

Abstract
Status epilepticus (SE) triggers abnormal expression of genes in the hippocampus, such as glutamate receptor subunit epsilon-2 (Grin2b/Nr2b) and brain-derived neurotrophic factor (Bdnf), that is thought to occur in temporal lobe epilepsy (TLE). We examined the underlying DNA methylation mechanisms and investigated whether these mechanisms contribute to the expression of these gene targets in the epileptic hippocampus. Experimental TLE was provoked by kainic acid-induced SE. Bisulfite sequencing analysis revealed increased Grin2b/Nr2b and decreased Bdnf DNA methylation levels that corresponded to decreased Grin2b/Nr2b and increased Bdnf mRNA and protein expression in the epileptic hippocampus. Blockade of DNA methyltransferase (DNMT) activity with zebularine decreased global DNA methylation levels and reduced Grin2b/Nr2b, but not Bdnf, DNA methylation levels. Interestingly, we found that DNMT blockade further decreased Grin2b/Nr2b mRNA expression whereas GRIN2B protein expression increased in the epileptic hippocampus, suggesting that a posttranscriptional mechanism may be involved. Using chromatin immunoprecipitation analysis we found that DNMT inhibition restored the decreases in AP2alpha transcription factor levels at the Grin2b/Nr2b promoter in the epileptic hippocampus. DNMT inhibition increased field excitatory postsynaptic potential in hippocampal slices isolated from epileptic rats. EEG monitoring confirmed that DNMT inhibition did not significantly alter disease course, but promoted the latency to seizure onset or SE. Thus, DNA methylation may be an early event triggered by SE that persists late into the epileptic hippocampus to contribute to gene expression changes in TLE.
Keywords: epigenetics, bdnf, Grin2b/Nr2b, epilepsy, seizures, DNA demethylation
Introduction
Epilepsy is a neurological disorder characterized by recurrent unprovoked seizures. In humans, temporal lobe epilepsy (TLE) is the most common adult form of epilepsy and can be triggered by an insult such as status epilepticus (SE) or prolonged seizure activity. SE results in changes in gene expression that are thought to subsequently lead to molecular and structural changes to produce the epileptic phenotype (Jones et al., 2001, Ma et al., 2009b, Kobow and Blumcke, 2011). However, to date, the underlying transcriptional mechanisms that orchestrate aberrant steady-state gene expression changes in TLE are still uncertain. Investigation of these underlying mechanisms is important because targeting them may help to mitigate or disrupt the epileptic phenotype.
DNA methylation is a potent epigenetic regulator of chromatin structure that controls persistent gene expression in the central nervous system (CNS) (Jiang et al., 2008, Lubin et al., 2008). DNA methylation in the CNS exists in two methylation forms: 5-methylcytosine (5-mC) formation is catalyzed by DNA methyltransferases (DNMTs) from 5-cytosine (5-C), and 5-hydroxymethylcytosine (5-hmC) formation is catalyzed by TET1 from 5-mC (Kriaucionis and Heintz, 2009, Munzel et al., 2010, Guo et al., 2011). DNA methylation is thought to occur primarily during neuronal development and differentiation, and to remain static thereafter. However, a number of studies have challenged this idea and recent evidence suggests that DNA methylation is in fact both a dynamic and persistent molecular process controlling gene transcription in post-mitotic neurons in the adult CNS (Levenson et al., 2006, Jiang et al., 2008, Nelson et al., 2008, Feng and Fan, 2009, Feng et al., 2010). Additionally, numerous studies have linked dysregulation of DNA methylation to several neurological disorders, including Rett syndrome, schizophrenia, and depression (Amir et al., 1999, Connor and Akbarian, 2008, Feng and Fan, 2009, Higuchi et al., 2011, Sales et al., 2011). Despite the link between DNA methylation and other neurological disorders, its role in aberrant transcriptional gene regulation in epilepsy disorders has not been fully explored (reviewed in Lubin, 2012). Therefore, we used kainic acid (KA)-induced SE as an experimental TLE model system to investigate the contribution of DNA methylation to gene expression changes in TLE. Specifically, we focused on a potential role for DNA methylation in the abnormal transcriptional regulation of the Glutamate receptor subunit epsilon-2 also known as N-methyl D-aspartate receptor subtype 2B (Grin2b/Nr2b) and brain-derived neurotrophic factor (Bdnf) genes in the epileptic hippocampus, both of which have been reported to accompany, and perhaps contribute to TLE (Mathern et al., 1998, Tongiorgi et al., 2004, Bovolenta et al., 2010, Ghasemi and Schachter, 2011).
The present study was undertaken to investigate whether DNA methylation contributes to Grin2b and Bdnf expression during the epileptogenic process triggered by SE. We found that SE triggered increases in DNA methylation levels at the Grin2b/Nr2b promoter and decreased DNA methylation levels at the Bdnf promoter with a positive correlation on Grin2b/Nr2b and Bdnf gene and protein expression levels in the epileptic hippocampus. We found that DNMT blockade had no effect on Bdnf DNA methylation. However, DNMT inhibition attenuated both global DNA methylation and Grin2b/Nr2b gene-specific DNA methylation levels, corresponding with increased binding of the AP2alpha transcription factor at the Grin2b/Nr2b promoter and increased GRIN2B/NR2B protein expression in the zebularine-treated epileptic hippocampus. Intriguingly, inhibiting DNMT activity during the initial SE insult further increased field excitatory postsynaptic potentials (fEPSP) in the zebularine-treated epileptic hippocampus. Results suggest that alterations of methylating and demethylating enzymes that facilitate DNA methylation mechanisms appear to be early and late events triggered by SE that serve to control hippocampal gene expression and subsequent protein expression in an experimental model of TLE.
Experimental Procedures
Animals
Adult Male Sprague Dawley rats (150-200 g) were used for all experiments. Animals were housed in a 12 hour (hr) light/dark cycle and allowed access to food and water ad libitum. Animals were handled for 3 to 5 days before use in experiments. All procedures were performed with the approval of the University of Alabama at Birmingham Institutional Animal Care and Use Committee and according to national guidelines and policies.
Kainate treatment
Animals were injected with kainic acid (KA) [15 mg/kg; (Tocris Cookson Inc., Ellisville, MO, USA)] or saline (vehicle) intraperitoneally (IP). The severity of behavioral seizures following KA injection was scored according to the Racine scale (Racine, 1972): score 1, mouth and face clonus and head nodding; score 2, clonic jerks of one forelimb; score 3, bilateral forelimb clonus; score 4, forelimb clonus and rearing; score 5, forelimb clonus with rearing and falling. The onset of status epilepticus (SE) was defined as the time from KA injection to the occurrence of continuous seizure activity (scores 4 or 5 in the Racine scale). One cohort of animals were sacrificed at 1 hour after the onset of SE while another cohort was sacrificed 6 weeks after the onset of SE with the control animals being sacrificed in parallel. A 3rd cohort of animals was sacrificed at 14 days after onset of SE. All control animals were handled in the same manner as the kainate treated animals, except for KA administration. All kainate treated animals used at the 14 day or 6 week time point had observable seizures. For tissue collection, the hippocampus was removed, oxygenated (95%/5% O2/CO2) in ice cold cutting solution (110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.5 mM CaCl2, 7 mM MgCl2, 5 mM glucose, 0.6 mM ascorbate), and then areas CA1, CA3, and the DG region were microdissected and frozen immediately on dry ice. The microdissection was performed with the aid of a dissecting microscope. The hippocampus was bisected with the dorsomedial half being divided into four pieces. Using anatomic landmarks, each piece was dissected into CA1, CA3, and the DG region. CA3 was dissected with a cut connecting the ends of the inner and outer blade of the DG. The DG and CA1 were dissected with a cut along the hippocampal fissure. The tissue was then stored in a −80°C freezer until RNA and DNA extractions were performed.
Intracerebralventricular (ICV) cannula implantation and drug treatment
For administration of the drugs to the entire brain, a unilateral 23 gauge single guide cannula (Plastics One) was implanted in each rat. Guide implantation was alternated between the left and right ventricles. The injector extended 1.5 mm beyond the guide. The stereotaxic coordinates used were: anteroposterior, −1.0 mm from bregma, ±1.2 mm lateral from the midline, and −3.5 mm from dura measured from the tip of the cannula guide (Lei et al., 2004, Lubin et al., 2007, Lubin and Sweatt, 2007). Animals were habituated to the dummy cannula removal and were allowed 5 days to recover and be handled before start of experimentation. Animals were then infused with 5 μL of zebularine (600 ng per μL in 10% DMSO) or vehicle (saline in 10% DMSO) 1 hour before KA injection. KA injections were performed as described previously. Animals were monitored throughout the process of entry into SE using the Racine scale as described previously.
Intra-CA1 cannula implantation and drug treatment
For administration of drugs into area CA1 of the hippocampus, a 26 gauge stainless steel guide cannula (Plastics One) was implanted bilaterally in each rat. The injector extended 1 mm beyond the guide to end in area CA1. The stereotaxic coordinates used were: anterior-posterior, −3.6 mm from bregma, ±1.7 mm lateral from the midline, and −2.6 mm from dura (Paxinos G, 1998, Lubin et al., 2008) measured from the tip of the cannula guide. Animals were habituated to the dummy cannula removal and were allowed 5 days to recover and be handled before start of experimentation. Following recovery, animals were then infused with 2 μL of zebularine (600 ng per μL in 10% DMSO) or vehicle (saline in 10% DMSO) 1 hour before KA injection. KA injections were performed as described previously. Animals were monitored using the Racine scale throughout the process of entry into SE, were then allowed to recover from SE, and returned to their holding room until they were sacrificed 14 days post-SE.
EEG Recordings
Electrodes for EEG recordings (Plastics One, Inc., Roanoke, VA) were trimmed to 1.75 mm in length and fitted into 3 holes so that they contacted the dura and the connector was flush with the skull. The ground wire was placed into the most caudal hole. Simultaneously, intra-ICV cannulas were implanted into the animals and experiments were carried out 5 days post-surgeries as described above in the section intracerebralventricular (ICV) cannula implantation and drug treatment. Following KA administration, animals were returned to their home cage. For EEG recordings, animals were transferred to individual housing in custom-designed and constructed plexiglass cages. EEG data was acquired using 8 Biopac Systems amplifiers and AcqKnowlege 4.1 EEG Acquisition and Reader Software (BIOPAC Systems, Inc.). Data was stored and analyzed in digital format. Each cage was also equipped with an IR Digital Color CCD camera (Lorex Technology, Inc.) and animals are recorded concurrent with EEG monitoring. All EEG data were analyzed manually by an observer blinded to experimental identity. Abnormalities in the recordings indicative of epileptic activity are aligned chronologically with the corresponding video in order to confirm seizures.
