

Abstract
Epidemiological studies corroborate a correlation between pesticide use and Parkinson’s disease (PD). Thiocarbamate and dithiocarbamate pesticides are widely used and produce neurotoxicity in the peripheral nervous system. Recent evidence from rodent studies suggests that these compounds also cause dopaminergic (DAergic) dysfunction and altered protein processing, two hallmarks of PD. However, DAergic neurotoxicity has yet to be documented. We assessed DAergic dysfunction in Caenorhabditis elegans (C. elegans) to investigate the ability of thiocarbamate pesticides to induce DAergic neurodegeneration. Acute treatment with either S-ethyl N,N-dipropylthiocarbamate (EPTC), molinate, or a common reactive intermediate of dithiocarbamate and thiocarbamate metabolism, S-methyl-N,N-diethylthiocarbamate (MeDETC), to gradual loss of DAergic cell morphology and structure over the course of 6 days in worms expressing green fluorescent protein (GFP) under a DAergic cell specific promoter. HPLC analysis revealed decreased DA content in the worms immediately following exposure to MeDETC, EPTC, and molinate. Additionally, worms treated with the three test compounds showed a drastic loss of DAergic-dependent behavior over a time course similar to changes in DAergic cell morphology. Alterations in the DAergic system were specific, as loss of cell structure and neurotransmitter content was not observed in cholinergic, glutamatergic, or GABAergic systems. Overall, our data suggest that thiocarbamate pesticides promote neurodegeneration and DAergic cell dysfunction in C. elegans, and may be an environmental risk factor for PD.
Keywords: C. elegans, molinate, EPTC, pesticide, dopamine, neurodegeneration
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease in the United States, affecting 1% of the population over the age of 55 (Lees et al., 2009). PD is characterized by the loss of selected catecholamenergic neurons in the central and peripheral nervous systems including the dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNpc). Loss of DAergic neurons in the SNpC gives rise to the motor symptoms observed in PD, such as bradykinesia, rigidity, resting tremors, and postural instability, with onset of symptoms occurring when ≥70% of these DAergic neurons are lost (Lees et al., 2009). Additionally, alterations in neurotransmitter systems other than dopamine (DA), such as glutamate, serotonin and acetylcholine, have been implicated in non-motor clinical symptoms in PD, including behavioral and cognitive decline (Caudle and Zhang, 2009; Politis and Loane, 2011; Yarnall et al., 2011). Mechanisms underlying the selective degeneration of the DAergic neurons leading to progressive motor disease are poorly understood, but complex interactions between an individual’s age, genetics and environment are thought to contribute to the etiology of PD. Because familial forms of PD account for only 10% of PD cases (Lees et al., 2009), environmental agents have gained attention as contributing factors in the etiology of PD. Among the environmental agents examined thus far, epidemiological studies have identified a strong correlation between development of PD and the use of pesticides (Ascherio et al., 2006; Hancock et al., 2008; Li et al., 2005; Tanner et al., 1999; van der Mark et al., 2012; Wang et al., 2011a).
Pesticides from a broad range of chemical classes have been implicated in the development of PD, including paraquat (a bipyridyl derivative herbicide), dieldrin (an organochloride insecticide), and rotenone (a rotenoid ester insecticide). The extent to which these compounds model human PD in rodents is variable. Both rotenone and paraquat exposure lead to DAergic neurodegeneration, increased oxidative stress, aggregation of α-synuclein, and motor deficits in rodents, while dieldrin and cyclodiene pesticides induce oxidative stress and promote aggregation of α-synuclein but do not cause motor deficits or loss of DAergic neurons (Betarbet et al., 2000; Kanthasamy et al., 2005; Karen et al., 2001; McCormack et al., 2005; Prasad et al., 2007; Richardson et al., 2006). These differences highlight some of the difficulties inherent when trying to select endpoints for testing environmental agents in animal models of neurodegenerative diseases, in that multiple biological processes can be targeted through divergent chemical properties of test agents that cannot be predicted a priori. This is important to consider, as there are a large number of chemical compounds applied as pesticides and numerous mixture combinations that can arise in fields that require evaluation. While much can be gleaned from in vivo rodent studies, the complexity of the mammalian brain and the time required for testing compounds and mixtures for DAergic neurodegeneration limit the utility of these studies for initial evaluations. For this reason model organisms such as Drosophila melanogastor and Ceanorhabditis elegans (C. elegans) are gaining wider attention for their simplified nervous systems and screening potential.
The nematode C. elegans is a powerful genetic model organism useful for exploring DAergic vulnerability to environmental contaminants and molecular mechanisms of PD. These worms possess all of the genes required for DA neurotransmission and contain homologues of most PD-associated genes. Visualization of DAergic neurons in strains expressing green fluorescent protein (GFP) under the dopamine transporter-1 (dat-1) promoter is possible due to the transparent bodies of these worms. DAergic neurons in C. elegans are sensitive to toxicants used to model PD in rodents and cell culture, such as 1-methyl-4-phenyl-pyridinium (MPP+) and 6-hydroxydopamine (6-OHDA) (Braungart et al., 2004; Nass et al., 2002). In the present study we used C. elegans to investigate the potential of thiocarbamate and dithiocarbamate pesticides to induce DAergic neurotoxicity.
Thiocarbamate and dithiocarbamate pesticides are widely used in the United States; these compounds repeatedly appear on the U.S. Environmental Protection Agency’s (US EPA) most commonly used pesticide list (Grube et al., 2011). Thiocarbamate and dithiocarbamate pesticides share many characteristics, including chemical and microbial degradation pathways, and metabolism in mammals (Mulkey, 2001; Szolar, 2007). Thiocarbamate pesticides are typically volatile and have short half-lives, reducing the chance of bioaccumulation in the food chain, while dithiocarbamate pesticides are more stable and present greater risk (Mulkey, 2001; Szolar, 2007). The major source of human exposure is usually through dermal contact, inhalation during application, and residues on food crops. Metabolism of thiocarbamates and dithiocarbamates produces electrophilic metabolites, that can covalently modify proteins and inhibit enzymes (Pentyala and Chetty, 1993; Savolainen and Hervonen, 1985; Staub et al., 1995), and has been associated with neurotoxicity in both rodents and humans (Bergouignan et al., 1988; Tonkin et al., 2004; Viquez et al., 2008). More specifically, previous studies have supported the ability of N,N-dialkyl dithiocarbamates to produce DAergic toxicity in vitro and degenerative change in the nigrostriatal pathway in vivo (Chou et al., 2008; Viquez et al., 2012). Covalent modification and inhibition of E1 ubiquitin activating enzyme was proposed to be a contributing mechanisms for this toxicity. Interestingly, the bioactivation pathway leading to the protein alkylating species derived from dithiocarbamates proceeds through an S-methyl thiocarbamate intermediate possessing the characteristic structure of thiocarbamate herbicides. This suggests that thiocarbamate herbicides may also be DAergic toxicants that are potentially more potent than the dithiocarbamates as a result of being located several steps down the activation pathway relative to dithiocarbamates.
