

Abstract
The protruding oxophilic central metal ion of ZrIV porphyrinoids facilitates axial coordination to the oxygen bearing functional groups on graphene oxide (GO) surfaces to result in new supramolecular photonic materials with high dye loading especially on edges and large defects. The reaction proceeds at room temperature with GO dispersed in tetrahydrofuran and GO films on glass. Since the ZrIV serves as a conduit, the photophysical properties of the dye sensitized GO derive from both the axially bound chromophores and the GO substrate. Self-organization of metalloporphyrinoids on GO mediated by axial coordination of group (IV) metal ions allows for direct sensitization of graphene and graphenic materials without requiring covalent chemistries with poorly conducting linkers.
Keywords: Graphene oxide, self-organization, zirconium, porphyrin and phthalocyanine, coordination chemistry
Introduction
The unique properties of graphene oxide (GO) have driven research on using GO in functional materials for diverse applications.1–4 GO is produced by a variety of scalable methods that result in similar, but not identical, products.5 Theories about the exact structure of GO have continuously evolved because of the non-homogenous nature of the material.6 A commonly accepted model (Lerf-Klinowski) describes a surface composed of a substituted network of sp2 and sp3 hybridized carbon atoms. Alcohol and epoxide functionalities are present on the basal planes with carboxylic groups along the edges and larger defect sites of the flakes, which range between tens of nanometers and tens of microns in edge length and 0.8–1.5 nm in thickness if fully exfoliated depending on the method of production.7
Interest in GO has focused on properties arising from its non-stoichiometric nature resulting in regions of variable oxygen content, controllable dispersibility in water and organic solvents,8 interesting photonic properties,9 and potential for use in biological systems such as for photodynamic therapeutics and for drug delivery.1,10–14 Recent reports demonstrate GO applications in nonlinear optics,15 as p or n type materials,16,17 as a functional surfactant,18 as antennae complexes to collect solar energy, and as efficient catalysts.19
Free base and metallotetraphenylporphyrins can be covalently bound to GO via amides20,21 and esters,22,23 or deposited by molecular beam24 to yield photonic materials. Cationic porphyrins electrostatically associate with GO and rGO, and GO can be appended with polyethylene glycols to make biocompatible materials.10,11 Large, planar phthalocyanines and their metalo derivatives associate with graphene and areas in reduced graphene oxide (rGO) via π-π interactions.25–27 Sensitized hybrid GO materials can serve as components of solar cell devices,28–30 and electrodes.31
We report herein the efficient self-organization and photonic properties of materials composed of 5,10,15,20-tetraphenylporphyrinato ZrIV, ZrIV(TPP), or phthalocyaninato ZrIV, ZrIV(Pc) (Figure 1) on graphene oxide, GO. The 7–8 coordinate oxophillic ZrIV ions protrude from one face of the ZrIV(TPP) and the ZrIV(Pc) so that they are available to strongly coordinate to the bidentate acetates or oxygen containing functional groups or on the surfaces and edges of GO. The supramolecular material is assembled from dyes with acetates as auxiliary ligands, ZrIV(TPP)Ac2 and Zr(Pc)Ac2, which are easily synthesized in good yields from commercially available precursors and have electronic spectra typical of metalloporphyrins and metallophthalocyanines, respectively.32,33 The large ionic radius of ZrIV, 0.72 Å, results in an unusual 0.9 Å to 1 Å displacement from the mean plane of the macrocycle nitrogen atoms, thereby enabling group IV metalloporphryrinoids to simultaneously bind oxygen groups on materials such as defects in polyoxometalates and to TiO2 surfaces used in dye sensitized solar cells.33–35 In contrast, ZnII resides in the mean plane of the Pc and TPP macrocycles and is weakly six coordinate, thus Zn(TPP) and Zn(Pc) (Figure 1) do not strongly interact with GO and are used as control dyes for comparison of the photonic properties of the dye-sensitized GO materials.
Figure 1.

Since the 8-coordinate ZrIV ion protrudes from one face of the 5,10,15,20-tetraphenylporphyrin (TPP) and phthalocyanine (Pc) the ZrIV axially coordinates GO by replacement of bidentate acetate groups with oxygen groups on the GO (bottom). The 4-coordinate Zn analogue very weakly accepts axial ligands and does not bind GO (top).
