

Summary
The Rho family of guanosine triphosphatases (GTPases) is composed of members of the Ras superfamily of proteins. They are GTP-bound molecules with a modest intrinsic GTPase activity that can be accelerated upon activation/localization of specialized guanine nucleotide exchange factors. Members of this family act as molecular switches and are required for coordinated cytoskeletal rearrangements that are crucial in a set of specialized functions of mammalian stem cells. These functions include self-renewal, adhesion, and migration. Mouse gene-targeting studies have provided convincing evidence of the indispensable and dispensable roles of individual members of the Rho GTPase family and the putative upstream and downstream mediators in stem cell-specific functions. The role of Rho GTPases and related signaling pathways previously seen in other cell types and organisms have been confirmed in mammalian hematopoietic stem cells (HSCs), and new signaling pathways and unexpected functions unique to HSCs have been identified and dissected. This review summarizes our current understanding of the role of Rho family of GTPases on HSC and progenitor activity through cytoskeleton-mediated signaling pathways, providing insight on relevant signaling pathways that regulate mammalian stem cell self-renewal, adhesion, and migration.
Keywords: Rac, Cdc42, Rho, stem cell, migration, self-renewal
Introduction to the Rho family of GTPases and their regulation
Rho GTPases (Ras homologue) belong to a family of regulatory small guanosine triphosphate (GTP) binding proteins in the Ras super family. In 1985, the Rho gene was first isolated from the abdominal ganglia of marine snail Aplysia californica. Subsequently, RhoA, RhoB, and RhoC isoforms were identified in humans (1). In mammals, the Rho GTPase family consists of 22 members, which are further subdivided into 5 subfamilies, Rho, Rac, Cdc42, Rnd, and RhoBTB, based on their sequence identity, domain structure, and function. Similar to their cousins in the Ras, Rab, Arf, and Ran families of small GTPases, most RhoGTPases are molecular switches that cycle between active GTP-bound and inactive guanosine diphosphate (GDP)-bound conformations (2) and control cytoskeleton organization and rearrangements. Their role in the formation of filopodia, lamellipodia, membrane ruffles, and stress fibers was described by Hall and others (3, 4). There are three regulators of RhoGTPase activity. First, the conversion from the inactive GDP-bound form to the active GTP-bound form is catalyzed by upstream guanine nucleotide exchange factors (GEFs)(5), and the activated Rho GTPases interact with a wide variety of effector proteins to a carry out downstream biological functions. Second, the intrinsic GTPase activity of Rho GTPases is stimulated by GTPase activating proteins (GAP) and results in the hydrolysis of the bound GTP to GDP and consequent inactivation (6). The third type of regulators of Rho GTPases, the guanine nucleotide dissociation inhibitors (GDIs), interact and stabilize the GDP-bound form to prevent spontaneous activation. The interaction of GDIs with the prenylated form of Rho GTPases plays a critical role in the regulation of the cytosolic versus membrane distribution of Rho-GDP, and also protects them from degradation (7). Out of the classically regulated RhoGTPases, the best studied in hematopoietic stem cells are members of the Rho, Rac, and Cdc42 families.
The human genome contains more than 60 GEFs and nearly 80 GAPs, far more than the number of their substrates (22 Rho GTPases). The fact that the number of regulators so far exceeds the substrates indicates that Rho GTPases are specifically and tightly regulated in a spatio-temporal manner (5, 6, 8). Dbl (diffuse B-cell lymphoma) was the first mammalian GEF homologue to be discovered and contains a 180 amino acid conserved region [Dbl homology (DH) domain] with a sequence that is similar to the yeast Cdc42 activator Cdc24 (9–11). The conserved DH domain of Rho GEF is necessary for its catalytic activity (12). RhoGEF is characterized by the presence of a DH domain followed by a plekstrin homology domain (PH domain), wherein the PH domain interacts with phosphorylated phosphoinositides (PIPs) and enhances the catalytic activity of the DH domain (13–15).
RhoGTPase activating proteins (RhoGAP) contain a 150 amino acid GAP domain, and the GAP domain is essential to stimulate the intrinsic GTPase activity of Rho GTPases (16). The breakpoint cluster region (Bcr) has been identified as the first RhoGAP isoform (17). With few exceptions, Rho GEFs and Rho GAPs are described as the activators and inhibitors, respectively, of one or more Rho GTPase isoforms. The unconventional Rho GEF of the Dock180 family lacks the DH-PH domain and requires additional cofactors for GDP/GTP-exchange in Rac GTPases (18, 19). The GAP domain of the p85 subunit of PI3K lacks GAP activity, therefore phosphatidylinositol-3-kinase is activated when p85 binds through its GAP domain with Cdc42 and Rac (20). Rho GAP n-chimaerin binds to Rac1 and Cdc42 and instead, acts as a positive regulator for lamellipodia and filopodia formation (21). In addition to GEF/GAP regulation isoprenylation, phosphorylation, oxidation of conserved cysteine residue, ubiquitination, transglutamination and AMPylation, the spatio-temporal location crucially regulates the expression and activity of the Rho GTPases (22–25). In addition, Rho GTPase expression has recently been shown to be regulated by various microRNAs (23). In the GTP-bound active state, RhoGTPases interact with more than 50 effector proteins that includes serine/threonine kinases, tyrosine kinases, lipases, oxidases, and scaffold proteins to regulate wide varieties of processes such as actin cytoskeletal rearrangement, microtubule dynamics, cell shape regulation, cell adhesion, intracellular signaling cascades, endocytosis, vesicle trafficking, G1 cell cycle progression, oncogenesis, gene transcription, enzymatic activities, cell polarity, and asymmetric cell division (2, 25–35).
Some members of the Rho GTPase family are not conventional and do not follow the same types of regulation. First, not all the Rho GTPases have intrinsic GTPase activity. The three Rnd isoforms and RhoH lack GTPase activity due to amino acid substitution in the GTPase domain, and exist in constitutively activated GTP bound forms (36). Since these proteins lack GEF/GAP regulation, it has been suggested that they are regulated by tissue specific differential expression (37, 38). Second, some members contain other structure domains that make them uniquely regulated. The Rho BTB members of the RhoGTPase family are unconventional because they contain a large C-terminal extension with two protein-protein interaction domains called BTB domains (from bric-à-brac, tramtrack, and broad complex in Drosophila melanogaster). In addition, Rho BTB family members also lack the C-terminal isoprenylation motif (CAAX) for the prenylation at the cysteine residue (39).
Basis of cytoskeleton regulation by Rho GTPases
Although the Rho gene was isolated for the first time in 1985, it was not until 1992 when two landmark publications by Ridley and colleagues (3, 4) opened a new avenue for cell biologists and biochemists to explore the role of RhoGTPases. Thereafter, Rho GTPases gained equal attention to their cousins in the Ras family (3, 4). Previously, the bacterial toxin C3 exotransferase was shown to inhibit RhoA activity by ADP rybosylation and induce the loss of actin stress fibers in Vero cells (40, 41). By using bacterial toxin C3 transferase exoenzyme (derived from Clostridium botulinum that inhibits GTPase activity of RhoA, RhoB and RhoC through ADP-ribosylation) and microinjecting RhoA, Rac1, and Rac1 dominant negative mutant in serum starved NIH 3T3 cells, Ridley and colleagues (3, 4, 42) established that RhoA induces the formation of actin stress fibers and focal adhesion assembly, and that Rac1 regulates the formation of membrane protrusions called lamelipodia by a localized increase in actin polymerization. Microinjection of Rac1 initially induces membrane ruffle followed by stress fiber formation which provided clues regarding cross talk between Rac1 and RhoA activation (4). They also demonstrated that lysophosphatidic acid (LPA), an external factor found in serum, induced RhoA activation in order to modulate the intracellular cytoskeletal rearrangement. Later Cdc42, another well studied member of the Rho GTPase family, was shown to induce the formation of spike-like extensions on the plasma membrane called filopodia (43, 44).
