

Abstract
Genetic alterations in specific driver genes lead to disruption of cellular pathways and are critical events in the instigation and progression of hepatocellular carcinoma. As a prerequisite for individualized cancer treatment, we sought to characterize the landscape of recurrent somatic mutations in hepatocellular carcinoma. We performed whole exome sequencing on 87 hepatocellular carcinomas and matched normal adjacent tissues to anaverage coverage of 59x. The overall mutation rate was roughly 2 mutations per Mb, with a median of 45 non-synonymous mutations that altered the amino acid sequence (range 2 to 381). We found recurrent mutations in several genes with high transcript levels: TP53 (18%), CTNNB1 (10%), KEAP1 (8%), C16orf62 (8%), MLL4(7%) and RAC2 (5%). Significantly affected gene families include the nucleotide-binding domain and leucine rich repeat containing family, calcium channel subunits, and histone methyltransferases. In particular, the MLL family of methyltransferases for histone H3 lysine 4 were mutated in 20% of tumors.
Conclusion
The NFE2L2-KEAP1 and MLL pathways are recurrently mutated in multiple cohorts of hepatocellular carcinoma.
Keywords: HCC, mutations, histone methyltransferases
INTRODUCTION
Hepatocarcinogenesis is instigated by copy number alterations, mutations and chromosomal rearrangements that activate oncogenes or inactivate tumor suppressors. Previous genetic characterization of hepatocellular carcinoma (HCC) has indicated significant heterogeneity among tumors, which has hampered the development of targeted therapy. Genomic and transcriptomic profiling studies have attempted to classify tumor molecular subgroups and have implicated several signaling pathways that are mutated in HCC (1–3). The Wnt β-catenin signaling pathway members, CTNNB1, AXIN1 and AXIN2 are collectively mutated in up to half of tumors. The most frequently mutated tumor suppressor is TP53, which has mutations in over 20% of tumors. Over half of hepatocellular carcinomas harbor gains of chromosome 1q and 8q, which include candidate oncogenes MCL1, SHC1, MYC and COPS5/JAB1, among hundreds of other genes(4–8).
To date, studies of the mutational spectrum of HCC have focused on a limited number of candidate genes. Advances in genome sequencing technologies have enabled the simultaneous analysis of thousands of expressed genes accelerating the search for additional novel and recurrently mutated genes (9–14). Recent studies have identified the ATP-dependent nucleosome remodeling enzymes, ARID1A and ARID2, to be mutated in approximately 15% and 5% of tumors, respectively (11–14). A regulator of the redox signaling pathway, NFE2L2, is mutated in 6% of tumors (12). Other genes mutated at greater than 3% frequency include RPS6KA3, IL6ST, NRAS, KRAS, PIK3CA, PTEN, SAMD9L, DMXL1 and NLRP1(11, 13, 15).
The genetic heterogeneity of HCC has complicated our understanding of the molecular basis of this disease. In order to further define theimportant recurrent and clinically actionablemutations in HCC, we embarked on a large-scale study of 87 tumors that was powered to detect mutated genes at a population prevalence of at least 10%. We hypothesized that multiple component genes of certain signaling pathways could be recurrently mutated in hepatocellular carcinoma.
EXPERIMENTAL PROCEDURES
Human subjects
Subjects were identified and informed consent was obtained from consecutive patients undergoing surgical resection for confirmed HCC at the University Health Network (Toronto, Canada) and the University of North Carolina. Five additional tumor specimens were procured from the Cooperative Human Tissue Network. All surgical specimens were processed according to established institutional HCC-tumor banking protocols. In brief, fresh surgical specimens underwent immediate gross pathologic examination by experienced liver pathologists. Samples were obtained from viable tumor and non-tumor liver >2 cm away from the primary lesion. Tumors with evidence of prior therapy, such as radiofrequency ablation or trans-arterial chemo-embolization were excluded. Tissue samples were bisected with half of the tissue stored in liquid nitrogen and the mirror sample retained for histologic confirmation. All tumor samples were histologically assessed for diagnosis and cellularity and non-tumor liver samples were confirmed to be free of tumor cells.