Herein, we determined the potential of a cyclic and archetypical N,N-dialkyl thiocarbamate herbicide, molinate and EPTC, respectively, to produce DAergic neurodegeneration in C. elegans. Molinate is a thiocarbamate herbicide commonly used worldwide in rice patty fields that has been banned by the US EPA due to its ability to cause peripheral neuropathies and reproductive toxicity (Cochran et al., 1997; Wickramaratne et al., 1998). EPTC was the 19th most commonly used pesticide in the United States in 1999, but its use is declining (Donaldson et al., 2002; Grube et al., 2011). An association between EPTC pesticide application and colon cancer and leukemia has been reported in male pesticide applicators in North Carolina and Iowa followed over 7–11 years (van Bemmel et al., 2008), suggesting the potential of EPTC to cause cumulative toxicities from chronic exposures. Additionally, selected metabolites of the N,N-dialkyldithiocarbamate DEDC were examined to evaluate the relevance of the bioactivation pathway of DEDC previously proposed to be responsible for nigrostriatal injury in vivo. General toxicity was assessed in the worms through determination of LD50 values and DAergic toxicity was determined through imaging of DAergic neurons, quantification of DA levels and measuring DA-dependent behavioral endpoints. To evaluate the selectivity of the test compounds for DAergic toxicity, morphological assessments of glutaminergic and cholinergic neurons were performed and the level of γ-aminobutyric acid (GABA) and glutamate were quantified. The data demonstrate that the S-alkyl thiocarbamate class of compounds exhibit substantial and selective DAergic tocxicity in this model. This finding supports further investigation into this class of compounds as a potential contributing risk factor for PD and suggests that metabolism of N,N-diaklydithiocarbamates to S-methyl thiocarbamate metabolites may contribute to the nigrostriatal injury in vivo and DAergic toxicity reported for these compounds in vitro.
Materials and Methods
Reagents
Unless otherwise stated all reagents were obtained from Sigma-Aldrich (St. Louis, MO). Sodium N,N-diethyldithiocarbamate (DEDC) was obtained from Alfa Aesar (Ward Hill, MA), while MeDETC, EPTC, molinate, and DETC-sulfoxide were synthesized as previously described (Zimmerman et al., 2004).
C. elegans strains and handling of the worms
C. elegans strains were handled and maintained at 20°C on Nematode Growth Medium (NGM) plates seeded with OP-50 strain of Escherichia coli, as previously described (Brenner, 1974). The following strains were used in this study: N2, BY200 (Pdat-1::GFP(vtIs1)), NC1307 (Pglr-1::GFP(rhIs2)), LX929 (Punc-17::GFP(vsIs48)), VP596 (dvls19[pAF15(gst-4::GFP::NLS)];vsls33[dop-3::RFP]) and CB1112 (cat-2(e1112)). All strains were provided by the Caenorhabditis Genetic Center (CGC; University of Minnesota), except for the NC1307 strain, which was provided by Dr. David Miller (Vanderbilt University), and the VP596 strain, provided by Dr. Keith P. Choe (University of Florida). Synchronous L1 populations were obtained by isolating embryos from gravid worms using a bleaching solution (1% NaOCl and 0.25 M NaOH), and segregating eggs from worm and bacterial debris by flotation on a sucrose gradient, as previously described (Stiernagle, 1999).
Dose-response curves and acute pesticide treatment
The lethal dose 50% (LD50) of EPTC, molinate, MeDETC, DEDC, DETC-sulfoxide, and ethyl isocyanate in C. elegans was determined by treating 5,000 synchronized L1 BY200 worms with doses ranging from 0.001 to 10 mM for 1 hour (h) in M9 liquid buffer at 25°C on a eppendorf tube rotator. Worms were also treated with 1% DMSO as a vehicle control. All exposures were carried out in triplicate and repeated 4 times. After treatment worms were washed 3 times with M9 buffer, transferred to OP-50-seeded NGM plates, and manually counted for lethality 24 h post treatment.
Lifespan Analysis
Aged L4 and healthy-looking worms previously treated with LD50 concentrations of MeDETC, EPTC, or molinate (40 per treatment, in duplicate) were transferred to new OP-50-seeded NGM plates. Worms were transferred to new plates every 2 days for feeding, and survival was assessed daily until all the worms died. Plotted curves represent averages of triplicate independent experiments.
Confocal microscopy and fluorescence quantification
To examine morphological changes in neurons as a result of test agent exposure, worms expressing GFP in DAergic, glutaminergic, or cholinergic neurons were visualized by confocal microscopy (BY200, NC1307, and LX929 strains, respectively). GFP-expressing worms were treated with LD50 concentrations of MeDETC, EPTC, or molinate and visualized 2, 4, and 6 days following exposure. 20–30 worms were mounted on 4% agarose pads in M9 and anaesthetized with 0.2% tricane and 0.02% tetramisole. Images were captured through Plan-Apochromat 20x objective on a LSM510 confocal microscope (Carl Zeiss MicroImaging, Inc) scanning every 200 nm for XZ sections. Images were processed with the Zeiss LSM Image Browser. Quantification of GFP fluorescence was performed using ImageJ 1.36 software as previously described (Gavet and Pines, 2010).
Activation of SKN-1, the worm homologue of nuclear factor (erythroid-derived-2)-like 2 (Nrf2), was measured using VP596 strain, which expresses GFP under the control of the promoter for the SKN-1 target glutathione S transferase 4 (gst-4). L1 VP596 worms were treated with LD50 concentrations of MeDETC, molinate, or EPTC for 1 h and visualized immediately, 1, 2, and 3 days following exposure. Confocal microscopy and GFP fluorescence quantification was performed as described above.
Neurotransmitter content measurement
Following a 1 h treatment with LD50 concentrations of molinate, EPTC, or MeDETC, 200,000 L1 worms were pelleted, the supernatant was removed, and the pellet was frozen in liquid nitrogen. The frozen pellet was re-suspended in lysis buffer and sonicated to disrupt cell membranes. DA, γ-aminobutyric acid (GABA), and glutamate levels were quantified by HPLC through services provided by the Center for Molecular Neuroscience Neurochemistry Core Laboratory at Vanderbilt University. Neurotransmitter levels were determined by a specific HPLC assays utilizing an Antec Decade II electrochemical detector (DA) or 474 scanning detector (GABA, glutamate). HPLC control and data acquisition were managed by Millennium 32 software. DA, GABA, and glutamate were quantified using internal standards after separation by HPLC and were reported as ng/mg protein. Protein levels were quantified using the BCA assay (Thermo Scientific, Sunnyvale, CA), following manufacturer’s instructions.
Basal slowing response
Assessment of DA-dependent behavior was performed using the basal slowing response assay, as previously described (Sawin et al., 2000). Briefly, following exposure to LD50 concentrations of MeDETC, EPTC, or molinate for 1 h, N2 L1 worms were seeded on OP-50-spread plates. BY200 worms could not be used for this assay as they contain the rol-6 gene as a balancer to the Pdat-1::GFP, causing a rolling phenotype. N2 worms were also pre-treated with either a nonlethal dose of dopamine chloride (10 mM) for 10 min, as previously described (Benedetto et al., 2010), or 2 mM N-acetyl-L-cysteine (NAC) for 1 h, followed by 5 washes in M9 and treatment with DMSO, or LD50 concentrations of MeDETC, molinate, or EPTC for 1 h. Three and 6 days post exposure worms were washed off the plates with S basal buffer and ~10 worms were transferred to either unseeded or OP-50-seeded NGM plates. Locomotor activity was assessed as the number of body-bends per 20 seconds (s). Worms deficient in cat-2 (homolog of mammalian tyrosine hydroxylase (TH)) were used as a positive control. Data is presented as change in body bends, which is calculated by subtracting the number of body bends of worms plated on OP-50-seeded plates from the number of body bends of worms plated on unseeded plates.
Glutathione quantification
Total intracellular glutathione (GSH) levels, i.e. reduced and oxidized GSH, were measured using the 5,5′-dithiobis-2-nitrobenzoic acid-glutathione disulfide reductase recycling method, as previously described (Rahman et al., 2006) in whole worm extracts from 50,000 BY200 worms. Worms were treated with either DMSO or LD50 concentrations of MeDETC, molinate, or EPTC for 1 h in the presence or absence of a 1 h pre-treatment with NAC (2 mM).
Statistics
All statistical analyses were performed using Prism 5 (Graphpad software). Dose-response lethality curves and LD50 determination were generated using a sigmoidal dose-response model with a top constraint at 100%. Statistical analysis of significance was carried out by one-way ANOVA followed by Dunnett’s post-hoc test for the neurotransmitter quantifications, basal slowing response, and GSH quantification. One-way ANOVA followed by Tukey’s post-hoc test was used for GFP fluorescence quantification. A two-tailed t-test was used to assess the statistical significance of the change in the basal slowing response between day 3 and 6. Gehan-Breslow-Wilcoxon Test was used to assess the statistical significance of the lifespan experiments. Values of P < 0.05 were considered statistically significant.