RhoA induces the actin stress fiber formation
The interaction of growth factors and extracellular matrix (ECM) adhesion molecules with cellular receptors leads to the activation of Rho, Rac, and Cdc42. The activated Rho GTPases interact with various effector proteins, which regulate the rearrangement of the actin microfilament system and results in the formation of the acto-myosin complex, protruding actin filament rich lamellipodia and spike-like projection of actin structures called filopodia (26, 45) (Fig. 1A). The GTP loaded RhoA interacts with and activates its well-known effector protein Rho kinase (ROCK), a serine threonine kinase. The activated ROCK phosphorylates the myosin light chain phosphatase (MLCP), which leads to the inactivation of MLCP and consequent increase in the level of p-MLC inside the cell (46, 47). Rho kinase can also directly phosphorylate the protein kinase N1 (PKN) family of serine/threonine kinases and eventually activate MLC kinase, potentially increasing the level of pMLC (48, 49). The pMLC is a positive regulator of the acto-myosin network. An increased level of pMLC inside the cell enhances acto-myosin contractile tension and ultimately induces the formation of actin stress fibers. Actin depolymerizing factor (ADF/cofilin) acts as a negative regulator of actin stress fibers by depolymerizing and severing the existing actin filaments. ADF/cofilin gets inactivated through LIM kinase (LIMK) mediated phosphorylation. Therefore, activated RhoA induces the actin stress formation by inhibiting ADF/cofilin through the ROCK mediated activation of LIM kinase (LIMK)(50, 51). RhoA GTPase also targets citron kinase at the cleavage furrow during cytokinesis. The activated citron kinase induces MLC phosphorylation that eventually increases acto-myosin contractile ring tension, which facilitates cell separation (52). In addition to the RhoA/ROCK/MLCP /pMLC, RhoA/ROCK/LIMK/pcofilin, and the RhoA/citron kinase/pMLC pathways, activated RhoA also induces actin polymerization through the activation of de novo actin polymerizing agent formin or mDia1 a mammalian homologue of Drosophila diaphanous (53). Formins/mDia are a family of diaphanous-related proteins known to stimulate actin polymerization by capping the barbed end of the actin filament (54, 55). The interaction of GTP-bound RhoA activates formin, exposing the FH2 domain (formin homology domain 2) for its interaction with the barbed end of actin filament, this interaction facilitates actin polymerization at the barbed end and results in a net elongation of actin filament (56).
Fig. 1. Schematic representation of the molecular pathways of the regulation of actin cytoskeleton remodeling through the interaction of activated RhoGTPases with effector proteins.
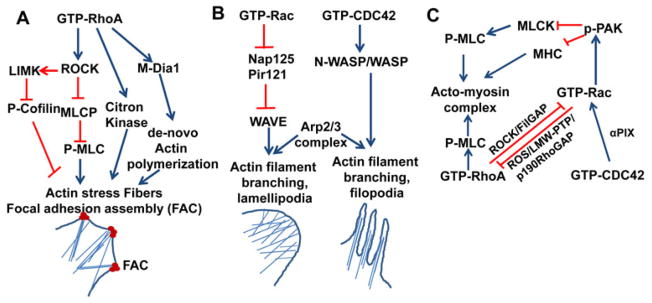
(A). Activated RhoA through effector protein ROCK phosphorylates and inhibits myosin light chain phosphatase (MLCK) activity that leads to the elevation of p-MLC level inside the cells. ROCK phosphorylates and activates LIM kinase (LIMK). LIMK in turn phosphorylates and inactivates cofilin. Increased levels of p-MLC and p-cofilin favor actin stress fiber formation. RhoA activated ROCK phosphorylates and activates citron kinase. Activated citron kinase phosphorylates MLC and increased the bundling of actin filaments at the cleavage furrow during cytokinesis. GTP coupled RhoA interacts and activates the auto inhibited formin or mDia1 that induces the de novo actin polymerization at the barbed end of F-actin. (B). Activated Rac and Cdc42 activate WAVE and WASP, respectively. Activated WAVE and WASP through their interaction with Arp2/3 complex initiate’s actin polymerization on the side of existing actin filament producing branched actin network that induces the formation of lamellipodia and filopodia at the leading edge of migrating cells. (C). Cross talk between Rho GTPases. Cdc42 through its interaction with α-PIX cooperates with Rac at the leading edge. Activated Rac induces ROS generation and ROS inhibits LMW-PTP, and thereby upregulates p190RhoGAP activity that in turn inhibits RhoA. Activated Rac through its effector p21-activated protein kinase (PAK) inhibits MLCK that results in decreased pMLC level. PAK phosphorylates and inactivates MHC. The combined effect of the reduction of pMLC level and phosphorylation of MHC negatively regulates the actin stress fiber formation. Rho/ROCK mediated activation of FilGAP inhibits Rac activity, as FilGAP has Rac specific GAP functional GAP domain. FAC, focal adhesion complex; ROCK, Rho Kinase; p-MLC, phosphorylated myosin light chain; MHC, myosin heavy chain; mDia1, Drosophila homologue of mammalian diaphanous related protein, α-PIX, PAK interacting exchange factor. Blue arrows denote activation pathways. Red blunt lines denote inhibitory pathways.
Rac and Cdc42 generates brached actin network at the leading edge
The molecular mechanism of the lamelipodial and filopodial membrane protrusion generated by Rac and Cdc42, respectively, through the Arp2/3 complex mediated initiation of actin polymerization and branching has been well established (57)(Fig. 1B). The Arp2/3 complex is an heptameric actin nucleation complex found in all eukaryotic cells which plays a decisive role in the nucleation and branching of existing actin filaments (57). Cdc42 activates Arp2/3 complex through its interaction with ubiquitously expressed N-WASP (Wiskott-Aldrich syndrome) and hematopoietic specific Wiskott-Aldrich syndrome protein (WASP) family proteins (58). The activation of WASP upon binding to Cdc42 induces the Arp2/3 mediated actin polymerization at the side of existing actin filaments and, thereby, generates the spike like projection called filopodia at the leading edge of a cell (59). Rac GTPase regulates the Arp2/3 complex mediated actin nucleation and polymerization through the activation of the WASP-family verprolin-homologous (WAVE) protein of the WASP family (59, 60). Unlike Cdc42, the activated Rac does not bind directly to WAVE/scar family of WASP proteins. The WAVE/scar proteins exist in an inactive complex along with Rac binding protein Nap125 and PIR121. The interaction of activated Rac GTPase with the inhibitors Nap125 and PIR121 releases WAVE enabling its interaction with the Arp2/3 complex to initiate actin polymerization and branching (61). Cdc42 and Rac also regulate actin branching in the area of protrusion through the interaction of PAK with the actin filament crosslinking protein, filamin (62). In addition to the Rac/Cdc42/WASP/WAVE/Arp2/3 complex pathway, Rac GTPases also activate phosphatidylinositol (P)I-5 kinase, which catalyzes the formation of PI-4,5-bisphosphate (PIP2) from PI-4-phosphate (PI4P) (63). PIP2 plays a critical role in uncapping the barbed end of the actin filament and induces actin polymerization at the barbed end (64).
RhoGTPase crosstalk modulates cytoskeleton rearrangement
The two significant publications by Ridley and Hall in 1992 (3, 4) described the potential cross communication between RhoA and Rac GTPases. Thereafter, the cross regulation of Rho GTPases has been studied extensively, particularly in migrating cells (Fig. 1C) (22). Cdc42 interacts with α-PIX (PAK interacting exchange factor), a Rac GEF, and thereby, cooperates with Rac GTPase in the formation of membrane protrusion at the leading edge of migrating cells (65). Through its interaction with NADPH oxidase complex, activated Rac induces reactive oxygen species (ROS) generation, and the resulting elevated ROS level inactivates the low molecular weight protein tyrosine phosphatase (LMW-PTP). The inactivation of LMW-PTP leads to the enhancement of p190 RhoGAP activity, which in turn negatively regulates RhoA activity at the leading edge of the migrating cells (66). Furthermore, RhoA activated ROCK phosphorylates and activates FilGAP, a filamin A binding Rho GAP protein having Rac specific GAP activity. The Rho/ROCK mediated activation of FilGAP cooperates with RhoA to antagonize the effect of Rac GTPase (67). This series of interactions indicates that in a migrating cell, the retracting tail has an elevated level of RhoA activity and the protrusive leading edge has increased levels of Rac and Cdc42 activities. Activation of Rac and RhoA has also been shown to differentially regulate the presence of peripheral membrane ruffles and pinocytic vesicles (68).
Rac GTPase negatively regulates the RhoA-induced stress fiber formation and focal adhesion assembly through its effector p21-activated kinases (PAK) at the leading edge. The activated PAK phosphorylates myosin light chain kinase (MLCK) and the phosphorylated MLCK is inactive towards phosphorylating myosin light chain resulting in reduced levels of pMLC inside the cells (69). At the same time, PAK also phosphorylates and inactivates myosin heavy chain and inhibits actin stress fibers formation (70). The reduced level of pMLC and the phosphorylation of myosin heavy chain both negatively regulate the acto-myosin contraction, leading to a loss of actin stress fibers and focal adhesion assembly.