DNA extraction and sequencing libraries
Tissue samples were thawed and weighed prior to homogenization. Genomic DNA and RNA was extracted with the Qiagen AllPrep kit (Valencia, CA) and quality was assessed on 2% agarose gel. Genomic DNA for exome capture and sequencing was prepared using either SureSelect Target Enrichment System for Illumina Paired-End Sequencing Library (Agilent Technologies, Santa Clara, CA) according to manufacturer’s instructions (protocol versions 1.2, May 2011 and 1.3.1, February 2012) or equivalent enzymes (New England Biolabs, Ipswich, MA) and Agilent’s protocol (version 2.0.1, May 2010). Briefly, 2–3 micrograms of genomic DNA were sheared on Covaris S220 (Woburn, MA) to 150–200 bp target size with the following instrument settings: 10% duty cycle; intensity 5; 200 cycles per burst; 6 cycles of 60 seconds each in frequency sweeping mode. Sheared fragments were ligated to Illumina’s adapters and enriched by 5–6 cycles of amplification. Five hundred nanograms of amplified libraries were incubated with Agilent’s whole exome capture oligos for 24h at 65°C. Hybridized fragments were captured with streptavidin-coated beads, eluted, and amplified by 10–12 cycles of PCR utilizing either one of 12 Agilent’s indexed primers or not indexed SureSelect GA PCR primers). Prepared libraries were pooled in batches of 4 and sequenced on Illumina HiSeq 2000 (San Diego, CA) instrument generating 100 bp paired end reads. Alternatively, individual libraries were sequenced on Illumina GAII instrument generating 76 bp paired end reads.
We enriched for protein coding regions with the Agilent AllExon version 1 sequence capture baits for 10 tumors and matched normals, and with the Nimblegen Human exome version 1 sequence capture baits for 5 tumors and matched normals. These captured libraries were sequenced on the Illumina Genome Analyzer II to an average of 4.8x and 5.8x, respectively. An additional 72 tumors and matched normals were captured on the Agilent AllExon version 4 sequence capture baits, and sequenced in pools of 4 samples with the IlluminaHiSeq 2000 to an average of 59x coverage (Supplementary Figure 1).
Bioinformatics sequence analysis
Sequences were mapped to the human genome (NCBI37/hg19), excluding alternative haplotype chromosomes, using the Bowtie 2 alignment algorithm (16). Alignments were refined using The Genome Analysis ToolKitto mark PCR duplicate reads and perform base quality recalibration(17, 18). Alignments from each tumor and matched normal were then analyzed by the MuTect algorithm(19, 20). In brief, MuTect includes a preprocessing step for sequence read qualities, a Bayesian classifier to assess the posterior probability of somatic mutations, and post-processing of candidate mutations. Somatic mutations were assigned to transcript and amino acid coordinates using the ANNOVAR software suite(21).
Availability of genetic sequencing data
Binary sequence alignment map files (BAM files) have been deposited in the National Center for Biotechnology Information dbGAP database.
Significantly mutated genes and pathways
The MutSig software version 1.5 identified the list of significantly mutated genes among 87 hepatocellular carcinomas (20)(https://confluence.broadinstitute.org/display/CGATools/MutSig). Genes that harbored a greater number of mutations than expected by chance were detected with a binomial test. For each gene, the observed number of mutations across the 87 tumors was compared to the expected number based on the background mutation rates and the covered bases in all samples. The binomial probabilities were adjusted to False Discovery Rate q-values with the Benjamini-Hochberg procedure and reported in Table 1.
Table 1.
Clinical characteristics of HCC samples.
| Patients N=87(%) | |
|---|---|
|
| |
| Male: Female | 61(77):18(23) |
|
| |
| Median Age (range) | 61 (16–83) |
|
| |
| Predisposing etiology* | |
| HBV | 38(43) |
| HCV | 19(21) |
| Alcohol | 10(11) |
| Hemochromatosis | 3(3) |
|
| |
| Cirrhosis | 49 (56) |
|
| |
| Tumor Number | |
| 1 | 69(78) |
| 2–3 | 15(17) |
| ≥4 | 4(5) |
|
| |
| Median tumor size (range) | 6.2 (2–18) |
|
| |
| Pathology Characteristics | |
| CK19 positive | 12 (14) |
| Fibrolamellar HCC | 2(2) |
| Tumor rupture | 2(2) |
|
| |
| Grade | |
| Well differentiated | 6(7) |
| Moderately differentiated | 66(75) |
| Poorly differentiated | 16(18) |
|
| |
| Edmonson-Steiner Grade | |
| 1 | 5(5) |
| 2 | 21(24) |
| 3 | 49(56) |
| 4 | 13(15) |
|
| |
| Vascular invasion | |
| Microvascular | 44(50) |
| Macrovascular (large vessel) | 12(14) |
-patients may have more than one predisposing condition
Gene families were downloaded in May 2012 from the HUGO Gene Nomenclature Committee database (22)(http://www.genenames.org/genefamilies/a-z). For each gene family, we tested for an enrichment of mutations in the genes within the family relative to the genes outside of the family. For each individual, we calculated the per-base mutation rate among the exons of the genes within the gene family and among the exons of the genes outside of the gene family. We then tested whether the average mutation rate within a gene family was higher than the average mutation rate for genes outside the family using a one-sided, paired t-test.