Direct attachment of the chromophore to the GO surface via the protruding metal ion results in a coplanar orientation of the chromophores relative to the GO surface and at an angle on the edges and large defects. Some plausible geometries derived from MM2 calculations are suggested in Figure 2, and are consistant with ZrIV(TPP) and ZrIV(Pc) bound to a defect site in a polyoxometalate.33 In contrast to covalent chemistry, this supramolecular chemistry approach increases the efficiency of dye loading, strongly couples the electronic properties of the two components, and preserves the properties of the substrate. The ZrIV serves as a direct conduit for electronic coupling of the dye and GO systems because of the mixing of the metal ion and chromophore orbitals.15,36 The photonic properties of this new hybrid material can be tuned by altering the photophysical properties of the dye, and represents an avenue for the development of a variety of sensitized graphenic materials.
Figure 2.

Three ways of binding of ZrIV(Pc) on a small region of GO are representative of the several possible binding modes The ZrIV(TPP) bind similarly. 3-dimensional renderings are approximated from MM2 calculations (top, left to right) for internal diol, side carboxylates, and side diols; grey=C, blue=N, red=O, dark grey=ZrIV, H left out for clarity.
Materials and methods
Atomic Force Microscopy (AFM) measurements were conducted with an Asylum Research Corp. (MFP-3D) or an Agilent 5500 instrument using standard n-type silicon cantilevers with reflective aluminum coating (MikroMasch NSC15-AlBS) with a nominal spring constant of 40 N/m and frequency of 325 kHz, at a scan rate of 15 μm/sec. UV-visible absorption spectroscopy was performed on a Cary 1-Bio UV-Visible spectrometer. Steady state fluorescence spectra were obtained on a HORIBA Jobin-Yvon FluoroLog-3 fluorometer. Quartz or optical glass cuvettes were used for all spectroscopic studies. All photophysical studies were carried out in distilled solvents. MM2 calculations were done in Chem3D. Experiments have been repeated multiple times by at least three different researchers.
Zn(Pc) is synthesized by simple click-type substitution of peripheral fluorine atoms on the commercially available perfluorophthalocyanine precursor with thioalkanes to improve solubility.37 Synthetic details of ZrIV dyes are reported.33 GO material was purchased from Graphene Supermarket who reports that the material is >60% single layer, flakes are 0.5–5 microns, and with 20% oxygen (w/w).
Optical Spectra
A dispersion of 0.1 mg/mL GO is made by sonicating single layer GO flakes in freshly distilled THF,38 and titrated in 40 μL aliquots into 3 mL solutions of 3 μM ZrIV(Pc)Ac2 or 2 μM ZrIV(TPP)Ac2 in THF. After 24 h to allow the dyes to bind the GO, the UV-visible and fluorescence emission spectra were recorded in 1x1 cm cuvettes (λex: 655 nm for Zn(Pc), 633 nm for ZrIV(Pc), 422 nm for Zn(TPP), and 417 nm for ZrIV(TPP)). Fluorescence quenching comparisons were recorded at consistent absorbance values.
Nanocomposites
GO was added to 100 μM solutions of ZrIV(Pc)Ac2 or ZrIV(TPP)Ac2 in THF to make a 0.1 mg/mL GO solution, and sonicated for 5 min. to ensure good dispersion. After allowing 48 h for equilibration at room temperature, samples were centrifuged at 13,000 RPM for 15 min. and washed twice with fresh THF to remove unbound dye molecules.
AFM
Samples for AFM were prepared either by dip coating ozone cleaned glass into the suspensions of the nanocomposites in THF or by centrifuging the suspension, dispersing the pellet into nanopure water by sonication, and spin coating onto ozone cleaned glass.
TEM
All data were collected at 200 kV on a Jeol 2100 transmission electron microscope equipped with EDAX at the eucentric point to ensure reproducibility of the measurements. An 8 μL drop of the above nanocomposite dispersion was placed on a 300 mesh carbon coated copper grid (TED Pella Inc.) and allowed to dry for 1 min. before imaging.
Films
GO in aqueous suspension (3 mg/mL) was spin coated onto piranha cleaned quartz slides and dried in an oven to make 15 nm thick continuous films. Films were then soaked in 0.1 mM solutions of dyes in CH2Cl2 for three days at room temperature in the dark to ensure equilibration. The films were rinsed extensively with clean CH2Cl2 to remove any unbound dye and the backs of the slides were cleaned repeatedly with MeOH before spectra were recorded.
