

Abstract
Lipid metabolism is of crucial importance for pathogens. Lipids serve as cellular building blocks, signalling molecules, energy stores, posttranslational modifiers, and pathogenesis factors. Parasites rely on a complex system of uptake and synthesis mechanisms to satisfy their lipid needs. The parameters of this system change dramatically as the parasite transits through the various stages of its life cycle. Here we discuss the tremendous recent advances that have been made in the understanding of the synthesis and uptake pathways for fatty acids and phospholipids in apicomplexan and kinetoplastid parasites, including Plasmodium, Toxoplasma, Cryptosporidium, Trypanosoma and Leishmania. Lipid synthesis differs in significant ways between parasites from both phyla and the human host. Parasites have acquired novel pathways through endosymbiosis, as in the case of the apicoplast, have dramatically reshaped substrate and product profiles, and have evolved specialized lipids to interact with or manipulate the host. These differences potentially provide opportunities for drug development. We outline the lipid pathways for key species in detail as they progress through the developmental cycle and highlight those that are of particular importance to the biology of the pathogens and/or are the most promising targets for parasite-specific treatment.
Keywords: Apicomplexa, Kinetoplastida, fatty acid synthesis, phospholipid synthesis, drugs, protozoa
1. Introduction
Diseases caused by protozoan parasites are among the most pressing concerns on the global health agenda. Within their ranks are not only some of the most widespread and important infectious diseases, but also many of the most sorely neglected [1]. In this review we will focus on two protozoan phyla in particular, Apicomplexa and Kinetoplastida, which stand out in their overall public health impact with respect to incidence and severity of the diseases they cause [2]. Not only are these the most important groups of parasites, they also are the best studied. Facile experimental model systems have been established for several members of both phyla. Driven by powerful genetics these models have yielded considerable insight into parasite metabolism. There are numerous significant metabolic differences between these parasites and their human host, and we will highlight these differences, as they afford opportunities for drug development. We will provide a brief introduction to the parasite models that are the focus of this article for readers unfamiliar with the organisms. We will describe the particular importance of lipid metabolism for pathogens to introduce the subsequent systematic and comprehensive discussion of the biosynthesis pathways of major lipid classes for both phyla.
1.1. Apicomplexan Parasites
Apicomplexans are eukaryotic pathogens named after the complex of secretory and cytoskeletal organelles located at the apical end of the parasite cell. Apicomplexa are intracellular parasites and the organelles of the apical complex are required for host cell invasion. Three genera represent a particular threat for human health and therefore will be the main focus of this review: Plasmodium, Toxoplasma and Cryptosporidium. Note, however, that there are numerous additional apicomplexans that are important veterinary pathogens.
Five species of Plasmodium collectively are the causative agents of malaria, a disease that claims the lives of about a million individuals every year. Most of these deaths are due to Plasmodium falciparum and occur in small children in sub-Saharan Africa. Plasmodium has a complex life cycle, and as we will see, profound metabolic and biochemical changes are associated with each life cycle transition. Infection of a human occurs through the bite of a mosquito, whereby a small number of sporozoites are injected with the saliva. The sporozoites travel to the liver and invade hepatocytes, where they massively proliferate. Merozoites are then released into the bloodstream, where they invade and replicate in red blood cells. It is this intraerythrocytic phase that is responsible for the disease. Control of malaria rests on the prevention of transmission (e.g. through treated bed nets) and drug treatment. The genetic malleability of the parasite and its propensity to evolve drug resistance [3] has long haunted malaria control. This famously includes the loss of chloroquine as an effective therapeutic. There is grave concern about the future of the current artemisinin-based treatment regimen with first signs of treatment failure emerging in South East Asia [4, 5]. A constantly evolving portfolio of new anti-malarials is required to keep up with the ever-changing parasites. Understanding parasite metabolism is an important cornerstone of this agenda.
Toxoplasma and Cryptosporidium were initially recognized as opportunistic infections in immunocompromised individuals and received particular attention as late stage manifestations of AIDS [6, 7]. The sexual phase of the Toxoplasma life cycle occurs in the intestinal tract of cats and results in the shedding of spore-like oocysts, which then become highly infectious to other animals and humans upon consumption of contaminated food or water. Within intermediate hosts, tachyzoites cause rapid systemic infection. The onset of immune control eliminates tachyzoites, but bradyzoites persist within tissue cysts for the life of the host. In immunocompromised adults, chronic infection is reactivated leading to Toxoplasma encephalitis. In addition, congenital toxoplasmosis is a significant concern during pregnancy. While anti-folate treatment is effective in controlling tachyzoites and acute disease, there are no drugs available to eliminate chronic infection, which is problematic in a variety of clinical settings [8]. Cryptosporidiosis is an acute enteric disease that typically is self-limiting. However, recent studies show that in particular in malnourished children the disease can be severe, protracted, and life threatening [9]. Cryptosporidium has a single host life cycle restricted to the intestinal epithelium, and oocysts are the only mode of transmission. Nitazoxanide was approved by the FDA for treatment but only shows moderate efficiency in immunocompetent children and produces no benefit in immunocompromised patients. Finding a reliable drug remains an important goal [10].
1.2. Kinetoplastid parasites
Kinetoplastids are flagellated protists that share a mitochondrial genome of unique organization and localization called the kinetoplast. There are numerous human pathogens among these early-branching eukaryotes, including African and American trypanosomes and many different species of Leishmania. All of these pathogens have insect and mammalian hosts. The different life cycle forms can be morphologically distinguished based on the position and length of the flagellum. More importantly, they also show remarkable metabolic differences. Stages from insect and mammalian hosts can be cultured axenically, which has greatly facilitated biochemical and genetic studies. Trypanosoma cruzi is the causative agent of Chagas disease, which is endemic in large parts of South America (note that although both the parasite and the vector are present in North America, transmission is rare). T. cruzi is transmitted by reduvid, or kissing bugs. Promastigote stages replicate within the bug’s midgut and infective metacyclic trypomastigotes are deposited with the feces onto the skin of the mammalian host. In the mammalian host, the parasite cycles between replicative intracellular amastigotes and trypomastigotes. Importantly, replicating amastigotes are free in the host cell cytoplasm providing intimate access to host metabolites. Chagas disease is characterized by chronic and progressive inflammatory tissue damage, in particular of the heart muscle. Treatment is available but is not consistently effective and suffers from significant adverse effects [11].
Trypanosoma vivax, T. congolense and T. brucei brucei cause Nagana, an important cattle disease that severely limits ranching and dairy production in many parts of Africa. T. brucei gambiense and rhodesiense are morphologically indistinguishable from the bovine subspecies and are the causative agents of Human African Trypanosomiasis, also known as sleeping sickness. All African trypanosomes are vectored by tsetse flies, where they are present as procyclic trypomastigotes, epimastigotes and metacyclic trypomastigotes. In the mammalian host, trypomastigotes replicate extracellularly in the bloodstream causing anemia and cachexia. In humans, the blood phase is ultimately followed by invasion of the central nervous system. Untreated infection is invariably fatal. Several drugs are available for treatment of Human African Trypanosomiasis and Nagana, but there are severe limitations. Some of the drugs have grave and even life threatening adverse effects, are limited in their potency once the parasites have entered the brain, or are only active against certain species or subspecies. Lastly, there is treatment failure and overt resistance to several drugs, in particular in veterinary practice [12–14].
Parasites of the genus Leishmania are transmitted by a variety of sand flies, where promastigotes replicate in the midgut and are mechanically introduced into the bite site. In the mammalian host, Leishmania parasites proliferate as intracellular amastigote in macrophages, and they do so within a fully matured and acidified phagolysosome [15]. A large number of different Leishmania species are responsible for a broad spectrum of disease manifestations, with currently 12 million people being infected worldwide. In its most benign form, leishmaniasis presents as a self-limiting skin ulcer at the bite site (cutaneous leishmaniasis). In contrast, progressive non-healing erosion of mucosal tissue in the vicinity of the bite site is characteristic for mucocutaneous forms. In the most severe visceral disease, parasites spread systemically causing hepatosplenomegaly, cachexia and immunosuppression. The etiology of these various forms appears to be closely linked to an unbalanced immune response to the chronic infection resulting in too much or too little inflammation and parasite control. Visceral leishmaniasis is fatal if not treated. Several drugs are available [16], but treatment of leishmaniasis remains challenging due to adverse effects, the requirement of lengthy regimens, limited drug availability and expenditure [17].
1.3. Lipids and Pathogens
Lipids are critical to the biology of all cells and organisms. They are the main structural elements of all biological membranes, they anchor glycoconjugates and many proteins to membranes, they serve as signaling molecules within and between cells, and they represent a highly efficient store and source of energy and reduction power. Lipids are of particular importance for pathogens, and some pathogens deliberately seek out lipid-rich host niches [18], or enhance the availability of lipids by manipulating the host [19, 20]. Intracellular pathogens have evolved sophisticated mechanisms to manipulate and tap into the lipid metabolism of their host cells. These include interference with vesicular and non-vesicular cellular lipid trafficking in viral [21], bacterial [22] and protozoal [23] pathogens. Within host cells, intracellular pathogens often develop in specialized vacuoles and the flow of lipids between host and pathogen-controlled membranous compartments is key to the pathogen’s ultimate success [15, 24–26].
Lipids are not only used by pathogens as food or structural building blocks, but are also important pathogenesis factors that allow the pathogen to evade immune responses, manipulate host processes, and cause disease. In many cases these are specialized lipids synthesized by the pathogen [27]. The best-characterized example in protozoan parasites is a class of specialized glycosylphosphatidylinositol (GPI) lipids. They include GPI-anchored lipophosphoglycans of Leishmania species, which are crucial for host specificity and survival of the parasite in the sand fly vector [28, 29], and are also thought to modulate the initial interaction with the mammalian host [30]. In African trypanosomes, GPI-anchored variant surface glycoproteins are at the heart of the antigenic variation mechanism used to enable chronic infection. In Plasmodium and Toxoplasma, precursor GPI lipids are believed to play important roles as toxins and immune modulators [31–33].
Due to the importance of specialized lipids in mycobacteria, lipid synthesis has been a major target of drug development for tuberculosis [34]. Such examples highlighting the important roles of lipids are also present in protozoan parasites. As we will describe in detail, kinetoplastids and apicomplexans rely on a number of mechanisms for lipid synthesis that are not found, or different from those used in the mammalian host. These include fatty acid synthesis in the mitochondrion and the plastid, specialized elongation and desaturation pathways, and differences in downstream pathways of phospholipid synthesis. The success of miltefosine as an orally available Leishmania drug is one important validation of lipid metabolism as a drug target. Lipid turnover in pathogens is complex and involves numerous mechanisms of uptake and synthesis. In the following we will systematically review fatty acid and phospholipid synthesis and uptake pathways in both apicomplexans and kinetoplastids.
2. Fatty acid synthesis in protozoan parasites
The genomes of protozoan parasites encode the enzymes for three distinct biochemical pathways involved in fatty acid synthesis. While there are significant differences between these pathways, the underlying chemistry and sequence of enzymatic reactions is highly conserved (Fig. 1). All pathways synthesize fatty acids by successive addition of two carbon units to a growing carboxylic acid chain that is held via the pantothenyl group of acyl carrier protein (ACP; see Table 1 for names of enzymes of fatty acid synthesis and modification and their corresponding genes in selected parasites) or coenzyme A (CoA). The length of this starter chain may vary. Fatty acid synthesis type I and II (FASI and FASII) typically produce fatty acids de novo, while the fatty acid elongation (FAE) pathway adds two carbon units to a typically much longer starter molecule. As detailed below, FASI and FASII differ in their architecture, FASI is expressed as a single very large polypeptide whereas FASII has multiple individual components (Fig. 1 B,C). The source of carbon to be added is malonyl-CoA, the activated form of a three carbon dicarboxylic acid. Malonyl-CoA is generated by acetyl-CoA carboxylase (ACC) from two molecules of acetyl-CoA (this step typically regulates the flux of fatty acid synthesis). Decarboxylative condensation of the malonyl substrate with the starter yields a chain elongated by two carbon units. The carbons are then fully reduced by the successive action of ketoacyl reductase, dehydratase and enoyl reductase. At this point the fatty acid can be elongated through condensation with another malonyl-CoA, or released by cleaving the thioester bond holding the carboxyl end.
Figure 1. Three mechanisms of fatty acid synthesis.

A, All fatty acid synthesis mechanisms follow a similar sequence of enzymatic reactions. A starter molecule is transferred to the phosphopantetheinyl group on ACP or CoA. The starter is then elongated by two carbon atoms and involving a four-reaction mechanism: decarboxylative condensation with a malonyl group by a synthase, reduction by a ketoreductase, dehydration by a dehydratase and reduction by an enoyl reductase. The product is released, or condensation with another malonyl group initiates the next round of elongation. B, Fatty acid synthase type I. All enzymes are domains of a single polypeptide (note that the apicomplexan FASI has a more complex multimodular architecture). C, Fatty acid synthase type II. All enzymes are encoded as individual proteins. D, Fatty acid elongation pathway. The system consists of enzymes encoded as individual proteins, acting on a CoA-bound starter molecule–typically a longer fatty acid (16 carbon or longer). ACP, acyl carrier protein; CoA, Coenzyme-A; FASI, fatty acid synthase type I pathway; FASII, fatty acid synthase type II pathway; FAE, fatty acid elongation pathway.
Table 1.
Names and EuPathDB gene identification numbers for enzymes of fatty acid synthesis and modification pathways in representative kinetoplastids and apicomplexan parasites.
| T. brucei | L. major | P. falciparum | T. gondii | ||
|---|---|---|---|---|---|
| ELO pathway | |||||
| ELO1 | Fatty acid elongase A | Tb927.7.4180 | Lmjf.14.0640 Lmjf.14.0650 Lmjf.14.0660 | PF3D7_0920000 | TGME49_042380 |
| ELO2 | Fatty acid elongase B | Tb927.7.4170 | Lmjf.14.0670 | PF3D7_0605900 | TGME49_005350 |
| ELO3 | Fatty acid elongase C | Tb927.7.4160 | Lmjf.14.0680 Lmjf.14.0690 | PF3D7_0109300 | TGME49_053880 |
| ELO4 | Fatty acid elongase D | Tb927.5.4530 | Lmjf.05.1170 | - | - |
| KCR | Ketoacyl CoA reductase | Tb927.5.1210 | LmjF.34.0010 | PF3D7_0422000 | TGME49_071890 |
| HCD | Hydroxyacyl CoA reductase | Tb927.10.10610 | LmjF.05.0280 | PF3D7_1331600 | TGME49_111290 |
| ECR | Enoyl CoA reductase | Tb927.3.1840 | LmjF.25.1770 | PF3D7_1135900 | TGME49_085240 |
| FASII pathway | |||||
| ACP | Acyl carrier protein | Tb927.1.2100 | LmjF.27.0290 | PF3D7_0208500 | TGME49_064080 |
| KAR | Ketoacyl ACP reductase | Tb927.2.5210 | LmjF27.2440 | PF3D7_0922900 | TGME49_017740 |
| HAD | Hydroxyacyl ACP dehydratase | Tb927.8.1440 | LmjF07.0430 LmjF07.0440 | PF3D7_1323000 | TGME49_121570 |
| EAR | Enoyl ACP reductase | Tb927.10.11930 | LmjF.04.0290 | PF3D7_0420200 | TGME49_051930 |
| PPTase | Phosphopantetheinyl transferase | Tb927.3.1840 | LmjF.25.1770 | PF3D7_0420200 | TGME49_003420 |
| FASI pathway | |||||
| FASI | Fatty acid synthase type I | - | - | - | TGME49_094820 |
| PPTase | Phosphopantetheinyl transferase | - | - | - | TGME49_014440 |
Not all fatty acid synthesis pathways are present in all apicomplexans. The FASII pathway has been thoroughly characterized in Toxoplasma and Plasmodium, but is absent in piroplasms (tick-transmitted apicomlexans, including Babesia [35] and Theileria [36] and Cryptosporidum [37]. A subgroup of Apicomplexa (the Coccidia and Cryptosporidium) also harbor a FAS I megasynthase.
The genomes of kinetoplastid parasites encode the enzymes of two pathways to synthesize fatty acids, a FASII pathway and a FAE pathway. Both pathways have been characterized in T. brucei using genetic and biochemical approaches. The genomes of other kinetoplastid parasites, namely T. cruzi and Leishmania major, encode clear homologs of the enzymes of these pathways, suggesting that fatty acid metabolism of these parasites may be similar to that of T. brucei. However, their roles have not been studied experimentally. Table 2 provides an overview of the distribution of the three pathways among well-characterized parasite species discussed in this review.
Table 2.
Comparison of fatty acid synthesis and modification pathways in apicomplexan and kinetoplastid parasites. The presence (+) or absence (−) of specific pathways is shown for well-studied model species.
| Species | FASII | FASI | PKS | FAE | ||
|---|---|---|---|---|---|---|
| Apicoplast | Mitochondrion | De novo | Elongation | |||
| Trypanosoma brucei | − | + | − | − | + | − |
| Trypanosoma cruzi | − | + | − | − | + | − |
| Leishmania major | − | + | − | − | + | − |
| Theileria annulata | − | − | − | − | − | − |
| Babesia bovis | − | − | − | − | − | − |
| Plasmodium falciparum | + | − | − | − | − | + |
| Toxoplasma gondii | + | − | + | + | − | + |
| Eimeria tenella | + | − | + | + | − | + |
| Cryptosporidium parvum | − | − | + | + | − | + |
2.1. The FASII pathway in apicomplexans
The FASII pathway is the stereotypical prokaryotic mechanism of fatty acid synthesis. Much of what we know about the pathway comes from studies in E. coli [38], however, it is widely distributed among bacteria and typically represents their main and often only source of fatty acids. All enzymes as well as the ACPs are expressed as individual polypeptides. In addition, the FASII pathway is found in some eukaryotes, where it is localized within organelles derived through endosymbiosis involving bacteria. Plants contain this pathway within the chloroplast, which is believed to be derived from a cyanobacterium [39] and represents the sole site of fatty acid de novo synthesis in plants. Recent work has also described a FASII pathway in mitochondria of certain eukaryotes, where its role is still unclear but likely includes the synthesis of lipoic acid [40]. In apicomplexan parasites the FASII pathway is found in the apicoplast, a plastid-like organelle that is derived from a red alga through secondary endosymbiosis [41]. Similar to plastids of plants and algae, the apicoplast is not only home to a FASII pathway but also to a non-mevalonate isoprenoid synthesis pathway and a portion of the heme pathway [41, 42]. The enzymes of the FASII pathway have been characterized in several apicomplexan species, with particular attention on P. falciparum, from which β-ketoacyl-ACP reductase [43], β-ketoacyl-ACP synthase [44] and β-ketoacyl-ACP dehydratase [45] have been expressed as recombinant enzymes and subjected to kinetic analyses. In addition, there is structural information available for P. falciparum 3-oxoacyl-ACP reductase [46] and β-hydroxyacyl ACP dehydratase [47].
Since the FASII pathway is not a major source of fatty acids for the mammalian host, it has been suggested that this pathway might represent a parasite-specific drug target against apicomplexan parasites. Consistent with this hypothesis, Plasmodium parasites were found to be sensitive to triclosan inhibition in vitro and in vivo [48]. Triclosan targets the enoyl-CoA reductase of the bacterial FASII pathway [49] and is a common ingredient of anti-bacterial soaps. Similarly, Toxoplasma and Babesia were reported to be susceptible to triclosan [50, 51], suggesting that enoyl-CoA reductase may represent a general target to inhibit growth of apicomplexan parasites and prompting further studies to characterize the Plasmodium enzyme and to find more potent inhibitors [52–54].
At around the same time, the genomes of several apicomplexans were sequenced, revealing that not all Apicomplexa have a FASII pathway. These include genera that apparently lost the apicoplast, such as Cryptosporidium, but also genera that still harbor an apicoplast [35–37]. Importantly, the FASII pathway and the presumptive triclosan target, enoyl-CoA reductase, are absent from Babesia and Theileria parasites, both of which are susceptible to triclosan at a dose comparable to the malaria parasite [35, 36, 55, 56]. This, together with the observation that some bacteria lacking enoyl-CoA reductase are susceptible to triclosan [57] and the lack of inhibition of enoyl-CoA reductase by triclosan in T. brucei [58], raised doubts about the specificity of the compound for the FASII pathway. These concerns were further heightened when new triclosan derivatives were tested. Careful structure-activity relationship studies revealed that the activity of triclosan against enoyl-CoA reductase did not correlate with the activity against parasites [59].
With the pharmacological support for an essential role for FASII weakened, genetic studies were conducted in Toxoplasma and Plasmodium. A conditional T. gondii mutant was constructed in the gene for FASII ACP. When apicoplast ACP expression was blocked in this mutant by exposure to anhydrous tetracycline, parasite growth in culture was significantly reduced, while continued suppression resulted in parasite death [60]. Furthermore, mice challenged with this mutant parasite were cured from a lethal infection by tetracycline treatment. When analyzed biochemically, these mutants showed a pronounced loss of lipoic acid production in the apicoplast. This was consistent with a loss of FASII activity, which is thought to provide octanoic acid-ACP, i.e. the precursor of apicoplast lipoic acid de novo synthesis [60–62]. In conclusion, Toxoplasma FASII is required for parasite development and pathogenesis.
The importance of FASII was revisited using genetic approaches in P. falciparum and two rodent malaria species [59, 63]. The studies demonstrated that the FASII enzymes are not expressed in the blood stages, which are the cause of clinical malaria. Furthermore the genes for several FASII enzymes including the presumptive triclosan target, enoyl-CoA reductase, could be deleted from the genome without impairing parasite growth in the blood stage or their development in the mosquito. Lastly, P. berghei enoyl-CoA reductase null mutants were as susceptible to triclosan as wild type parasites and the same was true for parasites expressing enoyl-CoA reductase carrying point mutations that commonly confer robust resistance to triclosan [59]. In conclusion, the anti-parasitic effect of triclosan is not due to inhibition of FASII, and FASII is not a valuable target for anti-malaria therapy to cure blood stage infection. Importantly however, the pathway was found to be essential for the later stage of parasite development in the liver phase of the infection [59, 63]. During liver infection Plasmodium generates thousands of merozoites per infected cell. This requires massive organelle and membrane biogenesis [64]. This extraordinary demand for lipid likely cannot be met by import from the host alone and as a consequence parasite lipid synthesis is indispensible. It is also conceivable that the Plasmodium FASII pathway may supply the parasite with specialized lipids that cannot be obtained from the liver cell [63].
Taken together, the studies in Toxoplasma and Plasmodium demonstrate that the importance of the FASII pathway is highly stage- and host cell-dependent. While Toxoplasma and the Plasmodium liver stage require FASII for survival, other stages and parasite species can apparently satisfy their needs by salvage of fatty acids from the host. This is consistent with findings in bacteria, where the importance of the FASII pathway is governed by the availability of exogenous fatty acids [65, 66].
While the requirement of the apicoplast FASII pathway is now well understood for life cycle progression of certain apicomplexans, its role in fatty acid metabolism of parasites has remained unclear. Is it a true engine of de novo synthesis of fatty acids, as in chloroplasts of plants, or does it have a more specialized and local role [67]? As pointed out above, biochemical analysis of FASII mutants in T. gondii suggested a link between FASII and lipoic acid synthesis [60, 61]. Lipoic acid in the apicoplast is required for the activity of the E2 subunit of pyruvate dehydrogenase, which in turn is required to supply the substrate for fatty acid synthesis, acetyl-CoA [68, 69]. In addition, lipoic acid is a cofactor for mitochondrial enzymes and, therefore, may be exported from the apicoplast to mitochondria. However, several studies have shown that mitochondrial lipoylation is independent of de novo synthesis of lipoic acid in the apicoplast but instead relies on lipoate salvage from the host [61, 70–72].