Results
Acute thiocarbamate toxicity to C. elegans
To determine whether C. elegans was a suitable in vivo model organism for investigating thiocarbamate neurotoxicity, we exposed the BY200 strain to increasing concentrations of the compounds for 1 h and generated dose-response survival curves (Figure 1). We treated worms with the dithiocarbamate DEDC, which has been implicated in DAergic dysfunction (Viquez et al., 2012), and the thiocarbamate herbicides EPTC and molinate (Figure 1A, Table 1). To evaluate the relevance of the previously proposed bioactivation pathways for N,N-dialkyl dithiocarbamates, we also treated worms with MeDETC, DETC-sulfoxide, and ethyl isocyanate (Figure 1B). MeDETC (LD50 = 0.114 mM) was found to be the most potent, followed by EPTC (LD50 = 0.209 mM), molinate (LD50 = 0.442 mM), DETC-sulfoxide (LD50 = 1.232 mM), DEDC (LD50 = 1.679 mM), and ethyl isocyanate (LD50 = 2.867 mM) (Table 1). The compounds that were the most toxic to C. elegans were the S-alkyl thiocarbamates. Based upon these data we used the two thiocarbamate herbicides, EPTC and molinate, and MeDETC, the most potent DEDC metabolite.
Figure 1.

Dose-response curves for selected thiocarbamate pesticides and intermediate metabolites. BY200 worms were treated for 1 h with increasing concentrations of (A) EPTC, molinate, and DEDC, or (B) the DEDC metabolites MeDETC, DETC-sulfoxide, and ethyl isocyanate. Data are expressed as means ± SEM from 4 independent experiments.
Table 1.
LD50 concentrations and structures of thiocarbamate pesticides and metabolites.
| Compound | Structure | LD50 (mM) |
|---|---|---|
| MeDETC |
|
0.114 |
| EPTC |
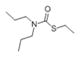
|
0.209 |
| Molinate |
|
0.442 |
| DETC-sulfoxide |
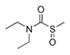
|
1.232 |
| DEDC |
|
1.679 |
| Ethyl isocyanate | CH3CH2-N=C=O | 2.867 |
Lifespan analysis was performed to further characterize toxicity to EPTC, molinate and MeDETC. Worms treated with LD50 concentrations of EPTC, molinate and MeDETC had significantly decreased lifespans as compared to DMSO control treated worms (Figure 2). From day 1 to around day 6 post exposure there was little difference in treated vs. untreated worms’ lifespan (Figure 2); however there was increased lethality following this lag time. Therefore subsequent neurotoxicity studies were conducted up to 6 days post exposure.
Figure 2.

Lifespan of C. elegans following treatment with EPTC, molinate, and MeDETC. BY200 worms were treated for 1 h with LD50 concentrations of (A) MeDETC, (B) EPTC, or (C) molinate or DMSO control, and worms were followed for their life time and counted for survival. Data are expressed as means ± SEM from 3 independent experiments. ***p<0.001,
Selective DAergic neurodegeneration following pesticide exposure
To investigate the propensity of EPTC, molinate and MeDETC to cause DAergic neurodegeneration, the BY200 strain, which expresses GFP under the control of the DAergic neuron specific DAT1 promoter, dat-1::GFP(vtIs1), was used to visualize DAergic neuron morphology. C. elegans hermaphrodites have a total of 8 DAergic neurons; 4 cephalic deirids (CEP) and 2 anterior deirids (ADE) in the head and 2 posterior deirids in the tail, all of which express dat-1. Worms were treated with LD50 concentrations of MeDETC, EPTC, molinate or DMSO vehicle control, and were visualized 2, 4, and 6 days after exposure. There were no significant changes in DA cell morphology or GFP expression prior to day 4 (Figure 3A and B). On the fourth and sixth days after exposure there were increased markers of neurodegeneration in the 4 CEP and 2 ADE head DAergic neurons (Figure 3A and B). Defects observed were primarily in neuron processes, resulting in discontinuous and punctuated GFP signals, as well as decreased size of neuronal bodies, and an overall loss of GFP intensity in worms treated with MeDETC, EPTC, or molinate as compared to DMSO control worms. Intensity of the GFP signal was quantified from the photomicrographs, revealing significant loss of GFP signals for worms treated with molinate, EPTC, and MeDETC on days 4 and 6 post treatment (Figure 3B).
Figure 3.
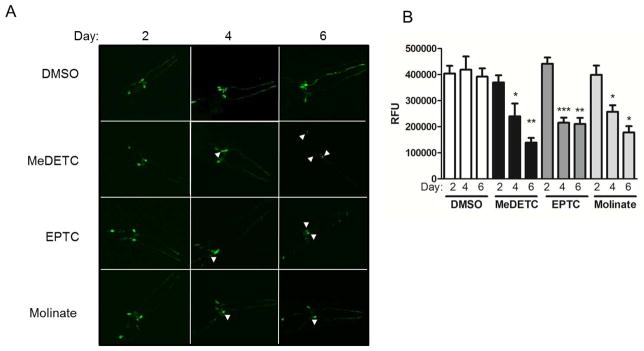
Time-dependent DAergic neurodegeneration following exposure to EPTC, molinate, and MeDETC. (A) Mechanosensory CEP and ADE neurons in BY200 (dat-1::GFP) expressing worms were visualized 2, 4, and 6 days post exposure to LD50 concentrations of MeDETC, EPTC, and molinate. Worms were also treated with DMSO as a vehicle control. White arrowheads indicate shrunken cell bodies. (B) Quantification of GFP fluorescence. Data are expressed as means ± SEM from 3 independent experiments. ***p<0.001, **p<0.01, *p<0.05.
The DAergic neurodegeneration following MeDETC, EPTC, and molinate was not observed in other neuronal cell types. Worms expressing GFP in cholinergic neurons (unc-17::GFP) or in glutaminergic neurons (glr-1::GFP) showed no changes in cell morphology in response to pesticide treatment as compared to DMSO treated worms (Figure 4A and 5A). This suggests that neurodegeneration in response to MeDETC, molinate, and EPTC is DAergic neuron selective. While unc-17 driven GFP florescence significantly increased over time, there was no difference between DMSO and pesticide treated worms (Figure 4B). There was no significant changes in GFP intensity in worms expressing glr-1::GFP.
Figure 4.
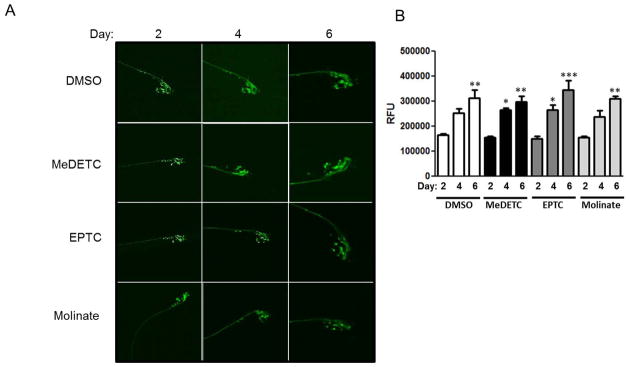
EPTC, molinate, and MeDETC do not cause degeneration of cholinergic neurons. (A) Worms expressing GFP in cholinergic neurons (unc-17::GFP) were visualized 2, 4, and 6 days post exposure to LD50 concentrations of MeDETC, EPTC, and molinate. Worms were also treated with DMSO as a vehicle control. (B) Quantification of GFP fluorescence. Data are expressed as means ± SEM from 3 independent experiments. ***p<0.001, **p<0.01, *p<0.05.
Figure 5.

