

Abstract
Purpose
Approximately 10–30 % of colorectal cancers exhibit somatic mutations in the phosphoinositide-3-kinase, catalytic, alpha polypeptide gene (PIK3CA). We evaluated the relationship between PIK3CA mutation status and demographic factors, lifestyle factors, and other tumor characteristics and the relationship between PIK3CA mutation status and colorectal cancer survival.
Methods
The population-based study included postmenopausal women with invasive colorectal cancer diagnosed between 1998 and 2002 in Western Washington State. Participants were interviewed, and tumor specimens were tested for PIK3CA mutations in exons 9 and 20 hotspots, KRAS exon 2 mutations, BRAF p.V600E mutation, and microsatellite instability. We used Cox regression to evaluate the association between PIK3CA mutation status and disease-specific and overall survival. Stratified analyses were conducted by KRAS mutation status.
Results
PIK3CA mutations were evident in approximately 13 % of cases (N=35). Women with PIK3CA-mutated colorectal cancer were significantly more likely than those with PIK3CA wild-type disease to be non-white, to have proximal colon cancer, and to have KRAS-mutated tumors (p<0.05). In Cox proportional hazards regression analyses, overall survival was poorer, although not statistically significantly so, for women with PIK3CA-mutated versus wild-type colorectal cancer (hazard ratio=1.74, 95 % confidence interval 0.86–3.50). This association between PIK3CA mutation status and survival was evident only when analyses were restricted to cases without somatic KRAS mutations (hazard ratio=2.94, 95 % confidence interval 1.12–7.73).
Conclusions
PIK3CA-mutated colorectal cancer appears to have a distinct epidemiologic profile that is of clinical significance. Women with PIK3CA-mutated colorectal cancer experience a poorer prognosis than those with PIK3CA wild-type disease.
Keywords: Colorectal cancer, PIK3CA, Survival, KRAS
Introduction
The phosphoinositide-3-kinase, catalytic, alpha polypeptide gene (PIK3CA) is mutated in approximately 10–30 % of all colorectal cancers [1-7]. Activating mutations in PIK3CA result in stimulation of the Akt pathway which, in turn, contributes to increased proliferation and tumor invasion [8, 9]. Although the PI3K/Akt pathway is likely to play a critical role in colorectal tumorigenesis and colorectal cancer progression, the prognostic significance of PIK3CA mutation status, and the descriptive epidemiology of PIK3CA-mutated colorectal cancer, has not yet been well characterized. Previous studies have suggested a possible relationship in colorectal cancer between somatic mutations in the PI3K/Akt and RAS/RAF/MAPK pathways, both of which are EGFR-dependent. In particular, PIK3CA mutations appear to be more common in KRAS-mutated than KRAS wild-type colorectal cancers [1-5]. The relationship between PIK3CA mutation status and other clinically relevant tumor characteristics, however, remains to be elucidated.
Using data from a population-based case–control study of incident invasive postmenopausal colorectal cancer [10], we evaluated differences in tumor characteristics, including KRAS mutation, BRAF mutation, microsatellite instability (MSI), and CpG island methylator phenotype (CIMP) status, as well as differences in patient characteristics and survival after diagnosis in women with PIK3CA-mutated versus PIK3CA wild-type colorectal cancer.
Methods
Study population
Details of the study population have been published elsewhere [10]. Briefly, eligible participants included women diagnosed with invasive colorectal cancer between January 1998 and June 2002 who, at the time of diagnosis, were aged 50–74 years and resided in Clallam, Grays Harbor, Island, Jefferson, Kitsap, Mason, San Juan, Skagit, Thurston, or Whatcom counties in Western Washington State. Women from three large additional counties (King, Pierce, and Snohomish) were also eligible for participation but were not included in the present analysis. All cases were identified through the population-based Surveillance, Epidemiology, and End Results (SEER) cancer registry serving Western Washington State. Study eligibility was limited to English speakers with a publicly available telephone number. Of 439 individuals contacted and identified as eligible, 44 (10 %) were deceased, 37 (8 %) were lost prior to interview, 3 (0.7 %) refused participation, and 2 (0.5 %) completed only a partial interview. In total, 80 % of eligible cases provided informed consent and were enrolled in the study (N=353). The present analysis was limited to enrolled cases for whom diagnostic tumor specimens could be obtained (N=279, 79 %), excluding two (0.7 %) cases for whom collected tumor specimens were not adequate for PIK3CA mutation testing.