The impact of FASII on overall lipid composition and metabolism has been determined in T. gondii. Studies on the apicoplast phosphate translocator revealed that FASII is linked to cytoplasmic glycolysis via phosphoenolpyruvate [69, 73]. When parasites are grown in the presence of 13C-labeled glucose, robust incorporation into fatty acids can be detected through gas chromatography/mass spectrometry-based isotopomer analysis. The bulk of parasite myristic and palmitic acid is labeled throughout the carbon chain in a fashion consistent with de novo synthesis from glucose. Importantly, this labeling is entirely lost when the FASII pathway is blocked by ablation of ACP [74]. Furthermore, as detailed below, fatty acids produced by FASII are apparently exported from the apicoplast and modified in the endoplasmic reticulum (ER) [74], demonstrating that the apicoplast FASII pathway represents a classical de novo synthesis machine that supplies a significant portion of fatty acids in Toxoplasma. In addition to the supply of bulk fatty acids, FASII may satisfy additional, and more specialized needs. In Plasmodium, the loss of the FASII pathway in liver stage parasites results in a lack of MSP1 (merozoite surface protein 1) [63, 75]. Palmitate and myristate have been shown to be the main lipid components of the MSP1 GPI anchor [76]. In addition, fatty acids are also used to directly modify proteins. In apicomplexans, protein myristoylation and palmitoylation has been shown to be critical for the targeting of a number of membrane-associated proteins [77]. This appears to be particularly important for the inner membrane complex, which not only gives the parasite cell shape and organization, but is also crucial for the gliding machinery the parasite uses to find and invade host cells [78–80].
In summary, the role the FASII pathway in apicomplexan parasites is much more complex and diverse than initially anticipated. Its value as a drug target depends on the parasite species and the life cycle stage under consideration. Parasites harboring this pathway utilize it for de novo synthesis of fatty acids as well as to supply the substrate for lipoic acid synthesis (Fig. 2). A recent study has used stable isotope labeling and organelle purification to follow FASII in P. falciparum [81]. Consistent with previous genetic studies, the authors do not detect FASII activity in the erythrocyte stage. However, they document robust activity when culturing parasites in minimal lipid media. This adds an additional layer of complexity in that it suggests that the parasites may metabolically adapt to the nutritional and physiological status of the host. This is consistent with the finding that the FASII genes are among those showing transcriptional modulation when comparing different patients with naturally acquired infections [82].
Figure 2. Apicomplexans can acquire fatty acids through a complex network of synthesis and uptake.
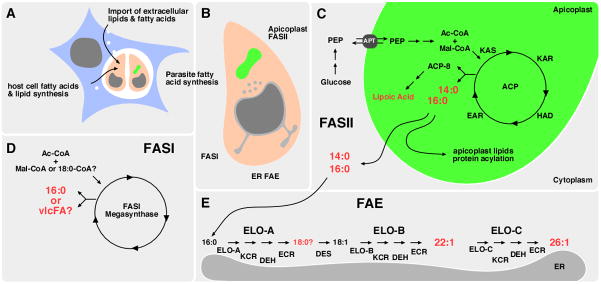
A, T. gondii is shown as a representative apicomplexan parasite (pink) and intracellular pathogen capable of fatty acid and lipid salvage from the host cell (blue). This process can intersect host cell import as well as synthesis routes. B, In addition, the parasite harbors three fatty acid synthesis pathways that are localized to different cellular compartments. C, Apicoplast (green)-localized FASII pathway produces significant amounts of myristic and palmitic acid, in addition to lipoic acid relying on cytoplasmic glycolysis for precursors. E, Apicomplexan parasites also maintain an ER-associated elongase system that synthesizes very long chain monounsaturated fatty acids, subsequently using the activity of ELO-B and ELO-C. D, Apicomplexan FASI remains largely uncharacterized. Its stage-specific expression pattern and localization are not established. It is also unclear whether this megasynthase synthesizes fatty acids de novo, like the FASI of humans, or acts as an elongase for saturated fatty acids, as demonstrated for FASI of C. parvum.
Major products are highlighted in red. Des, desaturase; PEP, phosphoenolpyruvate; Mal, malonate; Ac, acetate; vlcFA, very long chain fatty acids; Ac-CoA, acetyl-CoA; Mal-CoA, malonyl-CoA; ER, endoplasmic reticulum; KAS, ketoacyl-ACP synthase; KAR, ketoacyl-ACP reductase; HAD, hydroxyacyl-ACP dehydratase; EAR, enoyl-ACP reductase; ELO, elongase; KCR, ketoacyl-CoA reductase; DEH, acyl-CoA dehydratase; ECR, enoyl-CoA reductase. Other abbreviations are as in the legend of Fig. 1. Reproduced with minor modifications from [74].
2.2 The FASII pathway in kinetoplastids
The FASII pathway in kinetoplastid parasites appears similar to the FASII pathway from other organisms. An ACP holds the growing acyl chain while the chain is modified by a synthase, dehydratase and two reductases. The major difference between the apicomplexan and kinetoplastid FASII is their organellar localization. While the FASII pathway in apicomplexan parasites localizes to the apicoplast, the FASII pathway in kinetoplastids is found in the mitochondrion [83]. Mitochondrial localization of FASII has been confirmed in T. brucei by epitope tagging of the individual TbFASII enzymes. Three of the proteins, ACP, dehydratase and ketoacyl-ACP synthase, have been studied in detail. The presumptive dehydratase component of the TbFASII pathway complements the loss of the corresponding enzyme in yeast, i.e. it restores lipoic acid synthesis and respiration [84]. Genetic analysis in T. brucei shows that deletion or RNAi knockdown of ACP or ketoacyl-ACP synthase blocks growth of procyclic and bloodstream forms in culture [83], demonstrating that TbFASII is essential in both life cycle forms. Biochemical analyses using a cell free system and radioactive precursors demonstrate palmitate to be the final product of the TbFASII pathway. While capable of fatty acid production, the pathway shows moderate activity and contributes only about 10% of total parasite fatty acid synthesis activity [83]. This suggests that an alternate mechanism of synthesis may be the major contributor to the overall fatty acid pool in kinetoplastids. A second and maybe more important role for TbFASII appears to be the synthesis of octanoic acid ACP for lipoic acid production. It has been shown that ablation of the FASII pathway in T. brucei blocks lipoic acid synthesis [83], indicating that the TbFASII pathway is the main source of octanoic acid for lipoic acid synthesis.
Are there additional roles for FASII-synthesized fatty acids beyond lipoylation? Biochemical analyses indicate reduced phospholipid synthesis in TbFASII mutants [83], and loss of mitochondrial ACP decreases the levels of certain mitochondrial phospholipids. This change in phospholipid composition coincides with changes in morphology and membrane potential of the parasite mitochondrion. Loss of ACP also affects the respiratory complexes and significantly reduces cytochrome-mediated respiration [85]. A more recent report on TbFASII highlights the importance of the pathway for kinetoplastid DNA segregation in T. brucei bloodstream forms. This segregation defect is believed to be due to altered phospholipid composition of the mitochondrial membrane, resulting in disturbed assembly and structure of the tripartite assembly complex that is required for kDNA segregation [86].
The importance of the FASII pathway in other kinetoplastids has not been addressed yet. However, it has been demonstrated that expression of L. major dehydratase and ketoacyl reductase complements the respiration defect of yeast mutants for the corresponding enzymes (43). In addition, genome mining indicates the presence of additional FASII components in both L. major and T. cruzi (acyl carrier protein: LmjF.27.0290 and Tc00.1047053511867.140; ketoacyl synthase: LmjF.33.2720 and Tc00.1047053504157.20; enoyl-CoA reductase: LmjF.04.0290 and Tc00.1047053506627.20). All candidate proteins are predicted to localize to the mitochondrion, based on the presence of an N-terminal leader sequence. Together, these data suggest that the FASII pathway is likely conserved among kinetoplastids.
2.3. The FASI pathway in apicomplexans
Several apicomplexan parasites encode a FASI megasynthase. The overall chemistry of FASI is similar to that of FASII. The reaction sequence is initiated by loading an acyl starter moiety to the phosphopantetheinyl arm of the ACP domain, followed by elongation cycles catalyzed by a synthase, a dehydratase and two distinct reductase domains [87, 88]. In contrast to FASII, all activities and domains of FASI are part of a single polypeptide chain. FASI is the fatty acid synthesis mechanism typically found in eukaryotes, including humans and yeast, however, it has also been described in certain bacteria [89, 90]. Among apicomplexan parasites, candidate genes for the FASI pathway have been reported in Cryptosporidum [91], Eimeria [92], and Toxoplasma [67]. Bioinformatic and chemical analyses of apicomplexan FASI pathways suggest important differences compared to their mammalian counterparts. Structurally, mammalian FASI is a unimodular protein comprised of seven enzymatic domains. These enzymatic domains act sequentially for several cycles of elongation on an acyl moiety attached to ACP. Bioinformatic analysis of C. parvum FASI suggests that it is a multimodular protein where each module contains a set of multiple enzymatic domains [91]. The unusual architecture of apicomplexan FASI indicates that the mechanism of chain elongation is quite different from that of mammalian FASI. It is likely that the substrate is undergoing a single round of elongation in each module prior to being transferred to the ACP of the next module. The fate of the elongated product, in both mammals and apicomplexans, depends on the final domain of the enzyme. In mammals, a thioesterase domain releases the elongated product by hydrolytic cleavage [87], whereas in apicomplexan parasites, the thioesterase domain is replaced by a reductase domain, suggesting that the released product may be a fatty alcohol rather than a fatty acid [93].
The divergence of the apicomplexan FASI is not limited to its structural organization but may include its substrate specificity. Our current knowledge on the specificity of apicomplexan FASI is limited to data from the C. parvum enzyme, CpFASI. Biochemical experiments with recombinant CpFASI indicate that it may act as a fatty acid elongase rather than in de novo synthesis. Elongation versus de novo activity of FASI is defined by the substrate specificities of the starter and termination domains. The starter domain of CpFASI showed preference for long chain fatty acid substrates, with palmitoyl-CoA showing the highest activity in substrate competition assays [94], whereas the terminator domain showed preference for very long chain fatty acids, i.e. hexacosanoyl-CoA [93]. Expression of enzyme modules in E. coli is a remarkable idea to make these giant enzymes tractable, but may have limitations. Heterologous expression and the reliance on CoA-bound model substrates may modulate the specificity of the domains. Also, interaction with other enzymes in vivo may not be fully mimicked in an in vitro system. Nonetheless, these results strongly support the idea that CpFASI is involved in the production of very long chain fatty alcohols and, thus, is functionally different from mammalian FASI. Instead, CpFASI may resemble bacterial FASI (phylogenetic analyses appear to support a potential direct relationship via horizontal gene transfer). For example, it has been shown that FASI from Mycobacterium is capable of generating very long chain fatty acids using palmitate as substrate [95], and pathogenic mycobacteria use their FASI enzyme to elongate fatty acids imported from the host cell [96]. The resulting very long chain fatty acids can then be utilized for mycolic acid biosynthesis. It is likely that C. parvum FASI has a similar function, i.e. to elongate palmitate imported from the host cell to generate very long chain fatty acids.
Since the overall architecture of CpFASI is similar to FASI in Toxoplasma and Eimeria, it is likely that these proteins work in a similar fashion and perform a similar role. It is interesting to note that apicomplexan parasites harboring this pathway also harbor polyketide synthases (PKS) and form oocysts that are shed into the environment. These oocysts are surrounded and protected by an oocyst wall that confers remarkable chemical resistance. Oocysts are impervious to bleach and water chlorination, and the wall has a yet to be characterized lipid component. In contrast, apicomplexan parasites lacking FASI and PKS, like Plasmodium and Babesia [35, 36, 67], which are transmitted by arthropod vectors, do not form such environmentally stable oocysts. It is tempting to speculate that the products of the FASI enzyme could play an important role in oocyst formation, however, such a link has not been demonstrated experimentally.
PKS, like FASI, are multifunctional polypeptides and the apicomplexan enzymes appear to be type I PKS [97]. While the overall architecture of PKS enzymes resembles that of FASI enzymes, there are important differences. FASI fully reduces each added two-carbon unit. In contrast, PKS megasynthases typically lack certain domains, resulting in chains with multiple keto or hydroxyl groups [98]. These reactive groups can then be precursors for complex secondary products (this can but not always does involve additional enzymes). By varying the domain architecture, a myriad of compounds can be synthesized, including many molecules with importance in medical microbiology, such as toxins [99] and antibiotics [100]. PKS from C. parvum is the only characterized apicomplexan PKS so far, but its product is not known [101]. As for FASI, Zhu and colleagues used modular expression to study this enzyme. Interestingly, they found that the loading unit of PKS preferred very long fatty acids as substrates [102]. Dinoflagellates, a phylum of photosynthetic protists most closely related to apicomplexans, are well known to produce a variety of polyketide toxins (the typical cause of shellfish poisoning and red tide massive fish kills). These toxins appear to be the product of PKS-type enzymes and phylogenetic analysis has linked PKS from Cryptosporidium and some dinoflagellate PKS [103]. While initial studies in dinoflagellates have focused on toxins, it now appears that there may be additional roles for polyketides and related metabolites in the biology of these organisms [104]. Cysts are not restricted to apicomplexans but also found in dinoflagellates. It is tempting to speculate that FASI and two presumptive PKS megasynthases may collaborate to produce a special lipid of the oocyst wall and that such walls may predate apicomplexan parasitism. A similar collaboration between FASI and PKS has been reported for certain fungal [105] and bacterial [106] species.
2.4. The FAE pathway in apicomplexans
FAE represents the third mechanism of fatty acid synthesis in apicomplexan parasites. Similar to the FASII pathway, the FAE pathway operates using single monofunctional polypeptides to catalyze each elongation cycle. Every elongation cycle starts with a condensation reaction catalyzed by an elongase enzyme, however, in contrast to de novo synthesis systems, the FAE begins with a much longer starter (16:0 or longer). The product of this enzyme is modified sequentially by an enoyl reductase, a dehydratase and a ketoacyl reductase to produce a fatty acid that is elongated by a two carbon unit [107]. A major mechanistic difference that distinguishes the FAE pathway from the FASII pathway is the anchor molecule to which the growing acyl chain is attached. Unlike the FASII pathways that use ACPs, the FAE pathway employs CoA as an anchor molecule for fatty acid elongation [107, 108].
The FAE pathway is present in all eukaryotes. In plants [109], animals [110] and yeast [111], the pathway localizes to the ER and is important for proper growth and development [112, 113]. In apicomplexan parasites, elements of the FAE pathway were first characterized in C. parvum, which encodes a single fatty acid elongase localized in the parasitophorous vacuole membrane. This implies that the enzyme is secreted from the parasite and could act on substrates derived from the host cell. This may be part of a specialized uptake and modification process. It would be very interesting to localize additional components of the Cryptosporidium FAE machinery to further characterize this unusual compartmentalization. Biochemical studies on the recombinant enzyme showed that its preferred substrates are myristoyl-CoA and palmitoyl-CoA. Starting with myristoyl-CoA the enzyme can conduct up to two rounds of elongation generating stearic acid as the final product [114].
In contrast to C. parvum, in most organisms the FAE pathway employs multiple elongases with defined substrate and product length specificities that can act successively. While yeast contains three elongase genes [113], mammals contain six [107] and plants as many as twenty-one, highlighting the particular importance very long chain fatty acids, fatty alcohols and waxes play in the biology of plants [115]. Similar to S. cerevisiae, T. gondii and P. falciparum encode three fatty acid elongases [67]. Metabolic labeling experiments with radioactive myristate and palmitate suggest fatty acid elongation to be active in blood stage malaria parasites [116] and the T. gondii fatty acid elongases have been studied in detail [74]. In contrast to Cryptosporidium, the T. gondii pathway resides in the ER. The specific substrates and products of the T. gondii fatty acid elongases, named TgELO-A, TgELO-B and TgELO-C, have been established using a combination of conditional gene ablation and metabolomic analysis [74]. Stable isotope labeling in combination with gas chromatography/mass spectrometry analysis showed that TgELO-A is required to generate stearate from palmitate. Interestingly, TgELO-B and TgELO-C exhibit elongation activities exclusively towards monounsaturated fatty acids, with TgELO-B required to generate erucic acid (22:1) from oleic acid (18:1) and TgELOC to generate hexacosenoic acid (26:1) from erucic acid [74]. Chain length specificity for products has been previously reported for yeast and plant enzymes. However, how ELOs measure the actual chain lengths of their products was puzzling for some time. A seminal article by Denic and Weismann has elucidated this mechanism [111]. Fatty acid elongases in yeast are membrane proteins of the ER with their active sites exposed on the cytoplasmic surface. A lysine residue on the luminal side of a transmembrane helix is responsible for sensing the methyl end of the fully elongated fatty acid. Using mutational analyses, it was shown that the position of this lysine residue in the sixth transmembrane helix determines the length of the fatty acid released from a fatty acid elongase [111]. A precise residue responsible for the selectivity of T. gondii ELOs has not been identified yet.
The products of the FAE pathway are essential in mammals, plants and yeast. Stable isotope labeling shows that a significant proportion of fatty acids in T. gondii is the product of the FAE pathway [74]. Thus, it is reasonable to assume that these lipids also play an essential role in T. gondii. However, the loss of any individual elongase has no effect on parasite growth. Similarly, in yeast the loss of individual elongase enzymes is tolerated, whereas simultaneous disruption of two fatty acid elongases is lethal [113]. This has been attributed to overlapping specificity of the fatty acid elongases [113]. It is possible that the lack of growth defects in TgELO mutants is equally due to overlap. Our preliminary work showed that disruption of non-redundant enzyme components of the T. gondii FAE pathway results in strong inhibition of parasite growth, supporting such a model (S. Ramakrishnan and B. Striepen, unpublished results).
While we now know that apicomplexans use two different systems (FAE and FASI) to synthesize long chain fatty acids, it is not known what the products are used for and why they cannot be taken from the host. In plants [112] and yeast [117], loss of the FAE pathway affects phospholipid synthesis. Long chain fatty acids could be performing a similar role in apicomplexan parasites as well. However, such a link between phospholipid synthesis and fatty acid elongation in apicomplexan parasites has yet to be studied experimentally.
2.5. The FAE pathway in kinetoplastids
The kinetoplastid FAE pathway comprises of multiple elongases, dehydratases and reductases. The best-characterized components of this pathway are the four elongase enzymes of T. brucei. The T. brucei FAE resides in the membrane of the ER [118] (Fig. 3). A transgenic version of TbElo3 fused to GFP co-localized with blue-white DPX staining, a dye that preferentially accumulates in the ER [118]. While at first glance the enzymes and their localization in T. brucei appear to be quite similar to those in other eukaryotes, their functions are remarkably different. Unlike any other FAE pathway studied so far, the TbFAE pathway is capable of synthesizing fatty acids de novo. The key to this ability lies in the chain length specificity for the starter. While FAE pathways typically use a medium or long chain fatty acyl-CoA as primer, the TbFAE can use butyryl-CoA to prime the elongation process. The names of the elongase enzymes in the pathway (ELO1-4) refer to the order in which they act in the pathway. The specific role of each elongase enzyme was determined by analyzing ELO mutants in T. brucei bloodstream forms [118]. Fatty acid elongases are generally divided into two categories: elongases that act on saturated and monounsaturated fatty acids and elongases that act on polyunsaturated fatty acids (PUFAs). In the TbFAE pathway, ELO1-3 act on saturated fatty acids, whereas ELO4 is specific for PUFAs. Chain elongation for the TbFAE pathway starts with the action of TbELO1, which uses butyryl-CoA to generate decanoyl-CoA, which is then extended by TbELO2 to myristoyl-CoA. The identification of this synthesis route for myristoyl-CoA solved a long-standing conundrum. The membrane of T. brucei bloodstream forms is covered with a dense coat of variant surface glycoproteins, which allows the parasite to evade host immunity through antigenic variation. These proteins are attached to the plasma membrane via a GPI anchor, which represents the first fully characterized GPI structure in eukaryotes [119]. Its composition is unique in that the fatty acyl chains are composed of myristic acid exclusively [120], requiring an elaborate fatty acid remodeling process to ensure this exclusivity [121]. Myristic acid is in low abundance in the serum of the mammalian host and it was puzzling for a long time as to how the parasite satisfies its enormous demand for this fatty acid. Myristoyl-CoA synthesized by the peculiar FAE de novo pathway solved this puzzle. Unexpectedly, however, T. brucei bloodstream forms lacking the FAE pathway showed no growth defect in culture and were not attenuated in vivo in the rat model [118].
Figure 3. Kinetoplastid fatty acid synthesis occurs in the mitochondrion and the ER.

Kinetoplastids obtain fatty acids using synthesis and import mechanisms. A, Kinetoplastid parasites replicate extracellularly in the bloodstream of the mammalian host, red blood cells are also shown (red). B, Additionally, the parasite harbors two mechanisms of fatty acid synthesis that are localized in two different organelles (redrawn in part after [323]). C, A FASII pathway localizes to the mitochondrion (light violet), where it is required for the synthesis of lipoic acid and palmitic acid. D, Kinetoplastid parasites also harbor an ER-localized fatty acid elongase system. Unlike all other organisms, kinetoplastid FAE is used for de novo synthesis of fatty acids. The kinetoplastid FAE uses butyrate and malonate as substrates to generate myristate/stearate and adrenate as products. Major products are highlighted in red. Mal, malonate; Ac, acetate. Other abbreviations are as in the legend of Fig. 2.
Analysis of TbELO3 shows that this enzyme is capable of generating stearoyl-CoA from myristoyl-CoA. Importantly, this enzyme is only expressed in T. brucei procyclic forms, which synthesize surface proteins and GPI anchors that differ from bloodstream forms. The major acyl groups on procyclic GPI anchors are palmitate and stearate, and not myristate [122], suggesting that TbELO3 is the main provider of fatty acids for GPI anchors in procyclic trypanosomes. In contrast to the situation in T. brucei bloodstream forms, FAE appears to be essential for growth of procyclic parasites in culture. Altogether, these results show FAE in T. brucei to be the dominant source of fatty acids–even for shorter species like myristate. Thus, as in Apicomplexa, the importance of a fatty acid synthesis pathway for trypanosome survival depends on the life cycle stage and may be influenced by the availability of fatty acids in the host environment.
Components of the FAE pathway have also been detected in T. cruzi and L. major. A search of the respective genome sequences suggests the presence of candidate genes for ketoacyl-CoA reductases, dehydratases, and enoyl-CoA reductases in all kinetoplastid parasites. While T. brucei harbors four elongase genes, T. cruzi seems to contain five and L. major as many as fourteen. None of the encoded candidate enzymes has been studied experimentally. However, the elongases detected in T. cruzi and L. major are believed to be orthologs of TbELO1-3, or TbELO4, based on phylogenetic analysis [123]. The presence of additional elongase enzymes in L. major and T. cruzi may be related to the fact that the GPI anchors in these parasites have longer fatty acids [124] compared to those in T. brucei. In addition, the lipid portion of Leishmania lipophosphoglycan is composed of very long (24 and 26 carbon) alkyl chains that are likely derived from very long fatty acids [124].
2.6. Synthesis of unsaturated fatty acids
Unsaturation of fatty acids alters their physical and chemical properties. Incorporation of unsaturated fatty acids into membrane lipids can increase the overall flexibility and fluidity of membranes. The degree to which unsaturated fatty acids are used varies considerably between organisms. Unsaturated fatty acids are synthesized from saturated fatty acids by the action of desaturases [125]. Unsaturated fatty acids are classified into two main categories, monounsaturated fatty acids (MUFAs) and PUFAs. The synthesis of MUFAs occurs by introduction of a single double bond and is universally catalyzed by the enzyme stearoyl-CoA desaturase (SCD) [126–128]. SCD in the apicomplexan parasite P. falciparum was identified only recently [129]. The enzyme seems to be active in blood stage parasites [116] and is required for growth [129]. Data from other systems suggest that expression of SCD can be influenced by certain PUFAs [130]. Whether such an effect on SCD is responsible for PUFA-mediated inhibition of Plasmodium growth [131] has yet to be determined. Genome searches in apicomplexan parasites reveal the presence of SCD in Toxoplasma, Eimeria and Neospora, but not in Theileria, Babesia or Cryptosporidium. Heavy isotope labeling studies in T. gondii indicate that SCD likely acts downstream of FASII and ELO-A and upstream of ELO-B and ELO-C [74]. However, the absence of FASII activity in Plasmodium blood stages suggests that precursors for desaturation also may be salvaged from the host.