Probe Measurements
Devices were made by spin coating layers of GO (of 8 nm or 15 nm) onto ozone cleaned glass from an aqueous suspension. Sheet resistance was measured by Van der Pauw technique of the pristine GO layers, of an 8 nm GO film soaked in ZrIV(Pc) or ZrIV(TPP) (0.1 mM in CH2Cl2), or of an 8 nm GO film spin coated on top of a ca. 40 nm thick layer of ZrIV(TPP) or ZrIV(Pc) spin cast from chlorobenzene on ozone cleaned glass. Measurements were taken in the dark and under illumination.
Results and Discussion
GO dispersions in THF were added to solutions of ZrIV(Pc)Ac2 or ZrIV(TPP)Ac2 and allowed to react for 24 h to allow the maximal amount of the ZrIV(Pc) or ZrIV(TPP) to bind to GO. These are similar conditions used to bind the ZrIV metalloporphyrinoids to surface oxygens on nanoparticles of TiO2.34 The UV-visible spectral shifts and decreased intensity in the ZrIV(Pc) Q band (676 nm) and ZrIV(TPP) Soret (421 nm) after incubation are consistent with displacement of the acetate ligands by the oxygen functional groups on the GO and formation of the hybrid material (Figure 3).34 The intensity of the UV-visible spectra of the hybrid materials with ZrIV(Pc) or ZrIV(TPP) attached to GO monotonically decreases without changes in peak λmax as the solution is diluted, thus indicating that the solution is homogeneously dissolved and the zirconium dyes are strongly bound to the GO. Since the zinc species, Zn(Pc) and Zn(TPP), cannot accommodate further ligation and do not chemically bond GO, the electronic spectra are not altered upon addition of the GO dispersion.
Figure 3.

Top: the UV-visible and fluorescence spectra of the Zn dyes overlap with the spectra after addition of 120 μL of 0.1 mg/mL GO dispersed in THF; indicating that neither ZnPc nor ZnTPP interact with GO. Bottom: the significant changes in the UV-visible and fluorescence quenching of ZrIV(TPP)Ac2 and ZrIV(Pc)Ac2 after addition of 120 μL of 0.1 mg/mL GO dispersed in THF indicate exchange of the acetate ligands for oxygen functional groups on the GO and binding of the metalo dyes. λex = 417 nm for ZrIV(TPP) and Zn(TPP), λex = 633 nm for Zn(Pc) and ZrIV(Pc), in each case the optical density was <0.1.
Significant fluorescence quenching of the ZrIV dyes indicates charge injection from the chromophore excited state (HOMO −5.4 eV, LUMO −3.6 eV, band gap 1.8 eV)34 into the GO made possible by the large band gap of GO (ca. −5.6 eV – −4.0 eV).20,39–41 When binding is complete, the emission is quenched by 70% for the ZrIV(Pc) and 30% for the ZrIV(TPP), respectively. The quenching is consistent with electron transfer to the GO as found with covalently bound porphryinoids to graphene and GO.4 Note that the peripheral phenyl groups on TPP are nominally orthogonal to the macrocycle with dihedral angles 90±20°, thus the phenyl moieties on ZrIV(TPP) likely reduce binding to the surfaces of GO by steric hindrance. For Zn(TPP) and Zn(Pc) controls, the electronic absorption and emission spectra are unchanged by addition of the GO dispersion, indicating no interaction. Note that the large Stokes shift for the ZrIV(Pc) is typical for these compounds.
To investigate the properties of the material in the solid state, films of GO were spin cast onto quartz substrates from dispersions in water, soaked in 0.1 mM dye solutions in CH2Cl2 for 72 h, and rinsed thoroughly with clean solvent. An aqueous GO dispersion of 3 mg/mL yielded 15 nm thick continuous films while a 1 mg/mL dispersion yielded an 8 nm film. After soaking in solutions of the ZrTPP(ac)2 or ZrPc(ac)2 and rinsing well with fresh THF, fluorescence quenching by charge transfer from Zr containing dyes to GO is observed for these metal-coordinated films (Figure 4).42 UV-visible spectra demonstrate the presence of the coordinated dyes on the GO films by the absorbance from the ZrIV(TPP) Soret (421 nm) and ZrIV(Pc) Q (693 nm) bands (Figure 4). The small peaks from the control ZnII complexes are likely the result of dye molecules interacting with the GO in graphene like regions through π-π interactions.
Figure 4.