RNA Isolation from Tissue samples and Quantitative Real-time PCR
Total RNA was prepared using Qiagen AllPrep kit (Qiagen, Valencia, CA) and quality was assessed on 2% agarose gel. MVP™ Human Liver Total RNA pool (Agilent Technologies, Santa Clara, CA) was introduced as a standard control. cDNA was synthesized using 200ng random primers (Thermo Scientific, Rockford, IL) and 200U M-MLV Reverse Transcriptase (Life Technologies, Grand Island, NY) from 2μg of each total RNA sample according to the manufacturer’s instructions. All samples within an experiment were reverse transcribed under the same condition, the resulting cDNA diluted 1:5 in nuclease-free water and stored in aliquots at −20°C until used. Real-time PCR with SYBR green detection was performed in Light Cycler 480 System (Roche Applied Science, Indianapolis, IN) in a total reaction volume of 20 μL in 384-well plate. Each reaction contained 5μL of diluted cDNA, 500nM of each primer (as listed in Supplementary Table 1) and 1× LightCyclerR 480 SYBR Green I master mix. The real-time PCR running protocol consisted of (1) 5 min pre-incubation at 95°C; (2) amplification (10 s at 95°C; 10 s at 60°C; 15 s at 72°C); (3) melting curve (10 s at 95°C, 65–97°C at at 2.5°C/s –1 and a continuous fluorescent measurement); and (4) 10 s cooling at 40°C. Relative quantitative analysis was carried out according to the 2−ΔΔCt method(23).
Statistical analyses
Descriptive characteristics of genetic and clinical variables were reported as frequencies and percentages for categorical variables; continuous variable were reported as medians and range. Comparisons of frequencies between genetic and clinical variables were performed using chi-square and Fisher’s exact test, where appropriate. Survival analyses were performed using the Kaplan-Meier method. Univariate survival analysis was performed using the Log-rank tests and multivariate analyses conducted using Cox proportional hazards model.
RESULTS
Clinical characteristics of HCC specimens
Complete clinical data was available for 87 of 89 (98%) tumor samples and is shown in Table 1. Cases included a mix of predisposing disease etiologies including 43% and 21% of patients with hepatitis B and C, respectively. Nineteen cases had multifocal disease at the time of surgery, however only one tumor was submitted for analysis in each case. Positive staining for CK19 in >5% of cells was seen in 12 cases and 2 tumors were fibrolamellar HCC. Median follow-up of cases was 33.8 months (range 3–130 months). There were 3 (3.8%) perioperative deaths within 90 days. A total of 28/87 (32%) patients died with a mean overall survival of 80.6 months with a 5-year overall survival estimated at 76% by Kaplan-Meier analysis. During follow-up there were a total of 44 recurrences for a median disease-free survival of 39.1 months.
Significantly mutated genes in HCC cohort
In total, we found 5820 non-synonymous mutations and 433 nonsense mutations in these 87 tumors (average 66.1, range 4 to 362) or 2.5 mutations per Mb sequenced (Figure 1A). The somatic mutation rate is comparable to those reported in previous studies (11–14). The mutational bias for CpG to A/T transversions in hepatocellular carcinoma was consistent with previous studies (Figure 1B).
Figure 1. Mutation landscape of 87 hepatocellular carcinomas.

The matrix in the center of the figure represents individual mutations in patient samples, color-coded by type of mutation, for the significantly mutated genes. The rate of synonymous and non-synonymous mutations is displayed at the top of the matrix. The barplot on the left of the matrix shows the number of mutations in each gene. The percentages represent the fraction of tumors with at least one mutation in the specified gene. The barplot to the right of the matrix displays the q-values for the most significantly mutated genes. The purple boxplots below the matrix represent the distributions of allelic fractions observed in each sample. The plot at the bottom represents the base substitution distribution of individual samples, using the same categories that were used to calculate significance.
