1 with the Wilson Disease Protein Is Essential for Copper Homeostasis. Proc. Natl. Acad. Sci. USA. 1999;96:13363–13368. doi: 10.1073/pnas.96.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larin D, Mekios C, Das K, Ross B, Yang A-S, Gilliam TC. Characterization of the Interaction between the Wilson and Menkes Disease Proteins and the Cytoplasmic Copper Chaperone, Hah1p. J. Biol. Chem. 1999;274:28497–28504. doi: 10.1074/jbc.274.40.28497. [DOI] [PubMed] [Google Scholar]
- 60.Boal AK, Rosenzweig AC. Structural Biology of Copper Trafficking. Chem. Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yatsunyk LA, Rosenzweig AC. Copper(I) Binding and Transfer by the N-terminus of the Wilson Disease Protein. J. Biol. Chem. 2007;282:8622–8631. doi: 10.1074/jbc.M609533200. [DOI] [PubMed] [Google Scholar]
- 62.Xiao Z, Brose J, Schimo S, Ackland SM, La Fontaine S, Wedd AG. Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1, and related proteins: detection probes and affinity standards. J. Biol. Chem. 2011;286:11047–11055. doi: 10.1074/jbc.M110.213074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Palumaa P. Affinity gradients drive copper to cellular destinations. Nature. 2010;465:645–648. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- 64.Badarau A, Dennison C. Copper trafficking mechanism of CXXC-containing domains: insight from the pH-dependence of their Cu(I) affinities. J Am Chem Soc. 2011;133:2983–2988. doi: 10.1021/ja1091547. [DOI] [PubMed] [Google Scholar]
- 65.Xiao Z, Wedd AG. The challenges of determining metal-protein affinities. Nat. Prod. Rep. 2010;27:768–789. doi: 10.1039/b906690j. [DOI] [PubMed] [Google Scholar]
- 66.Banci L, Bertini I, Cantini F, Massagni C, Migliardi M, Rosato A. An NMR Study of the Interaction of N-terminal Cytoplasmic Tail of the Wilson Disease Protein with Copper(I)-Hah1. J. Biol. Chem. 2009;284:9354–9360. doi: 10.1074/jbc.M805981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wernimont AK, Huffman DL, Lamb AL, O'Halloran TV, Rosenzweig AC. Structural Basis for Copper Transfer by the Metallochaperone for the Menkes/Wilson Disease Proteins. Nat. Struct. Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 68.Arnesano F, Banci L, Bertini I, Bonvin MJJ. A Docking Approach to the Study of Copper Trafficking Proteins: Interactions between Metallochaperones and Soluble Domains of Copper ATPases. Structure. 2004;12:669–676. doi: 10.1016/j.str.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 69.Huffman DL, O'Halloran TV. Function, Structure, and Mechanism of Intracellular Copper Trafficking Proteins. Ann. Rev. Biochem. 2001;70:677–701. doi: 10.1146/annurev.biochem.70.1.677. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez-Granillo A, Crespo A, Estrin DA, Wittung-Stafshede P. Copper-Transfer Mechanism from the Human Chaperone Atox1 to a Metal-Binding Domain of Wilson Disease Protein. J. Phys. Chem. B. 2010;114:3698–3706. doi: 10.1021/jp911208z. [DOI] [PubMed] [Google Scholar]
- 71.Arguello JM, Eren E, Gonzalez-Guerrero M. The structure and function of heavy metal transport P1B-ATPases. Biometals. 2007;20:233–248. doi: 10.1007/s10534-006-9055-6. [DOI] [PubMed] [Google Scholar]
- 72.Arguello JM. Identification of ion-selectivity determinants in heavy-metal transport P1B-type ATPases. J Membr Biol. 2003;195:93–108. doi: 10.1007/s00232-003-2048-2. [DOI] [PubMed] [Google Scholar]
- 73.Huffman DL, O'Halloran TV. Energetics of Copper Trafficking between the Atx1 Metallochaperone and the Intracellular Copper Transporter Ccc2. J. Biol. Chem. 2000;275:18611–18614. doi: 10.1074/jbc.C000172200. [DOI] [PubMed] [Google Scholar]