The action of desaturases is not limited to the formation of MUFAs. Additional desaturases distinct from SCD are responsible for the synthesis of PUFAs. These types of desaturases have not been reported in apicomplexan parasites. The presence of polyunsaturated fatty acids in T. gondii [130] and the demonstration of linoleic acid in C. parvum [130] and Eimeria oocysts [130, 132] suggests that these parasites could have evolved a mechanism to import the required polyunsaturated fatty acids from the host.
Kinetoplastids are able to synthesize PUFAs and they harbor multiple desaturases that can act sequentially. The pathway begins with SCD, which has been characterized experimentally in T. brucei and is essential for both bloodstream and procyclic forms [133]. Genome analysis of Trypanosoma and Leishmania further reveals the presence of delta-12, delta-4 and omega-3 desaturases (the numbers indicate the position of desaturation with respect to the carboxy- (delta) or methyl- (omega) end of the chain respectively). In contrast, delta-6 and delta-5 desaturases are only found in Leishmania [134–136], indicating that Leishmania but not Trypanosoma have a complete pathway to synthesize C22 PUFAs. In trypanosomes, a PUFA-specific elongase (TbELO4) and a delta-4 desaturase may generate C22 PUFAs from host-derived C20 substrates. In addition to high amounts of C22 PUFAs, trypanosomes also contain high amounts of linoleic acid [137, 138]. The omega-3 desaturase present in both Leishmania and Trypanosoma is responsible for the synthesis of linoleic acid and has received particular attention. The absence of this enzyme in humans makes omega-3 desaturase a potential candidate for parasite-specific intervention [134, 139].
Taken together, these studies show that both apicomplexan and trypanosomatid parasites follow a common paradigm for generating MUFAs using SCD. In both parasite groups this enzyme is important for growth. However, the processes of obtaining PUFAs appear quite different. Apicomplexans parasites rely on fatty acid import whereas trypanosomatids are capable of synthesizing PUFAs.
2.7. Substrates and activation of FA synthesis pathways
The difference in the evolutionary origin and cellular localization of fatty acid synthesis pathways in apicomplexans is also reflected in more peripheral aspects. A first example is the provision of malonyl-CoA, the main substrate for fatty acid synthesis, a second is the activation of ACPs that are critical in both FAS systems. Apicomplexan parasites that harbor both the FASI and FASII mechanism seem to have evolved two independent systems for substrate provision and enzyme activation. The malonyl-CoA substrate is generated by the action of ACC. Two genes encoding ACC have been identified in T. gondii. Localization studies suggest that one of these enzymes is recruited to the apicoplast while the other is cytosolic [140, 141]. The apicoplast-localized ACC is probably required to generate malonyl-CoA for the apicoplast-based FASII pathway, whereas cytosolic ACC may supply malonyl-CoA for the FASI pathway. Plasmodium, which lacks the FASI pathway, and Cryptosporidium, which lacks the FASII pathway, harbor only single genes for ACC [67]. The idea that the apicoplast and the cytoplasm rely on independent pools of malonyl-CoA is also consistent with our recent stable isotope labeling experiments. We found that 13C-acetate is readily incorporated into the products of the cytoplasm- and ER-based FAE pathways (see below). In contract, carbon derived from 13C-glucose but not 13C-acetate is incorporated into fatty acids produced by the FASII pathway [74]. The apicoplast pathway is solely fed through the import of phosphoenolpyruvate [69] from the cytoplasm.
Unlike apicomplexans, kinetoplastids harbor only a single ACC. In T. brucei, the enzyme is cytosolic and its main role is to supply the FAE pathway. RNAi experiments demonstrate that TbACC is required in procyclic forms but dispensable in bloodstream forms, thus mirroring the findings for TbELO mutants [142, 143]. It is likely, yet has not been demonstrated experimentally, that malonyl-CoA generated by cytoplasmic TbACC also serves as substrate for the mitochondrial FASII pathway.
Two independent ACP activating enzymes are present in T. gondii [67, 144]. In both the FASI and FASII pathway, apo-ACP is activated to the holo-ACP form by addition of a phosphopantetheinyl moiety. This reaction is catalyzed by phosphopantetheinyl transferase [145]. The two phosphopantetheinyl transferases in T. gondii have not been characterized in detail yet. However, one of them carries a bipartite leader and is likely localized to the apicoplast and activates the FASII ACP. The second protein is related to the cytoplasmic enzyme previously characterized in Cryptosporidium [144] and is probably involved in FASI (and PKS) activation. How T. brucei ACP is activated remains unknown, a candidate phosphopantetheinyl transferase gene is not immediately evident in the genome.
3. Phospholipid synthesis
3.1. Phospholipids of Kinetoplastida and Apicomplexa
Growth and proliferation of parasites depends on their ability to recruit the building blocks for membrane assembly. According to the paradigm of parasitism, it has long been thought that Kinetoplastida and Apicomplexa scavenge lipids for membrane formation from their hosts. However, recent genetic, biochemical, and pharmacological data have made it clear that de novo lipid synthesis plays a crucial role for parasite viability at different life cycle stages.
Phospholipids are the major lipid components of biological membranes. They comprise of two categories, the glycerophospholipids consisting of hydrophilic head groups linked via phosphate to glycerol-bound fatty acid or fatty alcohol chains and the sphingophospholipids consisting of head groups linked via phosphate to ceramide. The composition of the different phospholipid classes, together with the acyl and alkyl chain composition, determines the biophysical properties of a given membrane. In addition to their roles as membrane constituents, phospholipids are involved in many other biological processes, such as lipid signaling, maintaining and regulating protein structure and function, formation of specialized membrane domains, and attachment of proteins to membranes.
3.1.1. Phospholipids of Kinetoplastida
African trypanosomes have a glycerophospholipid composition similar to that of other eukaryotic cells, with phosphatidylcholine (PC) (45–60% of total lipid phosphorus) and phosphatidylethanolamine (PE) (10–20%) representing the most abundant, and phosphatidylinositol (PI) (6–12%), phosphatidylserine (PS) (<4%), cardiolipin (CL) (2–4%) and phosphatidylglycerol (PG) (<2%) representing minor glycerophospholipid classes [146–150]. Similarly, in Leishmania, PC is the most abundant glycerophospholipid class (30–50%), followed by PE (10%) and PI (10%) [151], while PA, CL, and PG, as well as lyso-PE and lyso-PC, are present in trace amounts only [151–153]. Although the presence of PS in Leishmania parasites has been reported in several studies [151, 154, 155], recent analyses using mass spectrometry and serine labeling failed to detect this lipid in L. major promastigotes [156–158] (see also below). Interestingly, the glycerophospholipid classes in T. brucei and Leishmania consist of high amounts of ether-type molecular species [137, 147, 159]. In particular, PE in T. brucei procyclic and bloodstream forms [147, 159] and in L. major promastigotes [152] is composed mostly of alkenyl-acyl and alkyl-acyl glycerol, also called plasmenyl or plasmalogen and plasmanyl, respectively, molecular species. The most prominent glycerophospholipids of Leishmania promastigotes contain saturated and unsaturated C18 fatty acids [152], and most ether-type phospholipids in T. brucei contain C18:0 at the sn-1 and mainly unsaturated fatty acids (C18:2, C22:2) at the sn-2 position [147]. Phospholipid analysis in T. cruzi epimastigotes revealed PC as the most abundant phospholipid class (44%), followed by PE (28%), PI (12%), SM (4%), and small amounts of CL, PA, lyso-PC and PS [160].
The sphingophospholipid classes, sphingomyelin (SM), inositol phosphorylceramide (IPC) and ethanolamine phosphorylceramide (EPC), constitute 10–15% of total lipid phosphorus in T. brucei [146, 147, 161, 162]. Remarkably, due to stage-specific expression of the enzymes catalyzing the final reactions in SM, IPC and EPC synthesis, the relative amounts of these classes vary considerably between different T. brucei life cycle forms, with IPC being primarily present in procyclic, and EPC in bloodstream form parasites [162] (see also below). The presence of SM and IPC has also been demonstrated in T. cruzi epimastigotes [160, 163–165]. The ceramide portion of IPC consists primarily of a mixture of d18:0/16:0 dihydroceramide and d18:1/16:0 ceramide [164]. IPC also represents the major sphingophospholipid class in Leishmania [166, 167], with d16:1/18:0 IPC representing the major molecular species. The unusual composition of the long chain bases of Leishmania IPC suggests that the parasite preferentially uses myristoyl-CoA for ceramide synthesis, instead of palmitoyl-CoA as in T. brucei [162], T. cruzi [164], mammalian cells and yeast [168, 169]. Unlike trypanosomes, Leishmania parasites do not synthesize SM [166, 167].
3.1.2. Phospholipids of Apicomplexa
Membranes of Plasmodium are composed of several glycerophospholipid classes, but contain only small amounts of SM and cholesterol [170]. The main glycerophospholipid classes in isolated parasites include PC (40–50% of total lipid phosphorus), PE (35–45%) and PI (4–11%), while PS (<5%), CL (5%) and PA (<0.1%) represent minor classes [170–172]. Interestingly, upon parasite infection, the phospholipid content of erythrocytes was shown to increase 6-fold, indicating the high phospholipid biosynthetic capacity of the parasite [170]. In particular, compared to uninfected erythrocytes, plasma membranes from P. falciparum-infected erythrocytes contained more PC (38.7% versus 31.7%) and PI (2.1% versus 0.8%), but less SM (14.6% versus 28.0%) [172]. In addition, membranes of infected erythrocytes showed an altered phospholipid molecular species composition, with a shift in the fatty acyl chain composition towards that of the parasite, with increased amounts of palmitic, oleic and linoleic acid and a decrease in arachidonic acid [172]. Together, these results indicate that Plasmodium parasites are capable of modulating the membrane lipid composition of host cells.
Lipid analysis of isolated T. gondii tachyzoites revealed a 5-fold higher amount of phospholipid over cholesterol, with the major glycerophospholipid classes being PC (62% of total lipid phosphorus), PE (11%), PS (8%) and PI (6%), and SM representing the major sphingophospholipid class (8% of total lipid phosphorus) [173]. The most abundant fatty acids in tachyzoites are oleic acid > linoleic acid > palmitic acid [173]. In addition, metabolic labeling experiments demonstrated de novo synthesis of glycosphingolipids in T. gondii tachyzoites [174]. A more recent lipidomic analysis using electrospray ionization mass spectrometry confirmed the presence of hexosylceramide and dihexosylceramide and, in addition, revealed small amounts of PA and EPC, in addition to the above-mentioned phospholipid classes [175].
3.2. Substrate acquisition for de novo phospholipid biosynthesis
Despite the ability of trypanosomatids to scavenge lipids from their hosts via uptake of fatty acids and lyso-glycerophospholipids, or by receptor-mediated endocytosis [138, 176–178], the genomes of T. brucei, T. cruzi and Leishmania spp. contain candidate genes encoding enzymes involved in de novo phospholipid biosynthesis [179, 180]. Indeed, work in recent years has demonstrated that many lipid synthesis pathways known from other eukaryotes are also active in trypanosomatids. A prerequisite for de novo formation of glycerophospholipids is the availability of substrates for head group attachment, including choline, ethanolamine, myo-inositol and serine, which have to be taken up by parasites from their host environments.
Transport of choline has been reported in L. major promastigotes [181]. This study showed that choline uptake occurs with a Km in the low micromolar range, is Na+-dependent and inhibited by high concentrations of hemicholinium-3, suggesting that Leishmania express a transporter related to members of the mammalian CTL transporter family [182]. However, a gene encoding a choline transporter has not been identified yet. In contrast to Leishmania, T. brucei bloodstream forms were reported to meet their demand for the essential nutrient, choline, by uptake of lyso-PC from the medium or host plasma, while direct uptake of choline into parasites was negligible [138, 183]. However, in contrast to a previous report [184], we recently found that [3H]choline can be taken up by T. brucei procyclic and bloodstream forms in culture, with Km values in the low micromolar range, suggesting that T. brucei expresses a choline transporter [185].
Uptake of ethanolamine has been reported in Leishmania parasites and T. brucei bloodstream and procyclic forms, where it is used as precursor for the synthesis of PE [159, 186, 187], GPIs [187–189] and the unique ethanolamine phosphoglycerol modification of eEF1A (eukaryotic elongation factor 1A) [190]. However, an ethanolamine transporter has not been identified.
Although myo-inositol can be produced de novo from glucose (see below), uptake of myo-inositol has been described in L. donovani [191], T. cruzi [192], and T. brucei [193]. Detailed characterization of the transporters, including functional expression in Xenopus oocytes, demonstrated that myo-inositol uptake in L. donovani promastigotes and T. brucei procyclic forms is mediated via H+-coupled electrogenic cotransport [150, 191, 194]. Interestingly, down-regulation of the transporter in T. brucei blocked myo-inositol uptake and formation of PI and IPC, but had no effect on GPI production [150]. Together with a previous study [195], these results demonstrate that de novo synthesized myo-inositol and myo-inositol taken up from the environment are compartmentalized and used for the formation of different pools of inositol-containing lipids in T. brucei (see below). The observation that a hemagglutinin-tagged form of the T. brucei myo-inositol transporter localizes to the Golgi in procyclic forms suggests that bulk PI and IPC synthesis may occur in the Golgi [150], whereas PI production for GPI synthesis using endogenously produced myo-inositol occurs in the ER [191].
Uptake of exogenous lipids and lipid precursors has also been reported in apicomplexan parasites. In Plasmodium, incorporation of exogenous fatty acids [196–198] and lyso-PC [199] is necessary for parasite growth and to sustain phospholipid synthesis during the intraerythrocytic stage. In addition, PC is readily transferred from the erythrocyte membrane to intraerythrocytic parasites, indicating the presence of a selective PC transport mechanism providing PC from the host cell to the parasite [200]. Furthermore, it has been shown that P. falciparum efficiently take up ethanolamine and, to a lesser extent choline and myo-inositol, from the host erythrocyte [201, 202]. Choline transport into P. falciparum has been shown to be Na+-independent and mediated by an H+-motive force [202]. However, a choline transporter has not yet been identified. Interestingly, uptake of choline can be inhibited by a set of choline analogs, which are toxic for malaria parasites [202, 203]. At present, it is unclear if the anti-malarial activity of the compounds is related to their potency to block choline uptake or inhibit PC synthesis [204–206].
Similarly, intracellular T. gondii parasites are able to take up glycerophospholipid precursors, in particular choline for PC synthesis, but they also divert host cell PC for membrane formation [207]. In addition, uptake of choline, ethanolamine and serine with subsequent incorporation into phospholipid classes has also been reported in extracellular T. gondii parasites [208].
Finally, serine, which is used as head group for PS formation and as precursor for sphingolipid synthesis, is taken up by protozoan parasites via amino acid transporters. Although no serine-specific transporter has been identified, serine uptake from the environment and metabolism into membrane lipids has been demonstrated in Leishmania and T. brucei parasites [156, 159, 161, 186]. In addition, intraerythrocytic Plasmodium parasites use serine from the blood or from hemoglobin catabolism after import into the acidic food vacuole [209] for phospholipid formation.
3.3. Phospholipid synthesis pathways
The major routes for phospholipid synthesis in eukaryotes are summarized in Fig. 4; only those pathways that are active in Apicomplexa and Kinetoplastida are shown (for explanations see below).
Figure 4. Phospholipid synthesis pathways in kinetoplastids and apicomplexans.

Schematic overview on the major pathways for phospholipid synthesis in kinetoplastid and apicomplexan parasites. Metabolites taken up from the environment appear in a black box, major phospholipid classes are indicated by a green circle. 3KSR, 3-ketosphinganine reductase; CDS, cytidine diphosphate diacylglycerol synthase; CEPT, choline/ethanolamine phosphotransferase; CK, choline kinase; CLS, cardiolipin synthase; CT, choline-phosphate cytidylyltransferase; DAG, diacylglycerol; DHCD, dihydroceramide desaturase; DHCS, dihydroceramide synthase; EK, ethanolamine kinase; PMT, phosphoethanolamine N-methyltransferase; EPT, ethanolamine phosphotransferase; ET, ethanolamine-phosphate cytidylyltransferase; PEMT, PE N-methyltransferase; PGPP, PGP phosphatase; PGPS, PGP synthase; PIS, PI synthase; PSD, PS decarboxylase; PSS/PSS2, PS synthase/PS synthase-2; SAM, S-adenosyl methionine; SD, serine decarboxylase; SLS, sphingolipid synthase; SPL, sphingosine-1-phosphate lyase; SPT, serine palmitoyltransferase. * the substrate for prokaryotic-type CLS in protozoan parasites has not been confirmed experimentally.
3.3.1. Synthesis of PC, PE and PS
Eukaryotes possess several pathways for the synthesis of the two most abundant phospholipid classes, PC and PE. A major route involves formation of the CDP-activated precursors, CDP-choline and CDP-ethanolamine, and is known as the Kennedy pathway after its discovery more than 55 years ago by Kennedy and Weiss [210]. In the CDP-choline branch, choline is first phosphorylated by choline kinase (see Table 3 for names of enzymes of phospholipid synthesis and their corresponding genes in selected parasites) and then activated to CDP-choline by choline-phosphate cytidylyltransferase. The latter reaction often represents the rate-limiting step in PC synthesis by the Kennedy pathway [211]. The third step is mediated by choline phosphotransferase, transferring CDP-choline to diacyl glycerol or the corresponding ether-type glycerol species, to generate diacyl or alk(en)yl-acyl PC, respectively [211]. Since in most cells this enzyme shows dual specificity for CDP-choline and CDP-ethanolamine, i.e. the corresponding metabolite of the PE branch of the Kennedy pathway, it will be named choline/ethanolamine phosphotransferase (CEPT) throughout this review. In the CDP-ethanolamine branch of the pathway, ethanolamine is phosphorylated by ethanolamine kinase and activated by ethanolamine-phosphate cytidylyltransferase to CDP-ethanolamine [212, 213]. An alternative route for ethanolamine-phosphate formation involves degradation of sphingoid bases by sphingosine-1-phosphate lyase [214]. The last step of the CDP-ethanolamine pathway is catalyzed by ethanolamine phosphotransferase (EPT), leading to the formation of diacyl- and ether-type PE molecular species, using diacyl or alk(en)yl-acyl glycerol, respectively, as substrates [212, 213] (Fig. 4).
Table 3.
Names and EuPathDB gene identification numbers for enzymes of phospholipid synthesis pathways in representative kinetoplastids and apicomplexan parasites.
| T. brucei | L. major | P. falciparum | T. gondii | ||
|---|---|---|---|---|---|
| Phospholipid pathways | |||||
| 3KSR | 3-Ketosphinganine reductase | Tb927.10.4040 | LmjF.35.0330 | ? | ? |
| CEPT | Choline/ethanolamine phosphotransferase | Tb927.10.8900 | LmjF.36.5900 | PF3D7_0628300 | TGME49_261760 |
| CK | Choline kinase | Tb11.18.0017 | LmjF.27.1420 | PF3D7_1401800 | TGME49_320630 |
| CLS | Cardiolipin synthase | Tb927.4.2560 | LmjF.34.2110 | PF3D7_0609400 | TGME49_309940 |
| CT | Choline-phosphate cytidylyltransferase | Tb927.10.12810 | LmjF.18.1330 | PF3D7_1316600 | TGME49_216930 |
| DHCS | Dihydroceramide synthase | Tb927.8.7730 Tb927.4.4740 | LmjF.31.1780 | ? | ? |
| EK | Ethanolamine kinase | Tb927.5.1140 | LmjF.35.1470 | PF3D7_1124600 | TGVEG_081320 |
| EPT | Ethanolamine phosphotransferase | Tb927.10.13290 | LmjF.18.0810 | - | ? |
| ET | Ethanolamine-phosphate cytidylyltransferase | Tb11.01.5730 | LmjF.32.0890 | PF3D7_1347700 | TGME49_310280 |
| INO | myo-Inositol-3-phosphate synthase | Tb927.10.7110 | LmjF.14.1360 | PF3D7_0511800 | - |
| PEMT | Phosphatidylethanolamine N- methyltransferase | - | LmjF.31.2290 LmjF.31.3120 | - | - |
| PGPP | Phosphatidylglycerophosphate phosphatase | ? | ? | ? | ? |
| PGPS | Phosphatidylglycerophosphate synthase | Tb927.8.1720 | LmjF.07.0200 | PF3D7_0820200 | TGVEG_005460 |
| PIS | Phosphatidylinositol synthase | Tb09.160.0530 | LmjF.26.2480 | PF3D7_1315600 | TGME49_207710 |
| PMT | Phosphoethanolamine N- methyltransferase | - | - | PF3D7_1343000 | - |
| PSD | Phosphatidylserine decarboxylase | Tb09.211.1610 | LmjF.35.4590 | PF3D7_0927900 | TGME49_269920 TGME49_225550 |
| PSS | Phosphatidylserine synthase | - | - | PF3D7_1366800 | - |
| PSS2 | Phosphatidylserine synthase 2 | Tb927.7.3760 | LmjF.14.1200 | - | TGME49_261480 |
| SD | Serine decarboxylase | - | - | ? | - |
| SK | Sphingosine kinase | Tb927.7.1240 | LmjF.26.0710 | - | - |
| SLS | Sphingolipid synthase | Tb09.211.1000 Tb09.211.1010 Tb09.211.1020 Tb09.211.1030 | LmjF.35.4990 | PF3D7_0625000 PF3D7_0625100 | ? |
| SPL | Sphingosine-1-phosphate lyase | Tb927.6.3630 | LmjF.30.2350 | - | - |
| SPT | Serine palmitoyltransferase | Tb927.4.1020 | LmjF.34.3740 | PF3D7_1415700 | TGVEG_084380 |
Alternatively, PC can be generated by methylation of ethanolamine-phosphate by phosphoethanolamine N-methyltransferase and subsequent PC formation via the Kennedy pathway, or by methylation of PE involving PE N-methyltransferase [211]. In addition, PC can be formed by head group exchange with PS [211]. Furthermore, in most eukaryotes PE can be synthesized by irreversible decarboxylation of PS, or reversible head group exchange with PS [213, 215, 216] (Fig. 4).
Finally, PS in eukaryotes can be synthesized in single step reactions via head group exchange with PC and PE, catalyzed by PS synthase 1 and 2, respectively, or from serine and CDP-diacyl glycerol via prokaryotic-type PS synthase [213] (Fig. 4). The contributions of the different routes to PC, PE and PS formation greatly vary between eukaryotes, with certain routes being absent in some organisms.
PC synthesis in Kinetoplastida
The most abundant phospholipid class in T. brucei bloodstream and procyclic forms, PC, is synthesized via the CDP-choline branch of the Kennedy pathway (Figs 4, 5). The T. brucei enzyme catalyzing the first step in this pathway, choline/ethanolamine kinase, has been expressed as a recombinant protein in E. coli and characterized biochemically [217]. The purified enzyme shows dual specificity for choline and ethanolamine, with a 80-fold lower Km value for choline [217]. Expression of choline/ethanolamine kinase in T. brucei bloodstream forms has been reported to be essential (cited as unpublished data in [179]). Homologs of the T. brucei enzyme have been identified and annotated in the T. cruzi and Leishmania spp. genome databases. The second enzyme of the Kennedy pathway, choline-phosphate cytidylyltransferase, catalyzes the activation of choline-phosphate to CDP-choline. Candidate enzymes have been identified in all trypanosomatids, however, none of them has been characterized experimentally so far. The final reaction in the CDP-choline branch of the Kennedy pathway is mediated by CEPT and has been characterized in T. brucei procyclic forms [159]. The enzyme is essential for parasite growth in culture and shows dual specificity for CDP-choline and CDP-ethanolamine [159]. Homologs of CEPT have been identified and annotated in the T. cruzi and Leishmania spp. genome databases.
Figure 5. Common and unique phospholipid synthesis pathways in kinetoplastids and apicomplexans.

Schematic representation of lipid biosynthetic pathways that are common (black squares) or unique (colored squares) to selected apicomplexan and kinetoplastid parasites to indicate their potential as drug targets. The colored squares indicate experimental evidence of enzyme activity or the presence of a predicted gene in the genome of a given parasite species. The absence of a square indicates that no enzyme activity or candidate gene has been reported. Abbreviations are as in the legend of Fig. 4.