Electrophysiology
14 days following zebularine and KA treatment, animals were deeply anesthetized with isofluorane before being decapitated. The brains were then quickly removed and placed in ice-cold cutting solution containing (in mM) 135 N-methyl-D-glucamine, 1.5 KCl, 1.5 KH2PO4, 23 choline HCO3, 0.4 ascorbic acid, 0.5 CaCl2, 3.5 MgCl2, and 25 D-glucose. The solution was bubbled with 95% O2/5% CO2 to maintain a pH around 7.4. Coronal brain slices, including the hippocampus, were cut using a vibratome (Microm, Waldorf Germany). Slices rested for 40-60 minutes (min) at 37° C, and then were kept at room temperature until the experiments were performed. Slices were transferred to a recording chamber and continuously perfused (4 mL/min) with oxygenated recording solution containing (in mM) 124 NaCL, 2.5 KCl, 10 D-Glucose, 26 NaHCO3, 2.0 Ca2+, 2.0 Mg2+. Extracellular field potentials were recorded by lowering a glass electrode (3-5 megohms) filled with recording solution into the stratum radiatum of area CA1 of the hippocampus. Field responses were evoked via a single shock (190 μs) from a bipolar stimulating electrode placed within the Schaffer collateral pathway. Electrical signals were amplified with an Axoclamp 1A amplifer (Molecular Devices), digitized with a Digidata 1322A (Axon Instruments), and stored on a computer using clampex (Molecular Devices) for later analysis.
Real time PCR (Rt-PCR)
Total RNA was extracted from CA1, CA3, and DG subregions of the hippocampus using the AllPrep DNA/RNA Mini Kit by Qiagen. Total mRNA was converted to cDNA using the iSript cDNA synthesis kit (Bio-Rad) and cDNA was then pre-amplified at 95.0°C for 10 min, 95.0°C for 15 s (20 repeats), 60.0°C for 1 min, and, finally, held at 4.0°C. Real-Time PCR amplifications of the cDNA was performed on the iQ5 real-time PCR system (Bio-Rad) using the following protocol: 95.0°C for 3 min, then 95.0°C for 10 s, followed by 62.6°C for 30 s (50 repeats), 95.0°C for 1 min, 55.0°C for 1 min, followed by a melt curve starting at 55.0°C for 10 s (81 repeats), and then held at 4.0°C. Primers used to amplify specific cDNA regions of interest were the following: Apobec1 (sense 5’-GGGAACTTCGGAAAGAGACC-3’; antisense 5’– CCAGGACAGGAACCAGGTAA-3’), Bdnf exon IX (sense 5’–GAGAAGAGTGATGACCATCCT-3’; antisense 5’–TCACGTGCTCAAAAGTGTCAG-3’), Dnmt1 (sense 5’–CCACCACCAAGCTGGTCTAT-3’; antisense 5’–TACGGCCAAGTTAGGACACC-3’), Dnmt3a (sense 5’-GAGGGAACTGAGACCCCAC-3’; antisense 5’-CTGGAAGGTGAGTCTTGGCA-3’), Dnmt3b (sense 5’-GTTAATGGGAACTTCAGTGACCAA-3’; antisense 5’-CTGCGTGTAATTCAGAAGGCT-3’), Grin2b/Nr2b (sense 5’-AGGAACCAGGCTACATCAAAAA-3’; antisense 5’- TAGTGATCCCACTGCCATGTAG-3’), TET1 (sense 5’-TGTCACCTGTTGCATGGATT-3’: antisense 5’–TTGGATCTTGGCTTTCATCC-3’), Gadd45a (sense 5’-TCATTCGTGCTTTCTGTTGC-3’: antisense 5’-TCCCGGCAAAAACAAAATAAG-3’), Gadd45b (sense 5’-GCAACAGAAAGCACGAATGA-3’: antisense 5’-CCAAGCTGCTCAACGTGTAA-3’) and Apobec1 (sense 5’-GGGAACTTCGGAAAGAGACC-3’: antisense 5’-CCAGGACAGGAACCAGGTAA-3’). All genes were run in duplicate and compared to ribosomal 18s (r18s) (sense 5’-CGGCTACCACATCCAAGGAA-3’; antisense 5’–GCTGGAATTACCGCGGCT-3’). Expression of r18s remained unchanged across treatment groups. Cycle threshold (Ct) values were analyzed using the comparative Ct method to calculate differences in gene expression between samples (Livak and Schmittgen, 2001, Pfaffl, 2001). The same Dnmt3a and Dnmt3b mRNA primers were used as in (LaPlant et al., 2010) while the Bdnf exon IX and r18s primers were from (Lubin et al., 2008).
Determining total DNA 5-methylcytosine
Total 5-methylcytosine of each sample was determined using the MethylFlash Methylated DNA Quantification Kit (Colorimetric) by Epigentek. 100 ng of DNA was used per each reaction and each sample was run in duplicate.
Determining total DNA 5-hydroxymethylcytosine
Total 5-hydroxymethylcytosine of each sample was determined using MethylFlash Hydroxymethylated DNA Quantification Kit (Colorimetric) by Epigentek. 100 ng of DNA was used per each reaction and each sample was run in duplicate.
Direct Bisulfite DNA sequencing
1 μg of DNA was prepared for bisulfite modification using the EpiTect Bisulfite Kit by Qiagen. Bisulfite treated DNA was then amplified for a primer targeting 13 sites in cytosine phosphodiester guanine (CpG) island 3 (see Fig 3A) of the Grin2b/Nr2b promoter with the sense strand as 5’- TTTTTTAGGGGAGAGGTTGAGTAGC-3’; and the antisense strand as 5’- AATAAAACACACTAACACGCGCGTA-3’ with a product size of 220 base pairs. Bisulfite treated DNA was also amplified for a primer designed to target 12 CpG sites in the promoter region of Bdnf exon IX (see Fig. 4A) with the sense strand as 5’-GTGAATGGGTTTAGGGTAGGTT-3’; and the antisense strand as 5’–CCAACAAAAAAAACAAAAAAAACTC-3’ with a product size of 200 base pairs. The thermocycler protocol used to amplify both primers was as follows: 5 min at 95°C, 50 repeats at 95°C for 1 min, followed by 60°C for 1 min, followed by 72°C for 1 min, which was then followed by a final cycle of 5 min at 72°C and then terminated at 4°C. The PCR products were then cleaned using ExoSAP-IT (Affymetrix) and each sample was sequenced in duplicate using the reverse primer at the University of Alabama at Birmingham Genomics Core Facility of the Heflin Center for Human Genetics (http://www.heflingenetics.uab.edu). Using Chromas software to read the electropherogram, the percent methylation of the CpG sites was then determined by the ratio between peak values of guanine (G) and adenine (A) (G/(G +A)). In brief, percent methylation levels for each CpG site within the DNA amplicon was quantified by measuring the ratio between peak height values of cytosine (C) and thymine (T), yielding the basic equation for the methylation percentage to be (C/(C+T)*100). Note that this equation only applies in cases where the forward primer is used for DNA sequencing. If the reverse primer was used, the guanine (G) and adenine (A) peak heights were used instead, yielding the equation (G/(G+A)*100). In our present studies, sequencing was performed with the reverse primer because it results in a cleaner chromatogram and more consistent analysis of DNA methylation. An extended protocol of the direct bisulfite sequencing can be found in (Parrish et al., 2012). Western Blotting. For quantification of BDNF and NR2B, protein extracts (10μg) were separated on a 10% polyacrylamide gel with a 4% stacking gel. The proteins were transferred onto an Immobilon-FL membrane which then was probed with the following primary antibodies: (BDNF (1:1000, Santa Cruz. Cat. No. sc-546) and NR2B (1:1000, Antibodies Incorporated. Cat. No. 75-101). Secondary goat anti-rabbit or goat anti-mouse 800CW antibody was used for detection of the proteins using the Licor Odyssey system. All quantifications were normalized to Actin levels (1:1000, Abcam. Cat. No. ab1801).
Figure 3.

Increased Bdnf mRNA and protein expression in epilepsy correlates to decreased DNA methylation levels in the epileptic hippocampus. A) Diagram of the CpG sites analyzed in the Bdnf coding exon IX. The putative transcription factor binding sites encompassing CpG sites 2 and 4 are shown that are important for activity-dependent gene transcription. B1) Overall Bdnf DNA methylation levels were decreased in hippocampal areas CA1, CA3 and the DG region from epileptic animals. (CA1, t(5) = 3.22, p = 0.02, n = 6; CA3, t(5) = 7.18, p = 0.0008, n = 6; DG, t(5) = 4.79, p = 0.005, n = 6; student paired t-test, *p<0.05, **p<0.01, ***p<0.001). B2) BDNF DNA methylation analysis across 12 CpG sites at the Bdnf gene in area CA1 demonstrates site-specific CpG methylation changes during the epilepsy phase (Student paired t-test, *p<0.05, n=6). C) Bdnf mRNA levels were significantly increased in hippocampal areas CA1, CA3, and DG region from epileptic animals relative to controls (CA1, t(9) = 2.55, p = 0.03, n = 5-6; CA3, t(11) = 2.37, p = 0.04, n = 6-7; DG, t(14) = 2.50, p = 0.03, n = 6) D) BDNF protein was significantly increased in area CA1 of the epileptic animals relative to controls (t(6) = 2.68, p = 0.04, n = 4). Student unpaired t-test, *p<0.05. Error bars are SEM.
Figure 4.