At an average 15.9 months after diagnosis (median 2.5 months), participants completed a structured telephone interview in which they were asked to provide detailed information on a number of potential risk factors, including smoking history, body mass index, and use of selected medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
This study was approved by the Institutional Review Board of the Fred Hutchinson Cancer Research Center in accordance with assurances filed with and approved by the U.S. Department of Health and Human Services.
PIK3CA mutation testing and additional tumor characterization
DNA was extracted from paraffin-embedded formalin-fixed (FFPE) tumor tissue using the QIAamp DNA FFPE Tissue kit (QIAGEN, Germantown, MD, USA). For cases for whom tumor DNA was successfully extracted (N=277), pyrosequencing was used to detect mutations in PIK3CA in three hotspots: codons 542 and 545 in exon 9 and codon 1047 in exon 20. These hotspots account for approximately 80 % of all PIK3CA mutations [11, 12]. Pyrosequencing was performed using the PyroMark Q96-MD and Q24 systems (QIAGEN), with an optimized dispensation order to maximize the detection of known variants in the exons 9 and 20 hotspots. For quality control purposes, pyrosequencing was also conducted on three cell lines known to have mutations in these hotspot regions and any failed samples were repeated at least once. A subset of cases (N=20) were tested for PIK3CA mutations using both pyrosequencing and Sanger sequencing of hotspot regions for further assay validation; Sanger sequencing was performed using the BigDyeTerminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Life Technologies, Grand Island, NY, USA) and was run on a 3130xl DNA sequencer (Applied Biosystems, Foster City, CA, USA). Cases for whom testing repeatedly failed or test results were equivocal for mutations in any of these regions were classified as having unknown PIK3CA mutation status (N=2).
Extracted tumor DNA was also tested for mutations in KRAS and BRAF, CIMP, and MSI. With respect to KRAS, the coding sequence of KRAS exon 2 was amplified [13], and mutations in exon 2 were identified via forward and reverse sequencing of amplified tumor DNA [14]. Testing for the c.1799T>A (p.V600E) BRAF mutation was conducted using a fluorescent allele-specific PCR assay as described previously [15]. With respect to MSI status, testing was based on a 10-gene panel assayed in tumor DNA and in DNA extracted from normal surrounding tissue (BAT25, BAT26, BAT40, MYCL, D5S346, D17S250, ACTC, D18S55, D10S197, and BAT34C4) using a 3130xl DNA sequencer (Applied Biosystems) [10, 16]; tumors were classified as MSI-H if instability was observed in ≥30 % of markers and as MSS if instability was observed in <30 % of markers. CIMP status was determined using the MethyLight assay on a 7900HT sequence detection system (Applied Biosystems) for a five-gene panel (CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1) [17]. Based on this assay, cases were classified as CIMP high if at least three of the five markers had a percent methylated of reference (i.e., percent methylated DNA in tumor relative to a control reference sample) greater than 10; otherwise, they were classified as CIMP-negative [17]. Primers for all assays were obtained from Invitrogen (Life Technologies), and all qPCR probes and PCR reagents were obtained from Applied Biosystems (Life Technologies).
Tumor site and stage at diagnosis information were obtained from SEER. Tumors located in the cecum through the splenic flexure were grouped together as proximal colon cancers (ICD-O-3 codes C180, C182, C183, C184, and C185) [18]. Tumors in the descending (C186) and sigmoid colon (C187) were classified as distal colon cancer, and tumors in the rectosigmoid junction (C199) and rectum (C209) were grouped together as rectal cancer. Stage at diagnosis was classified according to the American Joint Committee on Cancer staging conventions (I, II, III, and IV) [19].
Survival information
Vital status was determined periodically via linkage to SEER and the National Death Index. For cases who died during study follow-up, information was obtained on the date and cause of death, classified according to ICD-10 conventions [20]. Deaths with an underlying cause attributed to ICD-10 codes C18.0-C20.0 or C26.0 (i.e., colorectal cancer) were classified as disease-specific mortality events.