In T. brucei and T. cruzi, the Kennedy pathway represents the only pathway for PC synthesis (Fig. 5). The respective genomes lack candidate genes encoding PE N-methyltransferases and (plant-like) phosphoethanolamine N-methyltransferases, catalyzing the methylation of PE to PC and ethanolamine-phosphate to choline-phosphate, respectively. The lack of PC formation from PE has been confirmed experimentally in T. brucei bloodstream and procyclic forms by labeling experiments using ethanolamine [186, 189] or serine [159]. These findings demonstrate that trypanosomes are auxotrophic for choline, i.e. they have to take up choline, or choline-containing metabolites, from the environment. In contrast, the Leishmania spp. genomes contain genes encoding candidate PE N-methyltransferases [180], suggesting that Leishmania parasites possess another route for PC synthesis. However, the corresponding enzymes or pathways have not been characterized experimentally.
PC synthesis in Apicomplexa
In apicomplexan parasites, synthesis of PC occurs via multiple routes (Figs 4, 5). The first enzyme of the CDP-choline branch of the Kennedy pathway, choline kinase, has been identified and characterized more than 25 years ago in P. falciparum and P. knowlesi [218–220] and, more recently, in the rodent malaria parasites, P. berghei and P. vinckei [221]. The enzymes catalyzing the two subsequent reactions in the Kennedy pathway, choline-phosphate cytidylyltransferase and CEPT, have been demonstrated in several Plasmodium species [221–223]. CEPT, which shows dual specificity towards CDP-choline and CDP-ethanolamine as substrates, has been studied in P. knowlesi-infected erythrocytes [218, 221], and corresponding genes are found in all Plasmodium genomes. In P. berghei, CEPT localizes to the ER [221]. Unsuccessful attempts to disrupt choline kinase, choline-phosphate cytidylyltransferase or CEPT suggest that PC synthesis by the CDP-choline pathway is essential in P. berghei [221].
P. falciparum is also able to use serine as substrate for PC formation, via decarboxylation to ethanolamine and phosphorylation to ethanolamine-phosphate, followed by three methylation reactions to form choline-phosphate [221] (Figs 4, 5). Subsequently, choline-phosphate is used to generate PC via the CDP-choline pathway [221, 224]. The formation of choline-phosphate is mediated by P. falciparum phosphoethanolamine N-methyltransferase, an enzyme that selectively methylates ethanolamine-phosphate, but not ethanolamine or PE [224, 225]. Interestingly, phosphoethanolamine N-methyltransferase activity is inhibited by the reaction product, phosphocholine, and the choline-phosphate analog, miltefosine [225], an anti-malarial that has been used to treat visceral and cutaneous Leishmaniasis [226–228]. Inhibition of PC formation by miltefosine has also been reported in L. donovani [229]. Disruption of the phosphoethanolamine N-methyltransferase gene in P. falciparum results in a complete loss of PC synthesis via serine decarboxylation and ethanolamine-phosphate methylation [230]. The P. falciparum enzyme has been localized to the Golgi [231]. Interestingly, the gene encoding phosphoethanolamine N-methyltransferase is absent from the genomes of the rodent malaria, P. berghei, P. yoelli and P. chabaudi ([171]; reviewed by [232]).
In contrast, Toxoplasma parasites lack candidate genes, and enzymatic activities, for phosphoethanolamine N-methyltransferase and PE N-methyltransferase [180, 208, 233] and, thus, depend on the CDP-choline pathway for PC synthesis (Fig. 5). In a recent report, T. gondii was shown to express a novel choline kinase harboring a unique hydrophobic N-terminus that is dispensable for enzyme activity but required for protein oligomerization [233]. Interestingly, expression of two additional choline kinase isoforms through a cryptic promoter compensated for the loss of choline kinase activity after conditional mutagenesis of the unusual choline kinase gene [233].
PE synthesis in Kinetoplastida
PE synthesis has been extensively studied in T. brucei [234] (Figs 4, 5). All three enzymes of the CDP-ethanolamine pathway have been identified and experimentally characterized. Down-regulation of ethanolamine kinase, ethanolamine-phosphate cytidylyltransferase and EPT using RNAi in T. brucei procyclic forms resulted in changes in mitochondrial morphology, formation of multinucleate cells [235], and growth arrest [159, 190], demonstrating that the CDP-ethanolamine branch of the Kennedy pathway is essential for parasite survival. In addition, depletion of ethanolamine-phosphate cytidylyltransferase was shown to cause a distinct defect in cytokinesis, reflected by parasites being unable to separate from each other during the final step of cytokinesis, suggesting that a lower PE content may inhibit membrane fusion and fission events [234, 235]. Ethanolamine-phosphate cytidylyltransferase is also essential for survival of T. brucei bloodstream forms [186]. Interestingly, EPT was shown to mediate the formation of alkenyl-acyl PE molecular species, while diacyl-type PE species are synthesized primarily by the dual-specificity enzyme, CEPT [159]. To date it is unclear if the role of EPT and CEPT in the synthesis of different PE sub-classes is the result of their substrate specificity for alkenyl-acyl glycerol and diacyl glycerol species, respectively, or of distinct sub-cellular localization. At present, cytosolic ethanolamine-phosphate cytidylyltransferase represents the only enzyme in PE synthesis that has been localized [186].
The T. cruzi and Leishmania genomes contain candidate genes for all three enzymes of the CDP-ethanolamine pathway [180, 236], indicating that this route for PE synthesis is present in other Kinetoplastida as well (Fig. 5). Depletion of EPT in L. major inhibits the synthesis of alkenyl-acyl PE molecular species (M. Pawlowic and K. Zhang, personal communication), indicating that EPT shows a similar specificity for ether-type PE formation in both T. brucei and Leishmania spp. Other enzymes of the CDP-ethanolamine pathway in Leishmania have not been characterized experimentally.
Alternatively, PE in kinetoplastids can be produced by decarboxylation of PS (Figs 4, 5). Metabolic labeling experiments have shown that PS decarboxylation activity is present in T. brucei procyclic [159] and bloodstream forms [186]. However, this reaction is unable to rescue a block in PE synthesis by the CDP-ethanolamine pathway [159, 186] and, thus, it is unclear at present if PS decarboxylation contributes to de novo PE formation (see below). The subcellular localization of PS decarboxylase in T. brucei has not been determined. Genes encoding predicted PS decarboxylases are also present in the genomes of T. cruzi and Leishmania spp. (Fig. 5), however, the roles of the enzymes have not been investigated.
A third pathway for PE synthesis in kinetoplastids involves ethanolamine-phosphate formation from sphingolipid degradation, with subsequent use in the CDP-ethanolamine pathway (Figs 4, 5). Sphingosine-1-phosphate lyase catalyzes the irreversible catabolism of phosphorylated sphingoid bases to produce ethanolamine-phosphate and a long chain fatty aldehyde [237]. L. major promastigotes, in which the gene encoding sphingosine-1-phosphate lyase was disrupted, were viable but defective in stationary phase differentiation and showed attenuated virulence [156]. Interestingly, parasite growth was restored by supplementing ethanolamine in the culture medium, indicating that a primary role of sphingolipid turnover in L. major promastigotes is to provide ethanolamine-phosphate for PE synthesis [156]. Gene orthologs of sphingosine-1-phosphate lyase are also present in the genomes of T. brucei and T. cruzi (Fig. 5), however, at present it is not known if ethanolamine-phosphate production via sphingosine-1-phosphate breakdown plays a similar role in trypanosomes. Our preliminary results using RNAi to down-regulate sphingosine-1-phosphate lyase expression in T. brucei indicate that the enzyme is not essential for parasite growth in culture (L. Farine and P. Bütikofer, unpublished results).
PE synthesis in Apicomplexa
The CDP-ethanolamine branch of the Kennedy pathway is also active in several Plasmodium species (Figs 1, 2). Ethanolamine kinase activity has been demonstrated in P. falciparum and P. knowlesi [220, 238], as well as rodent P. berghei and P. vinckei [221]. The second enzyme of the pathway, ethanolamine-phosphate cytidylyltransferase, represents the rate-limiting step in PE synthesis by the Kennedy pathway in P. falciparum and P. knowlesi [223]. In contrast to Trypanosoma and Toxoplasma, which contain separate genes encoding EPT and CEPT to catalyze the last steps in the two branches of the Kennedy pathway, the genome of Plasmodium contains a single gene encoding the dual-specificity enzyme, CEPT [180, 218, 221, 223, 239]. Attempts to disrupt the genes involved in PE synthesis by the Kennedy pathway in P. berghei failed, indicating that the pathway is likely essential for parasite survival in the blood stages [221].
PE synthesis by the Kennedy pathway has also been demonstrated in Toxoplasma (Fig. 5). A cytosolically located ethanolamine kinase in T. gondii phosphorylates ethanolamine only, whereas a choline kinase showing punctate intracellular distribution mediates phosphorylation of both choline and ethanolamine [233]. Similar to the situation in Plasmodium, the T. gondii choline kinase gene cannot be disrupted, unless using a conditional knock-out approach, indicating that the enzyme is essential for parasite viability [233]. Alternatively, PE in Plasmodium-infected erythrocytes can also be generated by PS decarboxylation [198], or from serine via decarboxylation to ethanolamine followed by synthesis by the CDP-ethanolamine pathway [223] (Figs 4, 5).
PS decarboxylases have been identified in P. falciparum [240] and P. knowlesi [241]. In P. falciparum, the enzyme was found to localize to the ER [240]. A gene encoding serine decarboxylase, a soluble and pyridoxylphosphate-dependent enzyme widely expressed in plants and algae [208, 233], has not been identified in the Plasmodium genome [180]. In T. gondii, PS decarboxylation represents a major contributor to PE formation [208, 242], establishing PS as an important precursor for PE in Toxoplasma (Fig. 5). Remarkably, T. gondii PS decarboxylase is a soluble enzyme localizing to dense granules or the parasitophorous vacuole of replicating parasites [242]. This is the first report of a soluble form of PS decarboxylase in any organism. Similar to Plasmodium, no serine decarboxylase gene has been detected in Toxoplasma. In contrast to Kinetoplastida, PE synthesis via ethanolamine-phosphate formation from sphingolipid degradation has not been reported in Apicomplexa (Fig. 5). A candidate gene encoding sphingosine-1-phosphate lyase has not been identified in the genomes of P. falciparum, T. annulata and T. gondii.
PS synthesis in Kinetoplastida
Synthesis of PS has been demonstrated in T. brucei procyclic [159] and bloodstream [186] forms using labeled serine as precursor. Bioinformatic analyses suggest the presence of a gene encoding PS synthase 2 in T. brucei (Figs 4, 5), however, the enzymatic reaction has not been characterized. A PS synthase 2 homolog is also found in the genomes of T. cruzi and Leishmania spp (Fig. 5). Interestingly, however, although the presence of PS in Leishmania has been suggested, in part based on indirect assays [155, 243, 244], other reports failed to detect PS in Leishmania promastigotes using several analytical methods, including mass spectrometry [153, 156, 158]. The findings that annexin V and anti-PS antibody binding is also observed in parasites lacking PS questions the specificity of these assays, which have been widely used to demonstrate PS exposure in the outer leaflet of the plasma membrane in eukaryotic cells, including T. brucei [245]. An overview on the methods and their limitations to detect PS exposure on the surface of eukaryotic cells has recently been published [246].
PS synthesis in Apicomplexa
In Plasmodium-infected erythrocytes, serine is used as substrate for PS synthesis via bacterial-type PS synthase [223, 247], while PS formation by head group exchange with PC or PE is absent (Figs 4, 5). In addition, in P. falciparum and P. knowlesi, serine can be decarboxylated to ethanolamine [223], making serine an important precursor for PE and PC formation [247]. The genes encoding PS synthase and serine decarboxylase have not been identified. In T. gondii, [3H]serine is rapidly incorporated into PS, which is then decarboxylated to PE [208]. In addition, small amounts of radioactivity can be detected in base-resistant minor lipid classes, tentatively identified as sphingolipids [248]. In contrast to Plasmodium, PS synthesis in T. gondii most likely occurs via head group exchange with PE [208]. In addition, direct decarboxylation of serine has not been reported in Toxoplasma (Fig. 5).
3.3.2. Synthesis of PI
PI contains a head group consisting of the cyclic polyalcohol, myo-inositol, which can be taken up from the environment via myo-inositol transporters (see above), or synthesized de novo from glucose. Biosynthesis of myo-inositol involves generation of myo-inositol–3-phosphate from glucose-6-phosphate by inositol–3-phosphate synthase, followed by dephosphorylation to myo-inositol by inositol–3-phosphate monophosphatase [249] (Fig. 4). The final step in PI synthesis, converting CDP-diacyl glycerol and myo-inositol to PI, is catalysed by PI synthase and is conserved in all eukaryotes [250] (Fig. 4). PI can then be used to generate the different phosphorylated forms of this phospholipid, many of which represent important signalling molecules in eukaryotic cells [249]. One particular form of phosphoinositide, phosphatidylinositol 3-phosphate, has recently been implicated in apicomplast biogenesis in Toxoplasma [251] and host targeting of P. falciparum proteins to the erythrocyte [252]. Phylogenetic analyses of microbial phosphoinositide kinases and phosphatases have revealed distinct amino acid sequence and catalytic characteristics from human homologs, suggesting they could be targeted for drug development against infectious diseases [253, 254]. In addition, PI represents the precursor for GPIs, prominent cell surface glycophospholipids and anchors for plasma membrane proteins, in particular in protozoan parasites [255–257].
PI synthesis in Kinetoplastida
The discovery that inositol–3-phosphate synthase is essential for viability of T. brucei bloodstream forms indicated that de novo myo-inositol synthesis is required for parasite survival and that myo-inositol taken up from the environment is unable to substitute for endogenous myo-inositol production [258] (Figs 4, 5). The observations that i) parasite GPI-anchored proteins are poorly labeled in vivo using exogenous [3H]inositol [150, 179], ii) de novo myo-inositol synthesis is required for GPI synthesis in the ER [195, 258], and iii) PI synthase in T. brucei bloodstream forms shows a dual localization in the ER and Golgi [193], lead to the hypothesis that two distinct cellular pools of myo-inositol exist in T. brucei [193]. This hypothesis was experimentally supported by recent observations in T. brucei procyclic forms showing that exogenous myo-inositol is essential for bulk PI and IPC production, but not GPI formation, and by the identification of a myo-inositol transporter located in the plasma membrane and Golgi [150]. Together, these finding suggest that de novo synthesis of myo-inositol for PI and GPI production is catalyzed by PI synthase in the ER, whereas uptake of exogenous myo-inositol into the Golgi is required for bulk PI formation by Golgi PI synthase.
Gene orthologs for myo-inositol–3-phosphate synthase and PI synthase have been identified in T. congolense, T. vivax, T. cruzi and Leishmania spp., suggesting that de novo synthesis of myo-inositol occurs in all kinetoplastids (Fig. 5). Similarly, myo-inositol uptake has been reported in several Leishmania species [191, 259], and orthologs of the T. brucei myo-inositol transporter have been found in most kinetoplastids [150].
PI synthesis in Apicomplexa
PI synthase activity has been detected in Plasmodium-infected erythrocytes [260], and the enzyme was characterized in P. falciparum and P. knowlesi [261]. Amino acid sequence comparison with predicted proteins suggests that PI synthase is also present in P. berghei, P. chabaudi and P. vivax [261] (Fig. 5). In T. gondii, two cDNA clones encoding functional PI synthases have been identified [262]. The two genes are expressed stage-specifically in tachyzoites and bradyzoites, with the tachyzoite-specific PI synthase sharing a high degree of identity to human, plant and yeast PI synthases [262]. PI synthases from both T. gondii and P. falciparum were able to complement PI synthase-deficient yeast mutants, demonstrating functional equivalence between PI synthases from different eukaryotes [261, 262] (Fig. 5).
Genes encoding putative myo-inositol–3-phosphate synthases have been annotated in the genomes of Plasmodium, but not T. gondii and T. annulata, suggesting that de novo synthesis of myo-inositol may be absent in the latter organisms (Fig. 5).
3.3.3. Synthesis of mitochondria-specific lipids
PG and CL are abundant glycerophospholipid classes in bacteria [263]. In eukaryotes, they are confined to the inner mitochondrial membrane, where they play important roles in maintaining protein structure and function [264]. The synthesis of PG and CL starts with PA, which is activated by CTP to CDP-diacyl glycerol [265]. After phosphatidyl group transfer to glycerol–3-phosphate, mediated by phosphatidylglycerophosphate (PGP) synthase, PGP is dephosphorylated to PG by PGP phosphatase (Figs 4, 5). In most prokaryotes, PG receives an additional phosphatidyl group from another PG to form CL, whereas in most eukaryotes, a phosphatidyl group from CDP-diacyl glycerol is transferred onto PG. The reactions are catalyzed by prokaryotic- and eukaryotic-type CL synthases, respectively, which can be distinguished based on their catalytic site motifs [265].
PG and CL synthesis in Kinetoplastida
Low levels of PG and CL have been demonstrated in T. brucei procyclic and bloodstream forms [137, 149] and Leishmania [151, 158, 229]. Very recently, a protein homologous to PGP synthase from mammalian cells and yeast has been identified and experimentally characterized in T. brucei [266]. RNAi-mediated down-regulation of PGP synthase activity resulted in decreased PG and CL levels, alterations in mitochondrial morphology, and growth arrest of procyclic and bloodstream forms in culture, indicating that PG and CL are indispensable for parasite viability in both life cycle forms [266] (Fig. 5).
Surprisingly, CL synthase in T. brucei was found to contain the characteristic prokaryotic-type, rather than eukaryotic-type, CL synthase motif consisting of two phospholipase D domains [149, 180]. Conditional gene knock-out of T. brucei CL synthase in procyclic forms demonstrated that it is essential for parasite survival in culture [149]. Ablation of CL synthase activity inhibited CL formation and resulted in impaired mitochondrial function [149]. Both PGP synthase and CL synthase localize to the mitochondrion, where they associate with high molecular mass protein complexes [149, 266]. It is possible that both enzymes are present in the same complex, forming a mitochondrial lipid synthesis complex.
Genes encoding homologous proteins to T. brucei PGP synthase and CL synthase are present in all kinetoplastids (Fig. 5). In contrast, a candidate gene encoding PGP phosphatase has not been identified in any protozoa (Fig. 5).
PG and CL synthesis in Apicomplexa
PG and CL synthesis has not been studied experimentally in apicomplexans. Predicted proteins sharing sequence similarities with mammalian and yeast PGP synthases and prokaryotic-type CL synthases are annotated in the genomes of Plasmodium, Theileria and Toxoplasma (Fig. 5). The putative enzymes show high homologies to the experimentally characterized PGP synthase and CL synthase of T. brucei [149].
The presence of prokaryotic-type CL synthases in Apicomplexa and Kinetoplastida indicates evolutionary conservation of the reaction mechanism into the eukaryotic kingdom [180]. The presence of CL synthases of bacterial origin in protozoan parasites validates this enzyme as potential drug target, however at present, functional studies addressing the essentiality of CL synthase in Apicomplexa are lacking.
3.3.4. Sphingophospholipids
The major higher-order sphingophospholipids include SM, IPC and EPC. In mammalian cells, SM is by far the most abundant sphingophospholipid class, whereas IPC is abundant in fungi and plants and EPC in Drosophila melanogaster [267].
Sphingophospholipid biosynthesis is initiated by condensation of L-serine and palmitoyl-CoA to 3-ketosphinganine by serine palmitoyltransferase, followed by reduction to dihydrosphingosine. Subsequently, ceramide synthase acylates dihydrosphingosine to dihydroceramide, which is then desaturated to ceramide, i.e. the common metabolite for the synthesis of sphingophospholipids (Fig. 4). The initial reactions in sphingolipid synthesis leading to ceramide formation occur in the ER [268]. After transport of ceramide to the Golgi, a set of sphingolipid synthases mediates transfer of polar head groups from glycerophospholipid donors to ceramide to produce sphingophospholipids. SM synthases catalyze transfer of phosphocholine using PC as substrate, whereas IPC and EPC synthases transfer phosphoinositol from PI and phosphoethanolamine from PE, respectively [267] (Fig. 4). Alternatively, ceramide can be modified by sugar residues to generate several glycosphingolipid classes.
Degradation of sphingolipids can occur via sphingosine-1-phosphate lyase, which breaks down phosphorylated sphingoid bases into ethanolamine phosphate and fatty aldehydes [214] (Fig. 4) (see also above).
Sphingolipid metabolism in Kinetoplastida
In contrast to mammals and plants, Leishmania parasites do not synthesize SM. Instead, the most abundant sphingophospholipid class in Leishmania is IPC, accounting for up to 10% of total membrane lipids [269]. Several enzymes involved in sphingophospholipid synthesis in Leishmania have been characterized experimentally (Fig. 5). The role of heterodimeric serine palmitoyltransferase, consisting of subunits Spt1 and Spt2, with Spt2 being the catalytic subunit, has been studied in L. major. Spt2 deletion mutants completely lacking ceramide and IPC were found to be viable, yet unable to differentiate into metacyclic forms [166]. In addition, a similar defect in differentiation into metacyclic forms has been observed in sphingosine-1-phosphate lyase knock-out strains [156]. These parasites are able to incorporate [3H]serine into ceramide and IPC, but show greatly reduced labeling of PE and PC. Surprisingly, supplementing the culture media with ethanolamine fully restored the defects, indicating that Leishmania use sphingolipid metabolism for bulk ethanolamine phosphate production for glycerophospholipid synthesis [156]. Furthermore, candidate genes encoding IPC synthases have been identified in all kinetoplastids [270] (Fig. 5). The L. major enzyme, which shows little sequence homology to fungal IPC synthases, was able to complement a yeast strain deficient in IPC synthase and showed activity in human cells, where it localizes to the Golgi [270]. Finally, Leishmania parasites were found to remodel sphingophospholipids acquired from the host to retain normal IPC levels [271].
In T. brucei procyclic forms, down-regulation of Spt2 expression by RNAi, or inhibition of Spt2 activity by myriocin, showed that sphingolipid metabolism is essential for parasite growth in culture [272] (Fig. 5). Spt2-depleted cells showed reduced IPC levels, dysfunctional cytokinesis and a defect in kinetoplast segregation, resulting in parasites with aberrant DNA content [272]. Addition of the downstream metabolite, 3-ketosphinganine [272], but not ethanolamine [161], was able to rescue the growth defect of Spt2 knock-down cells, indicating that unlike Leishmania, T. brucei uses sphingoid base synthesis not primarily for ethanolamine-phosphate production.
Remarkably, a sphingolipid synthase gene family consisting of four tandemly linked genes, TbSLS1–4, which are orthologs of L. major IPC synthase, is present in T. brucei [162]. Using RNAi against a sequence common to all four sphingolipid synthase genes demonstrated that sphingolipid synthesis is essential for growth of T. brucei bloodstream forms in culture [162]. Expression of the enzymes in a cell-free system revealed that TbSLS1 and TbSLS2 produce IPC and EPC, respectively, whereas TbSLS3 and TbSLS4 are bi-functional and generate SM and EPC [273]. In addition, expression of TbSLS4 in L. major leads to the production of SM and EPC, confirming its bi-functional activity [162]. Interestingly, sphingophospholipid synthesis in T. brucei is developmentally regulated, with IPC and SM being produced in procyclic forms, while bloodstream forms lack IPC but instead contain small amounts of EPC [162]. TbSLS4 was found to co-localize with galactosyltransferase, suggesting Golgi localization [162]. In addition, cell-free expression of the single T. cruzi sphingolipid synthase ortholog, TcSLS1, showed that it is a dedicated IPC synthase [273]. It is worth mentioning that, in contrast to T. brucei and Leishmania, IPC in T. cruzi is of special importance as it serves as membrane anchor for most GPI proteins and GPI lipids [274].