Grin2b/Nr2b and Bdnf DNA methylation levels in the hippocampus during kainic acid-induced SE. A1) Grin2b/Nr2b DNA methylation was significantly increased in hippocampal area CA1 of the hippocampus during SE compared to controls. No significant change in Grin2b/Nr2b DNA methylation levels were found in area CA3 and the DG region of the hippocampus during SE relative to controls (CA1, t(6) = 2.47, p = 0.04, n = 7; CA3, t(6) = 0.77, p = 0.47, n = 7; DG, t(6) = 0.57, p = 0.59, n = 7; student paired t-test, *p<0.05). A2) DNA Methylation analysis of 13 CpG dinucleotides at the Grin2b/Nr2b gene show significant increases in specific Grin2b/Nr2b DNA methylation at CpG sites in area CA1 during SE compared to controls (Student unpaired t-test, *p<0.05, **p<0.01, n=7-8). B1) No significant change in Bdnf DNA methylation levels were found in areas CA1 and CA3 and the DG region of the hippocampus during SE relative to control animals (CA1, t(6) = 0.98, p = 0.04, n = 7; CA3, t(6) = 1.77, p = 0.13, n = 7; DG, t(6) = 0.78, p = 0.46, n = 7; student paired t-test, p>0.05). B2) No significant changes were found at the Bdnf CpG sites assessed in area CA1 of the hippocampus during SE compared to controls (Student unpaired t-test, p>0.05, n=7-8). Student unpaired t-test, *p<0.05, **p<0.01. Error bars are SEM.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed as previously described (Gupta et al., 2010, Gupta-Agarwal et al., 2012). Briefly, micro-dissected tissue was held in ice-cold PBS solution containing protease inhibitors. The protein was crosslinked using 1% formaldehyde in PBS at 37°C for 10 min, followed by washes with ice-cold PBS containing protease inhibitors. Tissue homogenization was carried out in SDS lysis buffer (50 mM Tris, pH 8.1, 10 mM EDTA, 1% SDS) and subjected to shearing using a Diagenode Biorruptor on high power for 3x 10 cycles (30 sec ON/30 sec OFF) with sonication following every 10 cycles.. Extracts were precleared with 50% suspension of salmon sperm-saturated protein A overnight. Immunoprecipitations were carried out overnight at 4°C with the primary antibody (AP2alpha) or no antibody (control). Immune complexes were recovered with protein A followed up by consecutive washes with low salt buffer, high salt buffer, LiCl immune complex buffer, and Tris-EDTA (TE) buffer. The immunocomplex was extracted in 1X TE buffer and the protein-DNA crosslink was reverted by overnight treatment at 65°C. The samples were subjected to proteinase K digestion (100 μg; 2 h at 37°C), and DNA extraction via phenol/chloroform/isoamyl alcohol and precipitation using ethanol. All DNA samples were pre-amplified at 95.0°C for 10 mins, 20 repeats of 95.0°C 15 secs and 60.0°C for 1 min and held at 4.0°C. RT-q PCR was performed on the immunoprecipitated DNA using primers specific to the NR2B gene promoter with the sense strand as 5’-CTAGGGGAGAGGCTGAGCA-3’; and the antisense strand as 5’-GCGCGCACTCACACTCAC-3’ with a product size of 150 base pairs. All amplicon quantifications were normalized to input DNA.
Statistical Analysis
Relative mRNA fold changes for all genes and brain regions were analyzed using the comparative CT method (Livak and Schmittgen, 2001, Pfaffl, 2001). Differences in DNA methylation, mRNA expression, and protein expression were assessed using either a two-tailed unpaired t-test or a two-tailed paired t-test where appropriate. The field recording data was analyzed using a two-way ANOVA with a Bonferroni's post hoc test and analysis of covariance. GraphPad Prism was used for all statistical analysis of data. Significance was set at p≤0.05 for all analyses.
Results
Effect of status epilepticus on global 5-methylcytosine and 5-hydroxymethylcytosine DNA methylation levels in the hippocampus
Kainic acid (KA)-induced SE (15 mg/kg, IP) triggers early and late responses in hippocampal gene expression. Therefore, we first sought to characterize global DNA methylation levels in the hippocampus early at 1 hr into SE in one cohort of animals and late at 6 weeks post-SE in another cohort of animals (experimental design outlined in Fig. 1A). The hippocampus was isolated and sub-dissected into three major hippocampal subregions: CA1, CA3 and the dentate gyrus (DG) regions (Fig. 1A). Analysis of global 5-mC DNA methylation revealed no significant differences in 5-mC levels from all hippocampal regions assessed at 1 hr into SE compared to controls (Fig. 1B). However, at 6-weeks post-SE global 5-mC DNA methylation levels significantly increased in areas CA1 and CA3, and significantly decreased in the DG region of epileptic animals (Fig. 1B). Together, these results indicate that DNA methylation in the form of 5-mC is a molecular event that occurs in the hippocampus, developing over time after SE.
Figure 1.

KA-induced SE triggers changes in global hippocampal DNA methylation levels and expression of genes encoding for DNMT and TET methylating enzymes. A) Diagram of experimental setup and an illustration of the method for sub-dissection of hippocampal regions using the fissure markings of the hippocampus. Dotted lines indicate cuts made to dissect hippocampal regions: CA1, CA3, and the DG region. B) 5-mC DNA methylation levels remained unchanged in all hippocampal regions assessed during SE but were significantly increased later in areas CA1 and CA3 of the hippocampus from epileptic animals compared to controls. However, in the DG region of the hippocampus, 5-mC DNA methylation levels were significantly decreased following epilepsy onset (CA1 SE, t(13) = 0.68, p = 0.51, n = 7-8; CA1 epileptic, t(14) = 2.34, p = 0.03, n = 8; CA3 SE, t(13) = 1.17, p = 0.26, n = 7-8; CA3 epileptic, t(16) = 2.12, p = 0.05, n = 9; DG SE, t(13) = 0.94, p = 0.36, n = 7-8; DG epileptic, t(11) = 3.65, p = 0.003, n = 6-7). C) 5-hmC DNA methylation levels were not significantly altered in area CA1 of the hippocampus during SE or following the onset of epilepsy. 5-hmC DNA methylation levels were significantly decreased in hippocampal area CA3 during SE and later returned to baseline levels in the hippocampus of epileptic rats compared to controls. 5-hmC DNA methylation levels were unchanged in the DG region during SE but were significantly decreased in the DG region of the epileptic hippocampus relative to controls (CA1 SE, t(9) = 0.28, p = 0.78, n = 4-7; CA1 epileptic, t(10) = 0.01, p = 0.99, n = 5-7; CA3 SE, t(11) = 3.90, p = 0.003, n = 6-7; CA3 epileptic, t(9) = 0.93, p = 0.37, n = 5-6; DG SE, t(12) = 0.20, p = 0.84, n = 6-8; DG epileptic, t(10) = 3.15, p = 0.01, n = 5-7). D) DNMT1 mRNA levels were not significantly altered in hippocampal area CA1 during SE or in the epileptic hippocampus compared to controls. DNMT1 mRNA levels were significantly decreased in hippocampal area CA3 during SE, while no changes were observed in area CA3 of the epileptic hippocampus compared to controls. DNMT1 mRNA levels were significantly decreased in the DG region during SE, while no change occurred in the DG region of the epileptic hippocampus relative to controls (CA1 SE, t(13) = 0.92, p = 0.37, n = 7-8; CA1 epileptic, t(11) = 0.10, p = 0.93, n = 6-7; CA3 SE, t(13) = 2.28, p = 0.04, n = 7-8; CA3 epileptic, t(11) = 1.94, p = 0.08, n = 6-7; DG SE, t(13) = 3.14, p = 0.008, n = 7-8; DG epileptic, t(10) = 0.35, p = 0.73, n = 6). E) DNMT3a mRNA levels were significantly decreased in area CA1 at during SE and at the epileptic stage compared to controls. DNMT3a mRNA levels in area CA3 were unchanged during SE and significantly decreased later in the epileptic hippocampus compared to controls. DNMT3a mRNA levels were unchanged in the DG region during SE and in the epileptic hippocampus relative to controls (CA1 SE, t(13) = 2.40, p = 0.03, n = 7-8; CA1 epileptic, t(15) = 3.248, p = 0.005, n = 8-9; CA3 SE, t(12) = 0.40, p = 0.70, n = 6-8; CA3 epileptic, t(16) = 2.56, p = 0.02, n = 9; DG SE, t(13) = 0.60, p = 0.57, n = 7-8; DG epileptic, t(13) = 0.99, p = 0.34, n = 6-9). F) DNMT3b mRNA levels were unchanged in hippocampal area CA1 during SE and in area CA1 of the epileptic hippocampus compared to controls. DNMT3b mRNA levels were unchanged in area CA3 during SE while being significantly increased in area CA3 of the epileptic hippocampus compared to controls. DNMT3b mRNA levels remained unchanged in the DG region during SE and in the DG region of the epileptic hippocampus relative to controls (CA1 SE, t(13) = 0.80, p = 0.44, n = 7-8; CA1 epileptic, t(15) = 0.62, p = 0.54, n = 8-9; CA3 SE, t(12) = 0.37, p = 0.72, n = 6-8; CA3 epileptic, t(14) = 2.33, p = 0.04, n = 8; DG SE, t(13) = 0.69, p = 0.50, n = 7-8; DG epileptic, t(11) = 0.32, p = 0.76, n = 5-8). G) No changes in TET1 mRNA levels were observed in area CA1 during SE or in the epileptic hippocampus compared to controls. TET1 mRNA levels were significantly decreased in area CA3 and the DG region during SE relative to controls. (CA1 SE, t(13) = 0.14, p = 0.90, n = 7-8; CA1 epileptic, t(10) = 0.56, p = 0.59, n = 6; CA3 SE, t(13) = 2.50, p = 0.03, n = 7-8; CA3 epileptic, t(10) = 0.85, p = 0.42, n = 6; DG SE, t(13) = 3.14, p = 0.008, n = 7-8; DG epileptic, t(10) = 0.49, p = 0.63, n = 6). Student unpaired t-test, *p<0.05, **p<0.01. Error bars are SEM.
Because both 5-mC and 5-hmC contribute to genome-wide DNA methylation in neurons, we also assessed 5-hmC DNA methylation levels in the hippocampus early and late after SE. Global 5-hmC DNA methylation levels significantly decreased in area CA3 with no detectable changes in area CA1 and the DG region of the hippocampus early at 1 hr into SE (Fig 1C). At 6 weeks post-SE, 5-hmC DNA methylation levels significantly decreased in the DG region, but not in hippocampal areas CA1 and CA3 of epileptic rats compared to controls (Fig 1C). These results demonstrate that in addition to 5-mC, 5-hmC DNA methylation was triggered in the hippocampus early during SE and 5-hmC regulation persisted late into epilepsy. Moreover, while SE triggered late changes in both 5-mC and 5-hmC DNA methylation levels in the hippocampus, 5-mC DNA methylation was persistently regulated in all hippocampal subregions late post-SE, indicating that 5-mC may have served as the more dominant form of DNA methylation contributing to aberrant regulation of gene expression in the epileptic hippocampus.
Effect of status epilepticus on DNA methylating enzyme gene expression in the hippocampus
The observed global DNA methylation changes in the hippocampus triggered by SE prompted additional experiments to determine whether DNA methylating enzymes were altered in the hippocampus. The three activity-dependent DNMTs in the mammalian nervous system include DNMT1, DNMT3a, and DNMT3b. DNMT1 is primarily involved in the maintenance of established methylation patterns while DNMT3a and DNMT3b are typically involved in de novo DNA methylation (Brooks et al., 1996). DNMTs serve to catalyze the formation of 5-mC from 5-C through the methyl group donor, S-adenosyl methionine, and TET enzymes catalyze the formation of 5-hmC from 5-mC.