Statistical analysis
We compared PIK3CA-mutated and PIK3CA wild-type colorectal cancer cases with respect to the distribution of demographic and lifestyle factors and additional tumor characteristics using chi-square tests. We also used Cox proportional hazards regression to evaluate the association between PIK3CA mutation status and survival after colorectal cancer diagnosis. The time axis for analysis was defined as days since diagnosis; participants were left-censored until the date of study enrollment. We conducted separate analyses using an outcome of all-cause mortality (i.e., overall survival) and death due to colorectal cancer (i.e., disease-specific survival). In analyses of disease-specific survival, persons who died due to causes other than colorectal cancer were censored at the time of death. All regression models were adjusted for age (5-year categories). We also assessed potential confounding by stage at diagnosis (I, II, and III–IV). Proportional hazards assumptions were assessed by testing for a nonzero slope of the scaled Schoenfeld residuals on ranked failure times [21]. All analyses were conducted in Stata SE version 12.0 (College Park, TX, USA).
Results
PIK3CA mutations were detected in approximately 13 % of cases (N=35). Characteristics of the study population are presented in Table 1 by PIK3CA mutation status. Compared to PIK3CA wild-type cases, cases with PIK3CA-mutated colorectal cancer were significantly more likely to be non-white (23 versus 4 %, p<0.001) and were slightly younger at diagnosis. There were no differences in the distribution of smoking history, body mass index, or NSAID use by PIK3CA mutation status. With respect to tumor characteristics, there were also no significant differences in the distribution of stage at diagnosis, CIMP, MSI, or BRAF mutation status by PIK3CA mutation status. However, we did find that PIK3CA-mutated colorectal cancer was significantly more likely to be KRAS-mutated (51 versus 30 %, p=0.01) and exhibited a right-sided shift: compared to PIK3CA wild-type colorectal cancer, PIK3CA-mutated tumors were more likely to be located in the proximal colon and less likely to be located in the rectum (p=0.03).
Table 1.
Patient and tumor characteristics by PIK3CA mutation status
| PIK3CA wild type (N=240) | PIK3CA-mutated (N=35) | Chi-square p value | |
|---|---|---|---|
| Age at diagnosis | |||
| <60 | 69 (29) | 10 (29) | 0.11 |
| 60–69 | 99 (41) | 20 (57) | |
| 70–79 | 72 (30) | 5 (14) | |
| Mean (SD) | 64.0 (7.2) | 63.3 (6.7) | |
| Race | |||
| White | 230 (96) | 27 (77) | <0.001 |
| Non-white | 10 (4) | 8 (23) | |
| Cigarette smoking history | |||
| Never smoker | 118 (49) | 22 (63) | 0.32 |
| Former smoker | 86 (36) | 9 (26) | |
| Current smoker | 36 (15) | 4 (11) | |
| Body mass index (kg/m2) | |||
| <25.0 | 95 (40) | 13 (37) | 0.85 |
| 25.0–29.9 | 71 (30) | 12 (34) | |
| ≥30.0 | 74 (31) | 10 (29) | |
| Ever regularly used nonsteroidal anti-inflammatory drugs | |||
| No | 107 (45) | 20 (57) | 0.19 |
| Yes | 129 (55) | 15 (43) | |
| Missing | 4 | 0 | |
| Stage at diagnosis | |||
| I | 80 (33) | 8 (24) | 0.10 |
| II | 76 (32) | 18 (53) | |
| III | 61 (26) | 9 (24) | |
| IV | 21 (9) | 0 (0) | |
| Missing | 2 | 0 | |
| Tumor site | |||
| Proximal colon | 109 (46) | 21 (60) | 0.03 |
| Distal colon | 56 (24) | 11 (31) | |
| Rectum | 71 (30) | 3 (9) | |
| Missing | 4 | 0 | |
| CpG island methylator phenotype (CIMP) status | |||
| CIMP-negative | 153 (74) | 20 (67) | 0.43 |
| CIMP-positive | 55 (26) | 10 (33) | |
| Missing | 32 | 5 | |
| Microsatellite instability (MSI) | |||
| Microsatellite stable (MSS) | 131 (77) | 24 (89) | 0.16 |
| MSI-high | 39 (23) | 3 (11) | |
| Missing | 70 | 8 | |
| KRAS mutation status | |||
| KRAS wild type | 160 (70) | 17 (49) | 0.01 |