Sphingolipid metabolism in Apicomplexa
Labeling experiments using [3H]serine demonstrated de novo synthesis of SM and glycosphingolipids, but not IPC, in P. falciparum [248] (Fig. 5). More recently, using a combination of isotope labeling and mass spectrometry, several sulfoglycosphingolipid species were detected in intraerythrocytic stages of P. falciparum [275]. In T. gondii, de novo synthesis of ceramide, SM and complex glycosphingolipids has been demonstrated using [3H]serine and [3H]galactose labeling [174] (Fig. 5). The identification of an inositol-containing lipid resisting alkaline treatment suggested the presence of IPC in T. gondii [276], however, a recent lipidomic analysis using mass spectrometry found no evidence of IPC [175]. Instead, the analysis revealed the presence of EPC, which was estimated to account for approximately 2% of total polar lipid content [175]. The genomes of P. falciparum and T. gondii contain two and one, respectively, putative SM synthase genes. However, it is not known if the encoded synthases are capable of synthesizing sphingolipids other than SM.
3.4. Lipid composition of parasite organelles
It is well known that the lipid composition of the plasma membrane is different from that of intracellular organelles and, in addition, that each organelle has its own characteristic lipid profile [277]. This is related in part to the localization of the enzymes involved in lipid biosynthesis in different organelles and to the existence, or the absence, of lipid trafficking pathways between organelles [278, 279]. Reliable and detailed data on the lipid composition of eukaryotic organelles is scarce, mostly because it has been difficult to isolate organelles in sufficient amounts and with high enough purity, i.e. without contamination by other membranes, for lipid analysis. The situation in parasitic protozoa is no different. However, recent studies have reported the isolation of highly purified preparations of organelles from kinetoplastids and apicomplexans for proteomic studies [280–282]. This together with the latest improvements in lipid analysis, in particular in mass spectrometry, is expected to result in valuable new information on the lipid composition of parasite organelle membranes in the near future.
Lipids of subcellular compartments of Kinetoplastida
Analysis of the phospholipid composition of purified glycosomes from T. brucei bloodstream forms revealed the presence of only two phospholipids, PC and PE, with PC being twice as abundant as PE [283]. In contrast, purified glycosomes from T. cruzi epimastigotes showed the presence of all major phospholipid classes, PC, PE, PI, SM and PS [163]. Relative to total cell lipid, T. cruzi glycosomes contained increased levels of PC, PI and SM, as well as endogenously synthesized sterols [163]. In addition, mass spectrometric analysis of acidocalcisomes, acidic calcium-storage organelles found in many protozoan parasites [284], from T. cruzi epimastigotes showed the exclusive presence of PC, PE and PI molecular species [285]. Furthermore, labeling experiments using [3H]mannose revealed the presence of GPI lipids in isolated acidocalcisomes [285].
Although no direct lipid analyses have been performed, experiments measuring association of lipid-modified proteins with the flagellar membrane in T. cruzi [286], T. brucei [287] and L. major [288], together with laurdan microscopy to measure changes in liquid order of membrane domains in T. brucei [287], suggest that the lipid composition of the flagellum may be different, i.e. more raft-like, compared to the plasma membrane of the main cell body.
Lipids of subcellular compartments of Apicomplexa
Analysis of T. gondii tachyzoites revealed increased levels of PC (75% versus 62%) and PS (12% versus 8%) in an enriched pellicle fraction compared to total cells, whereas PE (7% versus 11%) and SM (1% versus 8%) levels were decreased [173]. The study also showed a decreased ratio of saturated to unsaturated fatty acids in phospholipids of enriched pellicle fractions compared to whole cells [173]. In addition, a study to determine the lipid composition of isolated rhoptries from T. gondii tachyzoites revealed PC as major phospholipid class (75% of total lipid), with small amounts of PE (9%), PA (6.5%) and lysophospholipids (1.8%), while PI, PS and SM were absent [289]. Remarkably, the cholesterol to phospholipid ratio in purified rhoptries was significantly increased compared to whole cells (1.48 versus 0.2), suggesting that the altered lipid composition may be important for host cell invasion [289].
4. Fatty acid and phospholipid synthesis pathways as drug targets
Because lipid biosynthetic pathways have long been considered of little importance for parasite survival, their potential as drug targets has not been systematically addressed. After a brief discussion of selected compounds known to affect lipid metabolism in Kinetoplastida and Apicomplexa, we highlight potentially interesting lipid pathways unique to certain members of these groups of parasites, or alternatively, are common to several parasite species but differ from the corresponding pathways in mammals.
4.1. Drugs targeting fatty acid synthesis
Drugs targeting FASII
Triclosan [49] and thiolactomycin [290, 291] are well studied inhibitors of type II FAS. Soon after the discovery of a FASII pathway in apicomplexan parasites, these drugs were tested for their efficacy against Plasmodium. Both triclosan [48, 50] and thiolactomycin [292] inhibit the growth of blood stage Plasmodium parasites. Additional drugs known to target bacterial fatty acid synthesis include epigallocatechin gallate [293], NAS-91 [294], and flavonoids [295], all of which were evaluated as anti-malarials. Green tea extracts containing epigallocatechin gallate, NAS-91 [45] or flavonoids [296] inhibit the growth of P. falciparum parasites in vitro. However, as detailed above, molecular analysis has shown the FASII pathway to be dispensable for growth of blood stage Plasmodium parasites. Therefore the anti-malarial activities of these drugs are likely due to mechanisms independent of FASII. This conclusion is further supported by the activity of epigallocatechin gallate against Babesia, an apicomplexan parasite lacking FASII [51, 297]. Similarly, NAS-91, which inhibits the dehydratase of mycobacterial FASII, was initially thought to kill Plasmodium by the same mechanism. However, a more recent study demonstrates that it may also inhibit mycobacterial SCD [298]. In contrast to FAS II, Plasmodium SCD is required for intraerythrocytic development of the parasite [129]. Thus, SCD rather than FASII dehydratase appears to be the likely target of NAS-91.
Thiolactomycin, which is thought to be a more specific FAS II inhibitor, as well as several compounds derived from thiolactomycin were found to inhibit growth of T. gondii [299], T. cruzi, T. brucei [300, 301], and L. donovani [301]. In T. gondii and T. brucei, where this has been studied in more detail, the activity of thiolactomycin is consistent with molecular data indicating that the FASII pathway is essential for growth [74, 83]. Similar to their activity in Plasmodium, green tea extracts containing catechins also inhibit T. cruzi [302]. Whether this effect was due to the activity of catechins against T. cruzi FASII pathway is not known.
Drugs targeting FASI and FAE
Cerulenin is a more general fatty acid synthesis inhibitor, whose activity is not limited to a particular type of FAS pathway. It is effective against human cancer cells by inhibiting mammalian FASI [303, 304]. Cerulenin has also been tested and proven effective in inhibiting C. parvum FASI [94]. In addition, in kinetoplastids lacking FASI but containing the FAE pathway, cerulenin has been suggested to be effective by inhibiting ELO-2, but not ELO-1 [118, 305]. However, cerulenin shows little activity against C. parvum fatty acid elongase [114]. A small number of specific inhibitors of fatty acid elongation has been identified. However, the activity of these inhibitors appears limited to a specific subtype of plant fatty acid elongases [115].
Drugs targeting FA desaturation
The identification of an active metabolism for unsaturated fatty acids in protozoan parasites has stimulated interest in desaturase inhibitors as anti-parasitic compounds. Isoxyl, which has been used for a long time in the treatment of tuberculosis, is known to inhibit bacterial SCD, and also inhibits growth of T. cruzi epimastigotes with an EC50 of 2 μM [306]. However, this drug typically has to be activated by a bacterial monooxygenase to be effective [307]. A homolog of this enzyme is not evident when searching the genomes of apicomplexans and kinetoplastids. Further studies are needed to establish the target and mode of activation of isoxyl in parasites. Methylsterculate, a methylester of sterculic acid, inhibits the intraerythrocytic growth of P. falciparum. The anti-malarial activity of methylsterculate can be reversed by oleic acid supplementation, strongly implicating P. falciparum SCD as the likely target of this drug [129].
Thiastearates (TS) are another class of compounds that target the synthesis of unsaturated fatty acids. 10-TS has been shown to inhibit T. cruzi SCD [306], whereas 12-TS and 13-TS prevent the conversion of oleate to linoleate and inhibit the growth of T. cruzi [306] and T. brucei [308]. The enzyme responsible for conversion of oleate to linoleate is delta-12 oleate desaturase. This enzyme is present in kinetoplastid parasites but absent in the human host, making it a strong candidate for a parasite-specific target. Note that based on bioinformatic analyses, this enzyme is absent in apicomplexan parasites, suggesting that 12-TS and 13-TS are likely not effective against Apicomplexa.
4.2. Drugs targeting phospholipid synthesis
Miltefosine (hexadecyl phosphocholine) represents the first orally active anti-leishmanial and one of only two compounds that have been developed and approved to treat visceral leishmaniasis in the last 25 years [309]. It is a member of a class of synthetic alkylphospholipids showing anti-proliferative effects on parasites and tumor cells, by interfering with lipid metabolism [310, 311]. Possible modes of action of miltefosine in Leishmania may involve inhibition of PE N-methyltransferase and CDP-phosphocholine cytidylyltransferase activity [229], or by blocking choline uptake from the environment [181], resulting in changes in parasite PC and PE contents. Similar effects have also been reported after miltefosine treatment of T. cruzi epimastigotes [312]. In addition, miltefosine also inhibits recombinant P. falciparum PE N-methyltransferase and shows anti-malarial activity on intraerythrocytic parasites with an IC50 of approximately 80 μM [225].
The observation that Leishmania and Trypanosoma synthesize IPC, a sphingophospholipid class typical for fungi, prompted a series of studies to investigate the effects of the anti-fungal drug, aureobasidin A, an inhibitor of fungal IPC synthase at sub-nanomolar concentrations [313], on Kinetoplastida. The results showed that although the drug at high concentrations showed moderate effects on proliferation of L. major promastigotes and on differentiation of T. cruzi amastigotes, it did not act on IPC formation in vivo or in vitro [270, 271, 314]. In addition, although it has been reported that aureobasidin A blocks the activity of TbSLS4 expressed in yeast and inhibits growth of T. brucei bloodstream forms in culture [315], the drug showed no effect on TbSLS4 expressed in a cell-free system [273]. Conversely, aureobasidin A treatment inhibited general sphingolipid synthesis and replication in T. gondii parasites lacking IPC [276], suggesting unspecific, i.e. off-target effects of the drug.
Sphingophospholipid synthesis has also been proposed as drug target against malaria parasites. Earlier work reported that 1-phenyl-2-palmitoylamino-3-N-morpholine-1-propanol blocks SM synthase activity and inhibits intraerythrocytic maturation of P. falciparum [316]. However, more recent work demonstrated that other well-characterized inhibitors of ceramide synthesis, such as fumonisin B1 and myriocin, showed no effect on intraerythrocytic parasite development, and only partly affected Plasmodium sphingolipid synthesis [248], suggesting that pathways other than de novo synthesis may provide Plasmodium parasites with sphingolipids. One such alternative route may involve hydrolysis of host sphingophospholipids and uptake of sphingolipid intermediates [316, 317].
Recently, a phospholipid analog affecting P. falciparum development and intraerythrocytic maturation through inhibition of PC synthesis via blocking CTP-phosphocholine cytidylyltransferase has been described [318]. Treatment of parasitized erythrocytes with the novel compound, PG12, lead to strongly reduced PC synthesis using choline or ethanolamine as substrates, indicating that both the CDP-choline and the serine-decarboxylase phosphoethanolamine N-methyltransferase pathways are affected by PG12 [318]. In a different study aimed at identifying inhibitors of P. falciparum phosphoethanolamine N-methyltransferase, two compounds were found to inhibit the enzyme both in vitro and in vivo, using a choline auxotroph yeast strain complemented with the plasmodial enzyme [319]. Finally, PC synthesis has been evaluated as drug target in Plasmodium and Trypanosoma by generating a library of compounds mimicking the choline structure. Some of these drugs showed strong anti-malarial activity, by interfering with the synthesis of PC via the Kennedy pathway [205, 206, 320, 321], and were able to cure malaria-infected monkeys [322]. More recently, a selection of these first-, second- and third-generation choline analogs were found to also have anti-leishmanial and anti-trypanosomal activity [184]. However, in contrast to their mode of action in Plasmodium, they did not affect lipid metabolism in L. major and T. brucei bloodstream forms but disrupted mitochondrial function.
Acknowledgments
Work on T. brucei lipid metabolism in the lab of P.B. was supported by Swiss National Science Foundation grant 31003A-130815. P.B. thanks B.J. Armstrong and A. Niederer for valuable input. Work on apicomplexan lipid metabolism was supported by grants from the National Institutes of Health (AI 64671 and AI 84415) to B.S. B.S. is a Georgia Research Alliance Distinguished Investigator and S.R. received a pre-doctoral fellowship from the American Heart Association.
Abbreviations
- ACC
acetyl-CoA carboxylase
- ACP
acyl carrier protein
- CEPT
CDP-choline/ethanolamine:diacylglycerol phosphotransferase
- CL
cardiolipin
- EPC
ethanolamine phosphorylceramide
- EPT
CDP-ethanolamine:diacylglycerol phosphotransferase
- ER
endoplasmic reticulum
- FAE
fatty acid elongation
- FASI
fatty acid synthase type I
- FASII
fatty acid synthase type II
- GPI
glycosylphosphatidylinositol
- IPC
inositol phosphorylceramide
- MSP1
merozoite surface protein-1
- MUFAs
monounsaturated fatty acids
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PGP
phosphatidylglycerophosphate
- PI
phosphatidylinositol
- PKS
polyketide synthase
- PS
phosphatidylserine
- PUFAs
polyunsaturated fatty acids
- RNAi
RNA interference
- SM
sphingomyelin
- SCD
stearoyl-CoA desaturase
- TS
thiastearates
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hotez PJ, Fenwick A, Savioli L, Molyneux DH. Rescuing the bottom billion through control of neglected tropical diseases. Lancet. 2009;373:1570–5. doi: 10.1016/S0140-6736(09)60233-6. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2013;380:2095–128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg DE, Siliciano RF, Jacobs WR., Jr Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell. 2012;148:1271–83. doi: 10.1016/j.cell.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–6. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fairhurst RM, Nayyar GM, Breman JG, Hallett R, Vennerstrom JL, Duong S, et al. Artemisinin-resistant malaria: research challenges, opportunities, and public health implications. The American journal of tropical medicine and hygiene. 2012;87:231–41. doi: 10.4269/ajtmh.2012.12-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 1992;15:211–22. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 7.Tzipori S, Ward H. Cryptosporidiosis: biology, pathogenesis and disease. Microbes and infection/Institut Pasteur. 2002;4:1047–58. doi: 10.1016/s1286-4579(02)01629-5. [DOI] [PubMed] [Google Scholar]
- 8.Striepen B. Drug Resistance and Emerging Targets in the Opportunistic Pathogens Toxoplasma gondii and Cryptosporidium parvum. In: Douglas L, Mayers SAL, Ouellette Marc, Sobel Jack D, editors. Antimicrobial Drug Resistance: Clinical and epidemiological aspects. Humana Press; 2009. pp. 605–19. [Google Scholar]
- 9.Shirley DA, Moonah SN, Kotloff KL. Burden of disease from cryptosporidiosis. Current opinion in infectious diseases. 2012;25:555–63. doi: 10.1097/QCO.0b013e328357e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cabada MM, White AC., Jr Treatment of cryptosporidiosis: do we know what we think we know? Current opinion in infectious diseases. 2010;23:494–9. doi: 10.1097/QCO.0b013e32833de052. [DOI] [PubMed] [Google Scholar]
- 11.Viotti R, Vigliano C, Lococo B, Alvarez MG, Petti M, Bertocchi G, et al. Side effects of benznidazole as treatment in chronic Chagas disease: fears and realities. Expert review of anti-infective therapy. 2009;7:157–63. doi: 10.1586/14787210.7.2.157. [DOI] [PubMed] [Google Scholar]
- 12.Liappis N, Schlebusch H, Knapp M. Reference values for the concentration of free triiodothyronine (FT3), triiodothyronine (TT3), free thyroxine (FT4), thyroxine (TT4), thyrotropin (TSH) and thyroxine-binding globulin (TBG) in umbilical cord blood. Method: luminescence-enhanced enzyme immunoassay. Klinische Padiatrie. 1992;204:34–6. doi: 10.1055/s-2007-1025319. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy PGE. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113:496–504. doi: 10.1172/JCI21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yun O, Priotto G, Tong J, Flevaud L, Chappuis F. NECT Is Next: Implementing the New Drug Combination Therapy for Trypanosoma brucei gambiense Sleeping Sickness. Plos Neglect Trop D. 2010:4. doi: 10.1371/journal.pntd.0000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naderer T, McConville MJ. Intracellular growth and pathogenesis of Leishmania parasites. Essays in biochemistry. 2011;51:81–95. doi: 10.1042/bse0510081. [DOI] [PubMed] [Google Scholar]
- 16.Ouellette M, Drummelsmith J, Papadopoulou B. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2004;7:257–66. doi: 10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Moore EM, Lockwood DN. Treatment of Visceral Leishmaniasis. Journal of Global Infectious Diseases. 2010;2:151–8. doi: 10.4103/0974-777X.62883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanowitz HB, Jelicks LA, Machado FS, Esper L, Qi X, Desruisseaux MS, et al. Adipose tissue, diabetes and Chagas disease. Advances in parasitology. 2011;76:235–50. doi: 10.1016/B978-0-12-385895-5.00010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell host & microbe. 2012;12:669–81. doi: 10.1016/j.chom.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 20.Meester IR-T, Geovanni Adrián, Solís-Soto Juan Manuel, Salinas-Carmona Mario César. The roles of lipid droplets in human infectious disease. Medicina Universitaria. 2011;13(53):207–16. [Google Scholar]
- 21.Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends in microbiology. 2011;19:368–75. doi: 10.1016/j.tim.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elwell CA, Engel JN. Lipid acquisition by intracellular Chlamydiae. Cellular microbiology. 2012;14:1010–8. doi: 10.1111/j.1462-5822.2012.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coppens I. Contribution of host lipids to Toxoplasma pathogenesis. Cellular microbiology. 2006;8:1–9. doi: 10.1111/j.1462-5822.2005.00647.x. [DOI] [PubMed] [Google Scholar]
- 24.Gilk SD. Role of lipids in Coxiella burnetii infection. Advances in experimental medicine and biology. 2012;984:199–213. doi: 10.1007/978-94-007-4315-1_10. [DOI] [PubMed] [Google Scholar]
- 25.Hubber A, Roy CR. Modulation of Host Cell Function by Legionella pneumophila Type IV Effectors. Annu Rev Cell Dev Bi. 2010;26:261–83. doi: 10.1146/annurev-cellbio-100109-104034. [DOI] [PubMed] [Google Scholar]
- 26.Lingelbach K, Joiner KA. The parasitophorous vacuole membrane surrounding Plasmodium and Toxoplasma: an unusual compartment in infected cells. Journal of cell science. 1998;111 ( Pt 11):1467–75. doi: 10.1242/jcs.111.11.1467. [DOI] [PubMed] [Google Scholar]
- 27.Ehrt S, Schnappinger D. Mycobacterium tuberculosis virulence: lipids inside and out. Nat Med. 2007;13:284–5. doi: 10.1038/nm0307-284. [DOI] [PubMed] [Google Scholar]
- 28.Dobson DE, Kamhawi S, Lawyer P, Turco SJ, Beverley SM, Sacks DL. Leishmania major survival in selective Phlebotomus papatasi sand fly vector requires a specific SCG-encoded lipophosphoglycan galactosylation pattern. PLoS pathogens. 2010;6:e1001185. doi: 10.1371/journal.ppat.1001185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sacks DL. Leishmania-sand fly interactions controlling species-specific vector competence. Cellular microbiology. 2001;3:189–96. doi: 10.1046/j.1462-5822.2001.00115.x. [DOI] [PubMed] [Google Scholar]
- 30.Spath GF, Garraway LA, Turco SJ, Beverley SM. The role(s) of lipophosphoglycan (LPG) in the establishment of Leishmania major infections in mammalian hosts. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9536–41. doi: 10.1073/pnas.1530604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schofield L, Hewitt MC, Evans K, Siomos MA, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature. 2002;418:785–9. doi: 10.1038/nature00937. [DOI] [PubMed] [Google Scholar]
- 32.Nebl T, De Veer MJ, Schofield L. Stimulation of innate immune responses by malarial glycosylphosphatidylinositol via pattern recognition receptors. Parasitology. 2005;130 (Suppl):S45–62. doi: 10.1017/S0031182005008152. [DOI] [PubMed] [Google Scholar]
- 33.Debierre-Grockiego F, Schwarz RT. Immunological reactions in response to apicomplexan glycosylphosphatidylinositols. Glycobiology. 2010;20:801–11. doi: 10.1093/glycob/cwq038. [DOI] [PubMed] [Google Scholar]
- 34.Bhatt A, Molle V, Besra GS, Jacobs WR, Kremer L. The Mycobacterium tuberculosis FAS-II condensing enzymes: their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol Microbiol. 2007;64:1442–54. doi: 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- 35.Brayton KA, Lau AO, Herndon DR, Hannick L, Kappmeyer LS, Berens SJ, et al. Genome sequence of Babesia bovis and comparative analysis of apicomplexan hemoprotozoa. PLoS pathogens. 2007;3:1401–13. doi: 10.1371/journal.ppat.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardner MJ, Bishop R, Shah T, de Villiers EP, Carlton JM, Hall N, et al. Genome sequence of Theileria parva, a bovine pathogen that transforms lymphocytes. Science. 2005;309:134–7. doi: 10.1126/science.1110439. [DOI] [PubMed] [Google Scholar]
- 37.Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–5. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 38.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annual review of biochemistry. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 39.McFadden GI. Endosymbiosis and evolution of the plant cell. Current opinion in plant biology. 1999;2:513–9. doi: 10.1016/s1369-5266(99)00025-4. [DOI] [PubMed] [Google Scholar]
- 40.Hiltunen JK, Chen Z, Haapalainen AM, Wierenga RK, Kastaniotis AJ. Mitochondrial fatty acid synthesis--an adopted set of enzymes making a pathway of major importance for the cellular metabolism. Progress in lipid research. 2010;49:27–45. doi: 10.1016/j.plipres.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Striepen B. The apicoplast: a red alga in human parasites. Essays in biochemistry. 2011;51:111–25. doi: 10.1042/bse0510111. [DOI] [PubMed] [Google Scholar]