EPTC, molinate, and MeDETC do not cause degeneration of glutaminergic neurons. (A) Worms expressing GFP in glutaminergic neurons (glr-1::GFP) were visualized 2, 4, and 6 days post exposure to LD50 concentrations of MeDETC, EPTC, and molinate. Worms were also treated with DMSO as a vehicle control. (B) Quantification of GFP fluorescence. Data are expressed as means ± SEM from 3 independent experiments.
Reduction in DA levels and DA-dependent behavior by thiocarbamates
As DAergic neuron morphology was significantly altered by MeDETC, EPTC, and molinate treatments, it was next investigated whether this effect was paralleled by selective loss of DA content in the worms. Levels of DA, GABA, and glutamate were measured in extracts from BY200 worms harvested immediately following treatment with LD50 concentrations of MeDETC, EPTC, and molinate. MeDETC, EPTC, and molinate exposure significantly decreased DA levels as compared to DMSO control (Figure 6A). There was no change in GABA or glutamate levels in worms treated with these compounds as compared to control (Figure 6B and C).
Figure 6.

Exposure to EPTC, molinate, and MeDETC decrease DA levels, but not glutamate or GABA levels in C. elegans. (A) DA, (B) GABA, and (C) glutamate levels were measured immediately following exposure to LD50 concentrations of MeDETC, EPTC, and molinate. Worms were also treated with DMSO as a vehicle control. Data are expressed as means ± SEM relative to DMSO controls from 3 independent experiments. **p<0.01, *p<0.05.
To determine whether the loss of DAergic morphology and reduction in DA levels results in a loss of DAergic-dependent function, we next analyzed the effect of MeDETC, EPTC, and molinate treatment on the basal slowing response. It has been previously shown that N2 worms significantly slow their movement on NGM plates containing a bacterial food source as compared to worms placed on unseeded NGM plates (Sawin et al., 2000). Worms deficient in CAT-2, the nematode homologue of TH, the rate-limiting enzyme for DA biosynthesis, do not show this behavior (Sawin et al., 2000). N2 worms treated with LD50 concentrations of MeDETC, EPTC, and molinate were assessed for the basal slowing response 3 days and 6 days after exposure. After 3 days of treatment, worms treated with DMSO showed a change in locomotor activity similar to Sawin et al (Figure 7A). Worms containing mutant cat-2 were used as a positive control, and showed a loss of basal slowing response behavior similar to Sawin et al (Figure 7A). Both EPTC and molinate significantly decreased the basal slowing response 3 and 6 days after exposure as compared to DMSO control (Figure 7A and B). MeDETC only decreased the basal slowing response after 6 days of treatment as compared to DMSO control (Figure 7A and B). Comparing the change in body bends of worms treated with MeDETC, molinate, or EPTC, there was significantly greater deficit in DA-dependent behavior on day 6 than on day 3 (Figure 7C), suggesting that the loss of DAergic behavior was progressive.
Figure 7.

Progressive loss of DAergic behavior following exposure to EPTC, molinate and MeDETC. Wild type N2 worms were treated with LD50 concentrations of MeDETC, EPTC, and molinate in the presence or absence of a 10 min pre-treatment with DA (10 mM), and the basal slowing response was measured (A) 3 and (B) 6 days following exposure. Worms expressing mutant cat-2 were used as a negative control. (C) Comparison of the change in the basal slowing response between worms treated with EPTC, molinate, and MeDETC for 3 and 6 days. Data are expressed as means ± SEM from 5 independent experiments. ***p<0.001, **p<0.01, *p<0.05.
As we observed a significant loss of DA content immediately following treatment with MeDETC, EPTC, and molinate, we next determined whether pre-treating worms with DA could restore DA-dependent behavior. N2 worms were pre-treated with 10 mM DA in M9 for 10 min, washed 5 times with M9, and then treated with LD50 concentrations of MeDETC, molinate, or EPTC. Basal slowing response was not significantly different between worms treated with MeDETC, molinate, or EPTC with or without the DA pre-treatment at either 3 or 6 days post treatment (Figure 7A and B). This suggests that loss of DA-dependent behavior following exposure may not be dependent on the initial loss of DA, but on protracted neurotoxicity of the thiocarbamates.
Increased oxidative stress following thiocarbamate exposure
Since oxidative stress has been implicated in carbamate pesticide-induced neurotoxicity, and increased oxidative stress and autoxidation of DA are implicated in DAergic neurodegeneration in PD (Chen et al., 2000; Jana et al., 2007; Khan et al., 2012; Valentine et al., 2009; Viquez et al., 2009), we investigated whether there was increased oxidative stress in worms treated with S-alkyl thiocarbamates by measuring total GSH levels. Worms treated with LD50 concentrations of MeDETC, EPTC, and molinate contained significantly less total GSH than DMSO treated control worms (Figure 8A). Reduction in total GSH levels could be prevented by pre-treating the worms with 2 mM NAC (Figure 8A). As a decrease in GSH levels following thiocarbamate treatment can lead to alteration of the redox potential of the cell and increased oxidative stress, we then examined whether these compounds activated SKN-1, the worm homologue of Nrf2. Transgenic worms expressing GFP under the gst-4 promoter were treated with LD50 concentrations of MeDETC, molinate, or EPTC, and GFP fluorescence was measured. Since gst-4 is a gene up-regulated by SKN-1 under conditions of oxidative stress, GFP fluorescence serves as an indicator of oxidative stress and SKN-1 activity (Choe et al., 2009; Link and Johnson, 2002). Worms treated with MeDETC, EPTC, or molinate showed increased GFP fluorescence as compared to DMSO treated worms immediately following exposure and continuing for 2 days (Figure 8B). DMSO treatment mildly increased GFP expression in C. elegans, which is not surprising as DMSO has been shown to induce Nrf2 in human umbilical vein endothelial cells (Liang et al., 2011). On the third day following exposure there was no significant difference in GFP fluorescence between DMSO or thiocarbamate treated worms. This suggests that the thiocarbamate-induced oxidative stress initiates an antioxidant response in worms.
Figure 8.

Oxidative stress is involved in thiocarbamate pesticide-induced loss of DA-dependent behavior. (A) Total GSH levels were measured at the end of a 1 h exposure to LD50 concentrations of MeDETC, EPTC, and molinate in the presence or absence of a 1 h pre-treatment with NAC (2 mM). Worms were also treated with DMSO as a vehicle control. (B) Photomicrographs and quantification of GFP fluorescence of transgenic worms expressing GFP under the gst-4 promoter treated with DMSO, MeDETC, EPTC, or molinate immediately (time 0), 1, 2, and 3 days following exposure. (C) Basal slowing response in N2 worms treated with LD50 concentrations of MeDETC, molinate, EPTC in the presence or absence of a 1 h pre-treatment with NAC (2mM). Worms expressing mutant cat-2 were used as a negative control. Data are expressed as means ± SEM relative to DMSO controls from 4 independent experiments. ***p<0.001, **p<0.01, *p<0.05. vs. DMSO treated worms. +++p<0.001, +p<0.05 vs. respective MeDETC, molinate, or EPTC treatment.
To determine whether oxidative stress may play a role in thiocarbamate-induced DAergic dysfunction, the basal slowing response was measured in worms 6 days following pre-treatment with 2 mM NAC and treatment with LD50 concentrations of MeDETC, molinate, and EPTC. Measuring the behavior 6 days post exposure was selected as there is the greatest loss of DA-dependent behavior from the thiocarbamates at this time point (Figure 7C). Pre-treatment with NAC significantly prevented the loss of DA-dependent behavior induced by MeDETC, molinate, and EPTC (Figure 8C). Treatment of NAC to worms deficient in CAT-2 did not significantly increase DA-dependent behavior, suggesting that the NAC treatment alone does not affect the basal slowing response.