Rho GTPases regulate microtubule dynamics and polarization
Microtubules are heteropolymers of α- and β-tubulin subunits and are highly polar in nature, with a minus end having depolymerizing activity usually anchored to centrosome and a plus end with both polymerizing and depolymerizing activity towards the cell cortex. Unlike in the actin cytoskeleton, the role of Rho GTPases in microtubule assembly is not very elaborate. The microtubular dynamic instability (polymerization versus depolymerization efficiency) at the plus end is a critical phenomenon for microtubule-associated functions and is regulated by plus end-binding proteins. The activation of Rho GTPases regulates the dynamic instability by interacting with the microtubule plus end-binding protein. The oncoprotein18 (OP18)/stathmin family of proteins enhances the depolymerization effect at the plus end. The phosphorylation and subsequent inactivation of OP18 by Cdc42/Rac GTPase effector PAK leads to the net elongation of the microtubule at the plus end (71). A Rac GEF of DOCK 180 family of proteins, DOCK-7, regulates the RacGTPase/PAK-mediated phosphorylation and subsequent inhibition of OP18 in the nascent axon, which is essential for microtubule-mediated axonal growth (72). ROCK-mediated phosphorylation and inactivation of collapsin response mediator protein 2 (CRMP-2), a positive regulator of microtubule assembly at the plus end in neurons, lead to microtubule destabilization and growth cone collapse (73). Cortical microtubule capture is regulated by the microtubule plus end-binding protein CLIP-170 (74). The CLIP-170 family proteins play a critical role in microtubule capture at the plus end. CLIP-170 interacts with IQGAP, a Cdc42/Rac1 effector scaffold protein, while concurrently interacting with the microtubule plus end. The activation of Cdc42/Rac1 at the leading edge increases the association of CLIP-170, IQGAP, and the microtubule plus end and regulates cell polarity (73). Adenomatous polyposis coli (APC), a microtubule binding protein, also interacts with IQGAP and forms a ternary complex with Rac1/Cdc42, IQGAP, and CLIP-170 at the leading edge of the migrating cells (75). The interaction of IQGAP with CLIP-170 and APC indicates the interconnection between the actin microfilament and the microtubule network in the maintenance of cell polarity and directional movement.
Rho GTPases in hematopoietic activity
Hematopoietic stem cells (HSCs) are the source of the billions of blood cells that are generated every day throughout the life, which is mediated through two critical features of HSCs, indefinite self-renewal and multi-lineage differentiation potential. The roles of Rho GTPases in the hematopoietic system were initially studied in mature hematopoietic cells such as neutrophils, macrophages, mast cells, and T cells. While a detailed explanation of the roles of RhoGTPases in differentiated hematopoietic cells is not an objective of this article, a brief summary of the most important findings on the Rho GTPase activity in hematopoietic cells relevant to HSC functions are described below. More information on specific functions of Rho GTPases in differentiated hematopoietic cells are summarized elsewhere (76).
During all 20th century, most of studies were carried out by using Rho GTPase inhibitors and/or by expressing ectopically or microinjecting the dominant negative and constitutively activated forms of the GTPases. The ADP ribosylation of RhoA by C3 transferase exoenzyme leads to defective actin polymerization of many hematopoietic cells, as shown at the leading edge of the formylpeptide (fLMP)-stimulated neutrophil, resulting in impaired motility (77–79). The inhibition of RhoA by C3 transferase also impairs macrophage motility, and integrin-mediated cell adhesion of leukocytes (80, 81). Using the ectopic expression of dominant negative and constitutively activated forms, Rac and Cdc42, respectively, have been implicated in the formation of lamellipodia and filopodia in macrophage (82, 83). Rac1 and Rac2 are important components and the critical regulators of the NADPH oxidase complex for superoxide production in phagocytic cells such as neutrophils and monocytes (84, 85). Although the activated forms of both Rac1 and Rac2 induce superoxide production, Rac2 has a many fold higher affinity for the NADPH oxidase complex than Rac1 (86). The Rac GTPase subfamily includes three highly homologous isoforms: Rac1, Rac2, and Rac3. Rac1 is the most studied and ubiquitously expressed Rac isoform, whereas Rac 2 expression is restricted to hematopoietic cells (87–89). Rac3 is also ubiquitously expressed with a predominant expression pattern in the central nervous system (90). Since a high degree of homology exists at the amino acid level among the three isoforms of Rac GTPases, the conclusions drawn from the studies that used ectopic expression or microinjection of dominant negative and constitutively activated forms could be misleading. These studies were followed by several elegant studies that illustrate the role of specific Rho GTPases through homologous recombination-mediated gene targeting in mice, either constitutively or conditionally. Robert et al. (91), using a Rac2-deficient murine model, demonstrated that Rac 2 regulates the chemotaxis and superoxide production of neutrophils. Neutrophils from Rac 2−/− mice had reduced chemotaxis and superoxide production associated with defective actin cytoskeleton remodeling and impaired chemoattractant-stimulated p42/44 and p38 mitogen-activated protein kinase (MAPK) activation. The reduced chemotaxis and superoxide formation is manifested in vivo in Rac2-deficient mice as reduced inflammatory peritoneal exudate formation and compromised host defense following pathogenic bacterial challenge. The mast cells derived from the bone marrow cells of Rac2−/− mice show disorganized actin cytoskeleton remodeling, which affects the adhesion of the mast cells to fibronectin, as well as movement and degranulation (92). Rac2 plays a crucial role in the survival and growth of mast cells through the growth factor-mediated activation of the PI3K/Akt pathway. The poor survival and proliferation of Rac2-deficient mast cells is accompanied with the upregulation of the Bcl2-associated pro-apoptotic protein BAD and the downregulation of the anti-apoptotic protein BclXL. Rac2 GTPase, a hematopoietic specific Rac isoform, plays a critical role in T-helper 1 (Th1) cell-mediated cellular immunity, where it regulates the interferon-γ (IFN-γ) production of Th1 cells through the activation of the nuclear factor-κB and p38-MAPK pathways (93). Applying the knowledge and technical tools used to dissect the roles of RhoGTPases in differentiated hematopoietic cells, we and others have been able to identify the mechanisms of control of specific functions of HSC/P through Rho GTPases.
Physiological and pharmacological control of HSC/P traffic through RhoGTPase activity
During ontogeny, HSCs are originated in the extra-embryonic yolk sac and para-aortic splanchnopleura/aorta gonad mesonephros (AGM) of the embryo proper, and then migrate to, lodge at, and proliferate in the fetal liver (94–98). During late gestation and early postnatal period, HSCs from the fetal liver migrate to the spleen and bone marrow (BM) niche, where they reside and produce all hematopoietic cells in adequate quantity throughout the life (99). The BM microenvironment plays a critical role in fine-tuning the balance between self-renewal and differentiation potential of HSCs (100–104). Although HSCs primarily reside in the BM microenvironment, peripheral blood (PB) also contains a minute fraction of HSCs (~1 circulating HSC per every 5,000 murine BM HSC/P) and progenitors (~1 circulating progenitor per 100 murine BM progenitors). At any given time, a dynamic flow of HSC/Ps exits the BM microenvironment and goes to the PB in a process called stem cell trafficking (105, 106). The traffic from BM to PB can be enhanced by cytokines, chemotherapy, and chemokine signaling antagonists, resulting in HSC/P mobilization. Pharmacological use of traffic enhancer drugs laid the foundation of harvesting transplantable HSC/Ps from the PB instead of BM for therapeutic use. The administration of cytokines, such as granulocyte colony-stimulating factor (G-CSF), stem cell factor (SCF), chemokine receptor inhibitors (e.g. AMD3100), epidermal growth factor receptor (EGFR) inhibitors, and cytotoxic drugs (e.g., cyclophosphamide) enhance the mobilization of HSC/Ps from the BM to the PB (106–109). After transplantation, intravenously injected HSC/Ps transmigrate from the PB to the medullary cavity of BM in a process called homing. The migration of HSC/Ps during ontogeny and in the adult life, the homing and engraftment in the BM microenvironment, and the self-renewal, proliferation, and differentiation potential are tightly regulated by both the intrinsic and environmental or extrinsic cues. The stromal derived factor (SDF-1α or CXCL12) produced by the BM stromal cells and BM endothelial cells, nestin-expressing perivascular mesenchymal stem cells (110), SCF produced and secreted by various cell types in the hematopoietic microenvironment, and the fibronectin in the ECM are all known to regulate HSC functions through their interaction with seven membrane span G-protein-coupled receptor CXCR4 (111–113), tyrosine kinase receptor c-Kit (114, 115), and adhesion receptor integrin (116–118), respectively, expressed on the HSCs. The interactions of these cytokines and adhesion molecules with their receptors on the HSC/Ps trigger the intrinsic signaling pathways, which in turn modulate the cytoskeletal rearrangement, migration, transcriptional activation, survival, and cell cycle progression. Rho GTPases are at the heart of these signaling pathways and regulate almost all HSC/P activities in one or more ways.
Rac GTPases regulate HSC/P homing, retention, and migration. Among the Rho family GTPases, the role of Rac and Cdc42 in HSC migration is well established. The interaction of CXCL12, SCF, EGF, and fibronectin with their respective receptors on the surface of the HSC/P, CXCR4, c-Kit, EGFR and integrin, triggers the activation of a GEF that in turn activates specific Rho GTPases (Fig. 2). Signal transduction through Rho GTPases translates into regulation of specific HSC/P functions. A summary of our current understanding of the specific activities of HSC/Ps regulated by Rho GTPases is described below and summarized in Fig. 3.