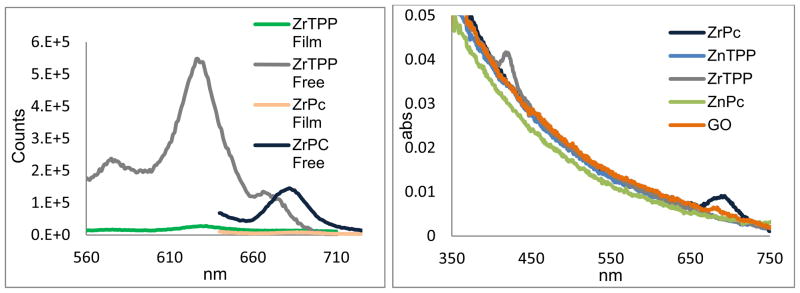
Left: fluorescence of ZrIV(Pc) and ZrIV(TPP) dyes bound to a ca. 15 nm thick film of GO quenched compared to solutions of the unbound dyes with the same optical density. λex = 417 nm for ZrIV(TPP) and 633 nm for ZrIV(Pc). Right: UV-visible spectra of the same GO films where control Zn dyes are not observed to bind the GO films.
The absence of significant shifts in the optical spectra indicates that the dye molecules are not aggregated. Optical data on the films suggests a surface density43 of ca. one chromophore per 20–50 nm2, assuming average flakes of 1 μm2 and dyes bound to only one face. The C:Zr mole ratio obtained by elemental analysis (31:0.006) of the ZrIV(TPP) and ZrIV(Pc) bound to GO indicated similar packing densities of ca. one per 40 nm2. Collectively, these data indicate that the high oxygen content of the GO facilitates tight binding and high surface coverage of the ZrIV dyes mediated by axial coordination.
In a separate experiment, ZrIV(Pc)Ac2 was added to a 0.1 mg/mL GO dispersion in THF to make solutions 100 μM in dye and ensure saturation of the GO surfaces with ZrIV chromophores. Suspensions were allowed to react ca. 72 h to reach equilibrium, whereupon the mixture was centrifuged, rinsed with clean THF to remove unbound dye, and dispersed again in THF by brief sonication to form nanocomposites of GO with ZrIV(Pc) coordinated to the protruding oxygen containing surface and edge groups. Ozone cleaned glass slides were dip coated into these dispersions and rinsed well with clean THF. AFM analyses of these composite films confirm that the large oxygen:carbon ratio of the GO allows for high surface density of the coordinatively bound chromophores. Sonication in THF selectively removes dye molecules coordinated on the basal plane of the GO while preserving those coordinated more strongly to the flake edges, where there is a great concentration of carboxylic groups44 (Figure 5). A single flake of GO is ca. 1.2 nm thick (Figure S8, S9),45,46 and single layer flakes of GO coated on both sides with ZrIV(Pc) are observed, corresponding to heights of ca. 2.8 nm. The total heights of the ZrIV(Pc)/GO composite correspond well to those predicted by adding the heights of the GO with the thickness of the dye obtained from crystal structures (Figure 5, S10).33 The AFM of control samples prepared by identical methods using Zn(Pc) and Zn(TPP) show GO ca. 1.1 nm flakes, and the absence of the zinc dyes.
Figure 5.

AFM image and height trace of ca. 2.8 nm high GO flake coated on both sides with ZrIV(Pc) cast onto an ozone cleaned glass coverslip. Higher dye loading on GO edge is visible.
TEM images of the same nanocomposites indicate a uniform coating of the GO surface with ZrIV(Pc) and EDAX spectra display characteristic bands for the presence of Zr (Figure 6, S7). No evidence of Zn is observed by EDAX when GO is mixed with Zn(Pc) under the same conditions.
Figure 6.

TEM images of GO flakes treated with Zn(Pc) (left) or ZrIV(Pc)Ac2 (right) and rinsed with clean solvent. Full surface coverage by coordinated ZrIV dye is indicated by EDAX spectra (see supporting information) and greater contrast, which also indicates greater dye loading on GO edge. The Zn dyes are completely removed by simple rinsing.