Discussion
Dithiocarbamate and thiocarbamate pesticides are highly used in the United States; however their effects on the central nervous system (CNS) are not fully characterized. Studies in rats treated with DEDC have shown altered protein processing and inhibition of the ubiquitination cascade, pathways implicated in PD (Viquez et al., 2012). However loss of DAergic neuron function or integrity was not previously reported. Therefore, we chose to address whether dithiocarbamate and thiocarbamate pesticides could cause DAergic neurodegeneration in C. elegans taking advantage of their simple nervous system, short lifespan and the ease of examining neuronal cell morphology and integrity in vivo with GFP tagged strains. Furthermore, we evaluated the contribution of S-alkyl thiocarbamate metabolites to the DAergic toxicity of N,N-diakly dithiocarbamates.
The adverse health effects of thiocarbamate and dithiocarbamate pesticides, including enzymatic inhibition, peripheral neuropathies, and reproductive toxicity, are believed to arise from the metabolism of the parental compounds to reactive electrophilic metabolites. Parental dithiocarbamate compounds are metabolized in a series of reactions by methyl transferases and cytochrome P450 enzymes to produce S-alkyl thiocarbamate metabolites, such as MeDETC (Scheme 1) (Madan and Faiman, 1995; Pike et al., 2001; Staub et al., 1995). MeDETC can be further metabolized to DETC-sulfoxide and ethyl isocyanate, which can covalently modify proteins through nucleophilic addition by sulfhydryl groups. In assessing general toxicity to C. elegans, MeDETC was the most potent compound tested, with an LD50 lower than the parental compounds tested, DEDC, molinate, and EPTC. The higher potency of MeDETC in C. elegans compared to the parental compounds agrees with previous data showing sulfoxide metabolites of molinate being more potent testicular toxins in rats (Jewell et al., 1998). Interestingly, the other metabolites tested were less toxic than MeDETC; both DETC-sulfoxide and ethyl isocyanate had LD50 concentrations higher than MeDETC (Figue 1 and Table 1). This may be due to the higher reactivity of DETC-sulfoxide and ethyl isocyanate with cysteine residues that could limit distribution to the nervous system. From these data, it can be inferred that other thiocarbamate and dithiocarbamate compounds that share the same metabolic pathway may also elicit similar toxic effects.
Scheme 1.

Recognized metabolic pathways of N,N-dialkyldithiocarbamates leading to the generation of reactive electrophilic species. N,N-dialkyldithiocarbamates are S-methylated and undergo oxidation to generate S-alkyl thiocarbamate metabolites that are homologues of commercial thiocarbamate herbicides. S-alkyl thiocarbamate metabolites can then be further oxidized to electrophilic S-alkyl thiocarbamate sulfoxide and sulfone metabolites capable of reacting with nucleophiles to form covalent adducts. Alternatively N,N-dialkyldithiocarbamate metabolites can be dealkylated and oxidatively metabolized to the N-monalkyl sulfoxide that can either react directly or undergo facile decomposition to an alkylisocyante prior to reacting with a nucleophile.
The C. elegans system has been used in studies examining the neurotoxicity of pesticides, such as rotenone, glyphosphate, mancozeb, and monocrotophos (Ali and Rajini, 2012; Anbalagan et al., 2013; Mocko et al., 2010; Negga et al., 2011; Negga et al., 2012), however, the effects of molinate and EPTC have not been evaluated in this organism. C. elegans expressing GFP under the dat-1 promoter were treated with the three compounds to visualize DAergic neuron morphology. A single exposure at the LD50 concentrations of EPTC, molinate, or MeDETC significantly altered DAergic cell morphology in the CEP and ADE head neurons of C. elegans over the course of 6 days following exposure (Figure 3A). Morphological changes in DAergic neurons included punctuated processes and shrinkage of cell bodies, which are similar to the neurodegeneration previously observed in worms treated with Mn or 6-OHDA (Benedetto et al., 2010; Nass et al., 2002). EPTC, molinate, and MeDETC were shown to decrease lifespan (Figure 2), however changes in DAergic cell morphology were observed at time points where there was no significant death as compared to control worms. The concentrations of molinate and EPTC used in our studies are higher than some of the reported environmental levels. Molinate levels in rice fields vary from 3.9 μg/l to 1042 μg/l (Castro et al., 2005; Quayle et al., 2006), however concentrations have not been directly measured during peak seasonal applications. It is also important to consider that toxicity to the DAergic system from pesticide exposure can occur over the course of years, giving rise to cumulative exposures higher than environmental levels. It is important to note that in comparison to compounds previously used in C. elegans to induce DAergic neurodegeneration, EPTC, molinate, and MeDETC are more potent. DAergic neurotoxicity has been observed with 10–50 mM 6-OHDA, 400 mM glyphosphate, and 8.2 mM mancozeb, while MeDETC produced DAergic neurodegeneration following a 0.114 mM treatment (Nass et al., 2002; Negga et al., 2011). As 6-OHDA and mancozeb are toxic to DAergic neurons in mammals, our data suggest that the thiocarbamate pesticides and metabolites may also be potent in mammalian systems.
Quantification of Pdat-1::GFP signal has been used as a secondary method to assess neurodegeneration in C. elegans (Negga et al., 2011; Negga et al., 2012). DAT is essential for proper DAergic neurotransmission, being responsible for the clearance of DA from synapses. Brain samples from PD patients have revealed that there is from 50–70% loss of the DAT (Seeman and Niznik, 1990). This can be viewed as a protective, compensatory response to the loss of DA. In our study, GFP intensity, which served as a proxy for dat-1 gene activity, was significantly diminished following treatment with EPTC, molinate, and MeDETC, not only suggesting that there is loss of DAergic morphology, but there may also be a loss of DAergic function (Figure 3B).
DAergic signaling and function was assessed by two parameters, DA content and the basal slowing response, a DA-dependent behavior. DA levels were significantly decreased in worms treated with EPTC, molinate, and MeDETC immediately following exposure, preceding the loss of DAergic cell morphology (Figure 6). Interestingly, rats treated with DEDC showed no change in DA levels, but had decreased TH protein levels in the striatum consistent with injury, possibly through loss of DAergic terminals and an up-regulation of TH activity through phosphorylation consistent with a compensatory response to maintain DA levels (Viquez et al., 2012). Comparisons between the rat and C. elegans are difficult, and differences between DA levels in the two animal models may be due to species differences, exposure protocols, or the rats were at an earlier stage of injury that could still be compensated and eventually would have decreased dopamine levels. Previous studies of Mn-containing dithiocarbamate pesticides in C. elegans have shown altered DA-cell morphology (Negga et al., 2011; Negga et al., 2012), however functional consequences have not been reported. Here, we observed that loss of DA-dependent behavior occurred concurrently with increased DA-ergic neurodegeneration. The DAergic neurons in C. elegans are mechanosensory neurons responsible for detecting and responding to changes in the environment (Goodman, 2006). The basal slowing response reflects the worm’s ability to sense the presence of food and adjust its locomotor activity to allow for consumption (Sawin et al., 2000). Both molinate and EPTC treatments resulted in significant decrease in the basal slowing response 3 days after exposure (Figure 7). Six days following exposure to EPTC, molinate, or MeDETC showed both a significant loss of DAergic cell morphology, GFP signal intensity, and decrease in the basal slowing response. Similar correlations between DA content, behavior, and morphology have been observed in C. elegans treated with the organophosphorous insecticide monocrotophos (Ali and Rajini, 2012). Interestingly, treatment of worms with excess DA prior to MeDETC, molinate, or EPTC did not prevent loss of DA-dependent behavior 3 or 6 days after exposure, suggesting that alterations in DAergic cell morphology and behavior may not be entirely due to an initial loss of DA but to time-dependent damage.