- 42.Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz MJ, Foth BJ, et al. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nature reviews Microbiology. 2004;2:203–16. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 43.Pillai S, Rajagopal C, Kapoor M, Kumar G, Gupta A, Surolia N. Functional characterization of beta-ketoacyl-ACP reductase (FabG) from Plasmodium falciparum. Biochemical and biophysical research communications. 2003;303:387–92. doi: 10.1016/s0006-291x(03)00321-8. [DOI] [PubMed] [Google Scholar]
- 44.Lack G, Homberger-Zizzari E, Folkers G, Scapozza L, Perozzo R. Recombinant expression and biochemical characterization of the unique elongating beta-ketoacyl-acyl carrier protein synthase involved in fatty acid biosynthesis of Plasmodium falciparum using natural and artificial substrates. The Journal of biological chemistry. 2006;281:9538–46. doi: 10.1074/jbc.M509119200. [DOI] [PubMed] [Google Scholar]
- 45.Sharma SK, Kapoor M, Ramya TN, Kumar S, Kumar G, Modak R, et al. Identification, characterization, and inhibition of Plasmodium falciparum beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ) The Journal of biological chemistry. 2003;278:45661–71. doi: 10.1074/jbc.M304283200. [DOI] [PubMed] [Google Scholar]
- 46.Wickramasinghe SR, Inglis KA, Urch JE, Muller S, van Aalten DM, Fairlamb AH. Kinetic, inhibition and structural studies on 3-oxoacyl-ACP reductase from Plasmodium falciparum, a key enzyme in fatty acid biosynthesis. The Biochemical journal. 2006;393:447–57. doi: 10.1042/BJ20050832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swarnamukhi PL, Sharma SK, Bajaj P, Surolia N, Surolia A, Suguna K. Crystal structure of dimeric FabZ of Plasmodium falciparum reveals conformational switching to active hexamers by peptide flips. FEBS letters. 2006;580:2653–60. doi: 10.1016/j.febslet.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 48.Surolia N, Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat Med. 2001;7:167–73. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 49.McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis. Nature. 1998;394:531–2. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- 50.McLeod R, Muench SP, Rafferty JB, Kyle DE, Mui EJ, Kirisits MJ, et al. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of apicomplexan Fab I. International journal for parasitology. 2001;31:109–13. doi: 10.1016/s0020-7519(01)00111-4. [DOI] [PubMed] [Google Scholar]
- 51.Bork S, Yokoyama N, Matsuo T, Claveria FG, Fujisaki K, Igarashi I. Growth inhibitory effect of triclosan on equine and bovine Babesia parasites. American Journal of Tropical Medicine and Hygiene. 2003;68:334–40. [PubMed] [Google Scholar]
- 52.Freundlich JS, Wang F, Tsai HC, Kuo M, Shieh HM, Anderson JW, et al. X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. The Journal of biological chemistry. 2007;282:25436–44. doi: 10.1074/jbc.M701813200. [DOI] [PubMed] [Google Scholar]
- 53.Muralidharan J, Suguna K, Surolia A, Surolia N. Exploring the interaction energies for the binding of hydroxydiphenyl ethers to enoyl-acyl carrier protein reductases. Journal of biomolecular structure & dynamics. 2003;20:589–94. doi: 10.1080/07391102.2003.10506875. [DOI] [PubMed] [Google Scholar]
- 54.Perozzo R, Kuo M, Sidhu A, Valiyaveettil JT, Bittman R, Jacobs WR, Jr, et al. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. The Journal of biological chemistry. 2002;277:13106–14. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 55.Bork S, Yokoyama N, Matsuo T, Claveria FG, Fujisaki K, Igarashi I. Growth inhibitory effect of triclosan on equine and bovine Babesia parasites. The American journal of tropical medicine and hygiene. 2003;68:334–40. [PubMed] [Google Scholar]
- 56.Lizundia R, Werling D, Langsley G, Ralph SA. Theileria apicoplast as a target for chemotherapy. Antimicrobial agents and chemotherapy. 2009;53:1213–7. doi: 10.1128/AAC.00126-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heath RJ, Rock CO. A triclosan-resistant bacterial enzyme. Nature. 2000;406:145–6. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 58.Paul KS, Bacchi CJ, Englund PT. Multiple triclosan targets in Trypanosoma brucei. Eukaryotic cell. 2004;3:855–61. doi: 10.1128/EC.3.4.855-861.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu M, Kumar TR, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell host & microbe. 2008;4:567–78. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazumdar J, EHW, Masek K, CAH, Striepen B. Apicoplast fatty acid synthesis is essential for organelle biogenesis and parasite survival in Toxoplasma gondii. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13192–7. doi: 10.1073/pnas.0603391103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crawford MJ, Thomsen-Zieger N, Ray M, Schachtner J, Roos DS, Seeber F. Toxoplasma gondii scavenges host-derived lipoic acid despite its de novo synthesis in the apicoplast. The EMBO journal. 2006;25:3214–22. doi: 10.1038/sj.emboj.7601189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wrenger C, Muller S. The human malaria parasite Plasmodium falciparum has distinct organelle-specific lipoylation pathways. Mol Microbiol. 2004;53:103–13. doi: 10.1111/j.1365-2958.2004.04112.x. [DOI] [PubMed] [Google Scholar]
- 63.Vaughan AM, O’Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, et al. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cellular microbiology. 2009;11:506–20. doi: 10.1111/j.1462-5822.2008.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stanway RR, Mueller N, Zobiak B, Graewe S, Froehlke U, Zessin PJ, et al. Organelle segregation into Plasmodium liver stage merozoites. Cellular microbiology. 2011;13:1768–82. doi: 10.1111/j.1462-5822.2011.01657.x. [DOI] [PubMed] [Google Scholar]
- 65.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458:83–6. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 66.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15378–83. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mazumdar J, Striepen B. Make it or take it: fatty acid metabolism of apicomplexan parasites. Eukaryotic cell. 2007;6:1727–35. doi: 10.1128/EC.00255-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foth BJ, Stimmler LM, Handman E, Crabb BS, Hodder AN, McFadden GI. The malaria parasite Plasmodium falciparum has only one pyruvate dehydrogenase complex, which is located in the apicoplast. Mol Microbiol. 2005;55:39–53. doi: 10.1111/j.1365-2958.2004.04407.x. [DOI] [PubMed] [Google Scholar]
- 69.Brooks CF, Johnsen H, van Dooren GG, Muthalagi M, Lin SS, Bohne W, et al. The toxoplasma apicoplast phosphate translocator links cytosolic and apicoplast metabolism and is essential for parasite survival. Cell host & microbe. 2010;7:62–73. doi: 10.1016/j.chom.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deschermeier C, Hecht LS, Bach F, Rutzel K, Stanway RR, Nagel A, et al. Mitochondrial lipoic acid scavenging is essential for Plasmodium berghei liver stage development. Cellular microbiology. 2012;14:416–30. doi: 10.1111/j.1462-5822.2011.01729.x. [DOI] [PubMed] [Google Scholar]
- 71.Gunther S, Wallace L, Patzewitz EM, McMillan PJ, Storm J, Wrenger C, et al. Apicoplast lipoic acid protein ligase B is not essential for Plasmodium falciparum. PLoS pathogens. 2007;3:e189. doi: 10.1371/journal.ppat.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gunther S, Matuschewski K, Muller S. Knockout studies reveal an important role of Plasmodium lipoic acid protein ligase A1 for asexual blood stage parasite survival. PloS one. 2009;4:e5510. doi: 10.1371/journal.pone.0005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lim L, Linka M, Mullin KA, Weber AP, McFadden GI. The carbon and energy sources of the non-photosynthetic plastid in the malaria parasite. FEBS letters. 2010;584:549–54. doi: 10.1016/j.febslet.2009.11.097. [DOI] [PubMed] [Google Scholar]
- 74.Ramakrishnan S, Docampo MD, Macrae JI, Pujol FM, Brooks CF, van Dooren GG, et al. Apicoplast and endoplasmic reticulum cooperate in fatty acid biosynthesis in apicomplexan parasite Toxoplasma gondii. The Journal of biological chemistry. 2012;287:4957–71. doi: 10.1074/jbc.M111.310144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pei Y, Tarun AS, Vaughan AM, Herman RW, Soliman JM, Erickson-Wayman A, et al. Plasmodium pyruvate dehydrogenase activity is only essential for the parasite’s progression from liver infection to blood infection. Mol Microbiol. 2010 doi: 10.1111/j.1365-2958.2009.07034.x. [DOI] [PubMed] [Google Scholar]
- 76.Gerold P, Schofield L, Blackman MJ, Holder AA, Schwarz RT. Structural analysis of the glycosyl-phosphatidylinositol membrane anchor of the merozoite surface proteins-1 and -2 of Plasmodium falciparum. Molecular and biochemical parasitology. 1996;75:131–43. doi: 10.1016/0166-6851(95)02518-9. [DOI] [PubMed] [Google Scholar]
- 77.Corvi MM, Alonso AM, Caballero MC. Protein palmitoylation and pathogenesis in apicomplexan parasites. Journal of biomedicine & biotechnology. 2012;2012:483969. doi: 10.1155/2012/483969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beck JR, Rodriguez-Fernandez IA, Cruz de Leon J, Huynh MH, Carruthers VB, Morrissette NS, et al. A novel family of Toxoplasma IMC proteins displays a hierarchical organization and functions in coordinating parasite division. PLoS pathogens. 2010;6:e1001094. doi: 10.1371/journal.ppat.1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D. Functional dissection of the apicomplexan glideosome molecular architecture. Cell host & microbe. 2010;8:343–57. doi: 10.1016/j.chom.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 80.Rees-Channer RR, Martin SR, Green JL, Bowyer PW, Grainger M, Molloy JE, et al. Dual acylation of the 45 kDa gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Molecular and biochemical parasitology. 2006;149:113–6. doi: 10.1016/j.molbiopara.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 81.Botte CY, Yamaryo-Botte Y, Rupasinghe TW, Mullin KA, Macrae JI, Spurck TP, et al. Atypical lipid composition in the purified relict plastid (apicoplast) of malaria parasites. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7506–11. doi: 10.1073/pnas.1301251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Daily JP, Scanfeld D, Pochet N, Le Roch K, Plouffe D, Kamal M, et al. Distinct physiological states of Plasmodium falciparum in malaria-infected patients. Nature. 2007;450:1091–5. doi: 10.1038/nature06311. [DOI] [PubMed] [Google Scholar]
- 83.Stephens JL, Lee SH, Paul KS, Englund PT. Mitochondrial fatty acid synthesis in Trypanosoma brucei. The Journal of biological chemistry. 2007;282:4427–36. doi: 10.1074/jbc.M609037200. [DOI] [PubMed] [Google Scholar]
- 84.Autio KJ, Guler JL, Kastaniotis AJ, Englund PT, Hiltunen JK. The 3-hydroxyacyl-ACP dehydratase of mitochondrial fatty acid synthesis in Trypanosoma brucei. FEBS letters. 2008;582:729–33. doi: 10.1016/j.febslet.2008.01.051. [DOI] [PubMed] [Google Scholar]
- 85.Guler JL, Kriegova E, Smith TK, Lukes J, Englund PT. Mitochondrial fatty acid synthesis is required for normal mitochondrial morphology and function in Trypanosoma brucei. Mol Microbiol. 2008;67:1125–42. doi: 10.1111/j.1365-2958.2008.06112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clayton AM, Guler JL, Povelones ML, Gluenz E, Gull K, Smith TK, et al. Depletion of mitochondrial acyl carrier protein in bloodstream-form Trypanosoma brucei causes a kinetoplast segregation defect. Eukaryotic cell. 2011;10:286–92. doi: 10.1128/EC.00290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28:4523–30. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 88.Smith S. The animal fatty acid synthase: one gene, one polypeptide, seven enzymes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1994;8:1248–59. [PubMed] [Google Scholar]
- 89.Fernandes ND, Kolattukudy PE. Cloning, sequencing and characterization of a fatty acid synthase-encoding gene from Mycobacterium tuberculosis var. bovis BCG. Gene. 1996;170:95–9. doi: 10.1016/0378-1119(95)00842-x. [DOI] [PubMed] [Google Scholar]
- 90.Kawaguchi A, Okuda S. Fatty acid synthetase from Brevibacterium ammoniagenes: formation of monounsaturated fatty acids by a multienzyme complex. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:3180–3. doi: 10.1073/pnas.74.8.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu G, Marchewka MJ, Woods KM, Upton SJ, Keithly JS. Molecular analysis of a Type I fatty acid synthase in Cryptosporidium parvum. Molecular and biochemical parasitology. 2000;105:253–60. doi: 10.1016/s0166-6851(99)00183-8. [DOI] [PubMed] [Google Scholar]
- 92.Lu JZ, Muench SP, Allary M, Campbell S, Roberts CW, Mui E, et al. Type I and type II fatty acid biosynthesis in Eimeria tenella: enoyl reductase activity and structure. Parasitology. 2007;134:1949–62. doi: 10.1017/S0031182007003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhu G, Shi X, Cai X. The reductase domain in a Type I fatty acid synthase from the apicomplexan Cryptosporidium parvum: restricted substrate preference towards very long chain fatty acyl thioesters. BMC biochemistry. 2010;11:46. doi: 10.1186/1471-2091-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu G, Li Y, Cai X, Millership JJ, Marchewka MJ, Keithly JS. Expression and functional characterization of a giant Type I fatty acid synthase (CpFAS1) gene from Cryptosporidium parvum. Molecular and biochemical parasitology. 2004;134:127–35. doi: 10.1016/j.molbiopara.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 95.Kikuchi S, Rainwater DL, Kolattukudy PE. Purification and characterization of an unusually large fatty acid synthase from Mycobacterium tuberculosis var. bovis BCG. Archives of biochemistry and biophysics. 1992;295:318–26. doi: 10.1016/0003-9861(92)90524-z. [DOI] [PubMed] [Google Scholar]
- 96.Wheeler PR, Bulmer K, Ratledge C. Enzymes for biosynthesis de novo and elongation of fatty acids in mycobacteria grown in host cells: is Mycobacterium leprae competent in fatty acid biosynthesis? Journal of general microbiology. 1990;136:211–7. doi: 10.1099/00221287-136-1-211. [DOI] [PubMed] [Google Scholar]
- 97.Hopwood DA. Genetic Contributions to Understanding Polyketide Synthases. Chemical reviews. 1997;97:2465–98. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- 98.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Tolerance and specificity of polyketide synthases. Annual review of biochemistry. 1999;68:219–53. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 99.George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, et al. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science. 1999;283:854–7. doi: 10.1126/science.283.5403.854. [DOI] [PubMed] [Google Scholar]
- 100.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Natural product reports. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 101.Zhu G, LaGier MJ, Stejskal F, Millership JJ, Cai X, Keithly JS. Cryptosporidium parvum: the first protist known to encode a putative polyketide synthase. Gene. 2002;298:79–89. doi: 10.1016/s0378-1119(02)00931-9. [DOI] [PubMed] [Google Scholar]
- 102.Fritzler JM, Zhu G. Functional characterization of the acyl-[acyl carrier protein] ligase in the Cryptosporidium parvum giant polyketide synthase. International journal for parasitology. 2007;37:307–16. doi: 10.1016/j.ijpara.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Monroe EA, Van Dolah FM. The toxic dinoflagellate Karenia brevis encodes novel type I-like polyketide synthases containing discrete catalytic domains. Protist. 2008;159:471–82. doi: 10.1016/j.protis.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 104.Salcedo T, Upadhyay RJ, Nagasaki K, Bhattacharya D. Dozens of toxin-related genes are expressed in a nontoxic strain of the dinoflagellate Heterocapsa circularisquama. Molecular biology and evolution. 2012;29:1503–6. doi: 10.1093/molbev/mss007. [DOI] [PubMed] [Google Scholar]
- 105.Watanabe CM, Wilson D, Linz JE, Townsend CA. Demonstration of the catalytic roles and evidence for the physical association of type I fatty acid synthases and a polyketide synthase in the biosynthesis of aflatoxin B1. Chemistry & biology. 1996;3:463–9. doi: 10.1016/s1074-5521(96)90094-0. [DOI] [PubMed] [Google Scholar]
- 106.Miyanaga A, Funa N, Awakawa T, Horinouchi S. Direct transfer of starter substrates from type I fatty acid synthase to type III polyketide synthases in phenolic lipid synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:871–6. doi: 10.1073/pnas.0709819105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jakobsson A, Westerberg R, Jacobsson A. Fatty acid elongases in mammals: their regulation and roles in metabolism. Progress in lipid research. 2006;45:237–49. doi: 10.1016/j.plipres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 108.Lassner MW, Lardizabal K, Metz JG. A jojoba beta-Ketoacyl-CoA synthase cDNA complements the canola fatty acid elongation mutation in transgenic plants. The Plant cell. 1996;8:281–92. doi: 10.1105/tpc.8.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kohlwein SD, Eder S, Oh CS, Martin CE, Gable K, Bacikova D, et al. Tsc13p is required for fatty acid elongation and localizes to a novel structure at the nuclear-vacuolar interface in Saccharomyces cerevisiae. Molecular and cellular biology. 2001;21:109–25. doi: 10.1128/MCB.21.1.109-125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cinti DL, Cook L, Nagi MN, Suneja SK. The fatty acid chain elongation system of mammalian endoplasmic reticulum. Progress in lipid research. 1992;31:1–51. doi: 10.1016/0163-7827(92)90014-a. [DOI] [PubMed] [Google Scholar]
- 111.Denic V, Weissman JS. A molecular caliper mechanism for determining very long-chain fatty acid length. Cell. 2007;130:663–77. doi: 10.1016/j.cell.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 112.Zheng H, Rowland O, Kunst L. Disruptions of the Arabidopsis Enoyl-CoA reductase gene reveal an essential role for very-long-chain fatty acid synthesis in cell expansion during plant morphogenesis. The Plant cell. 2005;17:1467–81. doi: 10.1105/tpc.104.030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oh CS, Toke DA, Mandala S, Martin CE. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. The Journal of biological chemistry. 1997;272:17376–84. doi: 10.1074/jbc.272.28.17376. [DOI] [PubMed] [Google Scholar]
- 114.Fritzler JM, Millership JJ, Zhu G. Cryptosporidium parvum long-chain fatty acid elongase. Eukaryotic cell. 2007;6:2018–28. doi: 10.1128/EC.00210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trenkamp S, Martin W, Tietjen K. Specific and differential inhibition of very-long-chain fatty acid elongases from Arabidopsis thaliana by different herbicides. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11903–8. doi: 10.1073/pnas.0404600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mi-Ichi F, Kita K, Mitamura T. Intraerythrocytic Plasmodium falciparum utilize a broad range of serum-derived fatty acids with limited modification for their growth. Parasitology. 2006;133:399–410. doi: 10.1017/S0031182006000540. [DOI] [PubMed] [Google Scholar]
- 117.Schneiter R, Hitomi M, Ivessa AS, Fasch EV, Kohlwein SD, Tartakoff AM. A yeast acetyl coenzyme A carboxylase mutant links very-long-chain fatty acid synthesis to the structure and function of the nuclear membrane-pore complex. Molecular and cellular biology. 1996;16:7161–72. doi: 10.1128/mcb.16.12.7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee SH, Stephens JL, Paul KS, Englund PT. Fatty acid synthesis by elongases in trypanosomes. Cell. 2006;126:691–9. doi: 10.1016/j.cell.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 119.Ferguson MA, Homans SW, Dwek RA, Rademacher TW. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science. 1988;239:753–9. doi: 10.1126/science.3340856. [DOI] [PubMed] [Google Scholar]
- 120.Ferguson MA, Cross GA. Myristylation of the membrane form of a Trypanosoma brucei variant surface glycoprotein. The Journal of biological chemistry. 1984;259:3011–5. [PubMed] [Google Scholar]
- 121.Masterson WJ, Raper J, Doering TL, Hart GW, Englund PT. Fatty acid remodeling: a novel reaction sequence in the biosynthesis of trypanosome glycosyl phosphatidylinositol membrane anchors. Cell. 1990;62:73–80. doi: 10.1016/0092-8674(90)90241-6. [DOI] [PubMed] [Google Scholar]
- 122.Field MC, Menon AK, Cross GA. A glycosylphosphatidylinositol protein anchor from procyclic stage Trypanosoma brucei: lipid structure and biosynthesis. The EMBO journal. 1991;10:2731–9. doi: 10.1002/j.1460-2075.1991.tb07821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee SH, Stephens JL, Englund PT. A fatty-acid synthesis mechanism specialized for parasitism. Nature reviews Microbiology. 2007;5:287–97. doi: 10.1038/nrmicro1617. [DOI] [PubMed] [Google Scholar]
- 124.Ferguson MA. The surface glycoconjugates of trypanosomatid parasites. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 1997;352:1295–302. doi: 10.1098/rstb.1997.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brett MT, MÜ Ller-Navarra DC. The role of highly unsaturated fatty acids in aquatic foodweb processes. Freshwater Biology. 1997;38:483–99. [Google Scholar]
- 126.Stukey JE, McDonough VM, Martin CE. The OLE1 gene of Saccharomyces cerevisiae encodes the delta 9 fatty acid desaturase and can be functionally replaced by the rat stearoyl-CoA desaturase gene. The Journal of biological chemistry. 1990;265:20144–9. [PubMed] [Google Scholar]
- 127.Kachroo A, Shanklin J, Whittle E, Lapchyk L, Hildebrand D, Kachroo P. The Arabidopsis stearoyl-acyl carrier protein-desaturase family and the contribution of leaf isoforms to oleic acid synthesis. Plant molecular biology. 2007;63:257–71. doi: 10.1007/s11103-006-9086-y. [DOI] [PubMed] [Google Scholar]
- 128.Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11482–6. doi: 10.1073/pnas.132384699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gratraud P, Huws E, Falkard B, Adjalley S, Fidock DA, Berry L, et al. Oleic acid biosynthesis in Plasmodium falciparum: characterization of the stearoyl-CoA desaturase and investigation as a potential therapeutic target. PloS one. 2009;4:e6889. doi: 10.1371/journal.pone.0006889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ntambi JM. Regulation of stearoyl-CoA desaturase by polyunsaturated fatty acids and cholesterol. Journal of lipid research. 1999;40:1549–58. [PubMed] [Google Scholar]
- 131.Kumaratilake LM, Robinson BS, Ferrante A, Poulos A. Antimalarial properties of n-3 and n-6 polyunsaturated fatty acids: in vitro effects on Plasmodium falciparum and in vivo effects on P. berghei. J Clin Invest. 1992;89:961–7. doi: 10.1172/JCI115678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mai K, Sharman PA, Walker RA, Katrib M, De Souza D, McConville MJ, et al. Oocyst wall formation and composition in coccidian parasites. Memorias do Instituto Oswaldo Cruz. 2009;104:281–9. doi: 10.1590/s0074-02762009000200022. [DOI] [PubMed] [Google Scholar]
- 133.Alloatti A, Gupta S, Gualdron-Lopez M, Nguewa PA, Altabe SG, Deumer G, et al. Stearoyl-CoA desaturase is an essential enzyme for the parasitic protist Trypanosoma brucei. Biochemical and biophysical research communications. 2011;412:286–90. doi: 10.1016/j.bbrc.2011.07.084. [DOI] [PubMed] [Google Scholar]
- 134.Tripodi KE, Buttigliero LV, Altabe SG, Uttaro AD. Functional characterization of front-end desaturases from trypanosomatids depicts the first polyunsaturated fatty acid biosynthetic pathway from a parasitic protozoan. The FEBS journal. 2006;273:271–80. doi: 10.1111/j.1742-4658.2005.05049.x. [DOI] [PubMed] [Google Scholar]
- 135.Uttaro AD. Biosynthesis of polyunsaturated fatty acids in lower eukaryotes. IUBMB life. 2006;58:563–71. doi: 10.1080/15216540600920899. [DOI] [PubMed] [Google Scholar]