When the ZrIV(Pc)/GO nanocomposites were centrifuged and redispersed in water, the nanocomposites precipitate rapidly, whereas the unfunctionalized GO remains suspended. This indicates a significantly increased hydrophobicity of the hybrid material relative to GO arising from the organic dye covering the face, and the lack of exposed oxygen species. Similar results are observed for the ZrIV(TPP) on GO. To assess the robustness of the ZrIV dye binding to the GO, the composite was sonicated (60 W, 30 minutes), and the solution was spin cast onto ozone cleaned glass coverslips. AFM of films of these samples display perforations of the graphene flakes resultant from removal of the oxophilic ZrIV(Pc) or ZrIV(TPP) (Figure S11). The perforation/degradation of the GO sheets also eliminates the fluorescence of the pristine GO flakes and no fluorescence was observed for the dyes.9,47 Control experiments with Zn(Pc) and GO displays the characteristic UV-visible spectrum and broad fluorescence of GO without perforations. In a separate control experiment GO dispersions in THF were reacted with ZrCl4. Large aggregates of metal ion cross-linked or intercalated GO sheets precipitate immediately. When dispersed by sonication into water no perforation or changes to the flakes are observed.
Four point probe measurements (Table S1) yielded sheet resistance values of 2.9×1010 and 3.6×109 Ω/□ for 8 nm and 15 nm thick GO films, respectively.48 For composite films, made by spin casting the ZrIV(dye) treated GO or dipping the cast GO into ZrIV(Pc) or ZrIV(TPP) solutions, the sheet resistance of 8 nm films decreased by ca. 50%. This indicates the ZrIV dyes can bridge some of the defects in the GO material, thereby increasing conductivity. Future work will investigate the properties of the dye/substrate interface as well as anticipated doping and photoconductivity applications.8,16,49,50
Layer-by-layer assembly is a simple technique to create functional materials with controlled size, morphology, and physical properties on different substrates.51 Films were made by sequential dipping of quartz slides with an 8 nm GO layer into solutions of Zn(Pc), ZrIV(Pc) or ZrCl4 (Figure 7, Figures S13–16). Initially, the hydroxyl groups on the GO flakes associate with the quartz substrate, likely by hydrogen bonding.52 The nonstoichiometric nature, oxidative debris, and varied size of the GO flakes result in imperfections and varied coverage in the initial layer, but these are filled in as the films develop in thickness. Zn(Pc) adheres to any hydrophobic basal plane regions of unoxidized sp2 hybridized carbon allowing for layer growth mediated by the GO, but with small amounts of the dye entrained within the layers. ZrIV(Pc) and ZrCl4 both coordinate to the oxygenated regions of the GO.8 Binding of the chromophores and the addition of new layers of GO is indicated by the linear increase absorbance from the dyes and GO in the UV-visible spectra of the films. (Figure S15). Though individual layers are incomplete, sequential dipping of the substrate into the GO and ZrIV dye solutions builds nanofilms with controlled thickness, controlled optical density, and controlled coverage. As observed in dye-coated GO dispersions, binding of the ZrIV dyes to the GO significantly diminishes the propensity of the GO to interact and results in only a gradual increase in film thickness as the substrate is sequentially dipped in the solutions. We observed that the bound dye molecules also serve as nucleation points for the formation of microcrystals of dye, suggesting that GO might serve as a good substrate to grow microcrystals of other species. The highly varied composition of the GO affords many possible binding modes and binding sites for the ZrIV dyes, for interactions between flakes of the GO, and for interactions with the substrate. The hydrophobic Zn dyes can interact with graphene like areas of the GO, while the oxophilic metal center can interact with oxygen functional groups on the GO and with the substrate.
Figure 7.

Representation of the possible intermolecular interactions between in multilayers of dye-bound GO made by layer-by-layer methods. Interactions between the GO and ZrIV(Pc) with the quartz substrate include: (a) π-π interactions between the starting ZrIV(Pc)Ac2, (b) coordination of the protruding ZrIV ion of the Pc to the oxygen groups on GO, (c) H-bonds between the substrate and the GO. The higher density of oxygen groups at the edges of GO result in greater binding of the ZrIV dyes, see Figure 5. Other variations of these interactions are also present, e.g. H-bond and π-π interactions between GO flakes, see Figure S13.
Conclusions
Self-organization of a composite film of ZrIV porphyrins or ZrIV phthalocyanines on GO is accomplished by the concomitant coordination of the metal ion to both the dye and the carboxylate groups decorating the edges of the GO sheets and the OH groups on the planar surfaces of GO. This supramolecular approach affords a material with high surface coverage of the dyes on the GO substrate and better electronic coupling of the two systems compared to porphyrinoid attached to GO by covalent chemistry. The hybrid material displays photonic properties derived from both the dyes and the GO.12 Reduced GO has enhanced electronic properties and the ZrIV dyes should bind to remaining oxygen containing defect sites.53 This type of direct sensitization will broaden the applications of GO and foster the creation of new composite functional materials using cost effective components and self-organization to create functional films.54–58 These studies also indicate that the GO may serve as a means to grow microcrystals of other species.