Fig. 2. The interaction stem cell factor (SCF), stromal derives factor 1α (SDF 1α), and adhesion molecule in the extracellular matrix (fibronectin) with their cellular receptor c-Kit, CXCR4, integrin, respectively, expressed on the hematopoietic stem cells (HSCs) triggers the activation of guanine nucleotide exchange factors.
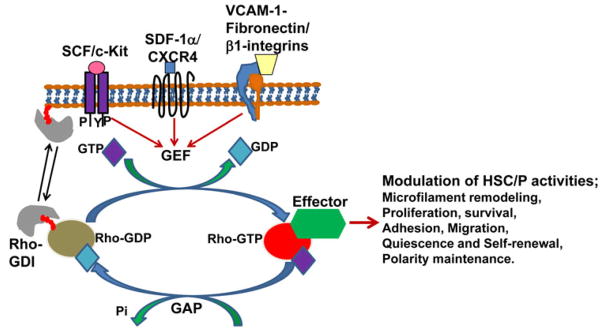
GEFs catalyze the exchange of GDP with GTP and activate Rho GTPase activity. The activated RhoGTPases through cytoskeletal remodeling and enzymatic pathway regulate the HSC/P activities such as quiescence, proliferation, survival, adhesion, migration, and self-renewal. GAPs inactivate Rho GTPase by stimulating the intrinsic GTPase activity to hydrolyze bound GTP. Rho GDI binds to the GDP bound and iso-prenylated form of Rho GTPase and regulates membrane versus cytosolic distribution of the latter. Red colored tail in Rho-GDP indicates iso-prenylation. GEF, guanine nucleotide exchange factor; GAP, GTPase activating protein; GDI, guanosine dissociation inhibitor.
Fig. 3. Roles of Rho GTPases in the regulation of HSC/P activities.

Rho GTPases such as Rac1, Rac2, Cdc42, RhoA, RhoH, and guanine nucleotide exchange factor Vav regulate hematopoietic stem and progenitor functions. Cdc42, Rac1, and RhoA control the self-renewal, and Cdc42 is required for cell cycle entry of quiescent HSCs. Rac1 and Cdc42 control proliferation, whereas Rac2 and Cdc42 regulate survival HSC/Ps. Rac1 and Cdc42 regulate HSC homing to the bone marrow microenvironment. Rac1, Rac2, and CDC42 control the retention of HSC in the bone marrow microenvironment. Vav1, a hematopoietic specific guanine nucleotide exchange factor, regulates localization of HSCs near nestin+ mesenchymal stem cells. The aged HSCs shows elevated level of Cdc42, and increased Cdc42 level is correlated with loss of the polarity and self-renewal activity. HSC/P, hematopoietic stem cells and progenitors.
Cxcl12-induced chemoattraction of HSC/Ps is mediated by both Rac1 and Rac2 and is at least partly mediated by the GEF Tiam1 (119). The integration of the SCF/c-Kit signaling to Rac1 and Rac2 is mediated through the GEF Vav, although the specific Vav (Vav1, Vav2, or Vav3) responsible for the signaling to Rac in hematopoietic cells remains unknown (120, 121). Rac GTPases also integrate signals from fibronectin/β1-integrins in HSC/Ps (122). Rac GTPases play a crucial role in homing, migration, interaction with the microenvironment, and in the long term engraftment potential of HSC/Ps. In fibroblasts and many other cell types, Rac GTPases are known to regulate the formation of membrane ruffle and lamellipodia mediated through actin cytoskeleton remodeling (2). In addition, Rac GTPases also activate a number of downstream kinase pathways in an actin-independent manner (2). Initial studies implicating the role of Rho GTPases in HSC/P activities used G-protein and Rho GTPase inhibitors. Pertussis toxin (PTX), a G-protein inhibitor, inhibits the Cxcl12 induced activation of Rac. However, it is ineffective towards Rac2 and Cdc42 activation. Using Clostridium difficile toxin B that inhibits Rho, Rac and Cdc42, it has been shown that Cxcl12 induced migratory potential of primitive hematopoietic cells is lower than the one exerted over more mature hematopoietic cells (123). However, these experiments lacked the ability to distinguish the effects on relatively high homologous Rac isoforms (92% homology between Rac 1 and Rac2) which, however, differ in crucial domains that modify their intracellular location (124). To establish the specific roles of the individual isoforms of Rho GTPases, our group and others have utilized homologous recombination-derived gene-targeted mutant mice deficient in Rac1, Rac2, Cdc42, RhoH, and specific GAP and GEF. In a Rac2−/− murine model, it has been shown that Rac2-deficient HSC/Ps are defective in actin cytoskeleton remodeling and α4β1-mediated adhesion to fibronectin or vascular cell adhesion protein 1 (VCAM 1)(125). The Rac2 −/− mice show increased numbers of circulating HSC/Ps in the PB, a reflection of reduced adhesion of HSC/P to the microenvironment. However Rac2−/− HSC/Ps show increased migration towards a Cxcl12 gradient, possibly through the compensatory up regulation of Rac1 and Cdc42 activities. This study suggested a critical role for Rac2 in the retention of HSC/Ps in the BM microenvironment and indicates the involvement of cross talk between Rac and Cdc42. Unlike Rac2-deficient mice, Rac1-deficient mice do not show defective mobilization of HSC/Ps. A group of publications from David Williams’ laboratory have dissected the signaling pathways regulated by Rac proteins. Sanchez-Aguilera et al. (126), in a gene targeted mouse model, have demonstrated that Vav1, an upstream GEF activator of Rac GTPases, plays a crucial role in the microenvironment near the location of nestin+ perivascular stromal cells. Vav1-deficient HSC/Ps show reduced Rac and Cdc42 activation and impaired short term and long term engraftment. Vav1−/− HSC/Ps are associated with reduced migration towards a Cxcl12 gradient and impaired circadian and pharmacologically induced mobilization. Downstream, failure of HSC homing, migration, assembly of the actin cytoskeleton, proliferation, and survival have been shown upon expression of dominant negative mutants of p21-activated kinase (Pak). Downregulation of PAK activity in HSC/P associated decreased phosphorylation of extracellular signal-regulated kinase (Erk), a mitogen-activated protein kinase (MAPK). Constitutive activation of Erk in HSC/P led to rescue of their proliferation in vitro and partial rescue of Pak-deficient HSC/P homing and engraftment in vivo. Using conditional knockout mice, Pak2−/− but not Pak1−/− HSPCs show reduced homing to the bone marrow and altered cell shape similar to Pak dominant negative mutant HSC/Ps and are completely defective in HSPCs engraftment, indicating that Pak2 plays a crucial role downstream Rac in HSC homing and migration (127).
An interesting signaling axis relevant in HSC migration/adhesion and controlled by Rac is the Rac/MAPK/Egr1/Klf5 axis (128, 129). Tyrosine kinase receptors, including c-kit (130) and EGFR (131), control HSC activity and signal through Rac (see above). Egr1 is a rapid response transcriptional regulator of stem cell migration that normally functions to promote HSC quiescence and retention in the niche, and its expression results from activation of multiple ligand/receptors including tyrosine kinase receptors and Wnt proteins. Egr1 expression is controlled by MAPK activation (132, 133) in response to EGFR signaling, which is a novel signaling pathway associated with HSC migration (109). Egr1 activity is crucial in the regulation of Klf5 expression, which is indispensable for adhesion, homing, lodging, and retention of HSC/P in the BM through Rab5-dependent post-translational regulation of β1/β2 integrins (129). A functional, integrative schema of how HSC migration could be controlled by extrinsic signals through Rac/Egr1/Klf5/β-integrins is found in Fig. 4A, B.
Fig. 4. Signaling pathways regulated by Rho GTPases controlling bone marrow HSC adhesion/homing.
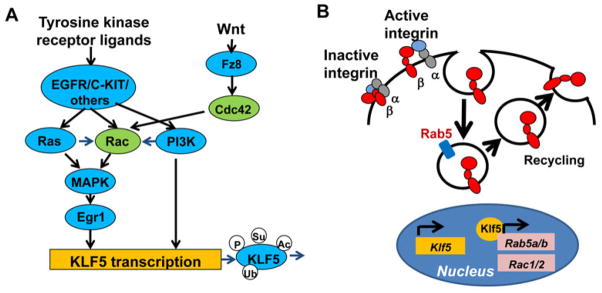
(A). Tyrosine kinase receptor ligands such as epidermal growth factor (EGF) and stem cell factor (SCF) through their interaction with their receptors (e.g. EGFR and c-Kit) activate small GTPases Rac and Ras in HSCs. Ras GTPase further activates Rac. Activated Ras and Rac, through MAPK pathway activation, induce the transcription of transcription factor Kruppel-like factor 5 (KLF5). EGF and SCF-mediated activation of receptor tyrosine kinase also induce KLF5 transcription through PI3K pathway activation. Activated PI3K also stimulates Rac activation. The Wnt ligand through its receptor Frizzled (Fz8) activates Cdc42. Cdc42 activates Rac and consequently leads to the induction of Klf5 transcription. Klf5 protein is post-translationally regulated through modifications such as phosphorylation, sumoylation, acetylation, and ubiquitination. (B). The transcription factor Klf5 regulates the expression of early endosome protein Rab5a and Rab5b, and as well, the expression of Rac. Rab 5, at the early endocytic compartment, regulates the endocytosis and recycling of active β1 and β2 integrin, which in turn regulate the adhesion and homing of HSC to the bone marrow.