- 74.Multhaup G, Strausak D, Bissig K-D, Solioz M. Interaction of the CopZ Copper Chaperone with the CopA Copper ATPase of Enterococcus hirae Assessed by Surface Plasmon Resonance. Biochem. Biophys. Res. Commun. 2001;288:172–177. doi: 10.1006/bbrc.2001.5757. [DOI] [PubMed] [Google Scholar]
- 75.Strausak D, Howies MK, Firth SD, Schlicksupp A, Pipkorn R, Multhaup G, Mercer JFB. Kinetic Analysis of the Interaction of the Copper Chaperone Atox1 with the Metal Binding Sites of the Menkes Protein. J. Biol. Chem. 2003;278:20821–20827. doi: 10.1074/jbc.M212437200. [DOI] [PubMed] [Google Scholar]
- 76.Arnesano F, Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Huffman DL, O'Halloran TV. Characterization of the Binding Interfaces between the Copper Chaperone Atx1 and the First Cytosolic Domain of Ccc2 ATPase. J. Biol. Chem. 2001;276:41365–41376. doi: 10.1074/jbc.M104807200. [DOI] [PubMed] [Google Scholar]
- 77.Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. The Atx1-Ccc2 Complex is a Metal-Mediated Protein-Protein Interaction. Nat. Chem. Biol. 2006;2:367–368. doi: 10.1038/nchembio797. [DOI] [PubMed] [Google Scholar]
- 78.Banci L, Bertini I, Cantini F, Chasapis CT, Hadjiliadis N, Rosato A. A NMR Study of the Interactions of a Three-Domain Construct of ATP7A with Copper(I) and Copper(I)-Hah1: The Interplay of Domains. J. Biol. Chem. 2005;280:38259–38263. doi: 10.1074/jbc.M506219200. [DOI] [PubMed] [Google Scholar]
- 79.Achila D, Banci L, Bertini I, Bunce J, Ciofi-Baffoni S, Huffman DL. Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc. Natl. Acad. Sci. USA. 2006;103:5729–5734. doi: 10.1073/pnas.0504472103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Banci L, Bertini I, Cantini F, Della-Malva N, Migliardi M, Rosato A. The Different Intermolecular Interactions of the Soluble Copper-Binding Domains of the Menkes Protein, ATP7A. J. Biol. Chem. 2007;282:23140–23146. doi: 10.1074/jbc.M700695200. [DOI] [PubMed] [Google Scholar]
- 81.Banci L, Bertini I, Francesca C, Rosenzweig AC, Yatsunyk LA. Metal Binding Domains 3 and 4 of the Wilson Disease Protein: Solution Structure and Interaction with the Copper(I) Chaperone Hah1. Biochemistry. 2008;47:7423–7429. doi: 10.1021/bi8004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Banci L, Bertini I, Calderone V, Della-Malva N, Felli IC, Neri S, Pavelkova A, Rosato A. Copper(I)-mediated protein-protein interactions result from suboptimal interaction surfaces. Biochem. J. 2009;422:37–42. doi: 10.1042/BJ20090422. [DOI] [PubMed] [Google Scholar]
- 83.Fatemi N, Korzhnev DM, Velyvis A, Sarkar B, Forman-Kay JD. NMR Characterization of Copper-Binding Domains 4–6 of ATP7B. Biochemistry. 2010;49:8468–8477. doi: 10.1021/bi1008535. [DOI] [PubMed] [Google Scholar]
- 84.Badarau A, Firbank SJ, McCarthy AA, Banfield MJ, Dennison C. Visualizing the Metal-Binding Versatility of Copper Trafficking Sites. Biochemistry. 2010;49:7798–7810. doi: 10.1021/bi101064w. [DOI] [PubMed] [Google Scholar]
- 85.Zimmerman M, Clarke O, Gulbis JM, Keizer DW, Jarvis RS, Cobbett CS, Hinds MG, Xiao Z, Wedd AG. Metal Binding Affinities of Arabidopsis Zinc and Copper Transporters: Selectivities Match the Relative, but Not the Absolute, Affinities of their Amino-Terminal Domains. Biochemistry. 2009;48:11640–11654. doi: 10.1021/bi901573b. [DOI] [PubMed] [Google Scholar]