- 136.Livore VI, Tripodi KE, Uttaro AD. Elongation of polyunsaturated fatty acids in trypanosomatids. The FEBS journal. 2007;274:264–74. doi: 10.1111/j.1742-4658.2006.05581.x. [DOI] [PubMed] [Google Scholar]
- 137.Richmond GS, Gibellini F, Young SA, Major L, Denton H, Lilley A, et al. Lipidomic analysis of bloodstream and procyclic form Trypanosoma brucei. Parasitology. 2010;137:1357–92. doi: 10.1017/S0031182010000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mellors A, Samad A. The acquisition of lipids by African trypanosomes. Parasitology Today. 1989;5:239–44. doi: 10.1016/0169-4758(89)90255-x. [DOI] [PubMed] [Google Scholar]
- 139.Maldonado RA, Kuniyoshi RK, Linss JG, Almeida IC. Trypanosoma cruzi oleate desaturase: molecular characterization and comparative analysis in other trypanosomatids. The Journal of parasitology. 2006;92:1064–74. doi: 10.1645/GE-845R.1. [DOI] [PubMed] [Google Scholar]
- 140.Jelenska J, Crawford MJ, Harb OS, Zuther E, Haselkorn R, Roos DS, et al. Subcellular localization of acetyl-CoA carboxylase in the apicomplexan parasite Toxoplasma gondii. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2723–8. doi: 10.1073/pnas.051629998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jelenska J, Sirikhachornkit A, Haselkorn R, Gornicki P. The carboxyltransferase activity of the apicoplast acetyl-CoA carboxylase of Toxoplasma gondii is the target of aryloxyphenoxypropionate inhibitors. The Journal of biological chemistry. 2002;277:23208–15. doi: 10.1074/jbc.M200455200. [DOI] [PubMed] [Google Scholar]
- 142.Vigueira PA, Paul KS. Trypanosoma brucei: inhibition of acetyl-CoA carboxylase by haloxyfop. Experimental parasitology. 2012;130:159–65. doi: 10.1016/j.exppara.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vigueira PA, Paul KS. Requirement for acetyl-CoA carboxylase in Trypanosoma brucei is dependent upon the growth environment. Mol Microbiol. 2011;80:117–32. doi: 10.1111/j.1365-2958.2011.07563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cai X, Herschap D, Zhu G. Functional characterization of an evolutionarily distinct phosphopantetheinyl transferase in the apicomplexan Cryptosporidium parvum. Eukaryotic cell. 2005;4:1211–20. doi: 10.1128/EC.4.7.1211-1220.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, et al. A new enzyme superfamily - the phosphopantetheinyl transferases. Chemistry & biology. 1996;3:923–36. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 146.Dixon H, Williamson J. The lipid composition of blood and culture forms of Trypanosoma lewisi and Trypanosoma rhodesiense compared with that of their environment. Comp Biochem Physiol. 1970;33:111–28. doi: 10.1016/0010-406x(70)90487-1. [DOI] [PubMed] [Google Scholar]
- 147.Patnaik PK, Field MC, Menon AK, Cross GA, Yee MC, Bütikofer P. Molecular species analysis of phospholipids from Trypanosoma brucei bloodstream and procyclic forms. Molecular and biochemical parasitology. 1993;58:97–105. doi: 10.1016/0166-6851(93)90094-e. [DOI] [PubMed] [Google Scholar]
- 148.Carroll M, McCrorie P. Lipid composition of bloodstream forms of Trypanosoma brucei brucei. Comp Biochem Physiol B. 1986;83:647–51. doi: 10.1016/0305-0491(86)90312-3. [DOI] [PubMed] [Google Scholar]
- 149.Serricchio M, Bütikofer P. An essential bacterial-type cardiolipin synthase mediates cardiolipin formation in a eukaryote. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E954–61. doi: 10.1073/pnas.1121528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gonzalez-Salgado A, Steinmann ME, Greganova E, Rauch M, Mäser P, Sigel E, et al. myo-Inositol uptake is essential for bulk inositol phospholipid but not glycosylphosphatidylinositol synthesis in Trypanosoma brucei. The Journal of biological chemistry. 2012;287:13313–23. doi: 10.1074/jbc.M112.344812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wassef MK, Fioretti TB, Dwyer DM. Lipid analyses of isolated surface membranes of Leishmania donovani promastigotes. Lipids. 1985;20:108–15. doi: 10.1007/BF02534216. [DOI] [PubMed] [Google Scholar]
- 152.Beach DH, Holz GG, Jr, Anekwe GE. Lipids of Leishmania promastigotes. The Journal of parasitology. 1979;65:201–16. [PubMed] [Google Scholar]
- 153.Zheng L, T’Kind R, Decuypere S, von Freyend SJ, Coombs GH, Watson DG. Profiling of lipids in Leishmania donovani using hydrophilic interaction chromatography in combination with Fourier transform mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:2074–82. doi: 10.1002/rcm.4618. [DOI] [PubMed] [Google Scholar]
- 154.Ramos RG, Libong D, Rakotomanga M, Gaudin K, Loiseau PM, Chaminade P. Comparison between charged aerosol detection and light scattering detection for the analysis of Leishmania membrane phospholipids. J Chromatogr A. 2008;1209:88–94. doi: 10.1016/j.chroma.2008.07.080. [DOI] [PubMed] [Google Scholar]
- 155.van Zandbergen G, Bollinger A, Wenzel A, Kamhawi S, Voll R, Klinger M, et al. Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13837–42. doi: 10.1073/pnas.0600843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang K, Pompey JM, Hsu FF, Key P, Bandhuvula P, Saba JD, et al. Redirection of sphingolipid metabolism toward de novo synthesis of ethanolamine in Leishmania. The EMBO journal. 2007;26:1094–104. doi: 10.1038/sj.emboj.7601565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zufferey R, Allen S, Barron T, Sullivan DR, Denny PW, Almeida IC, et al. Ether phospholipids and glycosylinositolphospholipids are not required for amastigote virulence or for inhibition of macrophage activation by Leishmania major. The Journal of biological chemistry. 2003;278:44708–18. doi: 10.1074/jbc.M308063200. [DOI] [PubMed] [Google Scholar]
- 158.Weingartner A, Kemmer G, Muller FD, Zampieri RA, Gonzaga dos Santos M, Schiller J, et al. Leishmania promastigotes lack phosphatidylserine but bind annexin V upon permeabilization or miltefosine treatment. PloS one. 2012;7:e42070. doi: 10.1371/journal.pone.0042070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Signorell A, Rauch M, Jelk J, Ferguson MA, Bütikofer P. Phosphatidylethanolamine in Trypanosoma brucei is organized in two separate pools and is synthesized exclusively by the Kennedy pathway. The Journal of biological chemistry. 2008;283:23636–44. doi: 10.1074/jbc.M803600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Oliveira MM, Timm SL, Costa SC. Lipid composition of Trypanosoma cruzi. Comp Biochem Physiol B. 1977;58:195–9. doi: 10.1016/0305-0491(77)90109-2. [DOI] [PubMed] [Google Scholar]
- 161.Sutterwala SS, Creswell CH, Sanyal S, Menon AK, Bangs JD. De novo sphingolipid synthesis is essential for viability, but not for transport of glycosylphosphatidylinositol-anchored proteins, in African trypanosomes. Eukaryotic cell. 2007;6:454–64. doi: 10.1128/EC.00283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sutterwala SS, Hsu FF, Sevova ES, Schwartz KJ, Zhang K, Key P, et al. Developmentally regulated sphingolipid synthesis in African trypanosomes. Mol Microbiol. 2008;70:281–96. doi: 10.1111/j.1365-2958.2008.06393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Quinones W, Urbina JA, Dubourdieu M, Luis Concepcion J. The glycosome membrane of Trypanosoma cruzi epimastigotes: protein and lipid composition. Experimental parasitology. 2004;106:135–49. doi: 10.1016/j.exppara.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 164.Bertello LE, Goncalvez MF, Colli W, de Lederkremer RM. Structural analysis of inositol phospholipids from Trypanosoma cruzi epimastigote forms. The Biochemical journal. 1995;310:255–61. doi: 10.1042/bj3100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Uhrig ML, Couto AS, Colli W, de Lederkremer RM. Characterization of inositolphospholipids in Trypanosoma cruzi trypomastigote forms. Biochim Biophys Acta. 1996;1300:233–9. doi: 10.1016/0005-2760(96)00021-5. [DOI] [PubMed] [Google Scholar]
- 166.Zhang K, Showalter M, Revollo J, Hsu FF, Turk J, Beverley SM. Sphingolipids are essential for differentiation but not growth in Leishmania. The EMBO journal. 2003;22:6016–26. doi: 10.1093/emboj/cdg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kaneshiro ES, Jayasimhulu K, Lester RL. Characterization of inositol lipids from Leishmania donovani promastigotes: identification of an inositol sphingophospholipid. Journal of lipid research. 1986;27:1294–303. [PubMed] [Google Scholar]
- 168.Williams RD, Wang E, Merrill AH., Jr Enzymology of long-chain base synthesis by liver: characterization of serine palmitoyltransferase in rat liver microsomes. Archives of biochemistry and biophysics. 1984;228:282–91. doi: 10.1016/0003-9861(84)90069-9. [DOI] [PubMed] [Google Scholar]
- 169.Pinto WJ, Wells GW, Lester RL. Characterization of enzymatic synthesis of sphingolipid long-chain bases in Saccharomyces cerevisiae: mutant strains exhibiting long-chain-base auxotrophy are deficient in serine palmitoyltransferase activity. J Bacteriol. 1992;174:2575–81. doi: 10.1128/jb.174.8.2575-2581.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vial HJ, Ancelin ML. Malarial lipids. An overview. Subcell Biochem. 1992;18:259–306. [PubMed] [Google Scholar]
- 171.Dechamps S, Shastri S, Wengelnik K, Vial HJ. Glycerophospholipid acquisition in Plasmodium - a puzzling assembly of biosynthetic pathways. International journal for parasitology. 2010;40:1347–65. doi: 10.1016/j.ijpara.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 172.Hsiao LL, Howard RJ, Aikawa M, Taraschi TF. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. The Biochemical journal. 1991;274 ( Pt 1):121–32. doi: 10.1042/bj2740121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Foussard F, Gallois Y, Girault A, Menez JF. Lipids and fatty acids of tachyzoites and purified pellicles of Toxoplasma gondii. Parasitol Res. 1991;77:475–7. doi: 10.1007/BF00928412. [DOI] [PubMed] [Google Scholar]
- 174.Azzouz N, Rauscher B, Gerold P, Cesbron-Delauw MF, Dubremetz JF, Schwarz RT. Evidence for de novo sphingolipid biosynthesis in Toxoplasma gondii. International journal for parasitology. 2002;32:677–84. doi: 10.1016/s0020-7519(02)00009-7. [DOI] [PubMed] [Google Scholar]
- 175.Welti R, Mui E, Sparks A, Wernimont S, Isaac G, Kirisits M, et al. Lipidomic analysis of Toxoplasma gondii reveals unusual polar lipids. Biochemistry. 2007;46:13882–90. doi: 10.1021/bi7011993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dixon H, Ginger CD, Williamson J. The lipid metabolism of blood and culture forms of Trypanosoma lewisi and Trypanosoma rhodesiense. Comp Biochem Physiol B. 1971;39:247–66. doi: 10.1016/0305-0491(71)90168-4. [DOI] [PubMed] [Google Scholar]
- 177.Coppens I, Baudhuin P, Opperdoes FR, Courtoy PJ. Receptors for the host low density lipoproteins on the hemoflagellate Trypanosoma brucei: purification and involvement in the growth of the parasite. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6753–7. doi: 10.1073/pnas.85.18.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Samad A, Licht B, Stalmach ME, Mellors A. Metabolism of phospholipids and lysophospholipids by Trypanosoma brucei. Molecular and biochemical parasitology. 1988;29:159–69. doi: 10.1016/0166-6851(88)90071-0. [DOI] [PubMed] [Google Scholar]
- 179.Smith TK, Bütikofer P. Lipid metabolism in Trypanosoma brucei. Molecular and biochemical parasitology. 2010;172:66–79. doi: 10.1016/j.molbiopara.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lykidis A. Comparative genomics and evolution of eukaryotic phospholipid biosynthesis. Progress in lipid research. 2007;46:171–99. doi: 10.1016/j.plipres.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 181.Zufferey R, Mamoun CB. Choline transport in Leishmania major promastigotes and its inhibition by choline and phosphocholine analogs. Molecular and biochemical parasitology. 2002;125:127–34. doi: 10.1016/s0166-6851(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 182.Lockman PR, Allen DD. The transport of choline. Drug Dev Ind Pharm. 2002;28:749–71. doi: 10.1081/ddc-120005622. [DOI] [PubMed] [Google Scholar]
- 183.Bowes AE, Samad AH, Jiang P, Weaver B, Mellors A. The acquisition of lysophosphatidylcholine by African trypanosomes. The Journal of biological chemistry. 1993;268:13885–92. [PubMed] [Google Scholar]
- 184.Ibrahim HM, Al-Salabi MI, El Sabbagh N, Quashie NB, Alkhaldi AA, Escale R, et al. Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J Antimicrob Chemother. 2011;66:111–25. doi: 10.1093/jac/dkq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.de Macêdo JP, Schmidt RS, Mäser P, Rentsch D, Vial HJ, Sigel E, et al. Characterization of choline uptake in Trypanosoma brucei procyclic and bloodstream forms. Molecular and biochemical parasitology. doi: 10.1016/j.molbiopara.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 186.Gibellini F, Hunter WN, Smith TK. The ethanolamine branch of the Kennedy pathway is essential in the bloodstream form of Trypanosoma brucei. Mol Microbiol. 2009;73:826–43. doi: 10.1111/j.1365-2958.2009.06764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rifkin MR, Fairlamb AH. Transport of ethanolamine and its incorporation into the variant surface glycoprotein of bloodstream forms of Trypanosoma brucei. Molecular and biochemical parasitology. 1985;15:245–56. doi: 10.1016/0166-6851(85)90088-x. [DOI] [PubMed] [Google Scholar]
- 188.Bütikofer P, Ruepp S, Boschung M, Roditi I. ‘GPEET’ procyclin is the major surface protein of procyclic culture forms of Trypanosoma brucei brucei strain 427. The Biochemical journal. 1997;326 ( Pt 2):415–23. doi: 10.1042/bj3260415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rifkin MR, Strobos CA, Fairlamb AH. Specificity of ethanolamine transport and its further metabolism in Trypanosoma brucei. The Journal of biological chemistry. 1995;270:16160–6. doi: 10.1074/jbc.270.27.16160. [DOI] [PubMed] [Google Scholar]
- 190.Signorell A, Jelk J, Rauch M, Bütikofer P. Phosphatidylethanolamine is the precursor of the ethanolamine phosphoglycerol moiety bound to eukaryotic elongation factor 1A. The Journal of biological chemistry. 2008;283:20320–9. doi: 10.1074/jbc.M802430200. [DOI] [PubMed] [Google Scholar]
- 191.Drew ME, Langford CK, Klamo EM, Russell DG, Kavanaugh MP, Landfear SM. Functional expression of a myo-inositol/H+ symporter from Leishmania donovani. Molecular and cellular biology. 1995;15:5508–15. doi: 10.1128/mcb.15.10.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Einicker-Lamas M, Almeida AC, Todorov AG, de Castro SL, Caruso-Neves C, Oliveira MM. Characterization of the myo-inositol transport system in Trypanosoma cruzi. Eur J Biochem. 2000;267:2533–7. doi: 10.1046/j.1432-1327.2000.01302.x. [DOI] [PubMed] [Google Scholar]
- 193.Martin KL, Smith TK. Phosphatidylinositol synthesis is essential in bloodstream form Trypanosoma brucei. The Biochemical journal. 2006;396:287–95. doi: 10.1042/BJ20051825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Seyfang A, Landfear SM. Four conserved cytoplasmic sequence motifs are important for transport function of the Leishmania inositol/H(+) symporter. The Journal of biological chemistry. 2000;275:5687–93. doi: 10.1074/jbc.275.8.5687. [DOI] [PubMed] [Google Scholar]
- 195.Martin KL, Smith TK. The glycosylphosphatidylinositol (GPI) biosynthetic pathway of bloodstream-form Trypanosoma brucei is dependent on the de novo synthesis of inositol. Mol Microbiol. 2006;61:89–105. doi: 10.1111/j.1365-2958.2006.05216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mitamura T, Hanada K, Ko-Mitamura EP, Nishijima M, Horii T. Serum factors governing intraerythrocytic development and cell cycle progression of Plasmodium falciparum. Parasitol Int. 2000;49:219–29. doi: 10.1016/s1383-5769(00)00048-9. [DOI] [PubMed] [Google Scholar]
- 197.Vial HJ, Thuet MJ, Broussal JL, Philippot JR. Phospholipid biosynthesis by Plasmodium knowlesi-infected erythrocytes: the incorporation of phospohlipid precursors and the identification of previously undetected metabolic pathways. The Journal of parasitology. 1982;68:379–91. [PubMed] [Google Scholar]
- 198.Vial HJ, Thuet MJ, Philippot JR. Phospholipid biosynthesis in synchronous Plasmodium falciparum cultures. J Protozool. 1982;29:258–63. doi: 10.1111/j.1550-7408.1982.tb04023.x. [DOI] [PubMed] [Google Scholar]
- 199.Asahi H, Kanazawa T, Hirayama N, Kajihara Y. Investigating serum factors promoting erythrocytic growth of Plasmodium falciparum. Experimental parasitology. 2005;109:7–15. doi: 10.1016/j.exppara.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 200.Simoes AP, Moll GN, Slotboom AJ, Roelofsen B, Op den Kamp JA. Selective internalization of choline-phospholipids in Plasmodium falciparum parasitized human erythrocytes. Biochim Biophys Acta. 1991;1063:45–50. doi: 10.1016/0005-2736(91)90351-8. [DOI] [PubMed] [Google Scholar]
- 201.Elabbadi N, Ancelin ML, Vial HJ. Use of radioactive ethanolamine incorporation into phospholipids to assess in vitro antimalarial activity by the semiautomated microdilution technique. Antimicrobial agents and chemotherapy. 1992;36:50–5. doi: 10.1128/aac.36.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Biagini GA, Pasini EM, Hughes R, De Koning HP, Vial HJ, O’Neill PM, et al. Characterization of the choline carrier of Plasmodium falciparum: a route for the selective delivery of novel antimalarial drugs. Blood. 2004;104:3372–7. doi: 10.1182/blood-2004-03-1084. [DOI] [PubMed] [Google Scholar]
- 203.Ancelin ML, Vial HJ. Quaternary ammonium compounds efficiently inhibit Plasmodium falciparum growth in vitro by impairment of choline transport. Antimicrobial agents and chemotherapy. 1986;29:814–20. doi: 10.1128/aac.29.5.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Roggero R, Zufferey R, Minca M, Richier E, Calas M, Vial H, et al. Unraveling the mode of action of the antimalarial choline analog G25 in Plasmodium falciparum and Saccharomyces cerevisiae. Antimicrobial agents and chemotherapy. 2004;48:2816–24. doi: 10.1128/AAC.48.8.2816-2824.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wein S, Maynadier M, Bordat Y, Perez J, Maheshwari S, Bette-Bobillo P, et al. Transport and pharmacodynamics of albitiazolium, an antimalarial drug candidate. British journal of pharmacology. 2012;166:2263–76. doi: 10.1111/j.1476-5381.2012.01966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ancelin ML, Calas M, Vidal-Sailhan V, Herbute S, Ringwald P, Vial HJ. Potent inhibitors of Plasmodium phospholipid metabolism with a broad spectrum of in vitro antimalarial activities. Antimicrobial agents and chemotherapy. 2003;47:2590–7. doi: 10.1128/AAC.47.8.2590-2597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Charron AJ, Sibley LD. Host cells: mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. Journal of cell science. 2002;115:3049–59. doi: 10.1242/jcs.115.15.3049. [DOI] [PubMed] [Google Scholar]
- 208.Gupta N, Zahn MM, Coppens I, Joiner KA, Voelker DR. Selective disruption of phosphatidylcholine metabolism of the intracellular parasite Toxoplasma gondii arrests its growth. The Journal of biological chemistry. 2005;280:16345–53. doi: 10.1074/jbc.M501523200. [DOI] [PubMed] [Google Scholar]
- 209.Francis SE, Sullivan DJ, Jr, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 210.Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. The Journal of biological chemistry. 1956;222:193–214. [PubMed] [Google Scholar]
- 211.Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. Journal of lipid research. 2008;49:1187–94. doi: 10.1194/jlr.R700019-JLR200. [DOI] [PubMed] [Google Scholar]
- 212.Vance JE. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: two metabolically related aminophospholipids. Journal of lipid research. 2008;49:1377–87. doi: 10.1194/jlr.R700020-JLR200. [DOI] [PubMed] [Google Scholar]
- 213.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 214.Serra M, Saba JD. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv Enzyme Regul. 2010;50:349–62. doi: 10.1016/j.advenzreg.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schuiki I, Daum G. Phosphatidylserine decarboxylases, key enzymes of lipid metabolism. IUBMB life. 2009;61:151–62. doi: 10.1002/iub.159. [DOI] [PubMed] [Google Scholar]
- 216.Suzuki TT, Kanfer JN. Purification and properties of an ethanolamine-serine base exchange enzyme of rat brain microsomes. The Journal of biological chemistry. 1985;260:1394–9. [PubMed] [Google Scholar]
- 217.Gibellini F, Hunter WN, Smith TK. Biochemical characterization of the initial steps of the Kennedy pathway in Trypanosoma brucei: the ethanolamine and choline kinases. The Biochemical journal. 2008;415:135–44. doi: 10.1042/BJ20080435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Vial HJ, Thuet MJ, Philippot JR. Cholinephosphotransferase and ethanolaminephosphotransferase activities in Plasmodium knowlesi-infected erythrocytes. Their use as parasite-specific markers. Biochim Biophys Acta. 1984;795:372–83. doi: 10.1016/0005-2760(84)90088-2. [DOI] [PubMed] [Google Scholar]
- 219.Ancelin ML, Vial HJ. Choline kinase activity in Plasmodium-infected erythrocytes: characterization and utilization as a parasite-specific marker in malarial fractionation studies. Biochim Biophys Acta. 1986;875:52–8. doi: 10.1016/0005-2760(86)90010-x. [DOI] [PubMed] [Google Scholar]
- 220.Ancelin ML, Vial HJ. Several lines of evidence demonstrating that Plasmodium falciparum, a parasitic organism, has distinct enzymes for the phosphorylation of choline and ethanolamine. FEBS letters. 1986;202:217–23. doi: 10.1016/0014-5793(86)80690-1. [DOI] [PubMed] [Google Scholar]
- 221.Dechamps S, Wengelnik K, Berry-Sterkers L, Cerdan R, Vial HJ, Gannoun-Zaki L. The Kennedy phospholipid biosynthesis pathways are refractory to genetic disruption in Plasmodium berghei and therefore appear essential in blood stages. Molecular and biochemical parasitology. 2010;173:69–80. doi: 10.1016/j.molbiopara.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 222.Ancelin ML, Vial HJ. Regulation of phosphatidylcholine biosynthesis in Plasmodium-infected erythrocytes. Biochim Biophys Acta. 1989;1001:82–9. doi: 10.1016/0005-2760(89)90310-x. [DOI] [PubMed] [Google Scholar]
- 223.Elabbadi N, Ancelin ML, Vial HJ. Phospholipid metabolism of serine in Plasmodium-infected erythrocytes involves phosphatidylserine and direct serine decarboxylation. The Biochemical journal. 1997;324 ( Pt 2):435–45. doi: 10.1042/bj3240435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pessi G, Choi JY, Reynolds JM, Voelker DR, Mamoun CB. In vivo evidence for the specificity of Plasmodium falciparum phosphoethanolamine methyltransferase and its coupling to the Kennedy pathway. The Journal of biological chemistry. 2005;280:12461–6. doi: 10.1074/jbc.M414626200. [DOI] [PubMed] [Google Scholar]
- 225.Pessi G, Kociubinski G, Mamoun CB. A pathway for phosphatidylcholine biosynthesis in Plasmodium falciparum involving phosphoethanolamine methylation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6206–11. doi: 10.1073/pnas.0307742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jha TK, Sundar S, Thakur CP, Bachmann P, Karbwang J, Fischer C, et al. Miltefosine, an oral agent, for the treatment of Indian visceral leishmaniasis. N Engl J Med. 1999;341:1795–800. doi: 10.1056/NEJM199912093412403. [DOI] [PubMed] [Google Scholar]
- 227.Sundar S, Makharia A, More DK, Agrawal G, Voss A, Fischer C, et al. Short-course of oral miltefosine for treatment of visceral leishmaniasis. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2000;31:1110–3. doi: 10.1086/318122. [DOI] [PubMed] [Google Scholar]
- 228.Soto J, Toledo J, Gutierrez P, Nicholls RS, Padilla J, Engel J, et al. Treatment of American cutaneous leishmaniasis with miltefosine, an oral agent. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2001;33:E57–61. doi: 10.1086/322689. [DOI] [PubMed] [Google Scholar]