Cdc42 and polarity in HSC activity
In many cell types, Cdc42 plays a vital role in directional migration by modulating the actin cytoskeleton at the leading edge (2). Cdc42 regulates the formation of filopodia in motile cells. Cdc42 also regulates the polarity of epithelial cells as well as stem cells during embryonic development and in adult life. In the hematopoietic system, Cdc42 critically regulates HSC/P trafficking and retention in the BM niche, as evidenced from studies using Cdc42 GAP−/− and Cdc42−/− murine models. Cdc42 GAP is a negative regulator of Cdc42, and therefore, HSC/Ps derived from Cdc42 GAP-deficient mice show elevated level of activated Cdc42. HSC/P lacking Cdc42 GAP exhibit disorganized F-actin structures, reduced adhesion, impaired directional migration, and defective short term and long term engraftment (134). Gene-targeted deletion of Cdc42 in mice leads to an increase in the HSC/P population in the BM due to enhanced cell cycle progression. However, the prototypic long term HSC (LT-HSC) population is reduced and is correlated with the impaired long term engraftment potential of Cdc42−/− HSC/Ps (134). Moreover, Cdc42 null HSC/P show gross defects in HSC/P activities, such as adhesion to fibronectin, migration in response to a Cxcl12 gradient, and homing to the BM endosteum niche. Xing et al. (135) have shown that the HSC/Ps in aged mice are hypersensitive to G-CSF-induced mobilization. The Cdc42 activity is elevated in aged HSCs when compared to young HSCs. Similarly to Cdc42GAP−/− mice, aged HSCs with enhanced Cdc42 activity show defective adhesion to the BM microenvironment. Also aged HSCs have a preferred myeloid commitment bias over a lymphoid lineage. Two significant publications have recently implicated Cdc42 in HSC self-renewal. First, Cdc42 has been found to be involved in a downstream noncanonical Wnt signaling pathway in the HSC niche (136). Flamingo (Fmi) and Frizzled (Fz) 8, members of noncanonical Wnt signaling, are expressed in HSC. Fmi regulates Fz8 distribution at the interface between HSCs and N-cadherin+ pre-osteoblasts, which favors the activation of the noncanonical Wnt signaling pathway. Under stress, this process shifts towards the predominance of canonical Wnt signaling. Non-canonical Wnt signaling mediated by Fz8 suppresses the signaling pathways induced by calcium release, nuclear factor of activating T cells (NFAT), and IFNγ through the activation of Cdc42, and antagonizes the canonical Wnt signaling in HSC. Second, increased Cdc42 activity in aged HSC is associated with the loss of cell polarity and a defective engraftment potential (137). The polarized distribution of tubulin and Cdc42 is impaired in HSC from aged mice. Pharmacological inhibition of Cdc42 with the rationally designed Cd42 inhibitor Casin reverses the loss of polarization and functionally rejuvenates aged HSCs (137).
Asymmetric cell division, a process where two cells of different fates are generated following division, plays a crucial role during embryonic development as well as in adult stem cell homeostasis (138)(Fig. 5A). The cell division is preceded by the asymmetric localization of polarity protein complexes such as apical PAR3-PAR6-aPKC complex, Crumb complex, and basal LgL/Dlg/Scribble complex (138). The role of Cdc42 in the establishment of asymmetry is well documented in invertebrates. Cdc42 interacts directly with PAR6, a scaffold protein of the apical polarity complex PAR3/PAR6/aPKC, through the CRIB domain, and activates the later (139–141). This is followed by PAR3/aPKC activation leading to the phosphorylation of Numb by aPKC, thereby causing the basal localization of Numb (140). The establishment of polarity by the Rho GTPases, in turn, determines positioning of the mitotic spindle in relation to the apico basal axis, and thereby ensures size asymmetry following cell division. Unlike in invertebrates or mammalian neural stem cells, asymmetric cell division in HSCs is poorly understood. In HSCs, centrosomic tubulin focalization is a major guide of pre-mitotic polarization (137). Wu et al. (142), using a Notch-driven enhanced green fluorescence protein (EGFP) reporter system in a real time microscopic imaging method, demonstrated that HSCs undergo both symmetric and asymmetric division and that this division is influenced by both intrinsic and extrinsic/environmental factors controlling Numb activation and localization. Two mammalian homologues of numb, numb and numblike, have also been proposed to physically interact with Notch1, inhibiting its function and participating in asymmetric cell division (142). Interestingly, two pathways of Numb activation, dependent on Musashi-2 and Hedgehog signaling, have been associated with chronic myelogenous leukemia stem cell proliferation (143, 144). A working hypothesis of the molecular mechanism of how Cdc42/PAR6 through PAR3/aPKC complex control HSC fate is described in Fig. 5B. This hypothesis remains to be tested in HSCs, since evidence has demonstrated that this complex may be dispensable. Using targeted animal models, Numb and Numb-like have been found to be dispensable in regulating mammalian HSC activity (145). Using an in vivo conditional gene-targeted murine model, our group has demonstrated that the constitutive or inducible deletion of atypical polarity complex proteins, aPKCζ and/or aPKCλ, does not affect the steady state or stress induced hematopoiesis (146). HSCs lacking aPKC show no defect in the tubulin polarization and all the HSC activities such as adhesion, migration, homing, and interaction with the microenvironment remain unaffected in absence of aPKC. Interestingly, loss of Lgl1, a component of the Scribble/Dlg/Lgl complex, results in increased HSC number in vivo without intrinsic change of the self-renewal capacity of each HSC (147). Loss of Lgl1 (147) and loss-of-function mutations of Lgl2 (148) have been associated to poor-prognosis myeloid leukemias.
Fig. 5. Mechanisms of symmetric versus asymmetric cell division in HSCs.
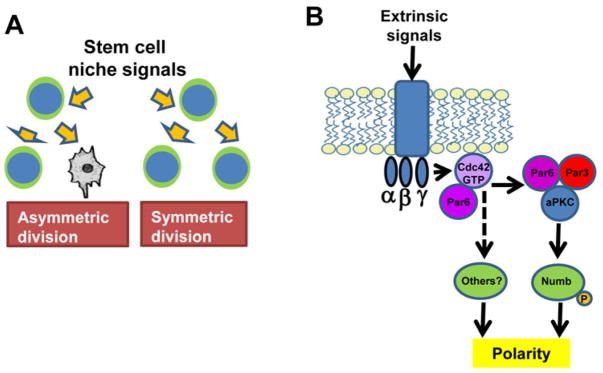
(A). Stem cells have the ability to self-renew and to differentiate into committed progenitors. Symmetric cell division results in a daughter cell that maintains self-renewal potential. Asymmetric cell division results in a daughter cell which loses stem cell self-renewal potential. The mechanisms that control the fate decision of asymmetric versus symmetric cell division depend on stem cell niche (microenvironment) derived signals. (B). Working hypothesis of the role of Cdc42/PAR6 in HSC fate. Cdc42 putatively interacts directly with PAR6 and activates PAR3/aPKC. Activated aPKC phosphorylates Numb, which differentially localizes to the basal side of the cell and the fate of daughter cells harboring Numb may potentially differ from their mother cells.
These observations indicate that further studies are needed to define conclusively the role of the polarity complex and asymmetric cell division in hematopoietic stem cell biology. Also the interaction of Cdc42 with its direct target PAR6, an adapter protein of the apical polarity complex, in HSCs remains unknown.
Self-renewal and long term engraftment potential of HSC/Ps Gu et al. (122) dissected the specific and overlapping role of Rac1 and Rac2 in primitive and mature hematopoietic cells. Although Rac1 and Rac2 are highly homologous proteins, they perform specific HSC/P functions. The Rac1-deficient HSC/Ps show severely impaired engraftment following transplantation into irradiated recipients, whereas the Rac 2−/− HSC/Ps show normal short term engraftment. Rac1−/− HSC/Ps has reduced proliferative potential associated with defective Erk1/2 activation, impaired growth factor-stimulated cyclin D1 induction, and p27KIP1 downregulation. However, Rac2 null HSC/Ps show defective integrin mediated adhesion to fibronectin in vitro, and this may explain the impaired BM retention and increased mobilization. Rac2−/− HSC/Ps show enhanced apoptosis due to defective growth factor-induced PI3K/Akt pathway activation. The Rac1 and Rac2 mutant phenotypes mentioned here become more severe in HSC/P lacking both Rac1 and Rac2, which suggests that Rac1 and Rac2 plays overlapping roles. The deletion of both Rac1 and Rac2 leads to a massive egress of HSC/P from BM, and re expression of Rac1, but not Rac2, in Rac1−/− and Rac2−/− HSC/Ps corrects the phenotypes. Other HSC/P activities such as proliferation, adhesion, and migration are severely impaired when both Rac1 and Rac2 are deleted. We found that the deletion of Rac1 after the successful engraftment of HSCs in an irradiated recipient does not affect the steady state hematopoiesis (149). The engraftment failure of Rac1−/− HSC/Ps is due to the impaired spatial localization to the BM endosteum niche, although homing to the medullary cavity remains unaffected. Also the interaction of Rac1−/− HSC/Ps with the BM microenvironment in vitro is markedly altered as evidenced by a decrease in cobblestone area forming cells (CAFC), a quantitative surrogate assay of stem cell activity that requires trans-stromal migration, proliferation, and survival in long-term BM stroma-dependent cultures. Rac2-deficient HSCs have normal short-term engraftment but display an abnormal interaction with the hematopoietic microenvironment, which leads to defective hematopoiesis in quantitative CAFC assays and long-term engraftment assays (150).