While there was overt DAergic dysfunction in C. elegans, there was no effect on glutamatergic, cholinergic, or GABAergic neurons. Both glutamate excitotoxicity and cholinergic dysfunction have been implicated in PD. Glutamate is the major fast excitatory neurotransmitter in the CNS, which in high concentrations can lead to an overload of calcium influx leading to necrotic or apoptotic cell death, termed excitotoxicity (Caudle and Zhang, 2009). DAergic neurons may be susceptible to excitotoxicity in PD due to impaired mitochondria and bioenergetics. Rats treated with rotenone, a mitochondrial complex I inhibitor, had a potentiated response to glutamate levels in the substantia nigra, increasing the cell’s vulnerability to excitotoxicity (Wu and Johnson, 2009). Therefore we examined whether there was an effect of thiocarbamates on glutaminergic neurons. Morphology of glutaminergic neurons did not change following exposure to MeDETC, EPTC, or molinate, nor were there differences in intracellular glutamate levels following thiocarbamate treatment in C. elegans (Figures 5 and 6). Similarly, we did not observe changes in intracellular GABA levels following EPTC, MeDETC, or molinate exposure (Figure 6). Cholinergic degeneration has been observed in the basal forebrain of PD patients and has been observed in vagal motorneurons of rats exposed to 6-OHDA (Zheng et al., 2011; Ziegler et al., 2012). In addition, dithiocarbamates are well characterized inhibitors of acetylcholinesterase (Pentyala and Chetty, 1993; Savolainen and Hervonen, 1985). We therefore investigated whether there was degeneration of cholinergic neurons of C. elegans. Exposure to MeDETC, EPTC, and molinate did not alter cholinergic cell morphology (Figure 4). Overall, the effects of MeDETC, molinate and EPTC in C. elegans were DA specific, suggesting these compounds may be effective tools in examining mechanisms of DAergic cell death.
PD is a complex disease involving motor and cognitive dysfunction resulting from as diverse mechanisms as DAergic neurodegeneration, mitochondrial dysfunction, oxidative stress, inflammation, and protein aggregation (Ebrahimi-Fakhari et al., 2012; Exner et al., 2012; Sutachan et al., 2012). While characterization of thiocarbamate-induced neurodegeneration was shown to be DA specific, mechanisms that led to selective loss of DAergic neurons in C. elegans remain to be fully characterized. DEDC has been shown to increase lipid peroxidation and induce the antioxidant enzymes superoxide dismutase 1 (SOD1), glutathione S-transferase–α (GST-α), and heme oxygenase-1 (HO-1) in sciatic nerves of rats (Valentine et al., 2010). Sodium methyldithiocarbamate has been shown to deplete intracellular GSH, induce antioxidant enzymes sulfiredoxin, peroxiredoxin, and isocitrate dehydrogenase, and alter the inflammatory cytokine profile of peripheral macrophages in mice (Pruett et al., 2009). Herein, we report that EPTC, molinate, and MeDETC decreased total GSH levels in C. elegans (Figure 8A). GSH depletion was fully reversible by pre-treating worms with NAC. These measurements were performed in whole worm extracts, and reflect the level of oxidative stress in every cell of the worm, including the glutaminergic and cholinergic neurons that were unaffected by thiocarbamate pesticide exposure. Important to note, many of the thiocabamate metabolites can react directly or be catalyzed by glutathione transferase to conjugate with GSH. Decreased glutathione levels will alter a cells’ thiol status, increasing vulnerability to oxidants. Our studies demonstrate that there is increased activation of SKN-1 following thiocarbamate exposure (Figure 8B), illustrating a protective mechanism by which worms respond to thiocarbamate pesticide-induced oxidative stress. It is well established that DAergic neurons are more susceptible to oxidative stress than other types of neurons. This is thought to be due, at least in part, to the autoxidation of DA to reactive quinones and semiquinones that can redox cycle and generate ROS, resulting in a vicious cycle of increased oxidative stress and cellular damage (Berman and Hastings, 1999; Jana et al., 2007). In our studies, pre-treatment of worms with NAC increased DA-dependent behavior in pesticide treated worms (Figure 8C), suggesting that oxidative stress may play an important role in thiocarbamate-induced DAergic neurodegeneration. Cellular targets of thiocarbamate-induced oxidative stress are currently unknown. Several of the proteins involved in familial PD are involved in oxidative stress pathways, such as DJ-1, PINK-1, and parkin (Cookson, 2012; Joselin et al., 2012; Wang et al., 2011b), or increase the cell’s vulnerability to oxidative stress, such as α-synuclein (Lastres-Becker et al., 2012; Wang et al., 2010). Whether thiocarbamate pesticides may interact with these PD-associated proteins remains to be determined.
In conclusion, our studies demonstrate that C. elegans are an effective tool for screening environmental agents for DAergic neurotoxicity. Using this model system we have characterized selective DAergic toxicity resulting from treatment of worms with thiocarbamate pesticides and dithiocarbamate metabolites. Our data demonstrate that thiocarbamates are potent DAergic toxicants, and support further investigation of this class of pesticides as a risk factor for development of PD. Additionally, we report a role for S-alkyl thiocarbamate metabolites in N,N-diakly dithiocarbamate-induced DAergic neurotoxicity. Further studies elucidating the underlying mechanisms of S-alkyl thiocarbamate DAergic toxicity will facilitate the development of structure activity relationships useful for predicting and prioritizing chemicals for assessing their potential as risk factors in PD.
Acknowledgments
This work was funded by the Center in Molecular Toxicology NIH grant P30 ES00267, R01 ES10563, R01 ES07331, R01 ES0199969, and the training program in Environmental Toxicology grant T32 ES007028. We would like to thank Dr. Daiana Silva Avila for technical assistance, Dr. Venkataraman Amarnath for his expertise in chemical synthesis, Dr. David Miller for the use of the NC1307 strain, and Dr. Keith P. Choe for use of the VP596 strain. Confocal images were obtained through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). Neurochemistry was performed by the Vanderbilt University Center for Molecular Neuroscience’s Neurochemistry Core Laboratory.
Abbreviations
- 6-OHDA
6-hydroxydopamine
- ADE
anterior deirid
- CEP
cephalic deirid
- CNS
central nervous system
- DA
dopamine
- DAergic
dopaminergic
- DAT1
dopamine transporter 1
- DEDC
N,N-diethyldithiocarbamate
- MeDETC
S-methyl-N,N-diethylthiocarbamate
- DMSO
dimethyl sulfoxide
- DOPAC
3,4-dihydroxyphenylacetic acid
- EPTC
S-ethyl N,N-dipropylthiocarbamate
- GABA
γ-aminobutyric acid
- GFP
green fluorescent protein
- GSH
glutathione
- GST-α
glutathione S-transferase-α
- h
hour
- HO-1
heme oxygenase-1
- MPP+
1-methyl-4-phenyl-pyridinium
- NAC
N-acetyl-Lcysteine
- NGM
nematode growth medium
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PD
Parkinson’s disease
- s
seconds
- SNpC
substantia nigr pars compacta
- SOD1
superoxide dismutase 1
- TH
tyrosine hydroxylase
- US EPA
United States Environmental Protection Agency
Footnotes
The authors would like to state that they do not have any conflicts of interest to declare.