We followed standard statistical analyses to discriminate driver mutations from random mutations (19, 24). We assumed that most of the mutations in cancer genomes represent background noise, while driver genes would be mutated more frequently than expected by chance. We used a binomial probability to estimate the expected number of mutations for each sample. This probability distribution corrects for gene length, due to the assumption that longer genes will be expected to accumulate more mutations by chance. We calculated the background mutation rate using all of the nonsynonymous and synonymous mutations determined in all 87 samples. For each mutated gene, we then calculated the binomial probability of observing at least N mutations, given the background mutation rate. The p-value was adjusted for multiple hypotheses using the Benjamini-Hochberg procedure for controlling False Discovery Rate (FDR). In this analysis, we identified 34genes that were significantly mutated from the discovery cohort, according to a False Discovery Rate cutoff of 10%.
The most frequently mutated genes in this cohort were the well-known oncogene CTNNB1 (10%) and tumor suppressor, TP53 (18%)(Table 2). CTNNB1 mutations and activation of the Wnt pathway have been associated with large (>3cm) tumors, poorly differentiated histology, tumor invasion and metastases as well as HCV-associated HCC. TP53 mutations have been associated with all predisposing etiologies with specific Ser249 mutations associated with Aflatoxin B exposure. KEAP1, encoding kelch-like ECH-associated protein 1, retains NFE2L2/NRF2 in the cytosol and regulates the Keap1-Nrf2 cell defense pathway (25). Previous studies have showed that the Keap1-Nrf2 signaling pathway mediates protective cellular responses to oxidative and xenobiotic damage (26, 27). The roles of IGSF3, ATAD3B and PCMTD1 have not been previously characterized in HCC.
Table 2.
Significantly mutated genes in 87 hepatocellular carcinomas
| Gene | Description | # patients | # sites | Mutation significance (FDR q-val) |
|---|---|---|---|---|
| CTNNB1 | Beta-catenin | 20 | 14 | <1 × 10−11 |
| TP53 | Tumor protein p53 | 17 | 14 | <1 × 10−11 |
| CPA2 | Carboxypeptidase A2 (pancreatic) | 8 | 2 | 3.22 × 10−6 |
| IGSF3 | Immunoglobulin superfamily, member 3 | 8 | 5 | 1.3 × 10−5 |
| KEAP1 | Kelch-like ECH-associated protein 1 | 7 | 7 | 4 × 10−4 |
| ATAD3B | ATPase, AAA domain containing 3B | 4 | 2 | 2.6 × 10−3 |
| PCMTD1 | protein-L-isoaspartate (D-aspartate) O- methyltransferase domain containing 1 | 3 | 2 | 8.3 × 10−3 |
| BRD9 | Bromodomain containing 9 | 5 | 2 | 0.017 |
| TTLL2 | Tubulin tyrosine ligase-like family, member 2 | 4 | 2 | 0.017 |
| TMEM170A | Transmembrane protein 170A | 3 | 1 | 0.037 |
| AR | Androgen receptor | 5 | 5 | 0.038 |
| TMEM51 | Transmembrane protein 51 | 3 | 1 | 0.042 |
| GJA1 | Gap junction protein, alpha 1, 43kDa | 3 | 3 | 0.049 |
Significant enrichment of mutations in histone methyltransferases
To further characterize the pattern of mutated genes and explore their significance of functional pathways in HCC, we analyzed mutations within known gene families (Table 3). Among 4 histone H3 lysine 4 methyltransferases of the MLL family, we validated 13 missense mutations by PCR and Sanger-based resequencing. We identified 2 tumors with MLL mutations, 4 tumors with MLL2 mutations, 1 tumor with MLL3 mutations and 6 tumors with MLL4 mutations (Figure 2A to 2D). Among the MLL gene family, the MLL2 and MLL4 genes seem to be the most likely driver genes in HCC. MLL4 encodes mixed lineage leukemia-4, one of the MLL family of histone H3 lysine-4 (H3K4)-specific methyl transferases. Notably, MLL4 is a recurrent hotspot for hepatitis B virus integration in nearly 12% of hepatocellular carcinoma genomes (28). MLL3 and MLL4 participate in transcriptional coactivator complexes and are necessary for the expression of p53 target genes in response to DNA damage (29). Knockdown of MLL4 reduces cell cycle progression and induces apoptosis (30).