| KRAS-mutated | 70 (30) | 18 (51) | |
| Missing | 10 | 0 | |
| BRAF mutation status | |||
| BRAF wild type | 188 (84) | 28 (80) | 0.60 |
| BRAF-mutated | 37 (16) | 7 (20) | |
| Missing | 15 | 0 | |
Over the course of study follow-up (mean=2 years, range 6 months–11 years), 35 % (N=97) of participants died (Fig. 1). Among those who died, 64 % (N=62) died due to CRC. Small numbers limited survival analyses; however, nonsignificant associations were suggestive of poorer survival in those with PIK3CA-mutated colorectal cancer, particularly with respect to overall survival [hazard ratio (HR)= 1.74, 95 % confidence interval (CI) 0.86–3.50 for overall survival and HR=1.25, 95 % CI 0.49–3.16 for disease-specific survival] (Table 2). Adjusting for stage at diagnosis, in addition to age at diagnosis, had an attenuating effect on point estimates. Further adjustment for all patient and tumor characteristics in Table 1 resulted in a stronger, but not statistically significant association between PIK3CA mutation status and overall survival (HR=1.82, 95 % CI 0.50–6.64), but attenuated the association with disease-specific survival (HR=1.08, 95 % CI 0.19–6.11). Adjustment for lifestyle factors and MSI status had the greatest impact on point estimates (results not shown). Although case numbers were limited, we found the association between PIK3CA mutation status and survival to be limited to women with KRAS wild-type disease: after excluding cases with KRAS-mutated colorectal cancer, age-adjusted point estimates for overall and disease-specific survival were elevated to 2.94 (95 % CI 1.12–7.73) and 2.13 (95 % CI 0.63–7.18), respectively.
Fig. 1.
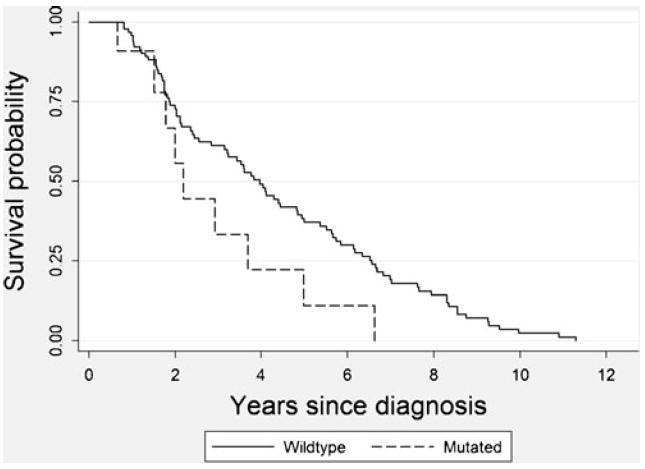
Unadjusted Kaplan–Meier survival curve comparing survival from all causes of death in postmenopausal women with PIK3CA wild-type colorectal cancer (N=240) (solid line) versus PIK3CA-mutated colorectal cancer (N=35) (dashed line). Proportional hazards regression analyses, adjusted for age and stage, provide further support for a poorer prognosis in those with PIK3CA-mutated disease, although point estimates are not statistically significant
Table 2.
Overall and disease-specific survival in relation to PIK3CA mutation status
| PIK3CA wild-type deaths/cases | PIK3CA-mutated deaths/cases | HR (95 % CI)a | HR (95 % CI)a,b | |
|---|---|---|---|---|
| All cases: | ||||
| Overall survival | 88/240 | 9/35 | 1.74 (0.86–3.50) | 1.44 (0.70–2.98) |
| Disease-specific survival | 57/240 | 5/35 | 1.25 (0.49–3.16) | 0.96 (0.37–2.47) |
| Excluding KRAS-mutated cases | ||||
| Overall survival | 60/170 | 5/17 | 2.94 (1.12–7.73) | 2.43 (0.88–6.70) |
| Disease-specific survival | 32/170 | 3/17 | 2.13 (0.63–7.18) | 1.44 (0.40–5.16) |
| Excluding KRAS wild-type cases | ||||
| Overall survival | 27/70 | 4/18 | 1.03 (0.35–3.04) | 0.57 (0.15–2.18) |
| Disease-specific survival | 23/70 | 2/18 | 0.64 (0.15–2.76) | 0.40 (0.07–2.22) |
Hazard ratio (HR) and 95 % confidence interval (CI) for survival in PIK3CA-mutated versus PIK3CA wild-type cases, adjusted for age at diagnosis
Also adjusted for stage at diagnosis (I, II, and III–IV)
Discussion
In this population-based cohort of women with incident invasive colorectal cancer, we found that PIK3CA-mutated tumors were distinct from PIK3CA wild-type tumors with respect to demographic and tumor characteristics and with respect to survival after diagnosis. In particular, we found that PIK3CA-mutated colorectal cancers were significantly more likely to be KRAS-mutated and that, among women with KRAS wild-type disease, the presence of a PIK3CA mutation was associated with significantly poorer survival. Unlike some previous studies, we found no association between PIK3CA mutation status and CIMP [1, 3] or BRAF mutation status [5].