- 86.Alvarez HM, Xue Y, Robinson CD, Canalizo-Hernández MA, Marvin RG, Kelly RA, Mondragón A, Penner-Hahn JE, O'Halloran TV. Tetrathiomolybdate Inhibits Copper Trafficking Proteins Through Metal Cluster Formation. Science. 2009;327:331–334. doi: 10.1126/science.1179907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rodriguez-Granillo A, Crespo A, Wittung-Stafshede P. Conformational Dynamics of Metal-Binding Domains in Wilson Disease Protein: Molecular Insights into Selective Copper Transfer. Biochemistry. 2009;48:5849–5863. doi: 10.1021/bi900235g. [DOI] [PubMed] [Google Scholar]
- 88.Hussain F, Rodriguez-Granillo A, Wittung-Stafshede P. Lysine-60 in Copper Chaperone Atox1 Plays an Essential Role in Adduct Formation with a Target Wilson Disease Domain. J. Am. Chem. Soc. 2009;131:16371–16373. doi: 10.1021/ja9058266. [DOI] [PubMed] [Google Scholar]
- 89.Rodriguez-Granillo A, Crespo A, Wittung-Stafshede P. Interdomain interactions modulate collective dynamics of the metal-binding domains in the Wilson disease protein. J. Phys. Chem. B. 2010;114:1836–1848. doi: 10.1021/jp909450x. [DOI] [PubMed] [Google Scholar]
- 90.Chiu DT, Wilson CF, Karlsson A, Danielsson A, Lundqvist A, Strömberg A, Ryttsén F, Davidson M, Nordholm S, Orwar O, Zare RN. Manipulating the biochemical nanoenvironment around single molecules contained within vesicles. Chem. Phys. 1999;247:133–139. [Google Scholar]
- 91.Boukobza E, Sonnenfeld A, Haran G. Immobilization in Surface-Tethered Lipid Vesicles as a New Tool for Single Biomolecule Spectroscopy. J. Phys. Chem. B. 2001;105:12165–12170. [Google Scholar]
- 92.Okumus B, Wilson TJ, Lilley DMJ, Ha T. Vesicle Encapsulation Studies Reveal that Single Molecule Ribozyme Heterogeneities Are Intrinsic. Biophys. J. 2004;87:2798–2806. doi: 10.1529/biophysj.104.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benitez JJ, Keller AM, Chen P. Nanovesicle Trapping for Studying Weak Protein Interactions by Single-Molecule FRET. Meth. Enzymol. 2010;472:41–60. doi: 10.1016/S0076-6879(10)72016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Benitez JJ, Keller AM, Ochieng P, Yatsunyk LA, Huffman DL, Rosenzweig AC, Chen P. Probing Real-time Transient Metallochaperone-Target Protein Interactions at the Single-Molecule Level with Nanovesicle Trapping. J. Am. Chem. Soc. 2008;130:2446–2447. doi: 10.1021/ja7107867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Benitez JJ, Keller AM, Huffman DL, Yatsunyk L, Rosenzweig AC, Chen P. Relating Dynamic Protein Interactions of Metallochaperones with Metal Transfer at the Single-Molecule Level. Faraday Discuss. 2011;148:71–82. doi: 10.1039/c004913a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keller AM, Benitez JJ, Klarin D, Zhong L, Goldfogel M, Yang F, Chen T-Y, Chen P. Dynamic Multi-Body Protein Interactions Suggest Versatile Pathways for Copper Trafficking. J. Am. Chem. Soc. 2012;134:8934–8943. doi: 10.1021/ja3018835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Portnoy ME, Rosenzweig AC, Rae TD, Huffman DL, O'Halloran TV, Culotta VC. Structure-Function Analyses of the ATX1 Metallochaperone. J. Biol. Chem. 1999;274:15041–15045. doi: 10.1074/jbc.274.21.15041. [DOI] [PubMed] [Google Scholar]
- 98.Lutsenko S, LeShane ES, Shinde U. Biochemical Basis of Regulation of Human Copper-Transporting ATPase. Arch. Biochem. Biophys. 2007;463:134–148. doi: 10.1016/j.abb.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gonzalez-Guerrero M, Arguello JM. Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5992–5997. doi: 10.1073/pnas.0711446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu CC, Rice WJ, Stokes DL. Structure of a copper pump suggests a regulatory role for its metal-binding domain. Structure. 2008;16:976–985. doi: 10.1016/j.str.