RhoH constitutes a distinct subtype of Rho GTPases that is GTPase-deficient and hematopoietic cell specific; it has been shown to modulate Rac1 signaling and function in primary HSC/Ps (151) and as such controls HSC migration and adhesion to the BM microenvironment. RhoH−/− HSC/Ps show increased chemotaxis in response to a Cxcl12 gradient. This migratory response is due to increased compensatory Rac1 activity and the translocation of the Rac1 protein to the cell membrane, where it colocalizes with cortical F-actin and lipid rafts. Conversely, overexpression of RhoH in HSC/P blocks the membrane translocation of Rac1 and impairs cortical F-actin assembly and chemotaxis.
Apparently controversial reports illustrate the role of RhoA. Ghiaur et al. (152) observed that the retrovirus-mediated ectopic expression of the dominant negative RhoA (RhoAN19) mutant enhanced stem cell self-renewal and engraftment potential. However, RhoA inhibition impaired the α4β1 and α5β1-mediated adhesion of HSC/P to fibronectin, and, as well, the migratory potential towards a Cxcl12 gradient. However in another study (153), elevated RhoA activity following the deletion of p190-B RhoGAP, a negative regulator of Rho GTPase, is advantageous for the self-renewal and long term engraftment potential of HSC/Ps. Recently, new conclusive data have arisen from the use of a RhoA conditional knockout mouse model (154). RhoA deficiency causes a multi-lineage hematopoietic failure associated with a blockage of hematopoiesis at the multipotent progenitor stage. RhoA−/− HSCs retain long-term engraftment potential but are defective in producing multipotent progenitors and differentiated blood cells. RhoA regulates actomyosin signaling, cytokinesis, and programmed necrosis of the hematopoietic progenitors. Loss of RhoA results in a mitotic failure of progenitors manifested by an accumulation of multinucleated cells due to failed cytokinesis and abscission of the cleavage furrow post-telophase and increased programmed necrosis.
Concluding remarks
Understanding the signals and modifications that regulate and alter stem cell activity is crucial for their manipulation and the development of novel therapies in the areas of transplantation and regenerative medicine. In the past decade, research into Rho GTPases shown that Rho GTPase activity is crucial for sensitive unique cytoskeletal functions that define stem cells, i.e. self-renewal, multipotential differentiation ability, migration, and adhesion, which eventually depend on a myriad of environmental cues that once integrated by the stem cells, results in a given functional output. The need for the rapid adaptation of stem cells to environmental cues requires the activation of fast acting signaling switches, represented by Rac, Cdc42, and Rho proteins. The use of genetic animal models has enabled us to dissect the relevant signaling pathways controlled by the Rho family of GTPases and identify rapidly acting cellular and molecular signaling pathways of regulation in stem cell self-renewal decisions, stem cell survival, and stem cell migration and adhesion to specific tissue environments. With the growing number of tools including novel in vivo forward and backward genetic approaches, sophisticated methods for single-cell genomics/proteomics/metabolomics and bioinformatic analysis, in vivo multiphoton microscopy, and in vivo fluorescence resonance energy transfer, and the use of state-of-the-art methods of stem cell reprogramming, the future will provide us a broader, more detailed picture on the role of these regulatory molecules in hematopoietic physiology and disease. The specific analysis of the cytoskeleton regulation of the growing field of cancer stem cells, resulting from disrupted hyerarchical organization and dysregulation of the physiological mechanisms of symmetric cell division, is likely to provide novel venues for therapeutic intervention.
References
- 1.Madaule P, Axel R. A novel ras-related gene family. Cell. 1985;41:31–40. doi: 10.1016/0092-8674(85)90058-3. [DOI] [PubMed] [Google Scholar]
- 2.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 3.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 4.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 6.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 7.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Tcherkezian J, Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol Cell. 2007;99:67–86. doi: 10.1042/BC20060086. [DOI] [PubMed] [Google Scholar]
- 9.Eva A, Aaronson SA. Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature. 1985;316:273–275. doi: 10.1038/316273a0. [DOI] [PubMed] [Google Scholar]
- 10.Bender A, Pringle JR. Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. Proc Natl Acad Sci U S A. 1989;86:9976–9980. doi: 10.1073/pnas.86.24.9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ron D, et al. A region of proto-dbl essential for its transforming activity shows sequence similarity to a yeast cell cycle gene, CDC24, and the human breakpoint cluster gene, bcr. New Biol. 1991;3:372–379. [PubMed] [Google Scholar]
- 12.Hart MJ, Eva A, Evans T, Aaronson SA, Cerione RA. Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature. 1991;354:311–314. doi: 10.1038/354311a0. [DOI] [PubMed] [Google Scholar]
- 13.Rebecchi MJ, Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson KM, et al. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell. 2000;6:373–384. doi: 10.1016/s1097-2765(00)00037-x. [DOI] [PubMed] [Google Scholar]
- 15.Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(Pt 1):1–18. [PMC free article] [PubMed] [Google Scholar]
- 16.Bax B. Domains of rasGAP and rhoGAP are related. Nature. 1998;392:447–448. doi: 10.1038/33040. [DOI] [PubMed] [Google Scholar]
- 17.Diekmann D, et al. Bcr encodes a GTPase-activating protein for p21rac. Nature. 1991;351:400–402. doi: 10.1038/351400a0. [DOI] [PubMed] [Google Scholar]
- 18.Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 19.Brugnera E, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 20.Zheng Y, Bagrodia S, Cerione RA. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J Biol Chem. 1994;269:18727–18730. [PubMed] [Google Scholar]
- 21.Kozma R, Ahmed S, Best A, Lim L. The GTPase-activating protein n-chimaerin cooperates with Rac1 and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol Cell Biol. 1996;16:5069–5080. doi: 10.1128/mcb.16.9.5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boulter E, Estrach S, Garcia-Mata R, Feral CC. Off the beaten paths: alternative and crosstalk regulation of Rho GTPases. FASEB J. 2012;26:469–479. doi: 10.1096/fj.11-192252. [DOI] [PubMed] [Google Scholar]
- 23.Liu M, Bi F, Zhou X, Zheng Y. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012;22:365–373. doi: 10.1016/j.tcb.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- 25.Wang HR, et al. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- 26.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 27.Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 28.Etienne-Manneville S. Cdc42--the centre of polarity. J Cell Sci. 2004;117:1291–1300. doi: 10.1242/jcs.01115. [DOI] [PubMed] [Google Scholar]
- 29.Joyce D, et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J Biol Chem. 1999;274:25245–25249. doi: 10.1074/jbc.274.36.25245. [DOI] [PubMed] [Google Scholar]
- 30.Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–957. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- 31.Coso OA, et al. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 32.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 33.Diekmann D, Abo A, Johnston C, Segal AW, Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science. 1994;265:531–533. doi: 10.1126/science.8036496. [DOI] [PubMed] [Google Scholar]
- 34.Takeya R, Sumimoto H. Molecular mechanism for activation of superoxide-producing NADPH oxidases. Mol Cells. 2003;16:271–277. [PubMed] [Google Scholar]
- 35.Cassimeris L. The oncoprotein 18/stathmin family of microtubule destabilizers. Curr Opin Cell Biol. 2002;14:18–24. doi: 10.1016/s0955-0674(01)00289-7. [DOI] [PubMed] [Google Scholar]
- 36.Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol. 1996;16:2689–2699. doi: 10.1128/mcb.16.6.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen SH, et al. Induced expression of Rnd3 is associated with transformation of polarized epithelial cells by the Raf-MEK-extracellular signal-regulated kinase pathway. Mol Cell Biol. 2000;20:9364–9375. doi: 10.1128/mcb.20.24.9364-9375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramos S, Khademi F, Somesh BP, Rivero F. Genomic organization and expression profile of the small GTPases of the RhoBTB family in human and mouse. Gene. 2002;298:147–157. doi: 10.1016/s0378-1119(02)00980-0. [DOI] [PubMed] [Google Scholar]
- 40.Chardin P, Boquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 1989;8:1087–1092. doi: 10.1002/j.1460-2075.1989.tb03477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol. 2005;3:397–410. doi: 10.1038/nrmicro1150. [DOI] [PubMed] [Google Scholar]