Our findings regarding the relationship between PIK3CA mutation status and KRAS mutation status, and the impact of this relationship on colorectal cancer survival, is consistent with at least two previous analyses [4, 7]. Ogino et al. [4] reported that the presence of a PIK3CA mutation was associated with significantly poorer disease-specific survival in cases with KRAS wild-type colon cancer (HR=3.8, 95 % CI 1.6–9.3) but reported no such association in cases with KRAS-mutated disease. In a cohort of patients with chemotherapy–refractory metastatic colorectal cancer, De Roock et al. [7] found that KRAS wild-type patients with a PIK3CA mutation in exon 20 had significantly poorer overall survival (HR=3.3, 95 % CI 1.5–7.5) and progression-free survival (HR=2.3, 95 % CI 4.7) than those without a PIK3CA exon 20 mutation; however, associations between PIK3CA mutation and survival outcomes were not significant when including patients with KRAS-mutated disease.
The basis for this suggestive pattern of interaction in the association between PIK3CA mutation status and colorectal cancer survival by KRAS mutation status is unclear. However, it is known that KRAS can activate PI3K signaling [22]; thus, it is plausible that the relative impact of a PIK3CA mutation on activation of the PI3K/Akt pathway could be lower in KRAS-mutated colorectal cancer with constitutively active KRAS. Thus, the prognostic significance of a PIK3CA mutation may be limited to the setting where the KRAS oncogene, and the downstream PI3K/AKT signaling pathway, has not been constitutively activated by a KRAS mutation.
The results presented here should be interpreted in the context of study limitations. In particular, due to small numbers, we were not able to conduct analyses distinguishing between cases with PIK3CA mutations in exon 9 (helical domain) versus exon 20 (catalytic domain). These mutations may have differing functional consequences [23] and have been suggested to be associated with slightly different molecular profiles [3] and prognosis [7]. Small numbers also precluded more detailed stratified analyses for survival outcomes. Additionally, despite the population-based nature of this study, the fact that the study was limited to postmenopausal women may impact upon the broader generalizability of findings presented here; however, our findings with respect to survival and the relationship between PIK3CA and KRAS mutation status are consistent with previous studies not limited to postmenopausal women [4, 7]. Lastly, in the absence of detailed treatment information, we were not able to adjust for received treatment in our survival analyses; however, at present, PIK3CA mutations are rarely tested for in clinical settings and, as such, it is unlikely that treatment would have differed according to PIK3CA mutation status.
Inhibition of the PI3K/Akt pathway is being explored as a possible therapeutic approach for colorectal cancer [24], such that testing for the presence of a somatic PIK3CA mutation may become more clinically relevant. The presence of a PIK3CA mutation has also been suggested to predict low response to widely used targeted anti-EGFR therapy, primarily in the absence of a somatic KRAS mutation [7]. All cases enrolled in the present study were diagnosed before anti-EGFR therapy was approved for the treatment of colorectal cancer; therefore, our results demonstrate that the relationship between PIK3CA and KRAS mutation status is significant beyond the setting of anti-EGFR therapy.
Point mutations in the helical (exon 9) and catalytic domains (exon 20) of PIK3CA have previously been hypothesized to contribute to colorectal tumorigenesis by activating the PI3K/Akt signaling pathway and promoting cellular proliferation [8, 9]. Despite small numbers, our results provide support for a role of PIK3CA mutations and, therefore, the PI3K/Akt pathway, in colorectal cancer survival as well. Our results also indicate a distinct clinicopathological profile of PIK3CA-mutated colorectal cancer, which includes an elevated prevalence of mutated KRAS and proximal location. We also observed a markedly higher prevalence of mutated PIK3CA in non-white study participants, which, to our knowledge, has not previously been reported and merits further investigation. Taken together with evidence from other small studies, our results provide support for the epidemiologic and clinical relevance of PIK3CA mutation status in colorectal cancer.