Supplementary Material
Acknowledgments
Funding Sources
Supported by the U.S. National Science Foundation (NSF) CHE-0847997 and 1213962 to CMD. Hunter College science infrastructure is supported by the NSF, the National the National Institute on Minority Health and Health Disparities (8G12 MD007599) from the National Institutes of Health, and the City University of New York.
We thank Jacopo Samson for his work with TEM and EDAX spectra, Ivana Radivojevic for the ZrIV dyes, and David Nissenbaum for help with figures.
ABBREVIATIONS
- GO
graphene oxide
- TPP
5,10,15,20-tetraphenylporphyrin
- Pc
phthalocyanine
- TEM
transmission electron microscopy
- AFM
atomic force microscopy
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. AFM, TEM, optical spectroscopy, control studies with Zn dyes, electronic measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Loh KP, Bao Q, Eda G, Chhowalla M. Nat Chem. 2010;2:1015–1024. doi: 10.1038/nchem.907. [DOI] [PubMed] [Google Scholar]
- 2.Chen D, Feng H, Li J. Chem Rev. 2012;112:6027–6053. doi: 10.1021/cr300115g. [DOI] [PubMed] [Google Scholar]
- 3.Wan X, Huang Y, Chen Y. Acc Chem Res. 2012;45:598–607. doi: 10.1021/ar200229q. [DOI] [PubMed] [Google Scholar]
- 4.Georgakilas V, Otyepka M, Bourlinos AB, Chandra V, Kim N, Kemp KC, Hobza P, Zboril R, Kim KS. Chem Rev. 2012;112:6156–6214. doi: 10.1021/cr3000412. [DOI] [PubMed] [Google Scholar]
- 5.(a) Dreyer DR, Park S, Bielawski CW, Ruoff RS. Chem Soc Rev. 2010;39:228–240. doi: 10.1039/b917103g. [DOI] [PubMed] [Google Scholar]; (b) Park S, Ruoff RS. Nature Nanotech. 2009;4:217–224. doi: 10.1038/nnano.2009.58. [DOI] [PubMed] [Google Scholar]
- 6.Rourke JP, Pandey PA, Moore JJ, Bates M, Kinloch IA, Young RJ, Wilson NR. Angew Chem Int Ed. 2011;50:3173–3177. doi: 10.1002/anie.201007520. [DOI] [PubMed] [Google Scholar]
- 7.Lerf A, He H, Forster M, Klinowski J. J Phys Chem B. 1998;102:4477–4482. [Google Scholar]
- 8.Kim J, Cote LJ, Kim F, Yuan W, Shull KR, Huang J. J Am Chem Soc. 2010;132:8180–8186. doi: 10.1021/ja102777p. [DOI] [PubMed] [Google Scholar]
- 9.Melucci M, Durso M, Zambianchi M, Treossi E, Xia ZY, Manet I, Giambastiani G, Ortolani L, Morandi V, De Angelis F, Palermo V. J Mat Chem. 2012;22:18237–18243. [Google Scholar]
- 10.Liu Z, Robinson JT, Sun X, Dai H. J Am Chem Soc. 2008;130:10876–10877. doi: 10.1021/ja803688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X, Liu Z, Welsher K, Robinson J, Goodwin A, Zaric S, Dai H. Nano Res. 2008;1:203–212. doi: 10.1007/s12274-008-8021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo Z, Vora PM, Mele EJ, Johnson ATC, Kikkawa JM. Appl Phys Let. 2009;94:111909. [Google Scholar]
- 13.Shih CJ, Lin S, Sharma R, Strano MS, Blankschtein D. Langmuir. 2011;28:235–241. doi: 10.1021/la203607w. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y, Zhang YM, Chen Y, Zhao D, Chen JT, Liu Y. Chem Eur J. 2012;18:4208–4215. doi: 10.1002/chem.201103445. [DOI] [PubMed] [Google Scholar]
- 15.Yao C, Yan LK, Guan W, Liu CG, Song P, Su ZM. Dalton Trans. 2010;39:7645–7649. doi: 10.1039/c002547j. [DOI] [PubMed] [Google Scholar]