We first characterized Dnmt1, Dnmt3a, Dnmt3b, and Tet1 gene expression levels in the hippocampus early at 1 hr into SE. Dnmt1 gene expression levels rapidly decreased in area CA3 and in the DG region of the hippocampus, with no significant changes observed in area CA1 early during SE (Fig. 1D). However, Dnmt3a gene expression levels were significantly decreased in area CA1, but not in area CA3 and the DG region at 1 hr into SE (Fig. 1E). We found no significant changes in Dnmt3b gene expression levels in all hippocampal subfields assessed at 1hr into SE (Fig. 1F). Together, these results suggest that SE triggers rapid and dynamic changes of select DNMT isoforms in the hippocampus.
In another cohort of animals, we characterized Dnmt gene expression levels in the hippocampus at 6 weeks post-SE, a time point at which animals had fully progressed into the epileptic stage. Dnmt1 gene expression levels showed no significant changes in all hippocampal subfields assessed from epileptic animals compared to controls (Fig. 1D). Dnmt3a gene expression levels were significantly decreased in areas CA1 and CA3, but not in the DG region of the hippocampus late post-SE (Fig. 1E). Additionally, Dnmt3b gene expression levels significantly increased in area CA3, but not in area CA1 and the DG region from epileptic animals (Fig. 1F). These data suggest that DNMT enzyme activity in the hippocampus is triggered by SE and further indicate that sufficient DNMT activity may have persisted long after SE to affect global DNA methylation levels in the epileptic hippocampus.
Similar to DNMTs, Tet1 gene expression levels were significantly decreased in the hippocampus at 1 hr into SE in area CA3 and the DG region (Fig. 1G). No significant changes were detected in Tet1 gene expression levels in area CA1 1 hr into SE (Fig. 1G). These findings are consistent with alterations in global 5-hmC DNA methylation levels in the hippocampus early during SE (See Fig. 1C). At 6 weeks post-SE, we found no significant alterations in Tet1 gene expression levels in the hippocampus of epileptic animals compared to controls (Fig. 1G). Together, these experiments demonstrate that Tet1 enzyme gene expression levels are differentially regulated in the hippocampus early during SE and return to baseline levels later in the epileptic hippocampus. Results further indicate that although TET1 enzyme activity may not have persisted beyond the 1 hr into SE time point, early changes in TET1 activity may be sufficient to set in motion an epigenetic process that modifies gene expression in an enduring manner that might contribute to the development of chronic seizures in TLE.
Grin2b/Nr2b and Bdnf DNA methylation levels in the epileptic hippocampus
Because global methylation patterns do not necessarily predict DNA methylation levels at a given gene, we next sought to further characterize DNA methylation levels at two gene targets known to be involved in epilepsy: Grin2b/Nr2b and Bdnf (Mathern et al., 1998, Tongiorgi et al., 2004, Bovolenta et al., 2010, Ghasemi and Schachter, 2011). It is important to note that there are no currently available sequencing detection methods to efficiently distinguish between 5-mC and 5-hmC forms of DNA methylation at gene-specific CpG sites. Therefore, the bisulfite sequencing assay employed for these studies allowed for the assessment of overall DNA methylation (5-mC and 5-hmC) levels at the Grin2b/Nr2b and Bdnf genes.
We first characterized Grin2b/Nr2b DNA methylation levels in the epileptic hippocampus using DNA sequencing primers designed against 13 CpG sites located within the regulatory region of the Grin2b/Nr2b gene (Fig. 2A). This region was selected for Grin2b/Nr2b DNA methylation sequencing analysis because it is rich in CpG sites and has previously been reported to undergo de novo DNA methylation changes in brain regions such as the visual cortex (Lee et al., 2008). We found that Grin2b/Nr2b DNA methylation levels were significantly increased in areas CA1 and CA3 of the epileptic hippocampus, with no significant changes observed in the DG region of epileptic animals compared to controls (Fig. 2B1). Upon close observation, we found that DNA methylation specifically increased at CpG sites 3, 5, 6 and 11 within the Grin2b/Nr2b gene, suggesting that these CpG sites may have contributed to overall Grin2b/Nr2b DNA methylation changes in area CA1 (Fig. 2B2). In concert with increased Grin2b/Nr2b DNA methylation levels, Grin2b/Nr2b mRNA levels decreased in areas CA1 and CA3 (Fig. 2C). Furthermore, we found that decreased Grin2b/Nr2b gene expression in area CA1 positively correlated with decreased GRIN2B/NR2B protein expression (Fig. 2D). These results strongly indicate that alterations in Grin2b/Nr2b DNA methylation levels may contribute to aberrant Grin2b/Nr2b gene expression in the epileptic hippocampus.
Figure 2.
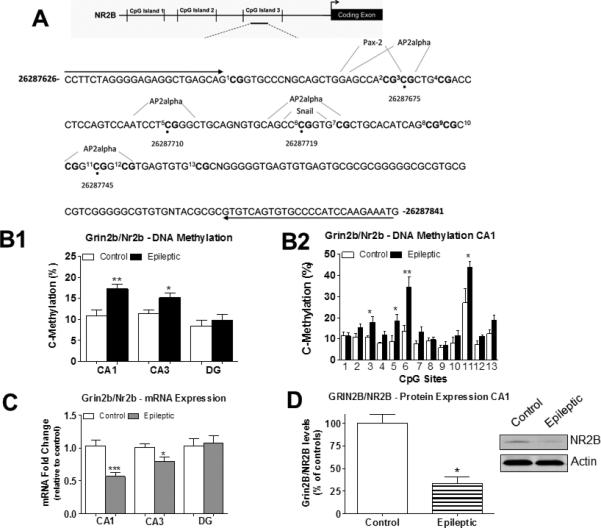
Grin2b/Nr2b DNA methylation levels are increased in the epileptic hippocampus and correlate to decreases in mRNA and protein expression. A) Diagram of three CpG Islands found upstream of the Grin2b/Nr2b transcription start site (TSS). Analysis of CpG sites were performed at CpG Island 3 found within the Grin2b/Nr2b gene. A schematic of the regulatory transcription factor binding sites that are important for activity-dependent transcription are shown encompassing CpG sites 3, 5, 6, and 11 within the Grin2b/Nr2b gene. B1) Grin2b/Nr2b DNA methylation levels at these CpG sites were significantly increased in hippocampal areas CA1 and CA3 from epileptic animals compared to controls. No significant change in Grin2b/Nr2b DNA methylation levels were found in the DG region from the epileptic animals relative to controls (CA1, t(5) = 4.29, p = 0.009, n = 6; CA3, t(5) = 2.90, p = 0.03, n = 6; DG, t(5) = 0.31, p = 0.77, n = 6; student paired t-test, *p<0.05, **p<0.01). B2) DNA methylation analysis across 12 CpG sites within the Grin2b/Nr2b gene demonstrates site-specific CpG methylation changes in area CA1 at the epileptic phase (Student paired t-test, *p<0.05, **p<0.01, n=6). C) Grin2b/Nr2b mRNA levels were found to be significantly decreased in hippocampal areas CA1 and CA3 from epileptic animals compared to controls. No significant changes were found in Grin2b/Nr2b mRNA levels in the DG region from epileptic animals relative to controls. (CA1, t(11) = 4.53, p = 0.009, n = 6-7; CA3, t(11) = 2.44, p = 0.03, n = 6; DG, t(11) = 0.28, p = 0.78, n = 6) D) Grin2b/Nr2b protein was significantly decreased in area CA1 of the epileptic animals relative to controls (t(6) = 5.754, p = 0.0022, n = 4) Student unpaired t-test, *p<0.05, **p<0.01, ***p<0.001. Error bars are SEM.
For Bdnf DNA methylation sequencing analysis, we selected a region that has been well characterized to be sensitive to activity-dependent neuronal responses (Lubin et al., 2008, Roth et al., 2009a, Gupta et al., 2010). Therefore, we analyzed DNA methylation levels across 12 CpG sites within the Bdnf gene (Fig. 3A) and found that Bdnf DNA methylation levels in this region were significantly decreased in all hippocampal subfields assessed from epileptic rats (Fig. 3B1). Specifically, we observed that the methylation status of CpG sites 1, 2, and 4 within the Bdnf gene was significantly decreased in area CA1 (Fig. 3B2), confirming that Bdnf DNA methylation alterations occur at this gene target in the epileptic hippocampus. Concurrent with Bdnf DNA demethylation, we found that Bdnf mRNA levels were significantly increased in areas CA1, CA3, and the DG region of epileptic animals compared to controls (Fig. 3C). We found that increased Bdnf gene expression corresponded with increased BDNF protein expression levels in area CA1 (Fig. 3D). These results suggest that, like Grin2b/Nr2b, Bdnf gene transcription may be mediated by DNA methylation mechanisms in the epileptic hippocampus.
Although our global DNA methylation and the gene-specific DNA methylation studies described above primarily support the involvement of DNA methylation mechanisms in the persistent steady-state regulation of genes in the epileptic hippocampus, we could not ignore the fact that gene-specific DNA methylation changes may still have occurred early at the 1 hr into SE time point. Indeed, we found that early during SE Grin2b/Nr2b gene-specific DNA methylation significantly increased in area CA1, but not in area CA3 and DG region of the hippocampus (Fig. 4A1, A2). Bdnf gene-specific DNA methylation levels showed no significant early SE-induced changes in the hippocampus (Fig. 4B1, B2). Together, these results underscore the idea that global DNA methylation changes do not necessarily reflect modifications at candidate genes. Furthermore, the fact that we found increased Grin2b/Nr2b DNA methylation levels both early and late in area CA1 of the hippocampus indicates that DNA methylation at this gene target may be sufficiently maintained in this hippocampal subregion during seizure progression.
Genes encoding for DNA demethylating proteins are altered in the epileptic hippocampus
It is well-established that in certain contexts, DNA methylation is dynamic, rather than fixed, requiring continuous upregulation and downregulation in the adult CNS. Therefore, we hypothesized that demethylation mechanisms might have served to regulate ongoing DNA methylation changes in the epileptic hippocampus. To test this hypothesis we assessed the gene expression profile of the growth arrest and DNA damage (Gadd45) isoforms: Gadd45a, Gadd45b, and the Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 1 (Apobec1) protein, all of which have previously been implicated in DNA demethylation in the hippocampus (reviewed in Barreto et al., 2007, Roth et al., 2009b, Guo et al., 2011, Lubin, 2011, Lubin et al., 2011, Puckett and Lubin, 2011).