- 229.Rakotomanga M, Blanc S, Gaudin K, Chaminade P, Loiseau PM. Miltefosine affects lipid metabolism in Leishmania donovani promastigotes. Antimicrobial agents and chemotherapy. 2007;51:1425–30. doi: 10.1128/AAC.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Witola WH, El Bissati K, Pessi G, Xie C, Roepe PD, Mamoun CB. Disruption of the Plasmodium falciparum PfPMT gene results in a complete loss of phosphatidylcholine biosynthesis via the serine-decarboxylase-phosphoethanolamine-methyltransferase pathway and severe growth and survival defects. The Journal of biological chemistry. 2008;283:27636–43. doi: 10.1074/jbc.M804360200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Witola WH, Pessi G, El Bissati K, Reynolds JM, Mamoun CB. Localization of the phosphoethanolamine methyltransferase of the human malaria parasite Plasmodium falciparum to the Golgi apparatus. The Journal of biological chemistry. 2006;281:21305–11. doi: 10.1074/jbc.M603260200. [DOI] [PubMed] [Google Scholar]
- 232.Bobenchik AM, Augagneur Y, Hao B, Hoch JC, Ben Mamoun C. Phosphoethanolamine methyltransferases in phosphocholine biosynthesis: functions and potential for antiparasite therapy. FEMS microbiology reviews. 2011;35:609–19. doi: 10.1111/j.1574-6976.2011.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sampels V, Hartmann A, Dietrich I, Coppens I, Sheiner L, Striepen B, et al. Conditional mutagenesis of a novel choline kinase demonstrates plasticity of phosphatidylcholine biogenesis and gene expression in Toxoplasma gondii. The Journal of biological chemistry. 2012;287:16289–99. doi: 10.1074/jbc.M112.347138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Farine L, Bütikofer P. The ins and outs of phosphatidylethanolamine synthesis in Trypanosoma brucei. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbalip.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 235.Signorell A, Gluenz E, Rettig J, Schneider A, Shaw MK, Gull K, et al. Perturbation of phosphatidylethanolamine synthesis affects mitochondrial morphology and cell-cycle progression in procyclic-form Trypanosoma brucei. Mol Microbiol. 2009;72:1068–79. doi: 10.1111/j.1365-2958.2009.06713.x. [DOI] [PubMed] [Google Scholar]
- 236.Zhang K, Beverley SM. Phospholipid and sphingolipid metabolism in Leishmania. Molecular and biochemical parasitology. 2010;170:55–64. doi: 10.1016/j.molbiopara.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Saba JD, Nara F, Bielawska A, Garrett S, Hannun YA. The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. The Journal of biological chemistry. 1997;272:26087–90. doi: 10.1074/jbc.272.42.26087. [DOI] [PubMed] [Google Scholar]
- 238.Alberge B, Gannoun-Zaki L, Bascunana C, Tran van Ba C, Vial H, Cerdan R. Comparison of the cellular and biochemical properties of Plasmodium falciparum choline and ethanolamine kinases. The Biochemical journal. 2010;425:149–58. doi: 10.1042/BJ20091119. [DOI] [PubMed] [Google Scholar]
- 239.Wunderlich F, Helwig M, Schillinger G, Vial H, Philippot J, Speth V. Isolation and characterization of parasites and host cell ghosts from erythrocytes infected with Plasmodium chabaudi. Molecular and biochemical parasitology. 1987;23:103–15. doi: 10.1016/0166-6851(87)90145-9. [DOI] [PubMed] [Google Scholar]
- 240.Baunaure F, Eldin P, Cathiard AM, Vial H. Characterization of a non-mitochondrial type I phosphatidylserine decarboxylase in Plasmodium falciparum. Mol Microbiol. 2004;51:33–46. doi: 10.1046/j.1365-2958.2003.03822.x. [DOI] [PubMed] [Google Scholar]
- 241.Choi JY, Augagneur Y, Ben Mamoun C, Voelker DR. Identification of gene encoding Plasmodium knowlesi phosphatidylserine decarboxylase by genetic complementation in yeast and characterization of in vitro maturation of encoded enzyme. The Journal of biological chemistry. 2012;287:222–32. doi: 10.1074/jbc.M111.313676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gupta N, Hartmann A, Lucius R, Voelker DR. The obligate intracellular parasite Toxoplasma gondii secretes a soluble phosphatidylserine decarboxylase. The Journal of biological chemistry. 2012;287:22938–47. doi: 10.1074/jbc.M112.373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wanderley JL, Pinto da Silva LH, Deolindo P, Soong L, Borges VM, Prates DB, et al. Cooperation between apoptotic and viable metacyclics enhances the pathogenesis of Leishmaniasis. PloS one. 2009;4:e5733. doi: 10.1371/journal.pone.0005733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wanderley JL, Barcinski MA. Apoptosis and apoptotic mimicry: the Leishmania connection. Cell Mol Life Sci. 2010;67:1653–9. doi: 10.1007/s00018-010-0291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Goldshmidt H, Matas D, Kabi A, Carmi S, Hope R, Michaeli S. Persistent ER stress induces the spliced leader RNA silencing pathway (SLS), leading to programmed cell death in Trypanosoma brucei. PLoS pathogens. 2010;6:e1000731. doi: 10.1371/journal.ppat.1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kay JG, Grinstein S. Sensing phosphatidylserine in cellular membranes. Sensors (Basel) 2011;11:1744–55. doi: 10.3390/s110201744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Dechamps S, Maynadier M, Wein S, Gannoun-Zaki L, Marechal E, Vial HJ. Rodent and nonrodent malaria parasites differ in their phospholipid metabolic pathways. Journal of lipid research. 2010;51:81–96. doi: 10.1194/jlr.M900166-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gerold P, Schwarz RT. Biosynthesis of glycosphingolipids de-novo by the human malaria parasite Plasmodium falciparum. Molecular and biochemical parasitology. 2001;112:29–37. doi: 10.1016/s0166-6851(00)00336-4. [DOI] [PubMed] [Google Scholar]
- 249.Michell RH. Inositol derivatives: evolution and functions. Nat Rev Mol Cell Biol. 2008;9:151–61. doi: 10.1038/nrm2334. [DOI] [PubMed] [Google Scholar]
- 250.Gardocki ME, Jani N, Lopes JM. Phosphatidylinositol biosynthesis: biochemistry and regulation. Biochim Biophys Acta. 2005;1735:89–100. doi: 10.1016/j.bbalip.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 251.Tawk L, Dubremetz JF, Montcourrier P, Chicanne G, Merezegue F, Richard V, et al. Phosphatidylinositol 3-monophosphate is involved in toxoplasma apicoplast biogenesis. PLoS pathogens. 2011;7:e1001286. doi: 10.1371/journal.ppat.1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Bhattacharjee S, Stahelin RV, Speicher KD, Speicher DW, Haldar K. Endoplasmic reticulum PI(3)P lipid binding targets malaria proteins to the host cell. Cell. 2012;148:201–12. doi: 10.1016/j.cell.2011.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Beresford NJ, Saville C, Bennett HJ, Roberts IS, Tabernero L. A new family of phosphoinositide phosphatases in microorganisms: identification and biochemical analysis. BMC genomics. 2010;11:457. doi: 10.1186/1471-2164-11-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Brown JR, Auger KR. Phylogenomics of phosphoinositide lipid kinases: perspectives on the evolution of second messenger signaling and drug discovery. BMC evolutionary biology. 2011;11:4. doi: 10.1186/1471-2148-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ferguson MA. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. Journal of cell science. 1999;112 ( Pt 17):2799–809. doi: 10.1242/jcs.112.17.2799. [DOI] [PubMed] [Google Scholar]
- 256.Gilson PR, Nebl T, Vukcevic D, Moritz RL, Sargeant T, Speed TP, et al. Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2006;5:1286–99. doi: 10.1074/mcp.M600035-MCP200. [DOI] [PubMed] [Google Scholar]
- 257.Wichroski MJ, Ward GE. Biosynthesis of glycosylphosphatidylinositol is essential to the survival of the protozoan parasite Toxoplasma gondii. Eukaryotic cell. 2003;2:1132–6. doi: 10.1128/EC.2.5.1132-1136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Martin KL, Smith TK. The myo-inositol-1-phosphate synthase gene is essential in Trypanosoma brucei. Biochem Soc Trans. 2005;33:983–5. doi: 10.1042/BST0330983. [DOI] [PubMed] [Google Scholar]
- 259.Seyfang A, Landfear SM. Substrate depletion upregulates uptake of myo-inositol, glucose and adenosine in Leishmania. Molecular and biochemical parasitology. 1999;104:121–30. doi: 10.1016/s0166-6851(99)00138-3. [DOI] [PubMed] [Google Scholar]
- 260.Elabbadi N, Ancelin ML, Vial HJ. Characterization of phosphatidylinositol synthase and evidence of a polyphosphoinositide cycle in Plasmodium-infected erythrocytes. Molecular and biochemical parasitology. 1994;63:179–92. doi: 10.1016/0166-6851(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 261.Wengelnik K, Vial HJ. Characterisation of the phosphatidylinositol synthase gene of Plasmodium species. Res Microbiol. 2007;158:51–9. doi: 10.1016/j.resmic.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 262.Seron K, Dzierszinski F, Tomavo S. Molecular cloning, functional complementation in Saccharomyces cerevisiae and enzymatic properties of phosphatidylinositol synthase from the protozoan parasite Toxoplasma gondii. Eur J Biochem. 2000;267:6571–9. doi: 10.1046/j.1432-1327.2000.01749.x. [DOI] [PubMed] [Google Scholar]
- 263.Cronan JE. Bacterial membrane lipids: where do we stand? Annu Rev Microbiol. 2003;57:203–24. doi: 10.1146/annurev.micro.57.030502.090851. [DOI] [PubMed] [Google Scholar]
- 264.Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta. 2009;1788:2084–91. doi: 10.1016/j.bbamem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. Journal of lipid research. 2008;49:1607–20. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Serricchio M, Bütikofer P. Phosphatidylglycerophosphate synthase associates with a mitochondrial inner membrane complex and is essential for growth of Trypanosoma brucei. Mol Microbiol. 2012 doi: 10.1111/mmi.12116. [DOI] [PubMed] [Google Scholar]
- 267.Tafesse FG, Ternes P, Holthuis JC. The multigenic sphingomyelin synthase family. The Journal of biological chemistry. 2006;281:29421–5. doi: 10.1074/jbc.R600021200. [DOI] [PubMed] [Google Scholar]
- 268.Mullen TD, Hannun YA, Obeid LM. Ceramide synthases at the centre of sphingolipid metabolism and biology. The Biochemical journal. 2012;441:789–802. doi: 10.1042/BJ20111626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang K, Bangs JD, Beverley SM. Sphingolipids in parasitic protozoa. Advances in experimental medicine and biology. 2010;688:238–48. doi: 10.1007/978-1-4419-6741-1_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Denny PW, Shams-Eldin H, Price HP, Smith DF, Schwarz RT. The protozoan inositol phosphorylceramide synthase: a novel drug target that defines a new class of sphingolipid synthase. The Journal of biological chemistry. 2006;281:28200–9. doi: 10.1074/jbc.M600796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Zhang K, Hsu FF, Scott DA, Docampo R, Turk J, Beverley SM. Leishmania salvage and remodelling of host sphingolipids in amastigote survival and acidocalcisome biogenesis. Mol Microbiol. 2005;55:1566–78. doi: 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Fridberg A, Olson CL, Nakayasu ES, Tyler KM, Almeida IC, Engman DM. Sphingolipid synthesis is necessary for kinetoplast segregation and cytokinesis in Trypanosoma brucei. Journal of cell science. 2008;121:522–35. doi: 10.1242/jcs.016741. [DOI] [PubMed] [Google Scholar]
- 273.Sevova ES, Goren MA, Schwartz KJ, Hsu FF, Turk J, Fox BG, et al. Cell-free synthesis and functional characterization of sphingolipid synthases from parasitic trypanosomatid protozoa. The Journal of biological chemistry. 2010;285:20580–7. doi: 10.1074/jbc.M110.127662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Nakayasu ES, Yashunsky DV, Nohara LL, Torrecilhas AC, Nikolaev AV, Almeida IC. GPIomics: global analysis of glycosylphosphatidylinositol-anchored molecules of Trypanosoma cruzi. Mol Syst Biol. 2009;5:261. doi: 10.1038/msb.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Landoni M, Duschak VG, Peres VJ, Nonami H, Erra-Balsells R, Katzin AM, et al. Plasmodium falciparum biosynthesizes sulfoglycosphingolipids. Molecular and biochemical parasitology. 2007;154:22–9. doi: 10.1016/j.molbiopara.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 276.Sonda S, Sala G, Ghidoni R, Hemphill A, Pieters J. Inhibitory effect of aureobasidin A on Toxoplasma gondii. Antimicrobial agents and chemotherapy. 2005;49:1794–801. doi: 10.1128/AAC.49.5.1794-1801.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Voelker DR. Genetic and biochemical analysis of non-vesicular lipid traffic. Annual review of biochemistry. 2009;78:827–56. doi: 10.1146/annurev.biochem.78.081307.112144. [DOI] [PubMed] [Google Scholar]
- 279.Lev S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nature reviews Molecular cell biology. 2010;11:739–50. doi: 10.1038/nrm2971. [DOI] [PubMed] [Google Scholar]
- 280.Acestor N, Panigrahi AK, Ogata Y, Anupama A, Stuart KD. Protein composition of Trypanosoma brucei mitochondrial membranes. Proteomics. 2009;9:5497–508. doi: 10.1002/pmic.200900354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Sam-Yellowe TY, Florens L, Wang T, Raine JD, Carucci DJ, Sinden R, et al. Proteome analysis of rhoptry-enriched fractions isolated from Plasmodium merozoites. J Proteome Res. 2004;3:995–1001. doi: 10.1021/pr049926m. [DOI] [PubMed] [Google Scholar]
- 282.Niemann M, Wiese S, Mani J, Chanfon A, Jackson C, Meisinger C, et al. Mitochondrial outer membrane proteome of Trypanosoma brucei reveals novel factors required to maintain mitochondrial morphology. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M112.023093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Opperdoes FR, Baudhuin P, Coppens I, De Roe C, Edwards SW, Weijers PJ, et al. Purification, morphometric analysis, and characterization of the glycosomes (microbodies) of the protozoan hemoflagellate Trypanosoma brucei. J Cell Biol. 1984;98:1178–84. doi: 10.1083/jcb.98.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Docampo R, de Souza W, Miranda K, Rohloff P, Moreno SN. Acidocalcisomes - conserved from bacteria to man. Nature reviews Microbiology. 2005;3:251–61. doi: 10.1038/nrmicro1097. [DOI] [PubMed] [Google Scholar]
- 285.Salto ML, Kuhlenschmidt T, Kuhlenschmidt M, de Lederkremer RM, Docampo R. Phospholipid and glycolipid composition of acidocalcisomes of Trypanosoma cruzi. Molecular and biochemical parasitology. 2008;158:120–30. doi: 10.1016/j.molbiopara.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Godsel LM, Engman DM. Flagellar protein localization mediated by a calcium-myristoyl/palmitoyl switch mechanism. The EMBO journal. 1999;18:2057–65. doi: 10.1093/emboj/18.8.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Tyler KM, Fridberg A, Toriello KM, Olson CL, Cieslak JA, Hazlett TL, et al. Flagellar membrane localization via association with lipid rafts. Journal of cell science. 2009;122:859–66. doi: 10.1242/jcs.037721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Tull D, Vince JE, Callaghan JM, Naderer T, Spurck T, McFadden GI, et al. SMP-1, a member of a new family of small myristoylated proteins in kinetoplastid parasites, is targeted to the flagellum membrane in Leishmania. Mol Biol Cell. 2004;15:4775–86. doi: 10.1091/mbc.E04-06-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Foussard F, Leriche MA, Dubremetz JF. Characterization of the lipid content of Toxoplasma gondii rhoptries. Parasitology. 1991;102(Pt 3):367–70. doi: 10.1017/s0031182000064313. [DOI] [PubMed] [Google Scholar]
- 290.Noto T, Miyakawa S, Oishi H, Endo H, Okazaki H. Thiolactomycin, a new antibiotic. III. In vitro antibacterial activity. The Journal of antibiotics. 1982;35:401–10. doi: 10.7164/antibiotics.35.401. [DOI] [PubMed] [Google Scholar]
- 291.Nishida I, Kawaguchi A, Yamada M. Effect of thiolactomycin on the individual enzymes of the fatty acid synthase system in Escherichia coli. Journal of biochemistry. 1986;99:1447–54. doi: 10.1093/oxfordjournals.jbchem.a135614. [DOI] [PubMed] [Google Scholar]
- 292.Waller RF, Keeling PJ, Donald RG, Striepen B, Handman E, Lang-Unnasch N, et al. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12352–7. doi: 10.1073/pnas.95.21.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Zhang YM, Rock CO. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. Journal of Biological Chemistry. 2004;279:30994–1001. doi: 10.1074/jbc.M403697200. [DOI] [PubMed] [Google Scholar]
- 294.Bhowruth V, Brown AK, Besra GS. Synthesis and biological evaluation of NAS-21 and NAS-91 analogues as potential inhibitors of the mycobacterial FAS-II dehydratase enzyme Rv0636. Microbiology (Reading, England) 2008;154:1866–75. doi: 10.1099/mic.0.2008/017434-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Brown AK, Papaemmanouil A, Bhowruth V, Bhatt A, Dover LG, Besra GS. Flavonoid inhibitors as novel antimycobacterial agents targeting Rv0636, a putative dehydratase enzyme involved in Mycobacterium tuberculosis fatty acid synthase II. Microbiology (Reading, England) 2007;153:3314–22. doi: 10.1099/mic.0.2007/009936-0. [DOI] [PubMed] [Google Scholar]
- 296.Tasdemir D, Lack G, Brun R, Ruedi P, Scapozza L, Perozzo R. Inhibition of Plasmodium falciparum fatty acid biosynthesis: evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. Journal of medicinal chemistry. 2006;49:3345–53. doi: 10.1021/jm0600545. [DOI] [PubMed] [Google Scholar]
- 297.AbouLaila M, Terkawi MA, Yokoyama N, Igarashi I. In vitro growth inhibitory effect of (-)-Epigallocatechin-3-gallate from green tea on the growth of equine Babesia parasites. Journal of Protozoology Research. 2011;21:30–5. [Google Scholar]
- 298.Gratraud P, Surolia N, Besra GS, Surolia A, Kremer L. Antimycobacterial Activity and Mechanism of Action of NAS-91. Antimicrobial agents and chemotherapy. 2008;52:1162–6. doi: 10.1128/AAC.00968-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Martins-Duarte ES, Jones SM, Gilbert IH, Atella GC, Souza Wd, Vommaro RC. Thiolactomycin analogues as potential anti-Toxoplasma gondii agents. Parasitol Int. 2009;58:411–5. doi: 10.1016/j.parint.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 300.Jones SM, Urch JE, Brun R, Harwood JL, Berry C, Gilbert IH. Analogues of thiolactomycin as potential anti-malarial and anti-trypanosomal agents. Bioorganic & medicinal chemistry. 2004;12:683–92. doi: 10.1016/j.bmc.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 301.Jones SM, Urch JE, Kaiser M, Brun R, Harwood JL, Berry C, et al. Analogues of thiolactomycin as potential antimalarial agents. Journal of medicinal chemistry. 2005;48:5932–41. doi: 10.1021/jm049067d. [DOI] [PubMed] [Google Scholar]
- 302.Paveto C, Guida MC, Esteva MI, Martino V, Coussio J, Flawia MM, et al. Anti-Trypanosoma cruzi activity of green tea (Camellia sinensis) catechins. Antimicrobial agents and chemotherapy. 2004;48:69–74. doi: 10.1128/AAC.48.1.69-74.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6379–83. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–8. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
- 305.Morita YS, Paul KS, Englund PT. Specialized fatty acid synthesis in African trypanosomes: myristate for GPI anchors. Science. 2000;288:140–3. doi: 10.1126/science.288.5463.140. [DOI] [PubMed] [Google Scholar]
- 306.Alloatti A, Testero SA, Uttaro AD. Chemical evaluation of fatty acid desaturases as drug targets in Trypanosoma cruzi. International journal for parasitology. 2009;39:985–93. doi: 10.1016/j.ijpara.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 307.Korduláková J, Janin YL, Liav A, Barilone N, Dos Vultos T, Rauzier J, et al. Isoxyl activation is required for bacteriostatic activity against Mycobacterium tuberculosis. Antimicrobial agents and chemotherapy. 2007;51:3824–9. doi: 10.1128/AAC.00433-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Alloatti A, Gupta S, Gualdrón-López M, Igoillo-Esteve M, Nguewa PA, Deumer G, et al. Genetic and Chemical Evaluation of Trypanosoma brucei Oleate Desaturase as a Candidate Drug Target. PloS one. 2010;5:1–10. doi: 10.1371/journal.pone.0014239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Dorlo TP, Balasegaram M, Beijnen JH, de Vries PJ. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67:2576–97. doi: 10.1093/jac/dks275. [DOI] [PubMed] [Google Scholar]
- 310.Jimenez-Lopez JM, Carrasco MP, Segovia JL, Marco C. Hexadecylphosphocholine inhibits phosphatidylcholine synthesis via both the methylation of phosphatidylethanolamine and CDP-choline pathways in HepG2 cells. Int J Biochem Cell Biol. 2004;36:153–61. doi: 10.1016/s1357-2725(03)00193-6. [DOI] [PubMed] [Google Scholar]
- 311.van Blitterswijk WJ, Verheij M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbalip.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 312.Lira R, Contreras LM, Rita RM, Urbina JA. Mechanism of action of anti-proliferative lysophospholipid analogues against the protozoan parasite Trypanosoma cruzi: potentiation of in vitro activity by the sterol biosynthesis inhibitor ketoconazole. J Antimicrob Chemother. 2001;47:537–46. doi: 10.1093/jac/47.5.537. [DOI] [PubMed] [Google Scholar]
- 313.Nagiec MM, Nagiec EE, Baltisberger JA, Wells GB, Lester RL, Dickson RC. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. The Journal of biological chemistry. 1997;272:9809–17. doi: 10.1074/jbc.272.15.9809. [DOI] [PubMed] [Google Scholar]
- 314.Figueiredo JM, Dias WB, Mendonca-Previato L, Previato JO, Heise N. Characterization of the inositol phosphorylceramide synthase activity from Trypanosoma cruzi. The Biochemical journal. 2005;387:519–29. doi: 10.1042/BJ20041842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Mina JG, Pan SY, Wansadhipathi NK, Bruce CR, Shams-Eldin H, Schwarz RT, et al. The Trypanosoma brucei sphingolipid synthase, an essential enzyme and drug target. Molecular and biochemical parasitology. 2009;168:16–23. doi: 10.1016/j.molbiopara.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 316.Lauer SA, Ghori N, Haldar K. Sphingolipid synthesis as a target for chemotherapy against malaria parasites. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9181–5. doi: 10.1073/pnas.92.20.9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Hanada K, Palacpac NM, Magistrado PA, Kurokawa K, Rai G, Sakata D, et al. Plasmodium falciparum phospholipase C hydrolyzing sphingomyelin and lysocholinephospholipids is a possible target for malaria chemotherapy. J Exp Med. 2002;195:23–34. doi: 10.1084/jem.20010724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Gonzalez-Bulnes P, Bobenchik AM, Augagneur Y, Cerdan R, Vial HJ, Llebaria A, et al. PG12, a phospholipid analog with potent antimalarial activity, inhibits Plasmodium falciparum CTP:phosphocholine cytidylyltransferase activity. The Journal of biological chemistry. 2011;286:28940–7. doi: 10.1074/jbc.M111.268946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Bobenchik AM, Choi JY, Mishra A, Rujan IN, Hao B, Voelker DR, et al. Identification of inhibitors of Plasmodium falciparum phosphoethanolamine methyltransferase using an enzyme-coupled transmethylation assay. BMC biochemistry. 2010;11:4. doi: 10.1186/1471-2091-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Ancelin ML, Calas M, Bompart J, Cordina G, Martin D, Ben Bari M, et al. Antimalarial activity of 77 phospholipid polar head analogs: close correlation between inhibition of phospholipid metabolism and in vitro Plasmodium falciparum growth. Blood. 1998;91:1426–37. [PubMed] [Google Scholar]
- 321.Calas M, Ancelin ML, Cordina G, Portefaix P, Piquet G, Vidal-Sailhan V, et al. Antimalarial activity of compounds interfering with Plasmodium falciparum phospholipid metabolism: comparison between mono- and bisquaternary ammonium salts. Journal of medicinal chemistry. 2000;43:505–16. doi: 10.1021/jm9911027. [DOI] [PubMed] [Google Scholar]
- 322.Wengelnik K, Vidal V, Ancelin ML, Cathiard AM, Morgat JL, Kocken CH, et al. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science. 2002;295:1311–4. doi: 10.1126/science.1067236. [DOI] [PubMed] [Google Scholar]
- 323.Vickerman K. The dyskinetoplasty mutation in Trypanosoma evansi and other flagellates. Protozoology. 1977;3:57–69. [Google Scholar]