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gourdon P, Liu XY, Skjorringe T, Morth JP, Moller LB, Pedersen BP, Nissen P. Crystal structure of a copper-transporting PIB-type ATPase. Nature. 2011;475:59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- 102.Leshane ES, Shinde U, Walker JM, Barry AN, Blackburn NJ, Ralle M, Lutsenko S. Interactions between copper-binding sites determine the redox status and conformation of the regulatory N-terminal domain of ATP7B. J. Biol. Chem. 2010;285:6327–6336. doi: 10.1074/jbc.M109.074633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsivkovskii R, MacArthur BC, Lutsenko S. The Lys1010-Lys1325 fragment of the Wilson's disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J. Biol. Chem. 2001;276:2234–2242. doi: 10.1074/jbc.M003238200. [DOI] [PubMed] [Google Scholar]
- 104.DiDonato M, Hsu HF, Narindrasorasak S, Que L, Jr., Sarkar B. Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry. 2000;39:1890–1896. doi: 10.1021/bi992222j. [DOI] [PubMed] [Google Scholar]
- 105.Walker JM, Huster D, Ralle M, Morgan CT, Blackburn NJ, Lutsenko S. The N-Terminal Metal-Binding Site 2 of the Wilson's Disease Protein Play a Key Role in the Transfer of Copper from Atox1. J. Biol. Chem. 2004;279:15376–15384. doi: 10.1074/jbc.M400053200. [DOI] [PubMed] [Google Scholar]
- 106.Rosenzweig AC, Argüello JM. Toward a Molecular Understanding of Metal Transport by P1B-Type ATPases. Curr. Top. Membr. 2012;69:113–136. doi: 10.1016/B978-0-12-394390-3.00005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen P, Andoy NM. Single-Molecule Fluorescence Studies from A Bioinorganic Perspective. Inorg. Chim. Acta. 2008;361:809–819. [Google Scholar]
- 108.Erker W, Lippitz M, Basche T, Decker H. Toward oxygen binding curves of single respiratory proteins. Micron. 2004;35:111–113. doi: 10.1016/j.micron.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 109.Erker W, Sdorra S, Basche T. Detection of Single Oxygen Molecules with Fluorescence-Labeled Hemocyanins. J. Am. Chem. Soc. 2005;127:14532–14533. doi: 10.1021/ja053720k. [DOI] [PubMed] [Google Scholar]
- 110.Schmauder R, Librizzi F, Canters GW, Schmidt T, Aartsma TJ. The Oxidation State of a Protein Observed Molecule-by-Molecule. ChemPhysChem. 2005;6:1381–1386. doi: 10.1002/cphc.200400628. [DOI] [PubMed] [Google Scholar]
- 111.Elmalk AT, Salverda JM, Tabares LC, Canters GW, Aartsma TJ. Probing redox proteins on a gold surface by single molecule fluorescence spectroscopy. J. Chem. Phys. 2012;136:235101. doi: 10.1063/1.4728107. [DOI] [PubMed] [Google Scholar]
- 112.Kinoshita M, Kamagata K, Maeda A, Goto Y, Komatsuzaki T, Takahashi S. Development of a technique for the investigation of folding dynamics of single proteins for extended time periods. Proc. Natl. Acad. Sci. USA. 2007;104:10453–10458. doi: 10.1073/pnas.0700267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kiel A, Kovacs J, Mokhir A, Krämer R, Herten DP. Direct monitoring of formation and dissociation of individual metal complexes by single-molecule fluorescence spectroscopy. Angew. Chem. Int. Ed. 2007;46:3363–3366. doi: 10.1002/anie.200604965. [DOI] [PubMed] [Google Scholar]