In the epileptic hippocampus, Gadd45a gene expression levels were significantly increased in area CA3 and the DG region, but not in area CA1 (Fig. 5A). Gadd45b gene expression levels were significantly increased in area CA1 and the DG region, while no significant changes were found in area CA3 of epileptic animals compared to controls (Fig. 5B). In addition, Apobec1 gene expression levels were significantly increased in all hippocampal regions of epileptic animals (Fig. 5C). Together, these results suggest that regulation of DNA demethylation via Gadd45 and Apobec1 proteins may contribute to dynamic DNA methylation changes in the epileptic hippocampus as the disease progresses.
Figure 5.

DNA demethylating enzymes are differentially expressed in the hippocampus following epilepsy onset. A) Gadd45a mRNA levels were significantly increased in hippocampal area CA3 and the DG region from epileptic animals compared to controls. No significant changes were found in area CA1 from epileptic animals compared to controls (CA1, t(15) = 1.32, p = 0.21, n = 8-9; CA3, t(14) = 2.34, p = 0.03, n = 8; DG, t(12) = 4.90, p = 0.0004, n = 6-8). B) Gadd45b mRNA levels were significantly increased in hippocampal area CA1 and the DG region from epileptic animals compared to controls. No significant changes were observed in hippocampal area CA3 from epileptic animals relative to controls (CA1, t(16) = 2.26, p = 0.04, n = 8,9; CA3, t(15) = 0.69, p = 0.50, n = 8; DG, t(15) = 4.38, p = 0.0005, n = 8). C) Apobec1 mRNA levels were significantly increased in hippocampal areas CA1, CA3, and the DG region from epileptic animals relative to controls. (CA1, t(9) = 2. 97, p = 0.02, n = 5,6; CA3, t(11) = 4.64, p = 0.0007, n = 6,7; DG, t(9) = 3.11, p = 0.01, n = 5,6) Student unpaired t-test, *p<0.05, ***p<0.001. Error bars are SEM.
DNMT inhibition prevents DNA methylation changes in the epileptic hippocampus
We next tested whether SE-mediated DNA methylation changes in the hippocampus were amenable to manipulation by blocking DNMT enzyme activity during KA-induced SE. We hypothesized that manipulating DNA methylation mechanisms in the hippocampus might alter specific aspects of the epileptic phenotype, such as gene expression and hippocampal network excitability. For these experiments, we chose to focus our studies on area CA1 of the hippocampus for the reasons that follow. First, area CA1 has been studied extensively as a brain region of increased seizure activity in the KA-induced SE experimental TLE model system (Meier et al., 1992, Smith and Dudek, 2001, Shao and Dudek, 2004). Second, a number of morphological and physiological changes occur in area CA1 that are hypothesized to contribute to the epileptic phenotype (Meier et al., 1992, Smith and Dudek, 2001, Shao and Dudek, 2004). Finally, several studies concerning activity-dependent DNA methylation regulation in the CNS have been performed in area CA1 of the hippocampus (Lubin et al., 2008, Gupta et al., 2010). Therefore, we sought to determine the effect of inhibiting DNA methylation in area CA1 of the hippocampus using our TLE model system.
To inhibit DNA methylation, we infused the potent and stable DNMT inhibitor zebularine (2 μl: 600 ng/μL in 10% DMSO) intra-CA1 1 hr before KA IP injections. This dosage of the DNMT inhibitor zebularine has previously been reported by us and others to effectively block DNA methylation in the hippocampus (Chwang et al., 2006, Lubin et al., 2008, Nelson et al., 2008, Munzel et al., 2010). Therefore, we examined whether zebularine treatment could block global DNA methylation, block DNA methylation at the Nr2b and Bdnf genes, and influenced the outcome of KA-induced SE. For these studies, we chose to collect hippocampal tissue at 14 days post-SE as compared to the 6-weeks post-SE time period where chronic seizures are quite severe in this TLE model system. Indeed, at 14 days post-SE area CA1 of the hippocampus has undergone many of the pro-epileptic events that lead to epilepsy (Smith and Dudek, 2001, Shao and Dudek, 2004).
At 14 days post-SE, hippocampal tissue was collected for biochemistry analysis (Fig. 6A). Treatment with the DNMT inhibitor had no effect on global DNA methylation levels in area CA1 of zebularine-treated controls compared to DMSO-treated non-epileptic controls (Fig. 6B). However, global DNA methylation levels decreased in area CA1 of zebularine-treated epileptic animals compared to DMSO-treated epileptic controls (Fig. 6C). In the hippocampus of zebularine-treated non-epileptic controls, we found that DNMT inhibition did not significantly affect Bdnf DNA methylation levels or BDNF gene expression (data not shown). Similarly, DNMT inhibition had no significant affect on Bdnf DNA methylation (Fig. 7A), gene expression (Fig. 7B), or BDNF protein expression (Fig. 7C) levels in the epileptic hippocampus. These results are in sharp contrast to our prior findings indicating that DNMT inhibition can alter Bdnf DNA methylation and gene expression in the normal hippocampus (Lubin et al., 2008). Thus, these data suggest that a single infusion of zebularine, administered once before KA-induced SE, was not sufficient to affect Bdnf DNA methylation and gene regulation in epilepsy assessed 14 days post-SE.
Figure 6.

Global DNA methylation levels decrease in the epileptic hippocampus following DNMT inhibition. A) Diagram of the experimental setup. B) DNMT inhibition did not alter global 5-mC DNA methylation levels in hippocampal area CA1 in zebularine-treated control animals compared to DMSO-treated control animals (t(11) = 1.16, p = 0.27, n = 6-7). C) DNMT inhibition significantly decreased global 5-mC DNA methylation levels in hippocampal area CA1 from zebularine-treated epileptic animals relative to DMSO-treated epileptic control animals (t(10) = 3.22, p = 0.009, n = 6). Student unpaired t-test, **p<0.01, n = 6. Error bars are SEM.
Figure 7.

Effect of DNMT inhibition on Bdnf and Grin2b/Nr2b DNA methylation, mRNA, and protein expression levels in the epileptic hippocampus. A) Bdnf DNA methylation levels in hippocampal area CA1 from zebularine-treated epileptic animals was not significantly altered compared to Bdnf DNA methylation levels from DMSO-treated epileptic controls (Student paired t-test, p>0.05, n = 6). B) Bdnf mRNA levels in hippocampal area CA1 from zebularine-treated epileptic animals was not significantly altered compared to DMSO-treated epileptic controls (t(9) = 0.91, p = 0.11, n = 6; student unpaired t-test). C) DNMT inhibition did not significantly alter BDNF protein expression in hippocampal area CA1 of zebularine-treated epileptic animals compared to DMSO-treated epileptic control. (t(5) = 0.89, p = 0.41, n = 3-4; Student unpaired t-test). D) DNMT inhibition significantly decreased Grin2b/Nr2b promoter DNA methylation levels in hippocampal area CA1 from zebularine-treated epileptic animals relative to DMSO-treated epileptic control animals (Student paired t-test, *p<0.05, n = 6). E) DNMT inhibition significantly decreased Grin2b/Nr2b mRNA expression levels in hippocampal area CA1 from zebularine-treated epileptic animals relative to DMSO-treated epileptic controls (t(9) = 3.26, p = 0.009, n = 6-7; student unpaired t-test, **p<0.01). F) DNMT inhibition significantly increased GRIN2B/NR2B protein expression in area CA1 of zebularine-treated epileptic animals relative to DMSO-treated epileptic control. (t(6) = 2.76, p = 0.03, n=4; Student unpaired t-test, *p<0.05). G) ChIP analysis revealed that Ap2alpha binding at the Grin2b/Nr2b promoter was significantly decreased in the DMSO-treated epileptic animals compared to DMSO-treated non-epileptic controls and compared to zebularine-treated epileptic animals (#,*p<0.05, n=4). Error bars are SEM.
In relation to Grin2b/Nr2b, DNMT inhibition had no significant affect on DNA methylation or mRNA expression levels in area CA1 of zebularine-treated non-epileptic controls compared to DMSO-treated non-epileptic controls (data not shown). However, we found that, treatment with zebularine before KA-induced SE significantly decreased Grin2b/Nr2b DNA methylation levels in the hippocampus of zebularine-treated epileptic animals compared to DMSO-treated epileptic controls (Fig. 7D). Interestingly, we found that DNMT inhibition further decreased Grin2b/Nr2b mRNA expression (Fig. 7E), which negatively correlated with increased GRIN2B/NR2B protein expression (Fig. 7F). To determine whether DNA methylation changes can affect the binding of transcription factors at the Grin2b/Nr2b gene promoter, we employed a chromatin immunoprecipitation (ChIP) assay using an antibody against the AP2alpha transcription factor and primers designed to amplify AP2alpha putative binding sites within the Grin2b/Nr2b promoter (See Fig. 2A). ChIP analysis revealed that AP2alpha was significantly decreased at the Grin2b/Nr2b gene promoter in the epileptic hippocampus, which was restored with DNMT inhibition (Fig. 7G). These results suggest that DNA methylation changes observed at the Grin2b/Nr2b promoter in the epileptic hippocampus were functionally significant, because it was accompanied by the full restoration of AP2alpha binding following DNMT inhibition. Thus, interfering with SE-induced DNA methylation can alter not only gene transcription but subsequent protein expression in the epileptic hippocampus assessed later at 14 days post-SE.
DNA methylation is required for homeostatic synaptic plasticity in the epileptic hippocampus
We next examined the effect of inhibiting DNA methylation at the synaptic plasticity level by performing fEPSP recordings in the stratum radiatum of area CA1 from epileptic rats at 14 days post-SE (Fig. 8A). We found that DNA methylation inhibition significantly increased the fEPSP slope in area CA1 dendrites from epileptic hippocampal sections (Fig. 8B). We observed no significant difference in the fEPSP slope obtained from area CA1 dendrites in DMSO-treated or zebularine-treated non-epileptic control hippocampal slices (Fig. 8B). Additionally, we plotted the average fEPSP slope against the average fiber volley amplitude for each stimulation intensity and drew a straight line to each data set using linear regression and found that the zebularine-treated epileptic group had a significantly steeper slope (1.767 ± 0.2899) compared to the epileptic control group (0.7327 ± 0.08164) (Fig. 8C). These results indicate that increased DNA methylation may be necessary to calm seizure activity in the epileptic hippocampus, as DNMT inhibition further increased fEPSPs in area CA1.
Figure 8.