- 42.Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–1952. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- 45.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 46.Kimura K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 47.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amano M, et al. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science. 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe G, et al. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science. 1996;271:645–648. doi: 10.1126/science.271.5249.645. [DOI] [PubMed] [Google Scholar]
- 50.Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 51.Maekawa M, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 52.Madaule P, et al. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–494. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe N, et al. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chesarone MA, Goode BL. Actin nucleation and elongation factors: mechanisms and interplay. Curr Opin Cell Biol. 2009;21:28–37. doi: 10.1016/j.ceb.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- 56.Kursula P, et al. High-resolution structural analysis of mammalian profilin 2a complex formation with two physiological ligands: the formin homology 1 domain of mDia1 and the proline-rich domain of VASP. J Mol Biol. 2008;375:270–290. doi: 10.1016/j.jmb.2007.10.050. [DOI] [PubMed] [Google Scholar]
- 57.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 58.Rohatgi R, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 59.Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- 60.Machesky LM, Insall RH. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- 61.Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- 62.Vadlamudi RK, et al. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002;4:681–690. doi: 10.1038/ncb838. [DOI] [PubMed] [Google Scholar]
- 63.Carpenter CL, Tolias KF, Couvillon AC, Hartwig JH. Signal transduction pathways involving the small G proteins rac and Cdc42 and phosphoinositide kinases. Adv Enzyme Regul. 1997;37:377–390. doi: 10.1016/s0065-2571(96)00005-2. [DOI] [PubMed] [Google Scholar]
- 64.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 65.Baird D, Feng Q, Cerione RA. The Cool-2/alpha-Pix protein mediates a Cdc42-Rac signaling cascade. Curr Biol. 2005;15:1–10. doi: 10.1016/j.cub.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 66.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 67.Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol. 2006;8:803–814. doi: 10.1038/ncb1437. [DOI] [PubMed] [Google Scholar]
- 68.Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- 69.Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science. 1999;283:2083–2085. doi: 10.1126/science.283.5410.2083. [DOI] [PubMed] [Google Scholar]
- 70.van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat Cell Biol. 1999;1:242–248. doi: 10.1038/12068. [DOI] [PubMed] [Google Scholar]
- 71.Daub H, Gevaert K, Vandekerckhove J, Sobel A, Hall A. Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J Biol Chem. 2001;276:1677–1680. doi: 10.1074/jbc.C000635200. [DOI] [PubMed] [Google Scholar]
- 72.Watabe-Uchida M, John KA, Janas JA, Newey SE, Van Aelst L. The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron. 2006;51:727–739. doi: 10.1016/j.neuron.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 73.Fukata M, et al. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell. 2002;109:873–885. doi: 10.1016/s0092-8674(02)00800-0. [DOI] [PubMed] [Google Scholar]
- 74.Wu X, Jung G, Hammer JA., 3rd Functions of unconventional myosins. Curr Op Cell Biol. 2000;12:42–51. doi: 10.1016/s0955-0674(99)00055-1. [DOI] [PubMed] [Google Scholar]
- 75.Watanabe T, et al. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7:871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 76.Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, Zheng Y. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936–947. doi: 10.1182/blood-2009-09-198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koch G, Norgauer J, Aktories K. ADP-ribosylation of the GTP-binding protein Rho by Clostridium limosum exoenzyme affects basal, but not N-formyl-peptide-stimulated, actin polymerization in human myeloid leukaemic (HL60) cells. Biochem J. 1994;299 (Pt 3):775–779. doi: 10.1042/bj2990775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ehrengruber MU, Boquet P, Coates TD, Deranleau DA. ADP-ribosylation of Rho enhances actin polymerization-coupled shape oscillations in human neutrophils. FEBS Lett. 1995;372:161–164. doi: 10.1016/0014-5793(95)00880-i. [DOI] [PubMed] [Google Scholar]
- 79.Stasia MJ, Jouan A, Bourmeyster N, Boquet P, Vignais PV. ADP-ribosylation of a small size GTP-binding protein in bovine neutrophils by the C3 exoenzyme of Clostridium botulinum and effect on the cell motility. Biochem Biophys Res Commun. 1991;180:615–622. doi: 10.1016/s0006-291x(05)81110-6. [DOI] [PubMed] [Google Scholar]
- 80.Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110(Pt 6):707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 81.Jones GE, Allen WE, Ridley AJ. The Rho GTPases in macrophage motility and chemotaxis. Cell Adhes Commun. 1998;6:237–245. doi: 10.3109/15419069809004479. [DOI] [PubMed] [Google Scholar]
- 82.Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cox D, Chang P, Zhang Q, Reddy PG, Bokoch GM, Greenberg S. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J Exp Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–670. doi: 10.1038/353668a0. [DOI] [PubMed] [Google Scholar]
- 85.Knaus UG, Heyworth PG, Evans T, Curnutte JT, Bokoch GM. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science. 1991;254:1512–1515. doi: 10.1126/science.1660188. [DOI] [PubMed] [Google Scholar]
- 86.Dorseuil O, Reibel L, Bokoch GM, Camonis J, Gacon G. The Rac target NADPH oxidase p67phox interacts preferentially with Rac2 rather than Rac1. J Biol Chem. 1996;271:83–88. doi: 10.1074/jbc.271.1.83. [DOI] [PubMed] [Google Scholar]
- 87.Shirsat NV, Pignolo RJ, Kreider BL, Rovera G. A member of the ras gene superfamily is expressed specifically in T, B and myeloid hemopoietic cells. Oncogene. 1990;5:769–772. [PubMed] [Google Scholar]
- 88.Moll J, Sansig G, Fattori E, van der Putten H. The murine rac1 gene: cDNA cloning, tissue distribution and regulated expression of rac1 mRNA by disassembly of actin microfilaments. Oncogene. 1991;6:863–866. [PubMed] [Google Scholar]
- 89.Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- 90.Haataja L, Groffen J, Heisterkamp N. Characterization of RAC3, a novel member of the Rho family. J Biol Chem. 1997;272:20384–20388. doi: 10.1074/jbc.272.33.20384. [DOI] [PubMed] [Google Scholar]
- 91.Roberts AW, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 92.Yang FC, et al. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity. 2000;12:557–568. doi: 10.1016/s1074-7613(00)80207-1. [DOI] [PubMed] [Google Scholar]
- 93.Li B, et al. Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation. Science. 2000;288:2219–2222. doi: 10.1126/science.288.5474.2219. [DOI] [PubMed] [Google Scholar]
- 94.Moore MA, Metcalf D. Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol. 1970;18:279–296. doi: 10.1111/j.1365-2141.1970.tb01443.x. [DOI] [PubMed] [Google Scholar]
- 95.Dieterlen-Lievre F, Martin C. Diffuse intraembryonic hemopoiesis in normal and chimeric avian development. Dev Biol. 1981;88:180–191. doi: 10.1016/0012-1606(81)90228-1. [DOI] [PubMed] [Google Scholar]
- 96.Muller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity. 1994;1:291–301. doi: 10.1016/1074-7613(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 97.Yoder MC, Hiatt K. Engraftment of embryonic hematopoietic cells in conditioned newborn recipients. Blood. 1997;89:2176–2183. [PubMed] [Google Scholar]
- 98.Kumaravelu P, et al. Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development. 2002;129:4891–4899. doi: 10.1242/dev.129.21.4891. [DOI] [PubMed] [Google Scholar]
- 99.Mikkola HK, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006;133:3733–3744. doi: 10.1242/dev.02568. [DOI] [PubMed] [Google Scholar]
- 100.Gong JK. Endosteal marrow: a rich source of hematopoietic stem cells. Science. 1978;199:1443–1445. doi: 10.1126/science.75570. [DOI] [PubMed] [Google Scholar]
- 101.Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- 102.Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol. 2005;21:605–631. doi: 10.1146/annurev.cellbio.21.012704.131525. [DOI] [PubMed] [Google Scholar]
- 103.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Williams DA, Cancelas JA. Leukaemia: niche retreats for stem cells. Nature. 2006;444:827–828. doi: 10.1038/444827a. [DOI] [PubMed] [Google Scholar]
- 105.Kessinger A, Sharp JG. The whys and hows of hematopoietic progenitor and stem cell mobilization. Bone Marrow Transplant. 2003;31:319–329. doi: 10.1038/sj.bmt.1703837. [DOI] [PubMed] [Google Scholar]
- 106.Cancelas JA, et al. Circulating stem cell collection in lymphoma and myeloma after mobilization with cyclophosphamide and granulocyte colony-stimulating factor for autologous transplantation. Vox Sang. 1994;67:362–367. doi: 10.1111/j.1423-0410.1994.tb01274.x. [DOI] [PubMed] [Google Scholar]
- 107.Briddell RA, Hartley CA, Smith KA, McNiece IK. Recombinant rat stem cell factor synergizes with recombinant human granulocyte colony-stimulating factor in vivo in mice to mobilize peripheral blood progenitor cells that have enhanced repopulating potential. Blood. 1993;82:1720–1723. [PubMed] [Google Scholar]