Table 3.
Significantly mutated gene families in 87 hepatocellular carcinomas
| Symbol | Family description | # genes | Mutation rate in family | t-test (rate) |
|---|---|---|---|---|
| NLR | Nucleotide-binding domain and leucine rich repeat containing | 22 | 2.68E-06 | 0.0033 |
| MLL | Mixed lineage leukemia histone methyltransferases | 5 | 2.85E-06 | 0.0037 |
| CACN | Calcium channel subunits | 26 | 2.17E-06 | 0.0041 |
| KMT | Chromatin-modifying enzymes/K-methyltransferases | 26 | 2.13E-06 | 0.0060 |
Figure 2. Protein domain structure of histone methyltransferases.

Mutations are displayed at the protein coding residues, and protein domains are indicated by the colored blocks.
Transcript levels of recurrently mutated genes
We further sought to confirm expression level signatures of 13 recurrently mutated genes in tumor and liver samples used for sequencing analysis. Total RNA was extracted from 49 tumor samples, 8 non-tumor liver samples from HCC patients and normal liver reference RNA. Among the tumors selected for expression analysis, 39 had mutations in recurrently altered genes, 10 tumors lacked mutations in the genes of interest. In the non-tumor liver specimens of HCC cases overexpression of several genes was observed including TP53 (6/8 samples), CTNNB1 (4/8), ATAD3B (5/8), PCMTD1 (5/8), BRD9 (4/8), TTLL2 (4/8), TMEM170A (4/8), TMEM51 (5/8) and GJA1 (3/8), while IGSF3 was underexpressed in 5/8 samples(Figure 3). Among samples with a confirmed TP53 mutation, the gene was overexpressed in 5/9 and underexpresed in 3/9 samples. CTNNB1 was overexpressed in 7/9 tumors with mutations in this gene. KEAP1 expression levels were similar in non-tumor liver samples compared to reference controls, but decreased expression was seen in 4/6 tumors with KEAP1 mutations. Increased expression of genes in samples harbouring mutations was observed for CPA2 (3/6 samples), ATAD3B (1/1), PCMTD (1/1), BRD9 (5/6), TTLL2 (2/4), TMEM170A (2/2), TMEM51 (3/3) and GJA1(2/2).
Figure 3. Expression levels of mutated genes in hepatocellular carcinomas.


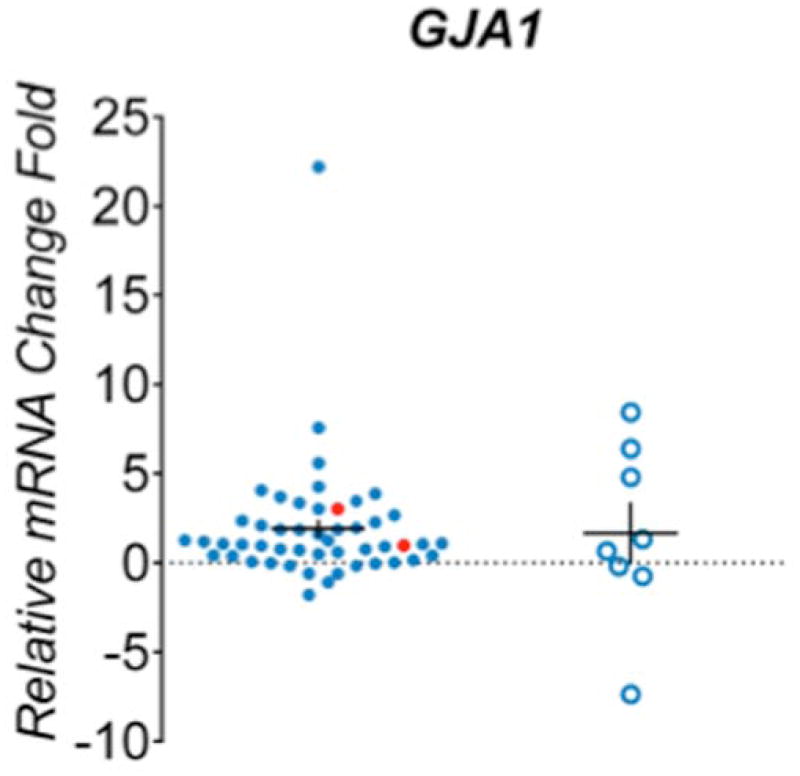
Real-time PCR analysis of 13 genes differentially expressed among 49 HCC tumors(blue dot) and 8 adjacent tissues (blue circle). Samples confirmed with target gene mutation were highlighted in red. The mRNA expression level was normalized to zero based on normal human liver samples. The vertical axis indicates the relative increase (+) or decrease (−) fold-change of each tumor or adjacent sample compared with normal human liver.