DNMT inhibition enhances excitability in the hippocampus and alters seizure onset, but not epilepsy outcome following SE. A) Diagram of the experimental setup. B) DNMT inhibition significantly increased fEPSP slope in CA1 dendrites after epilepsy onset (ANOVA, p<0.05 n=8-16). C) fEPSP slope plotted against fiber volley amplitude. Straight lines were plotted to each data set using linear regression. The zebularine-treated epileptic group showed a significantly steeper slope compared to the DMSO-treated epileptic control group (ANOVA, p<0.05 n=8-16). D) Example traces from DMSO-treated epileptic control (right) and zebularine-treated epileptic (left) hippocampal slices. E) DNMT inhibition significantly decreased latency to forelimb clonus following KA administration. (t(14) = 2.54, p = 0.02, n = 8). Student unpaired t-test, *p<0.05. F) EEG monitoring of epileptic animals revealed that DNMT inhibition did not alter the average number of interictal spikes or the number of seizures following SE (n = 2-3). Error bars are SEM.
We next examined whether DNMT inhibition prior to SE influenced the outcome of KA-induced SE. We found that DNA methylation blockade with zebularine decreased the latency to forelimb clonus in epileptic animals based on Racine scores (Fig. 8E). In experimental TLE, interictal-like activity typically precedes the first spontaneous seizure (Staley and Dudek, 2006, Pitkanen et al., 2007) while providing a measure of the natural history of the resulting epilepsy phenotype (White et al., 2010). In both groups of animals, electroencephalography (EEG) recordings revealed that numbers of interictal bursts were indistinguishable between the epileptic and zebularine-treated epileptic animals (Fig. 8F, left table). During EEG recordings, spontaneous seizures emerged concurrently in both controls and zebularine-treated epileptic groups (Fig. 8F right table). Together, these results suggest that interfering with DNA methylation prior to KA-induced SE increased latency to seizure onset but did not appear to have modified disease progression.
Discussion
In the studies described above, we used an experimental TLE model system to examine DNA methylation-related chromatin remodeling mechanisms. In addition, the contribution of these mechanisms to hippocampal Grin2b/Nr2b and Bdnf gene and protein expression changes in TLE were studied. First, we found that alterations in net global DNA methylation levels in the hippocampus were triggered early by KA-induced SE and remained persistently regulated late into epilepsy. DNA methylation triggered by SE was accompanied by early and late gene expression changes of several DNA methylating enzymes in the epileptic hippocampus. Analysis of DNA sequences revealed altered DNA methylation levels at specific CpG sites around the Grin2b/Nr2b and Bdnf genes in the hippocampus of epileptic animals. Furthermore, interfering with SE-induced DNA methylation changes with a DNMT inhibitor prior to SE effectively prevented the later observed changes in global and Grin2b/Nr2b DNA methylation levels, attenuated Grin2b/Nr2b mRNA expression, and increased GRIN2B/NR2B protein expression in the epileptic hippocampus. Early interference of DNMT enzyme activity altered DNA methylation levels triggered by SE and further exacerbated neuronal excitability in the epileptic hippocampus. These data highlight for the first time that DNA methylation-related chromatin remodeling in the hippocampus is triggered by SE and may be of particular importance for regulating transcriptional homeostasis in TLE.
Epileptogenesis can be defined as a process occurring during the latency period between the insult and occurrence of spontaneous seizures. Brain insults triggered by SE produce dynamic and enduring abnormal expression of hundreds of genes in the hippocampus that proceed through the different stages of epileptogenesis. Collectively, the observation that DNA methylation-related chromatin remodeling is altered around the Grin2b/Nr2b and Bdnf genes in the epileptic hippocampus suggests that DNA methylation is a transcriptional mechanism that is not only dynamically regulated at specific gene targets during the early phases of epileptogenesis, but may persistently affect a number of genes throughout the progression of epilepsy. These gene-specific DNA methylation changes are consistent with other studies in human TLE specimens that demonstrate increased DNA methylation levels at other genes such as reelin, assessed in the hippocampus (Kobow et al., 2009). Thus, epigenetic regulation of gene transcription by alterations in DNA methylation represents a provocative candidate mechanism for post-injury gene expression changes in epilepsy. Moreover, epilepsy is linked to changes in neuronal networks triggered by neurogenesis and several comorbidities including memory impairments. Thus, epigenetic DNA methylation-mediated gene expression changes in the hippocampus could be associated with epileptogenic circuitry reorganization and comorbidity modification in epileptogenesis.
The differential regulation of DNA methylation patterns observed in hippocampal subfields during SE and at the epileptic stage suggests alternative roles for DNA methylation mechanisms depending on the hippocampal region assessed. For example, we could not ignore the fact that not all hippocampal regions showed increased global DNA methylation changes in response to epilepsy onset (See Figure 1). We observed significant decreases in global DNA methylation levels in the DG region of the hippocampus from epileptic rats. This result raises the question of why DNA methylation would be decreased in the DG region of the epileptic brain. One commonly observed characteristic of epilepsy in animal models and in humans is an association with increased neurogenesis in the DG region (Parent et al., 1997, Wenzel et al., 2000, Parent, 2007). Thus, regulation of DNA methylation in the DG may control regulation of gene expression changes that contributes to increased neurogenesis in epilepsy disorders.
The gene transcription effect of DNA methylation-related chromatin remodeling in other subregions of the epileptic hippocampus may explain some aspects of cognitive impairments associated with epilepsy. For example, manipulation of histone acetylation levels in area CA1 via treatment with histone/lysine deacetylase (H/KDAC) inhibitors has been shown to reverse memory decline in KA-treated rats (Jessberger et al., 2007). Interestingly, preventing DNA methylation changes in area CA1 of the hippocampus during the process of memory consolidation interfered with long-term fear memory (Lubin et al., 2008). Thus, global alterations in DNA methylation levels in area CA1 of the hippocampus following SE may contribute to memory deficits associated with epilepsy disorders (reviewed in Lubin, 2012).
Research studies have demonstrated that the NMDA Grin2b/Nr2b subunit plays a critical role in the development of epilepsy (Moddel et al., 2005, Bandyopadhyay and Hablitz, 2006, Auzmendi et al., 2009, Sun et al., 2009). The DNA methylation levels observed around theGrin2b/Nr2b gene were significantly increased in association with decreased Grin2b/Nr2b mRNA levels and GRIN2B/NR2B protein expression in the epileptic hippocampus. These results suggest that Grin2b/Nr2b DNA methylation may have served as a mechanism to reduce the contribution of GRIN2B/NR2B to cellular excitability in the epileptic hippocampus. Within the DNA region studied in the epileptic hippocampus, we observed significant increases in DNA methylation levels at CpG sites 3, 5, 6, and 11 within the Grin2b/Nr2b gene (See Figure 2B, 2C). Encompassing these four critical CpG sites are consensus sequences for the AP2alpha TF (See Figure 2A). Furthermore, the decreased AP2alpha TF binding at the Grin2b/Nr2b gene in the epileptic hippocampus was restored with DNMT inhibition (Figure 8G). Therefore, increased Grin2b/Nr2b DNA methylation altered AP2alpha TF binding and recruitment of the transcriptional machinery at the Grin2b/Nr2b gene that further highlights the importance of DNA methylation at specific CpG sites targeted in this study.
Similar to Grin2b/Nr2b, we evaluated the contribution of DNA methylation to abnormal Bdnf gene expression in the epileptic hippocampus triggered by SE. Dysregulation of the Bdnf gene in the hippocampus has been shown to play a major role in epilepsy (Scharfman et al., 1999, Binder et al., 2001, Reibel et al., 2001, Binder, 2004, Koyama and Ikegaya, 2005). Furthermore, our prior reports indicate that in the normal hippocampus, DNA methylation-related chromatin remodeling is necessary for activity-dependent regulation of Bdnf gene transcription (Lubin et al., 2008, Roth et al., 2011). We obtained evidence that Bdnf DNA methylation levels were decreased in the epileptic hippocampus, which is in good agreement with prior findings demonstrating that Bdnf DNA from cortical cultures is demethylated in response to membrane depolarization by potassium chloride (Martinowich et al., 2003). In addition, recent research findings indicate that increased BDNF expression in the hippocampus can attenuate the epileptic phenotype (Tongiorgi et al., 2004, Bovolenta et al., 2010). Therefore, decreases in Bdnf DNA methylation levels may serve as a transcriptional mechanism to increase Bdnf mRNA and protein levels within the hippocampus and perhaps to attenuate seizure severity.
As with the Grin2b/Nr2b gene, the Bdnf DNA region that we assessed contained CpG sites that were specifically altered by methylation in the epileptic hippocampus (See Figure 4B2). Encompassing CpG site 2 within the Bdnf gene is a putative GATA-3 TF binding site. The GATA TF family interacts with H/KDACs to regulate expression of genes (Ozawa et al., 2001, Chen et al., 2006). Also of note, encompassing CpG sites 3, 4, and 5 within the Bdnf gene is a cAMP response element-binding (CREB) TF binding element (See Figure 4A). The binding of the CREB TF is a well-established transcriptional mechanism for activity-dependent regulation of the Bdnf gene upon membrane depolarization in hippocampal neurons (Martinowich et al., 2003). In addition, decreased DNA methylation levels were observed at CpG site 4 within the Bdnf gene containing a putative bZIP910 DNA binding element, which can serve to recruit the CREB TF to regulate activity-dependent Bdnf gene transcription in neurons (Halterman et al., 2010). Therefore, the decrease in DNA methylation levels we observed around the Bdnf gene in the epileptic hippocampus may allow for CREB-mediated transcriptional regulation of Bdnf mRNA levels in our rodent TLE model.
The formation of 5-mC DNA methylation is mediated by a family of DNMTs, which is abundantly expressed in the CNS (Fan et al., 2001, Feng and Fan, 2009). In the present study, we have obtained evidence of early and late SE-induced changes in Dnmt1, Dnmt3a, and Dnmt3b DNA methylating enzyme gene expression in the hippocampus. Interestingly, we observed altered Dnmt1 and Dnmt3b gene expression levels early during SE yet found no significant changes in global 5-mC DNA methylation levels in the hippocampus at this time point (see Figure 1C). These findings indicate that perhaps DNMT enzyme activity early during SE may not have reached sufficient levels to affect 5-mC DNA methylation on the global scale. Additionally, we found that DNMT activity in CA1 during SE was only represented by a decrease in DNMT3a gene expression; however, we observed net increases in global DNA methylation levels in this hippocampal region. Intriguingly, prior studies have demonstrated that upon increased chromatin structure remodeling a negative feedback mechanism exists to downregulate epigenetic enzyme activity in the hippocampus (Gupta-Agarwal et al., 2012). Therefore, an alternate explanation for activity-dependent Dnmt gene regulation in our study is that DNMT enzyme activity may be gene targets for increased DNA methylation to downregulate Dnmt gene expression in the hippocampus, as has been previously demonstrated in other mammalian cell systems (reviewed in Kinney and Pradhan, 2011).