Acknowledgments
This work was supported by the National Cancer Institute, National Institutes of Health, United States Department of Health and Human Services (R01CA076366 to P.A.N.). A.I.P. was supported by a training grant from the National Cancer Institute (R25CA094880). The contribution of P.A.N. was also supported by the National Cancer Institute (K05CA152715 to P.A.N.). We wish to gratefully acknowledge the study participants and staff who made this research possible. We also thank Michelle A. Wurscher (Molecular Epidemiology Lab) and Cassie Sather (Genomics Shared Resource) at Fred Hutchinson Cancer Research Center for their technical expertise.
Footnotes
Conflict of interest The authors declare that they have no conflicts of interest.
Contributor Information
Amanda I. Phipps, Email: aphipps@fhcrc.org, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., M4-C308, Seattle, WA 98109, USA.
Karen W. Makar, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., M4-B402, Seattle, WA 98109, USA
Polly A. Newcomb, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., M4-B402, Seattle, WA 98109, USA
References
- 1.Nosho K, Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Zepf D, et al. PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia. 2008;10:534–541. doi: 10.1593/neo.08336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, et al. Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res. 2012;18:2257–2268. doi: 10.1158/1078-0432.CCR-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitehall VL, Rickman C, Bond CE, Ramsnes I, Greco SA, Umapathy A, et al. Oncogenic PIK3CA mutations in colorectal cancers and polyps. Int J Cancer. 2012;131:813–820. doi: 10.1002/ijc.26440. [DOI] [PubMed] [Google Scholar]
- 4.Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009;27:1477–1484. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S, Jr, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649–1654. doi: 10.1016/j.ejca.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 6.Li HT, Lu YY, An YX, Wang X, Zhao QC. KRAS, BRAF and PIK3CA mutations in human colorectal cancer: relationship with metastatic colorectal cancer. Oncol rep. 2011;25:1691–1697. doi: 10.3892/or.2011.1217. [DOI] [PubMed] [Google Scholar]
- 7.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 8.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 9.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newcomb PA, Zheng Y, Chia VM, Morimoto LM, Doria-Rose VP, Templeton A, Thibodeau SN, Potter JD. Estrogen plus progestin use, microsatellite instability, and the risk of colorectal cancer in women. Cancer Res. 2007;67:7534–7539. doi: 10.1158/0008-5472.CAN-06-4275. [DOI] [PubMed] [Google Scholar]
- 11.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009;8:1352–1358. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker CL, Vaughn CP, Samowitz WS. A PIK3CA pyrosequencing-based assay that excludes pseudogene interference. J Mol Diagn. 2012;14:56–60. doi: 10.1016/j.jmoldx.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Oliner K, Juan T, Suggs S, Wolf M, Sarosi I, Freeman DJ, et al. A comparability study of 5 commercial KRAS tests. Diagn Pathol. 2010;5:23. doi: 10.1186/1746-1596-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alsop K, Mead L, Smith LD, Royce SG, Tesoriero AA, Young JP, et al. Low somatic K-ras mutation frequency in colorectal cancer diagnosed under the age of 45 years. Eur J Cancer. 2006;42:1357–1361. doi: 10.1016/j.ejca.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 15.Buchanan DD, Sweet K, Drini M, Jenkins MA, Win AK, English DR, et al. Risk factors for colorectal cancer in patients with multiple serrated polyps: a cross-sectional case series from genetics clinics. PLoS One. 2010;5:e11636. doi: 10.1371/journal.pone.0011636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 17.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 18.World Health Organization. International classification of diseases for oncology. WHO; Geneva: 2000. [Google Scholar]
- 19.Greene FL, Page DL, Fleming ID, Fritz AG, Balch CM, Haller DG, Morrow M. AJCC cancer staging handbook. 6. Springer; New York: 2002. [Google Scholar]
- 20.World Health Organization. International classification of diseases. WHO; Geneva: 2007. [Google Scholar]
- 21.Therneau TM, Grambsch PM. Modeling survival data: extending the Cox model. Springer; New York: 2000. [Google Scholar]
- 22.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 23.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrido-Laguna I, Hong DS, Janku F, Nguyen LM, Falchook GS, Fu S, et al. KRASness and PIK3CAness in patients with advanced colorectal cancer: outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS One. 2012;7:e38033. doi: 10.1371/journal.pone.0038033. [DOI] [PMC free article] [PubMed] [Google Scholar]