References
- Ali SJ, Rajini PS. Elicitation of dopaminergic features of Parkinson’s disease in C. elegans by monocrotophos, an organophosphorous insecticide. CNS Neurol Disord Drug Targets. 2012 doi: 10.2174/1871527311211080008. [DOI] [PubMed] [Google Scholar]
- Anbalagan C, Lafayette I, Antoniou-Kourounioti M, Gutierrez C, Martin JR, Chowdhuri DK, De Pomerai DI. Use of transgenic GFP reporter strains of the nematode Caenorhabditis elegans to investigate the patterns of stress responses induced by pesticides and by organic extracts from agricultural soils. Ecotoxicology. 2013;22:72–85. doi: 10.1007/s10646-012-1004-2. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ. Pesticide exposure and risk for Parkinson’s disease. Ann Neurol. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. Extracellular dopamine potentiates mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergouignan FX, Vital C, Henry P, Eschapasse P. Disulfiram neuropathy. J Neurol. 1988;235:382–383. doi: 10.1007/BF00314241. [DOI] [PubMed] [Google Scholar]
- Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem. 1999;73:1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Braungart E, Gerlach M, Riederer P, Baumeister R, Hoener MC. Caenorhabditis elegans MPP+ model of Parkinson’s disease for high-throughput drug screenings. Neurodegener Dis. 2004;1:175–183. doi: 10.1159/000080983. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M, Silva-Ferreira AC, Manaia CM, Nunes OC. A case study of molinate application in a Portuguese rice field: herbicide dissipation and proposal of a clean-up methodology. Chemosphere. 2005;59:1059–1065. doi: 10.1016/j.chemosphere.2004.11.041. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Zhang J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp Neurol. 2009;220:230–233. doi: 10.1016/j.expneurol.2009.09.027. [DOI] [PubMed] [Google Scholar]
- Chen SH, Liu SH, Liang YC, Lin JK, Lin-Shiau SY. Death signaling pathway induced by pyrrolidine dithiocarbamate-Cu(2+) complex in the cultured rat cortical astrocytes. Glia. 2000;31:249–261. doi: 10.1002/1098-1136(200009)31:3<249::aid-glia60>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Choe KP, Przybysz AJ, Strange K. The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol Cell Biol. 2009;29:2704–2715. doi: 10.1128/MCB.01811-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou AP, Maidment N, Klintenberg R, Casida JE, Li S, Fitzmaurice AG, Fernagut PO, Mortazavi F, Chesselet MF, Bronstein JM. Ziram causes dopaminergic cell damage by inhibiting E1 ligase of the proteasome. J Biol Chem. 2008;283:34696–34703. doi: 10.1074/jbc.M802210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran RC, Formoli TA, Pfeifer KF, Aldous CN. Characterization of risks associated with the use of molinate. Regul Toxicol Pharmacol. 1997;25:146–157. doi: 10.1006/rtph.1997.1082. [DOI] [PubMed] [Google Scholar]
- Cookson MR. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb Perspect Med. 2012;2:a009415. doi: 10.1101/cshperspect.a009415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson D, Kiely T, Grube A U.S.E.P. Agency, editor. Pesticide Industry Sales and Usage 1998 and 1999 Market Estimates. Washington, D.C: 2002. [Google Scholar]
- Ebrahimi-Fakhari D, Wahlster L, McLean PJ. Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol. 2012;124:153–172. doi: 10.1007/s00401-012-1004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. Embo J. 2012;31:3038–3062. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MB. WormBook. 2006. Mechanosensation; pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube A, Donaldson D, Kiely T, Wu L U.S.E.P. Agency, editor. Pesticides Industry Sales and Usage 2006 and 2007 Market Estimates. Washington, D.C: 2011. [Google Scholar]
- Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, Stacy MA, Scott BL, Vance JM, Scott WK. Pesticide exposure and risk of Parkinson’s disease: a family-based case-control study. BMC Neurol. 2008;8:6. doi: 10.1186/1471-2377-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana S, Maiti AK, Bagh MB, Banerjee K, Das A, Roy A, Chakrabarti S. Dopamine but not 3,4-dihydroxy phenylacetic acid (DOPAC) inhibits brain respiratory chain activity by autoxidation and mitochondria catalyzed oxidation to quinone products: implications in Parkinson’s disease. Brain Res. 2007;1139:195–200. doi: 10.1016/j.brainres.2006.09.100. [DOI] [PubMed] [Google Scholar]
- Jewell WT, Hess RA, Miller MG. Testicular toxicity of molinate in the rat: metabolic activation via sulfoxidation. Toxicol Appl Pharmacol. 1998;149:159–166. doi: 10.1006/taap.1998.8380. [DOI] [PubMed] [Google Scholar]
- Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, Shen J, Slack RS, Park DS. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet. 2012;21:4888–4903. doi: 10.1093/hmg/dds325. [DOI] [PubMed] [Google Scholar]
- Kanthasamy AG, Kitazawa M, Kanthasamy A, Anantharam V. Dieldrin-induced neurotoxicity: relevance to Parkinson’s disease pathogenesis. Neurotoxicology. 2005;26:701–719. doi: 10.1016/j.neuro.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Karen DJ, Li W, Harp PR, Gillette JS, Bloomquist JR. Striatal dopaminergic pathways as a target for the insecticides permethrin and chlorpyrifos. Neurotoxicology. 2001;22:811–817. doi: 10.1016/s0161-813x(01)00063-8. [DOI] [PubMed] [Google Scholar]
- Khan MS, Tabrez S, Priyadarshini M, Priyamvada S, Khan MM. Targeting Parkinson’s - tyrosine hydroxylase and oxidative stress as points of interventions. CNS Neurol Disord Drug Targets. 2012;11:369–380. doi: 10.2174/187152712800792848. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rabano A, Kirik D, Cuadrado A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Li AA, Mink PJ, McIntosh LJ, Teta MJ, Finley B. Evaluation of epidemiologic and animal data associating pesticides with Parkinson’s disease. J Occup Environ Med. 2005;47:1059–1087. doi: 10.1097/01.jom.0000174294.58575.3e. [DOI] [PubMed] [Google Scholar]
- Liang C, Xue Z, Cang J, Wang H, Li P. Dimethyl sulfoxide induces heme oxygenase-1 expression via JNKs and Nrf2 pathways in human umbilical vein endothelial cells. Mol Cell Biochem. 2011;355:109–115. doi: 10.1007/s11010-011-0844-z. [DOI] [PubMed] [Google Scholar]
- Link CD, Johnson CJ. Reporter transgenes for study of oxidant stress in Caenorhabditis elegans. Methods Enzymol. 2002;353:497–505. doi: 10.1016/s0076-6879(02)53072-x. [DOI] [PubMed] [Google Scholar]
- Madan A, Faiman MD. Characterization of diethyldithiocarbamate methyl ester sulfine as an intermediate in the bioactivation of disulfiram. J Pharmacol Exp Ther. 1995;272:775–780. [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J Neurochem. 2005;93:1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- Mocko JB, Kern A, Moosmann B, Behl C, Hajieva P. Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson’s disease. Neurobiol Dis. 2010;40:120–129. doi: 10.1016/j.nbd.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Mulkey ME U.S.E.P. Agency, editor. The Grouping of a Series of Dithiocarbamate Pesticides Based on a Common Mechanism of Toxicity. Washington, DC: 2001. [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negga R, Rudd DA, Davis NS, Justice AN, Hatfield HE, Valente AL, Fields AS, Fitsanakis VA. Exposure to Mn/Zn ethylene-bis-dithiocarbamate and glyphosate pesticides leads to neurodegeneration in Caenorhabditis elegans. Neurotoxicology. 2011;32:331–341. doi: 10.1016/j.neuro.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negga R, Stuart JA, Machen ML, Salva J, Lizek AJ, Richardson SJ, Osborne AS, Mirallas O, McVey KA, Fitsanakis VA. Exposure to glyphosate- and/or Mn/Zn-ethylene-bis-dithiocarbamate-containing pesticides leads to degeneration of gamma-aminobutyric acid and dopamine neurons in Caenorhabditis elegans. Neurotox Res. 2012;21:281–290. doi: 10.1007/s12640-011-9274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentyala SN, Chetty CS. Comparative study on the changes in AChE and ATPase activities in neonate and adult rat brains under thiobencarb stress. J Appl Toxicol. 1993;13:39–42. doi: 10.1002/jat.2550130109. [DOI] [PubMed] [Google Scholar]