In support of our study, recent research has demonstrated that DNMT protein expression levels were altered in neocortical tissue from human patients with intractable TLE (Zhu et al., 2012a). Specifically, Zhu and colleagues found that DNMT1 and DNMT3a protein expression levels were primarily altered in neurons, suggesting that active DNA methylation in the epileptic brain occurs in neurons, but not glia cells (Zhu et al., 2012a). These prior studies in the neocortex, along with our present work in the hippocampus, suggest that DNA methylation-mediated gene expression plays a major role in TLE. Thus, the transcriptional mechanisms affected by DNA methylation-related chromatin remodeling that contribute to gene expression changes in the epileptic brain merits further investigation in both rodent models of TLE and in human TLE.
DNA demethylation may occur in conjunction with conversion of 5-mC to 5-hmC DNA methylation. The Gadd45 isoforms, Gadd45a and Gadd45b are hypothesized to mediate active DNA demethylation in post-mitotic neurons (reviewed in Barreto et al., 2007, Roth et al., 2009b, Guo et al., 2011, Lubin, 2011, Lubin et al., 2011, Puckett and Lubin, 2011). Demethylation mechanisms in adult neurons involve activity of the Apobec1 proteins that aid in the DNA demethylation process via cytosine deamination (Matsuoka et al., 1999, Morganti-Kossmann et al., 2010). The formation of 5-hmC may be dependently linked to alterations in the expression of the Gadd45 and Apobec proteins (Ma et al., 2009a, Ma et al., 2009b, Guo et al., 2011). In addition, the formation of 5-hmC from 5-mC is mediated by the TET enzyme family (Figure 1). In the present study, we found alterations in gene expression for Gadd45 isoforms and Apobec1 proteins, indicating active DNA demethylation mechanisms in the epileptic hippocampus (See Figure 5). These data are consistent with a role for activity-dependent DNA methylation in epilepsy and underscores the importance of investigating how manipulation of the balance of DNA methylation and demethylation levels at specific genes may alter chronic seizures in TLE.
The effect of manipulating DNA methylation with DNMT inhibitors such as zebularine or 5-Aza-2-deoxycytidin has just begun to be investigated in the CNS (Nelson et al., 2008, Feng and Fan, 2009). However, manipulation of DNA methylation on symptoms of epilepsy has not been previously explored. We found that interfering with SE-induced DNA methylation via DNMT inhibition with zebularine significantly decreased global DNA methylation levels, reduced Grin2b/Nr2b DNA methylation, attenuated Grin2b/Nr2b mRNA levels and further increased GRIN2B/NR2B protein expression in the epileptic hippocampus. Intriguingly, the observed decrease in Bdnf DNA methylation in the epileptic hippocampus was not affected by DNMT inhibition. These results suggest that DNA methylation was necessary, but not sufficient, for Bdnf activation. If this is the case, strong induction of DNA methylation at the Bdnf gene could result from sustained DNA methylation levels and activation of positive regulators of TFs regulated by increased Ca2+ signaling in epilepsy. Future research directions should address the interactive role of DNA methylation and histone modifications in the regulation of gene transcription in epilepsy, as the combined role of these two mechanisms at a given gene dictates transcriptional outcome in response to brain insults. Alternatively, our findings may indicate that the effect of DNMT inhibition may be specific to some genes while not affecting others in the epileptic hippocampus. Nevertheless, the effect of DNMT inhibition on Grin2b/Nr2b gene expression and subsequent GRIN2B/NR2B protein expression demonstrates a molecular consequence of inhibiting DNA methylation in the epileptic hippocampus. In addition, the decrease in Grin2b/Nr2b gene expression and the increase in GRIN2B/NR2B protein expression with DNMT inhibition is not surprising as several studies have shown that in some cases, increased DNA methylation results in the upregulation of gene expression (Chahrour et al., 2008, Gupta et al., 2010). Furthermore, increases in GRIN2B/NR2B subunit containing NMDA receptors result in longer durations in activity and downregulation of Grin2b/Nr2b gene expression (Wang et al., 2004). Thus, our finding that DNA methylation contributes to Grin2b/Nr2b and not bdnf gene expression changes in epilepsy underscores the complexity of DNA methylation-related chromatin remodeling of gene transcription mechanisms in response to seizure progression.
Pathological increased network activity is a hallmark of TLE. Investigating how DNA methylation mechanisms that regulate gene expression alter network activity in epilepsy would be informative. Interestingly, we found that DNMT inhibition prior to SE increased fEPSPs in the epileptic hippocampus assessed 14 days post-SE (See Figure 8), suggesting that DNA methylation changes induced by SE may serve as a compensatory mechanism to stabilize gene expression changes that contribute to increased latency to behavioral seizure onset based on Racine scores. However, EEG recordings revealed that a single infusion of zebularine to inhibit DNMT activity prior to SE did not significantly alter epilepsy progression. As such, the epileptic hippocampus may have co-opted the DNA methylation machinery to control seizure severity early on. Nonetheless, our present study still supports the hypothesis that increased DNA methylation level in the epileptic hippocampus may be a protective transcriptional mechanism. In addition, it remains to be determined whether intermittent or continuous DNMT inhibition can affect epileptiform activity; a future study that we are currently exploring. Identification of circumstances under which DNMT inhibitors might control chronic seizures may require a selective time-window and brain-permeant DNMT inhibitors.
Molecular and cellular alterations have been well documented to contribute to the different stages of epileptogenesis, from the initial insult phase to the late phases wherein spontaneous recurrent seizures begin to develop and progress through epilepsy. However, the mechanisms that drive many of these molecular and cellular changes that occur during epileptogenesis remain elusive. In addition, the question of how the temporal waves of molecular alterations ultimately contribute to spontaneous seizure development is not well defined (Loscher and Brandt, 2010, Pitkanen and Lukasiuk, 2011). In this regard, early SE-induced changes in DNA methylation may transiently or persistently affect gene expression patterns that represent potential biomarkers for key phases of the epileptogenesis process that remains to be explored in future anti-epileptogenesis studies.
Transcriptional mechanisms such as DNA methylation are a viable candidate molecular process to consider for the control of temporal waves of gene expression during epileptogenesis. Our present study indicates that DNA methylation can transiently control gene expression for minutes to hours or persistently affect gene expression for days to months. However, it remains to be determined whether DNA methylation occurring early during SE or at the epileptic phase is pro or anti-epileptic. In the DNMT inhibitor studies we found that blockade of SE-induced DNA methylation changes exacerbated the epileptic phenotype, suggesting that DNA methylation-related chromatin remodeling mechanisms play a vital role in seizure control. Furthermore, our data suggest that preventing these DNA methylation changes early during insult might likely result in a worsened epileptic state such as an increase in hippocampal neuronal excitability. Therefore, increasing DNA methylation in epilepsy may be a viable target for therapeutic intervention in human TLE.
In summary, our data suggest that activity-dependent DNA methylation in the epileptic hippocampus is both an early and late event triggered by SE. Here, we provide evidence that SE induced global and gene-specific DNA methylation changes in the hippocampus, suggesting that DNA methylation mechanisms may serve as a molecular process for altered gene transcription in epilepsy. Moreover, altered DNA methylation levels at the Grin2b/Nr2b and Bdnf genes in response to SE was differentially affected by seizures in a manner well correlated with their mRNA and protein expression levels. The gene-specificity of this modification is likely achieved by transcriptional regulatory elements within the Grin2b/Nr2b and Bdnf genes, and the activity of their regulating transcription factors induced by seizures. Indeed, a single methylated CpG site can prevent TF binding that are critical for activity-dependent gene transcription in neurons (Lubin et al., 2007, McClelland et al., 2011, Zhu et al., 2012b). Finally, as epigenetic mechanisms continue to be linked to seizure disorders, increasing our understanding of how DNA methylation-related chromatin remodeling influences epilepsy onset will require additional work. Future studies are necessary to determine the effect of promoting DNA methylation levels on seizure activity. Such studies will provide further insights into the role of DNA methylation mechanisms in the development and maintenance of steady-state expression of genes in epilepsies that may lead to new and selective therapeutic interventions.
Highlights.
■ Grin2b/Nr2b and Bdnf DNA methylation is altered in the epileptic hippocampus.
■ Grin2b/Nr2b and Bdnf gene and protein expression correlated with DNA methylation.
■ SE triggered early and late increases in DNA methylating and demethylating enzymes.
■ DNMT inhibition altered SE-induced hippocampal methylation and synaptic plasticity.
■ DNMT inhibition restored decreases in AP2alpha binding at the Nr2b gene promoter.