Clinical characteristics of HCC according to mutation status
HCC arising from hepatitis C infection demonstrated a significantly higher rate of CTNNB1 mutations (62.5% vs 37.5%, p=0.038) confirming earlier reports associating mutations in b-catenin with HCV. There was also a trend toward higher rates of microvascular invasion in HCC with MLL gene mutations (67% vs 45%, p=0.11). Otherwise, there were no significant associations between individual gene or gene family mutations and clinical variables assessed.
Mutations in TP53 were associated with significantly higher rate of recurrence (89% vs 40%, p=0.006) and shorter disease-free survival (median DFS: 7.9 vs 42.9 months, p=0.001) (Figure 4). There was a trend towards decreased overall survival status among TP53-mutatedtumors (median OS: 26.0 vs 83.2 months, p=0.1) (Supplementary Figure 2). Tumors harboring mutations in the MLL family were associated with a trend toward earlier recurrence with a median disease free survival of 28.9 months for MLL mutation carriers compared to 45.8 months for cases without MLL mutations (p=0.22) and may be associated with a more aggressive disease phenotype(Supplementary Figure 3). A trend towards lower rates of recurrence (12.5% vs 49.3%, p=0.060) and prolonged disease free survival (p=0.23) was observed in cases with CTNNB1 mutations but did not reach statistical significance due to limited power. The presence of CPA2 and KEAP1 mutations were associated with decreased disease-free survival however these analyses lacked sufficient statistical power.
Figure 4.

Disease-free survival according to p53 mutation status.
Univariate analysis of disease-free survival for all clinical and genetic variables identified tumor size (p=0.042), multifocality (p=0.077), and p53 mutation status (p=0.001) as significant or borderline significant predictors of DFS(Table 4). Conditional multivariable survival analysis demonstrated that p53 mutation status was the only independent predictor of disease-free survival with a hazard ratio (HR)=4.245 (95%CI: 1.86- 9.70 (p=0.02)). Tumor multifocality was the only independent predictor of overall survival.
Table 4.
Predictors of disease-free survival (DFS)
| Median DFS months (95%CI) | Univariate Analysis1 | Multivariate analysis HR2 (95%CI), p-value | |
|---|---|---|---|
|
| |||
| P53 | P=0.001 | ||
| No mutation | 42.9 (14–72) | 4.2 (1.9- 9.7), p=0.02 | |
| Mutation (+) | 7.9 (7.2–8.5) | ||
|
| |||
| MLL | P=0.38 | ||
| No mutation | 42.9 (6.1–79.8) | ||
| Mutation (+) | 23.1 (3.9–42.2) | ||
|
| |||
| B-catenin | P=0.51 | ||
| No mutation | 27.1 (4.9–49.3) | ||
| Mutation (+) | 43.0 (1–99.1) | ||
|
| |||
| CPA2 | P=0.25 | ||
| No mutation | 42.9 (20.4–65.5) | ||
| Mutation (+) | 15.3 (5.3–25.2) | ||
|
| |||
| Age | P=0.16 | ||
|
| |||
| Tumour Number | P=0.08 | ||
| Solitary | 44.3 (10.3–78.3) | 2.0(0.97–4.1), p=0.06 | |
| Multifocal | 15.4 (3.0–27.3) | ||
|
| |||
| Tumour Size | P=0.04 | 1.04(0.9–1.08), p=0.08 | |
|
| |||
| HBV | P=1.0 | ||
| Negative | 27.1 (1–53.4) | ||
| Positive | 44.3 (32.1–56.5) | ||
|
| |||
| HCV | P=0.91 | ||
| Negative | 39.0 (15.2–62.9) | ||
| Positive | 27.1 (10–44.2) | ||
|
| |||
| Tumour Grade | P=0.83 | ||
| Well diff | 39.0 (5.2–72.8) | ||
| Moderately diff | 32.9 (14.5–71.5) | ||
| Poorly diff | 21.5 (9.7–33.2) | ||
|
| |||
| Microvascular Inv. | P=0.19 | ||
| Negative | 44.3 (12.8–75.7) | ||
| Positive | 21.8 (9.6–33.9) | ||
Log-rank test
Hazard ratio by Cox proportional hazards model
DISCUSSION
Hepatocellular carcinoma is a genetically heterogeneous disease; this molecular diversity has led many groups to attempt to characterize HCC to improve our understanding of the genes and pathways involved in the etiology of this disease. The goal of genomic and transcriptomic profiling efforts in HCC is to develop a molecular classification of hepatocellular carcinoma that identifies characteristic driver genes that either predict prognosis or eventually could be developed as targets for tailored therapies. This study of 87 matched tumor-normal pairs more than doubles the number of HCC characterized by whole exome sequencing, to a total of 158 tumors. Due to limited sample sizes (ranging from 10 to 27 tumors), it should not be surprising that these studies have not yielded many overlapping genes. Indeed, larger sample cohorts with clinical follow-up data will be required to discern the prognostic significance of recurrently mutated genes.