- 16.Arnold MS, Zimmerman JD, Renshaw CK, Xu X, Lunt RR, Austin CM, Forrest SR. Nano Let. 2009;9:3354–3358. doi: 10.1021/nl901637u. [DOI] [PubMed] [Google Scholar]
- 17.Tung VC, Huang JH, Tevis I, Kim F, Kim J, Chu CW, Stupp SI, Huang J. J Am Chem Soc. 2011;133:4940–4947. doi: 10.1021/ja1103734. [DOI] [PubMed] [Google Scholar]
- 18.Tung VC, Kim J, Cote LJ, Huang J. J Am Chem Soc. 2011;133:9262–9265. doi: 10.1021/ja203464n. [DOI] [PubMed] [Google Scholar]
- 19.Yan JM, Wang ZL, Wang HL, Jiang Q. J Mat Chem. 2012;22:10990–10993. [Google Scholar]
- 20.Xu Y, Liu Z, Zhang X, Wang Y, Tian J, Huang Y, Ma Y, Zhang X, Chen Y. Adv Mater. 2009;21:1275–1279. [Google Scholar]
- 21.Karousis N, Sandanayaka ASD, Hasobe T, Economopoulos SP, Sarantopoulou E, Tagmatarchis N. J Mat Chem. 2011;21:109–117. [Google Scholar]
- 22.Bala Murali Krishna M, Venkatramaiah N, Venkatesan R, Narayana Rao D. J Mat Chem. 2012;22:3059–3068. [Google Scholar]
- 23.Melucci M, Treossi E, Ortolani L, Giambastiani G, Morandi V, Klar P, Casiraghi C, Samori P, Palermo V. J Mat Chem. 2010;20:9052–9060. [Google Scholar]
- 24.Pandey PA, Rochford LA, Keeble DS, Rourke JP, Jones TS, Beanland R, Wilson NR. Chem Mat. 2012;24:1365–1370. [Google Scholar]
- 25.Chunder A, Pal T, Khondaker SI, Zhai L. J Phys Chem C. 2010;114:15129–15135. [Google Scholar]
- 26.Malig J, Jux N, Kiessling D, Cid JJ, Vázquez P, Torres T, Guldi DM. Angew Chem Int Ed. 2011;50:3561–3565. doi: 10.1002/anie.201003364. [DOI] [PubMed] [Google Scholar]
- 27.(a) Cárdenas-Jirón GI, Leon-Plata P, Cortes-Arriagada D, Seminario JMJ. Phys Chem C. 2011;115:16052–16062. [Google Scholar]; (b) Zhang XF, Xi Q. Carbon. 2011;49:3842–3850. [Google Scholar]
- 28.Wang X, Zhi L, Mullen K. Nano Let. 2007;8:323–327. doi: 10.1021/nl072838r. [DOI] [PubMed] [Google Scholar]
- 29.Zhong S, Zhong JQ, Mao HY, Wang R, Wang Y, Qi DC, Loh KP, Wee ATS, Chen ZK, Chen W. ACS Appl Mater Interfaces. 2012;4:3134–3140. doi: 10.1021/am300887j. [DOI] [PubMed] [Google Scholar]
- 30.Park H, Howden RM, Barr MC, Bulović V, Gleason K, Kong J. ACS Nano. 2012;6:6370–6377. doi: 10.1021/nn301901v. [DOI] [PubMed] [Google Scholar]
- 31.Poursaberi T, Hassanisadi M. J Porphyrins Phthalocyanines. 2012;16:1140–1147. [Google Scholar]
- 32.Brand H, Arnold J. Coord Chem Rev. 1995;140:137–168. [Google Scholar]
- 33.(a) Falber A, Burton-Pye BP, Radivojevic I, Todaro L, Saleh R, Francesconi LC, Drain CM. Eur J Inorg Chem. 2009;2009:2459–2466. doi: 10.1002/ejic.200900284. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Radivojevic I, Burton-Pye BP, Saleh RR, Ithisuphalap K, Francesconi LC, Drain CM. RSC Adv. 2013;3:2174–2177. [Google Scholar]
- 34.Radivojevic I, Bazzan G, Burton-Pye BP, Ithisuphalap K, Saleh R, Durstock MF, Francesconi LC, Drain CM. J Phys Chem C. 2012;116:15867–15877. doi: 10.1021/jp301853d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radivojevic I, Burton-Pye BP, Saleh R, Ithisuphalap K, Francesconi LC, Drain CM. RSC Adv. 2013;3:2174–2177. [Google Scholar]
- 36.Liao MS, Scheiner S. J Chem Phys. 2002:117. [Google Scholar]