- Pike MG, Mays DC, Macomber DW, Lipsky JJ. Metabolism of a disulfiram metabolite, S-methyl N,N-diethyldithiocarbamate, by flavin monooxygenase in human renal microsomes. Drug Metab Dispos. 2001;29:127–132. [PubMed] [Google Scholar]
- Politis M, Loane C. Serotonergic dysfunction in Parkinson’s disease and its relevance to disability. Scientific World Journal. 2011;11:1726–1734. doi: 10.1100/2011/172893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K, Winnik B, Thiruchelvam MJ, Buckley B, Mirochnitchenko O, Richfield EK. Prolonged toxicokinetics and toxicodynamics of paraquat in mouse brain. Environ Health Perspect. 2007;115:1448–1453. doi: 10.1289/ehp.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruett SB, Cheng B, Fan R, Tan W, Sebastian T. Oxidative stress and sodium methyldithiocarbamate-induced modulation of the macrophage response to lipopolysaccharide in vivo. Toxicol Sci. 2009;109:237–246. doi: 10.1093/toxsci/kfp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle WC, Oliver DP, Zrna S. Field dissipation and environmental hazard assessment of clomazone, molinate, and thiobencarb in Australian rice culture. J Agric Food Chem. 2006;54:7213–7220. doi: 10.1021/jf061107u. [DOI] [PubMed] [Google Scholar]
- Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- Richardson JR, Caudle WM, Wang M, Dean ED, Pennell KD, Miller GW. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson’s disease. Faseb J. 2006;20:1695–1697. doi: 10.1096/fj.06-5864fje. [DOI] [PubMed] [Google Scholar]
- Savolainen K, Hervonen H. Dithiocarbamate fungicides decrease histochemical reactivity of cholinesterases in the gut wall of the rat. Arch Toxicol Suppl. 1985;8:272–276. doi: 10.1007/978-3-642-69928-3_41. [DOI] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Seeman P, Niznik HB. Dopamine receptors and transporters in Parkinson’s disease and schizophrenia. Faseb J. 1990;4:2737–2744. doi: 10.1096/fasebj.4.10.2197154. [DOI] [PubMed] [Google Scholar]
- Staub RE, Sparks SE, Quistad GB, Casida JE. S-methylation as a bioactivation mechanism for mono- and dithiocarbamate pesticides as aldehyde dehydrogenase inhibitors. Chem Res Toxicol. 1995;8:1063–1069. doi: 10.1021/tx00050a010. [DOI] [PubMed] [Google Scholar]
- Stiernagle T. Maintenance of C. elegans. In: Hope IA, editor. C elegans: A Practical Approach. New York, NY: Oxford University Press; 1999. [Google Scholar]
- Sutachan JJ, Casas Z, Albarracin SL, Stab BR, 2nd, Samudio I, Gonzalez J, Morales L, Barreto GE. Cellular and molecular mechanisms of antioxidants in Parkinson’s disease. Nutr Neurosci. 2012;15:120–126. doi: 10.1179/1476830511Y.0000000033. [DOI] [PubMed] [Google Scholar]
- Szolar OH. Environmental and pharmaceutical analysis of dithiocarbamates. Anal Chim Acta. 2007;582:191–200. doi: 10.1016/j.aca.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, Langston JW. Parkinson disease in twins: an etiologic study. Jama. 1999;281:341–346. doi: 10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- Tonkin EG, Valentine HL, Milatovic DM, Valentine WM. N,N-diethyldithiocarbamate produces copper accumulation, lipid peroxidation, and myelin injury in rat peripheral nerve. Toxicol Sci. 2004;81:160–171. doi: 10.1093/toxsci/kfh190. [DOI] [PubMed] [Google Scholar]
- Valentine HL, Viquez OM, Amarnath K, Amarnath V, Zyskowski J, Kassa EN, Valentine WM. Nitrogen substituent polarity influences dithiocarbamate-mediated lipid oxidation, nerve copper accumulation, and myelin injury. Chem Res Toxicol. 2009;22:218–226. doi: 10.1021/tx8003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine HL, Viquez OM, Valentine WM. Peripheral nerve and brain differ in their capacity to resolve N,N-diethyldithiocarbamate-mediated elevations in copper and oxidative injury. Toxicology. 2010;274:10–17. doi: 10.1016/j.tox.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bemmel DM, Visvanathan K, Beane Freeman LE, Coble J, Hoppin JA, Alavanja MC. S-ethyl-N,N-dipropylthiocarbamate exposure and cancer incidence among male pesticide applicators in the agricultural health study: a prospective cohort. Environ Health Perspect. 2008;116:1541–1546. doi: 10.1289/ehp.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Mark M, Brouwer M, Kromhout H, Nijssen P, Huss A, Vermeulen R. Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results. Environ Health Perspect. 2012;120:340–347. doi: 10.1289/ehp.1103881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez OM, Caito SW, McDonald WH, Friedman DB, Valentine WM. Electrophilic adduction of ubiquitin activating enzyme E1 by N,N-diethyldithiocarbamate inhibits ubiquitin activation and is accompanied by striatal injury in the rat. Chem Res Toxicol. 2012;25:2310–2321. doi: 10.1021/tx300198h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez OM, Lai B, Ahn JH, Does MD, Valentine HL, Valentine WM. N,N-diethyldithiocarbamate promotes oxidative stress prior to myelin structural changes and increases myelin copper content. Toxicol Appl Pharmacol. 2009;239:71–79. doi: 10.1016/j.taap.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez OM, Valentine HL, Amarnath K, Milatovic D, Valentine WM. Copper accumulation and lipid oxidation precede inflammation and myelin lesions in N,N-diethyldithiocarbamate peripheral myelinopathy. Toxicol Appl Pharmacol. 2008;229:77–85. doi: 10.1016/j.taap.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Costello S, Cockburn M, Zhang X, Bronstein J, Ritz B. Parkinson’s disease risk from ambient exposure to pesticides. Eur J Epidemiol. 2011a;26:547–555. doi: 10.1007/s10654-011-9574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu L, Zhang L, Peng Y, Zhou F. Redox reactions of the alpha-synuclein-Cu(2+) complex and their effects on neuronal cell viability. Biochemistry. 2010;49:8134–8142. doi: 10.1021/bi1010909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Chou AH, Wu AS, Chen SY, Weng YH, Kao YC, Yeh TH, Chu PJ, Lu CS. PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim Biophys Acta. 2011b;1812:674–684. doi: 10.1016/j.bbadis.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Wickramaratne GA, Foster JR, Ellis MK, Tomenson JA. Molinate: rodent reproductive toxicity and its relevance to humans--a review. Regul Toxicol Pharmacol. 1998;27:112–118. doi: 10.1006/rtph.1998.1200. [DOI] [PubMed] [Google Scholar]
- Wu YN, Johnson SW. Rotenone reduces Mg2+-dependent block of NMDA currents in substantia nigra dopamine neurons. Neurotoxicology. 2009;30:320–325. doi: 10.1016/j.neuro.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Yarnall A, Rochester L, Burn DJ. The interplay of cholinergic function, attention, and falls in Parkinson’s disease. Mov Disord. 2011;26:2496–2503. doi: 10.1002/mds.23932. [DOI] [PubMed] [Google Scholar]
- Zheng LF, Wang ZY, Li XF, Song J, Hong F, Lian H, Wang Q, Feng XY, Tang YY, Zhang Y, et al. Reduced expression of choline acetyltransferase in vagal motoneurons and gastric motor dysfunction in a 6-OHDA rat model of Parkinson’s disease. Brain Res. 2011;1420:59–67. doi: 10.1016/j.brainres.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Ziegler DA, Wonderlick JS, Ashourian P, Hansen LA, Young JC, Murphy AJ, Koppuzha CK, Growdon JH, Corkin S. Substantia Nigra Volume Loss Before Basal Forebrain Degeneration in Early Parkinson Disease. Arch Neurol. 2012:1–7. doi: 10.1001/jamaneurol.2013.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman LJ, Valentine HL, Valentine WM. Characterization of S-(N,N-Dialkylaminocarbonyl)cysteine Adducts and Enzyme Inhibition Produced by Thiocarbamate Herbicides in the Rat. Chem Res Toxicol. 2004;17:258–267. doi: 10.1021/tx034209c. [DOI] [PubMed] [Google Scholar]