- 108.Devine SM, et al. Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non-Hodgkin’s lymphoma. J Clin Oncol. 2004;22:1095–1102. doi: 10.1200/JCO.2004.07.131. [DOI] [PubMed] [Google Scholar]
- 109.Ryan MA, et al. Pharmacological inhibition of EGFR signaling enhances G-CSF-induced hematopoietic stem cell mobilization. Nat Med. 2010;16:1141–1146. doi: 10.1038/nm.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mendez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peled A, et al. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peled A, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 113.Peled A, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289–3296. [PubMed] [Google Scholar]
- 114.Driessen RL, Johnston HM, Nilsson SK. Membrane-bound stem cell factor is a key regulator in the initial lodgment of stem cells within the endosteal marrow region. Exp Hematol. 2003;31:1284–1291. doi: 10.1016/j.exphem.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 115.Miyazawa K, Williams DA, Gotoh A, Nishimaki J, Broxmeyer HE, Toyama K. Membrane-bound Steel factor induces more persistent tyrosine kinase activation and longer life span of c-kit gene-encoded protein than its soluble form. Blood. 1995;85:641–649. [PubMed] [Google Scholar]
- 116.Williams DA, Rios M, Stephens C, Patel VP. Fibronectin and VLA-4 in haematopoietic stem cell-microenvironment interactions. Nature. 1991;352:438–441. doi: 10.1038/352438a0. [DOI] [PubMed] [Google Scholar]
- 117.Papayannopoulou T, Craddock C, Nakamoto B, Priestley GV, Wolf NS. The VLA4/VCAM-1 adhesion pathway defines contrasting mechanisms of lodgement of transplanted murine hemopoietic progenitors between bone marrow and spleen. Proc Natl Acad Sci U S A. 1995;92:9647–9651. doi: 10.1073/pnas.92.21.9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Scott LM, Priestley GV, Papayannopoulou T. Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol. 2003;23:9349–9360. doi: 10.1128/MCB.23.24.9349-9360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.del Pozo MA, Vicente-Manzanares M, Tejedor R, Serrador JM, Sanchez-Madrid F. Rho GTPases control migration and polarization of adhesion molecules and cytoskeletal ERM components in T lymphocytes. Eur J Immunol. 1999;29:3609–3620. doi: 10.1002/(SICI)1521-4141(199911)29:11<3609::AID-IMMU3609>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 120.Taylor ML, Metcalfe DD. Kit signal transduction. Hematol Oncol Clin North Am. 2000;14:517–535. doi: 10.1016/s0889-8588(05)70294-x. [DOI] [PubMed] [Google Scholar]
- 121.Alai M, Mui AL, Cutler RL, Bustelo XR, Barbacid M, Krystal G. Steel factor stimulates the tyrosine phosphorylation of the proto-oncogene product, p95vav, in human hemopoietic cells. J Biol Chem. 1992;267:18021–18025. [PubMed] [Google Scholar]
- 122.Gu Y, et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–449. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- 123.Whetton AD, Lu Y, Pierce A, Carney L, Spooncer E. Lysophospholipids synergistically promote primitive hematopoietic cell chemotaxis via a mechanism involving Vav 1. Blood. 2003;102:2798–2802. doi: 10.1182/blood-2002-12-3635. [DOI] [PubMed] [Google Scholar]
- 124.Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, Williams DA. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat Immunol. 2004;5:744–751. doi: 10.1038/ni1081. [DOI] [PubMed] [Google Scholar]
- 125.Yang FC, et al. Rac and Cdc42 GTPases control hematopoietic stem cell shape, adhesion, migration, and mobilization. Proc Natl Acad Sci U S A. 2001;98:5614–5618. doi: 10.1073/pnas.101546898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sanchez-Aguilera A, et al. Guanine nucleotide exchange factor Vav1 regulates perivascular homing and bone marrow retention of hematopoietic stem and progenitor cells. Proc Natl Acad Sci U S A. 2011;108:9607–9612. doi: 10.1073/pnas.1102018108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dorrance AM, et al. The Rac GTPase effector p21-activated kinase is essential for hematopoietic stem/progenitor cell migration and engraftment. Blood. 2013;121:2474–2482. doi: 10.1182/blood-2012-10-460709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Min IM, Pietramaggiori G, Kim FS, Passegue E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. 2008;2:380–391. doi: 10.1016/j.stem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 129.Taniguchi Ishikawa E, et al. Klf5 controls bone marrow homing of stem cells and progenitors through Rab5-mediated beta1/beta2-integrin trafficking. Nat Commun. 2013;4:1660. doi: 10.1038/ncomms2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zsebo KM, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213–224. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 131.Doan PL, et al. Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat Med. 2013;19:295–304. doi: 10.1038/nm.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tarcic G, et al. EGR1 and the ERK-ERF axis drive mammary cell migration in response to EGF. FASEB J. 2012;26:1582–1592. doi: 10.1096/fj.11-194654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Harada T, Morooka T, Ogawa S, Nishida E. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol. 2001;3:453–459. doi: 10.1038/35074516. [DOI] [PubMed] [Google Scholar]
- 134.Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. 2007;104:5091–5096. doi: 10.1073/pnas.0610819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xing Z, et al. Increased hematopoietic stem cell mobilization in aged mice. Blood. 2006;108:2190–2197. doi: 10.1182/blood-2005-12-010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sugimura R, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150:351–365. doi: 10.1016/j.cell.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Florian MC, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10:520–530. doi: 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–860. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Plant PJ, et al. A polarity complex of mPar-6 and atypical PKC binds, phosphorylates and regulates mammalian Lgl. Nat Cell Biol. 2003;5:301–308. doi: 10.1038/ncb948. [DOI] [PubMed] [Google Scholar]
- 140.Atwood SX, Chabu C, Penkert RR, Doe CQ, Prehoda KE. Cdc42 acts downstream of Bazooka to regulate neuroblast polarity through Par-6 aPKC. J Cell Sci. 2007;120:3200–3206. doi: 10.1242/jcs.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wodarz A, Ramrath A, Grimm A, Knust E. Drosophila atypical protein kinase C associates with Bazooka and controls polarity of epithelia and neuroblasts. J Cell Biol. 2000;150:1361–1374. doi: 10.1083/jcb.150.6.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu M, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541–554. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ito T, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010;466:765–768. doi: 10.1038/nature09171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhao C, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wilson A, et al. Normal hemopoiesis and lymphopoiesis in the combined absence of numb and numblike. J Immunol. 2007;178:6746–6751. doi: 10.4049/jimmunol.178.11.6746. [DOI] [PubMed] [Google Scholar]
- 146.Sengupta A, et al. Atypical protein kinase C (aPKCzeta and aPKClambda) is dispensable for mammalian hematopoietic stem cell activity and blood formation. Proc Natl Acad Sci U S A. 2011;108:9957–9962. doi: 10.1073/pnas.1103132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chae HD, Lee KE, Williams DA, Gu Y. Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood. 2008;111:2597–2605. doi: 10.1182/blood-2007-06-093237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Beekman R, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 2012;119:5071–5077. doi: 10.1182/blood-2012-01-406116. [DOI] [PubMed] [Google Scholar]
- 149.Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11:886–891. doi: 10.1038/nm1274. [DOI] [PubMed] [Google Scholar]
- 150.Jansen M, Yang FC, Cancelas JA, Bailey JR, Williams DA. Rac2-deficient hematopoietic stem cells show defective interaction with the hematopoietic microenvironment and long-term engraftment failure. Stem Cells. 2005;23:335–346. doi: 10.1634/stemcells.2004-0216. [DOI] [PubMed] [Google Scholar]
- 151.Chae HD, Lee KE, Williams DA, Gu Y. Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood. 2008;111:2597–2605. doi: 10.1182/blood-2007-06-093237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ghiaur G, Lee A, Bailey J, Cancelas JA, Zheng Y, Williams DA. Inhibition of RhoA GTPase activity enhances hematopoietic stem and progenitor cell proliferation and engraftment. Blood. 2006;108:2087–2094. doi: 10.1182/blood-2006-02-001560. [DOI] [PubMed] [Google Scholar]
- 153.Xu H, et al. Loss of the Rho GTPase activating protein p190-B enhances hematopoietic stem cell engraftment potential. Blood. 2009;114:3557–3566. doi: 10.1182/blood-2009-02-205815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhou X, et al. RhoA GTPase Controls Cytokinesis and Programmed Necrosis of Hematopoietic Progenitors. J Exp Med. 2013 doi: 10.1084/jem.20122348. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]