- 37.Varotto A, Nam C-Y, Radivojevic I, Tomé PCJ, Cavaleiro JAS, Black CT, Drain CM. J Am Chem Soc. 2010;132:2552–2554. doi: 10.1021/ja907851x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paredes JI, Villar-Rodil S, Martínez-Alonso A, Tascón JMD. Langmuir. 2008;24:10560–10564. doi: 10.1021/la801744a. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Xia J, Zhao Q, Liu L, Zhang Z. Small. 2010;6:537–544. doi: 10.1002/smll.200901680. [DOI] [PubMed] [Google Scholar]
- 40.Yamaguchi H, Murakami K, Eda G, Fujita T, Guan P, Wang W, Gong C, Boisse J, Miller S, Acik M, Cho K, Chabal YJ, Chen M, Wakaya F, Takai M, Chhowalla M. ACS Nano. 2011;5:4945–4952. doi: 10.1021/nn201043a. [DOI] [PubMed] [Google Scholar]
- 41.Zhu Y, Li X, Cai Q, Sun Z, Casillas G, Jose-Yacaman M, Verduzco R, Tour JM. J Am Chem Soc. 2012;134:11774–11780. doi: 10.1021/ja304471x. [DOI] [PubMed] [Google Scholar]
- 42.Treossi E, Melucci M, Liscio A, Gazzano M, Samorì P, Palermo V. J Am Chem Soc. 2009;131:15576–15577. doi: 10.1021/ja9055382. [DOI] [PubMed] [Google Scholar]
- 43.Bazzan G, Smith W, Francesconi LC, Drain CM. Langmuir. 2008;24:3244–3249. doi: 10.1021/la7031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quintana M, Montellano A, del Rio Castillo AE, Tendeloo GV, Bittencourt C, Prato M. Chem Com. 2011;47:9330–9332. doi: 10.1039/c1cc13254g. [DOI] [PubMed] [Google Scholar]
- 45.Becerril HA, Mao J, Liu Z, Stoltenberg RM, Bao Z, Chen Y. ACS Nano. 2008;2:463–470. doi: 10.1021/nn700375n. [DOI] [PubMed] [Google Scholar]
- 46.Li D, Muller MB, Gilje S, Kaner RB, Wallace GG. Nat Nano. 2008;3:101–105. doi: 10.1038/nnano.2007.451. [DOI] [PubMed] [Google Scholar]
- 47.Thomas HR, Valles C, Young RJ, Kinloch IA, Wilson NR, Rourke JP. J Mat Chem C. 2013;1:338–342. [Google Scholar]
- 48.Eda G, Fanchini G, Chhowalla M. Nat Nanotech. 2008;3:270–274. doi: 10.1038/nnano.2008.83. [DOI] [PubMed] [Google Scholar]
- 49.Jin M, Jeong HK, Yu WJ, Bae DJ, Kang BR, Lee YH. J Phys D: Appl Phys. 2009;42:135109. [Google Scholar]
- 50.Chang C-H, Fan X, Li L-J, Kuo J-L. J Phys Chem C. 2012 [Google Scholar]
- 51.Kovtyukhova NI, Ollivier PJ, Martin BR, Mallouk TE, Chizhik SA, Buzaneva EV, Gorchinskiy AD. Chem Mater. 1999;11:771–778. [Google Scholar]
- 52.Zhao X, Zhang Q, Hao Y, Li Y, Fang Y, Chen D. Macromolecules. 2010;43:9411–9416. [Google Scholar]
- 53.Luo D, Zhang G, Liu J, Sun X. J Phys Chem C. 2011;115:11327–11335. [Google Scholar]
- 54.Beletskaya I, Tyurin VS, Tsivadze AY, Guilard R, Stern C. Chem Rev. 2009;109:1659–1713. doi: 10.1021/cr800247a. [DOI] [PubMed] [Google Scholar]
- 55.Drain CM, Varotto A, Radivojevic I. Chem Rev. 2009;109:1630–1658. doi: 10.1021/cr8002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radivojevic I, Varotto A, Farley C, Drain CM. Energy Environm Sci. 2010;3:1897–1909. [Google Scholar]
- 57.Drain CM, Bazzan G, Milic T, Vinodu M, Goeltz JC. Israel J Chem. 2005;45:255–269. [Google Scholar]
- 58.Jurow MJ, Hageman BA, DiMasi E, Nam CY, Pabon C, Black CT, Drain CM. J Mater Chem A. 2013;1:1557–1565. doi: 10.1039/C2TA00415A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.