- 114.Kuznetsova S, Zauner G, Aartsma T, Engelkamp H, Hatzakis N, Rowan AE, Nolte RJM, Christianen PCM, Canters GW. The Enzyme Mechanism of Nitrite Reductase Studied at Single-Molecule Level. Proc. Natl. Acad. Sci. USA. 2008;105:3250–3255. doi: 10.1073/pnas.0707736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goldsmith RH, Tabares LC, Kostrz D, Dennison C, Aartsma TJ, Canters GW, Moerner WE. Redox cycling and kinetic analysis of single molecules of solution-phase nitrite reductase. Proc. Nat. Acad. Sci. USA. 2011;108:17269–17274. doi: 10.1073/pnas.1113572108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pugh RA, Honda M, Spies M. Ensemble and single-molecule fluorescence-based assays to monitor DNA binding, translocation, and unwinding by iron–sulfur cluster containing helicases. Methods. 2010;51:313–321. doi: 10.1016/j.ymeth.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Edman L, FiSldes-Papp Z, Wennmalm S, Rigler R. The fluctuating enzyme: a single molecule approach. Chem. Phys. 1999;247:11–22. [Google Scholar]
- 118.Edman L, Rigler R. Memory landscapes of single-enzyme molecules. Proc. Natl. Acad. Sci. USA. 2000;97:8266–8271. doi: 10.1073/pnas.130589397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cogdell RJ, Gall A, Kohler J. The Architecture and Function of the Light-Harvesting Apparatus of Purple Bacteria: From Single Molecules to in vivo Membranes. Q. Rev. Biophys. 2006;39:227–324. doi: 10.1017/S0033583506004434. [DOI] [PubMed] [Google Scholar]
- 120.Saga Y, Tamiaki H. Fluorescence Spectroscopy of Single Photosynthetic Light-Harvesting Supramolecular Systems. Cell Biochem. Biophys. 2004;40:149–165. doi: 10.1385/CBB:40:2:149. [DOI] [PubMed] [Google Scholar]
- 121.Rutkauskas D, Cogdell RJ, van Grondelle R. Conformational Relaxation of Single Bacterial Light-Harvesting Complexes. Biochemistry. 2006;45:1082–1086. doi: 10.1021/bi0524159. [DOI] [PubMed] [Google Scholar]
- 122.Loos D, Cotlet M, de Schryver F, Habuchi S, Hofkens J. Single-Molecule Spectroscopy Selectively Probes Donor and Acceptor Chromophores in the Phycobiliprotein Allophcocyanin. Biophys. J. 2004;87:2598–2608. doi: 10.1529/biophysj.104.046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.de Ruijter WP, Oellerich S, Segura JM, Lawless AM, Papiz M, Aartsma TJ. Observation of the Energy-Level Structure of the Low-Light Adapted B800 LH4 Complex by Single-Molecule Spectroscopy. Biophys. J. 2004;87:3413–3420. doi: 10.1529/biophysj.104.044719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tietz C, Jelezko F, Gerken U, Schuler S, Schubert A, Rogl H, Wrachtrup J. Single Molecule Spectroscopy on the Light-Harvesting Complex II of Higher Plants. Biophys. J. 2001;81:556–562. doi: 10.1016/S0006-3495(01)75722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van Oijen AM, Ketelaars M, Kohler J, Aartsma TJ, Schmidt J. Unraveling the Electronic Structure of Individual Photosynthetic Pigment-Protein Complexes. Science. 1999;285:400–402. doi: 10.1126/science.285.5426.400. [DOI] [PubMed] [Google Scholar]
- 126.Bopp MA, Jia Y, Li L, Cogdell RJ, Hochastrasser RM. Fluorescence and Photobleaching Dynamics of Single Light-Harvesting Complexes. Proc. Natl. Acad. Sci. USA. 1997;94:10630–10635. doi: 10.1073/pnas.94.20.10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. The Fluorescent Toolbox for Assessing Protein Location and Function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 128.Haugland RP. The Handbook: A Guide to Fluorescent Probes and Labeling Technologies. Invitrogen Corp; 2005. [Google Scholar]
- 129.Ha T. Single-Molecule Fluorescence Resonance Energy Transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- 130.Hansen AG, Zhang J, Christensen HEM, Welinder AC, Wackerbarth H, Ulstrup J. Electron transfer and redox metalloenzyme catalysis at the single-molecule level. Isr. J. Chem. 2004;44:89–100. [Google Scholar]