An interesting emerging consensus from these HCC sequencing studies is the prevalence of mutations in chromatin regulatory enzymes. In particular, several studies have reported mutations in the SWI/SNF-related, ATP-dependent nucleosome remodelers: ARID1A and ARID2(11–14). We only detected 2 mutations in ARID1A(2%) and 1 mutation in ARID2 (1%), despite over 20x coverage of these genomic regions. However, our study concurs with recent reports of mutations in the MLL family of histone H3 lysine4 methyltransferases, which can also be disrupted by genomic integration of hepatitis B virus(14, 28). The clinical characteristics of tumors harboring MLL gene mutations suggest that inactivation of the MLL gene family may be associated with an aggressive tumor phenotype. However, we have not evaluated the functional impact of these mutations on histone methylation. As more data on the MLL gene family are collected, further studies could assess how the most frequent mutations may impair the enzymatic function or recruitment of these enzymes. Further work is needed to elaborate how disrupted chromatin regulators cooperate with alterations in known signaling pathways – such as the Wnt/beta-catenin pathway or Myc targets – in tumor progression, cellular differentiation, or gene expression.
Woo and colleagues had previously demonstrated worse overall survival associated with p53 mutations in a cohort of predominantly Chinese HCC patients with hepatitis B virus etiology (31). This study complements those findings by demonstrating the prognostic value of HCC in a North American series of patients of mixed etiology (HBV/HCV). Combined, this data demonstrates that p53 is associated with recurrence and disease-free survival, oncologic outcomes that reflect an aspect of tumor biology, as well as overall survival, which includes death from both HCC and the underlying liver disease. The observation of p53 as an independent prognostic factor with an ability to predict outcomes in addition to tumor size and number may have important clinical implications in predicting outcomes for patients prior to treatment such as resection or transplantation.
Sorafenib represents the first molecularly targeted therapy for HCC, and the vast majority of HCC clinical trials are currently evaluating the efficacy of tyrosine kinase inhibitors (32, 33). However, the combined analysis of whole exome sequencing from 158 tumors reveals that no single protein kinase that is mutated at more than 5% frequency in hepatocellular carcinoma(9–14). This scarcity of kinase mutations suggests that HCC might be rarely susceptible to the dramatic responses to kinase inhibitors that are observed in other cancer types(34, 35). In contrast, the frequent mutation of MLLhistone methyltransferases – as well as ARID ATP-dependent nucleosome remodeling enzymes identified in previous studies – suggests that epigenetic regulatory enzymes may represent important target genes in HCC.
Since most studies to date have been conducted on surgically resected tumors, we have little knowledge about the genetic alterations that occur in either very early lesions treated with ablative modalities as well as later stages of HCC progression that are not amenable to surgical treatment. Our understanding of tumor evolution could be improved by more sensitive technologies that could sequence genomic DNA from core biopsy specimens. Another confounding issue with genomic profiling is the high rate of intra-tumor heterogeneity. Indeed, a pioneering study demonstrated considerable clonal heterogeneity within a single tumor lesion, with allelic frequencies as low as 13% (10).