Acknowledgements
We thank members of the Lubin laboratory including Swati Gupta for their thoughtful comments regarding the manuscript. We would also like to thank Drs. M. Crowley and M. Han and the University of Alabama at Birmingham Heflin Genomics Core Facility for assistance in DNA sequencing. This work was supported by the National Institute of Mental Health (MH082106, MH097909), The Epilepsy Foundation, Civitan International, and the Evelyn F McKnight Brain Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Financial Interests
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Auzmendi J, Gonzalez N, Girardi E. The NMDAR subunit NR2B expression is modified in hippocampus after repetitive seizures. Neurochem Res. 2009;34:819–826. doi: 10.1007/s11064-008-9828-0. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Hablitz JJ. NR2B antagonists restrict spatiotemporal spread of activity in a rat model of cortical dysplasia. Epilepsy Res. 2006;72:127–139. doi: 10.1016/j.eplepsyres.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- Binder DK. The role of BDNF in epilepsy and other diseases of the mature nervous system. Advances in experimental medicine and biology. 2004;548:34–56. doi: 10.1007/978-1-4757-6376-8_3. [DOI] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends in neurosciences. 2001;24:47–53. doi: 10.1016/s0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- Bovolenta R, Zucchini S, Paradiso B, Rodi D, Merigo F, Navarro Mora G, Osculati F, Berto E, Marconi P, Marzola A, Fabene PF, Simonato M. Hippocampal FGF-2 and BDNF overexpression attenuates epileptogenesis-associated neuroinflammation and reduces spontaneous recurrent seizures. J Neuroinflammation. 2010;7:81. doi: 10.1186/1742-2094-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Osada H, Santamaria-Babi LF, Kannagi R. Interaction of GATA-3/T-bet transcription factors regulates expression of sialyl Lewis X homing receptors on Th1/Th2 lymphocytes. Proc Natl Acad Sci U S A. 2006;103:16894–16899. doi: 10.1073/pnas.0607926103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor CM, Akbarian S. DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics : official journal of the DNA Methylation Society. 2008;3:55–58. doi: 10.4161/epi.3.2.5938. [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. 2011;22:617–640. doi: 10.1016/j.yebeh.2011.07.024. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, Lubin FD. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci. 2012;32:5440–5453. doi: 10.1523/JNEUROSCI.0147-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. The Journal of biological chemistry. 2010;285:21329–21340. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi F, Uchida S, Yamagata H, Otsuki K, Hobara T, Abe N, Shibata T, Watanabe Y. State-dependent changes in the expression of DNA methyltransferases in mood disorder patients. J Psychiatr Res. 2011;45:1295–1300. doi: 10.1016/j.jpsychires.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Nakashima K, Clemenson GD, Jr., Mejia E, Mathews E, Ure K, Ogawa S, Sinton CM, Gage FH, Hsieh J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J Neurosci. 2007;27:5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, Kumar A, Nestler EJ, Akbarian S, Beckel-Mitchener AC. Epigenetics in the nervous system. J Neurosci. 2008;28:11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Kinney SR, Pradhan S. Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Progress in molecular biology and translational science. 2011;101:311–333. doi: 10.1016/B978-0-12-387685-0.00009-3. [DOI] [PubMed] [Google Scholar]
- Kobow K, Blumcke I. The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia. 2011;52(Suppl 4):15–19. doi: 10.1111/j.1528-1167.2011.03145.x. [DOI] [PubMed] [Google Scholar]
- Kobow K, Jeske I, Hildebrandt M, Hauke J, Hahnen E, Buslei R, Buchfelder M, Weigel D, Stefan H, Kasper B, Pauli E, Blumcke I. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. Journal of neuropathology and experimental neurology. 2009;68:356–364. doi: 10.1097/NEN.0b013e31819ba737. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus. Neuroscientist. 2005;11:282–287. doi: 10.1177/1073858405278266. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, 3rd, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iniguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolanos CA, Kabbaj M, Xiao G, Neve RL, Hurd YL, Oosting RS, Fan G, Morrison JH, Nestler EJ. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim W, Ham BJ, Chen W, Bear MF, Yoon BJ. Activity-dependent NR2B expression is mediated by MeCP2-dependent epigenetic regulation. Biochemical and biophysical research communications. 2008;377:930–934. doi: 10.1016/j.bbrc.2008.10.082. [DOI] [PubMed] [Google Scholar]
- Lei LG, Sun S, Gao YJ, Zhao ZQ, Zhang YQ. NMDA receptors in the anterior cingulate cortex mediate pain-related aversion. Exp Neurol. 2004;189:413–421. doi: 10.1016/j.expneurol.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. The Journal of biological chemistry. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. 2010;62:668–700. doi: 10.1124/pr.110.003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD. Epigenetic gene regulation in the adult mammalian brain: multiple roles in memory formation. Neurobiol Learn Mem. 2011;96:68–78. doi: 10.1016/j.nlm.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD. Epileptogenesis: can the science of epigenetics give us answers? Epilepsy currents / American Epilepsy Society. 2012;12:105–110. doi: 10.5698/1535-7511-12.3.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Gupta S, Parrish RR, Grissom NM, Davis RL. Epigenetic mechanisms: critical contributors to long-term memory formation. Neuroscientist. 2011;17:616–632. doi: 10.1177/1073858411386967. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Ren Y, Xu X, Anderson AE. Nuclear factor-kappa B regulates seizure threshold and gene transcription following convulsant stimulation. J Neurochem. 2007;103:1381–1395. doi: 10.1111/j.1471-4159.2007.04863.x. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The IkappaB Kinase Regulates Chromatin Structure during Reconsolidation of Conditioned Fear Memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell cycle. 2009a;8:1526–1531. doi: 10.4161/cc.8.10.8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009b;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Leite JP, Kornblum HI, Mendoza D, Lozada A, Bertram EH., 3rd Hippocampal AMPA and NMDA mRNA levels and subunit immunoreactivity in human temporal lobe epilepsy patients and a rodent model of chronic mesial limbic epilepsy. Epilepsy Res. 1998;32:154–171. doi: 10.1016/s0920-1211(98)00048-5. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Kitamura Y, Okazaki M, Terai K, Taniguchi T. Kainic acid-induced activation of nuclear factor-kappaB in rat hippocampus. Exp Brain Res. 1999;124:215–222. doi: 10.1007/s002210050616. [DOI] [PubMed] [Google Scholar]
- McClelland S, Flynn C, Dube C, Richichi C, Zha Q, Ghestem A, Esclapez M, Bernard C, Baram TZ. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Annals of neurology. 2011;70:454–464. doi: 10.1002/ana.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier CL, Obenaus A, Dudek FE. Persistent hyperexcitability in isolated hippocampal CA1 of kainate-lesioned rats. J Neurophysiol. 1992;68:2120–2127. doi: 10.1152/jn.1992.68.6.2120. [DOI] [PubMed] [Google Scholar]
- Moddel G, Jacobson B, Ying Z, Janigro D, Bingaman W, Gonzalez-Martinez J, Kellinghaus C, Prayson RA, Najm IM. The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res. 2005;1046:10–23. doi: 10.1016/j.brainres.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Yan E, Bye N. Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury. 2010;41(Suppl 1):S10–13. doi: 10.1016/j.injury.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Munzel M, Globisch D, Bruckl T, Wagner M, Welzmiller V, Michalakis S, Muller M, Biel M, Carell T. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem Int Ed Engl. 2010;49:5375–5377. doi: 10.1002/anie.201002033. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa Y, Towatari M, Tsuzuki S, Hayakawa F, Maeda T, Miyata Y, Tanimoto M, Saito H. Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood. 2001;98:2116–2123. doi: 10.1182/blood.v98.7.2116. [DOI] [PubMed] [Google Scholar]
- Parent JM. Adult neurogenesis in the intact and epileptic dentate gyrus. Progress in brain research. 2007;163:529–540. doi: 10.1016/S0079-6123(07)63028-3. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish RR, Day JJ, Lubin FD. Direct bisulfite sequencing for examination of DNA methylation with gene and nucleotide resolution from brain tissues. 2012 doi: 10.1002/0471142301.ns0724s60. Current protocols in neuroscience / editorial board, Jacqueline N Crawley [et al] Chapter 7:Unit 7 24.
- Paxinos GWC. The rat brain in stereotaxic coordinates. 1998.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Kharatishvili I, Karhunen H, Lukasiuk K, Immonen R, Nairismagi J, Grohn O, Nissinen J. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl 2):13–20. doi: 10.1111/j.1528-1167.2007.01063.x. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet neurology. 2011;10:173–186. doi: 10.1016/S1474-4422(10)70310-0. [DOI] [PubMed] [Google Scholar]
- Puckett RE, Lubin FD. Epigenetic mechanisms in experience-driven memory formation and behavior. Epigenomics. 2011;3:649–664. doi: 10.2217/epi.11.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalography and clinical neurophysiology. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Reibel S, Depaulis A, Larmet Y. BDNF and epilepsy--the bad could turn out to be good. Trends in neurosciences. 2001;24:318–319. doi: 10.1016/s0166-2236(00)01869-5. [DOI] [PubMed] [Google Scholar]
- Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biological psychiatry. 2009a;65:760–769. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Lubin FD, Sodhi M, Kleinman JE. Epigenetic mechanisms in schizophrenia. Biochimica et biophysica acta. 2009b;1790:869–877. doi: 10.1016/j.bbagen.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res. 2011;45:919–926. doi: 10.1016/j.jpsychires.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales AJ, Biojone C, Terceti MS, Guimaraes FS, Gomes MV, Joca SR. Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. British journal of pharmacology. 2011;164:1711–1721. doi: 10.1111/j.1476-5381.2011.01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Actions of brain-derived neurotrophic factor in slices from rats with spontaneous seizures and mossy fiber sprouting in the dentate gyrus. J Neurosci. 1999;19:5619–5631. doi: 10.1523/JNEUROSCI.19-13-05619.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Increased excitatory synaptic activity and local connectivity of hippocampal CA1 pyramidal cells in rats with kainate-induced epilepsy. J Neurophysiol. 2004;92:1366–1373. doi: 10.1152/jn.00131.2004. [DOI] [PubMed] [Google Scholar]
- Smith BN, Dudek FE. Short- and long-term changes in CA1 network excitability after kainate treatment in rats. J Neurophysiol. 2001;85:1–9. doi: 10.1152/jn.2001.85.1.1. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Dudek FE. Interictal spikes and epileptogenesis. Epilepsy currents / American Epilepsy Society. 2006;6:199–202. doi: 10.1111/j.1535-7511.2006.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QJ, Duan RS, Wang AH, Shang W, Zhang T, Zhang XQ, Chi ZF. Alterations of NR2B and PSD-95 expression in hippocampus of kainic acid-exposed rats with behavioural deficits. Behav Brain Res. 2009;201:292–299. doi: 10.1016/j.bbr.2009.02.027. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Armellin M, Giulianini PG, Bregola G, Zucchini S, Paradiso B, Steward O, Cattaneo A, Simonato M. Brain-derived neurotrophic factor mRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J Neurosci. 2004;24:6842–6852. doi: 10.1523/JNEUROSCI.5471-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Hong CJ, Yen TY, Huang HY, Ou Y, Huang TN, Jung WG, Kuo TY, Sheng M, Wang TF, Hsueh YP. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron. 2004;42:113–128. doi: 10.1016/s0896-6273(04)00139-4. [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Woolley CS, Robbins CA, Schwartzkroin PA. Kainic acid-induced mossy fiber sprouting and synapse formation in the dentate gyrus of rats. Hippocampus. 2000;10:244–260. doi: 10.1002/1098-1063(2000)10:3<244::AID-HIPO5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- White A, Williams PA, Hellier JL, Clark S, Edward Dudek F, Staley KJ. EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia. 2010;51:371–383. doi: 10.1111/j.1528-1167.2009.02339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Wang L, Zhang Y, Zhao FH, Luo J, Xiao Z, Chen GJ, Wang XF. Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. Journal of molecular neuroscience : MN. 2012a;46:420–426. doi: 10.1007/s12031-011-9602-7. [DOI] [PubMed] [Google Scholar]
- Zhu X, Han X, Blendy JA, Porter BE. Decreased CREB levels suppress epilepsy. Neurobiol Dis. 2012b;45:253–263. doi: 10.1016/j.nbd.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]