- 131.Zhang J, Chi Q, Hansen AG, Jensen PS, Salvatore P, Ulstrup J. Interfacial electrochemical electron transfer in biology – Towards the level of the single molecule. FEBS Lett. 2012;586:526–535. doi: 10.1016/j.febslet.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 132.Alessandrini A, Corni S, Facci P. Unraveling Single Metalloprotein Electron Transfer by Scanning Probe Techniques. Phys. Chem. Chem. Phys. 2006;8:4383–4397. doi: 10.1039/b607021c. [DOI] [PubMed] [Google Scholar]
- 133.Bonanni B, Andolfi L, Bizzarri AR, Cannistraro S. Functional Metalloproteins Integrated with Conductive Substrates: Detecting Single Molecules and Sensing Individual Recognition Events. J. Phys. Chem. B. 2007;111:5062–5075. doi: 10.1021/jp070035m. [DOI] [PubMed] [Google Scholar]
- 134.Wigginton NS, Rosso KM, Hochella MF., Jr. Mechanisms of Electron Transfer in Two Decaheme Cytochromes from a Metal-Reducing Bacterium. J. Phys. Chem. B. 2007;111:12857–12864. doi: 10.1021/jp0718698. [DOI] [PubMed] [Google Scholar]
- 135.Pia EAD, Macdonald JE, Elliott M, Jones DD. Direct Binding of a Redox Protein for Single-Molecule Electron Transfer Measurements. Small. 2012;8:2341–2344. doi: 10.1002/smll.201102416. [DOI] [PubMed] [Google Scholar]
- 136.Bonanni B, Kamruzzahan ASM, Bizzarri AR, Rank C, Gruber HJ, Hinterdorfer P, Cannistraro S. Single Molecule Recognition between Cytochrome C 551 and Gold-Immobilized Azurin by Force Spectroscopy. Biophys J. 2005;89:2783–2791. doi: 10.1529/biophysj.105.064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bonanni B, Bizzarri AR, Cannistraro S. Optimized Biorecognition of Cytochrome c 551 and Azurin Immobilized on Thiol-Terminated Monolayers Assembled on Au(111) Substrates. J. Phys. Chem. B. 2006;110:14574–14580. doi: 10.1021/jp0610315. [DOI] [PubMed] [Google Scholar]
- 138.Tarantaa M, Bizzarria AR, Cannistraro S. Probing the interaction between p53 and the bacterial protein azurin by single molecule force spectroscopy. J. Mol. Recognit. 2008;21:63–70. doi: 10.1002/jmr.869. [DOI] [PubMed] [Google Scholar]
- 139.Bizzarri AR, Santini S, Coppari E, Bucciantini M, Agostino SD, Yamada T, Beattie CW, Cannistraro S. Interaction of an anticancer peptide fragment of azurin with p53 and its isolated domains studied by atomic force spectroscopy. Int. J. Nanomed. 2011;6:3011–3019. doi: 10.2147/IJN.S26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yersin A, Osada T, Ikai A. Exploring Transferrin-Receptor Interactions at the Single-Molecule Level. Biophys J. 2008;94:230–240. doi: 10.1529/biophysj.107.114637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ikuta K, Yersin A, Ikai A, Aisen P, Kohgo Y. Characterization of the Interaction between Diferric Transferrin and Transferrin Receptor 2 by Functional Assays and Atomic Force Microscopy. J. Mol. Biol. 2010;397:375–384. doi: 10.1016/j.jmb.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zheng P, Li H. Direct Measurements of the Mechanical Stability of Zinc-Thiolate Bonds in Rubredoxin by Single-Molecule Atomic Force Microscopy. Biophys. J. 2011;101:1467–1473. doi: 10.1016/j.bpj.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zheng P, Takayama S.-i. J., Mauk AG, Li H. Hydrogen Bond Strength Modulates the Mechanical Strength of Ferric-Thiolate Bonds in Rubredoxin. J. Am. Chem. Soc. 2013;134:4124–4131. doi: 10.1021/ja2078812. [DOI] [PubMed] [Google Scholar]
- 144.Zhou X, Andoy NM, Liu G, Choudhary E, Han K-S, Shen H, Chen P. Quantitative Super-resolution Imaging Uncovers Reactivity Patterns on Single Nanocatalysts. Nature Nanotech. 2012;7:237–241. doi: 10.1038/nnano.2012.18. [DOI] [PubMed] [Google Scholar]
- 145.Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 147.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature Meth. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]