In this series, we present the whole-exome sequencing analysis of a large diverse series of HCC tumors and matching normal liver tissue. Our results support the genetic heterogeneity of HCC in that most genes were mutated in few (<20%) of the samples analyzed, however analysis of gene families have indicated potentially important pathways, including MLL and NFE2L2-KEAP1, that are altered in subsets of tumors. Overexpression of several genes of interest were observed in tumors with identified mutations but also in adjacent non-tumor liver samples, which suggests a role of these genes in the pre-malignant “field-effect” that is seen in the unaffected liver of HCC patients (36). We observed phenotypic differences in HCC according to gene mutation status including p53 mutation status as an independent predictor of recurrence-free survival. Several other genes of interest demonstrated trends in time and risk of recurrence; these observations were limited by sample size and require further investigation in larger studies. The lack of correlation between traditional prognostic features such as tumor size, number and vascular invasion indicates that mutational profiling may enhance our ability to develop more predictive models of tumor behavior. Further investigation is required to enhance our understanding of the full breadth of gene mutations in HCC and identify clinically-relevant genes and pathways that can enhance ourunder standing of hepatocarcinogenesis and develop individualized therapy based on HCC genetic signatures.
Supplementary Material
Acknowledgments
Financial Support
Funding was provided by the National Institutes of Health (U24CA143845 to G.G.), and the University Cancer Research Fund (D.Y.C.). D.Y.C. is the recipient of an Alfred P. Sloan Research Fellowship.
We wish to thank Bert O’Neil for a critical revision of this manuscript.
List of Abbreviations
- DFS
Disease-free survival
- HCC
hepatocellular carcinoma
References
- 1.Nault JC, Zucman-Rossi J. Genetics of hepatobiliary carcinogenesis. Semin Liver Dis. 2011;31:173–187. doi: 10.1055/s-0031-1276646. [DOI] [PubMed] [Google Scholar]
- 2.Breuhahn K, Gores G, Schirmacher P. Strategies for hepatocellular carcinoma therapy and diagnostics: lessons learned from high throughput and profiling approaches. Hepatology. 2011;53:2112–2121. doi: 10.1002/hep.24313. [DOI] [PubMed] [Google Scholar]
- 3.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010;52:921–929. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moinzadeh P, Breuhahn K, Stutzer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade - results of an explorative CGH meta-analysis. Br J Cancer. 2005;92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patil MA, Gutgemann I, Zhang J, Ho C, Cheung ST, Ginzinger D, Li R, et al. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and Jab1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis. 2005;26:2050–2057. doi: 10.1093/carcin/bgi178. [DOI] [PubMed] [Google Scholar]
- 6.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, Cozza A, et al. Focal VEGFA gains and molecular classification of hepatocellular carcinoma. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, et al. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol. 2006;44:151–157. doi: 10.1016/j.jhep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Chan TH, Guan XY. Chromosome 1q21 amplification and oncogenes in hepatocellular carcinoma. Acta Pharmacol Sin. 2010;31:1165–1171. doi: 10.1038/aps.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 10.Tao Y, Ruan J, Yeh SH, Lu X, Wang Y, Zhai W, Cai J, et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proc Natl Acad Sci U S A. 2011;108:12042–12047. doi: 10.1073/pnas.1108715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–829. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 15.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seal RL, Gordon SM, Lush MJ, Wright MW, Bruford EA. genenames. org: the HGNC resources in 2011. Nucleic Acids Res. 2011;39:D514–519. doi: 10.1093/nar/gkq892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R, Foster BJ, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285:16782–16788. doi: 10.1074/jbc.M109.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li CQ, Kim MY, Godoy LC, Thiantanawat A, Trudel LJ, Wogan GN. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc Natl Acad Sci U S A. 2009;106:14547–14551. doi: 10.1073/pnas.0907539106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 28.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci U S A. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ansari KI, Kasiri S, Mishra BP, Mandal SS. Mixed lineage leukaemia-4 regulates cell-cycle progression and cell viability and its depletion suppresses growth of xenografted tumor in vivo. Br J Cancer. 2012;107:315–324. doi: 10.1038/bjc.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo HG, Wang XW, Budhu A, Kim YH, Kwon SM, Tang ZY, Sun Z, et al. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology. 2011;140:1063–1070. doi: 10.1053/j.gastro.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–1426. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu AX. Molecularly targeted therapy for advanced hepatocellular carcinoma in 2012: current status and future perspectives. Semin Oncol. 2012;39:493–502. doi: 10.1053/j.seminoncol.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 35.McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, et al. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012;30:1628–1634. doi: 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.