

Abstract
Endemic Burkitt lymphoma (eBL) is associated with Epstein–Barr virus (EBV) and Plasmodium falciparum coinfections. Malaria appears to dysregulate immunity that would otherwise control EBV, thereby contributing to eBL etiology. Juxtaposed to human genetic variants associated with protection from malaria, it has been hypothesized that such variants could decrease eBL susceptibility, historically referred to as “the protective hypothesis.” Past studies attempting to link sickle cell trait (HbAS), which is known to be protective against malaria, with protection from eBL were contradictory and underpowered. Therefore, using a case–control study design, we examined HbAS frequency in 306 Kenyan children diagnosed with eBL compared to 537 geographically defined and ethnically matched controls. We found 23.8% HbAS for eBL patients, which was not significantly different compared to 27.0% HbAS for controls [odds ratio (OR) = 0.85; 95% confidence interval (CI) 0.61–1.17; p-value = 0.33]. Even though cellular EBV titers, indicative of the number of latently infected B cells, were significantly higher (p-value < 0.0003) in children residing in malaria holoendemic compared to hypoendemic areas, levels were not associated with HbAS genotype. Combined, this suggests that although HbAS protects against severe malaria and hyperparasitemia, it is not associated with viral control or eBL protection. However, based on receiver operating characteristic curves factors that enable the establishment of EBV persistence, in contrast to those involved in EBV lytic reactivation, may have utility as an eBL precursor biomarker. This has implications for future human genetic association studies to consider variants influencing control over EBV in addition to malaria as risk factors for eBL.
What's new?
Although the hypothesis dates back to 1970, studies of the “protective effect” of sickle cell trait (HbAS) on the development of endemic Burkitt lymphoma (eBL) have led to contradictory conclusions. This new study analyzed an unprecedented number of eBL cases and highlighted the importance of matching controls on ethnicity as well as malaria endemicity. The findings contribute to resolving the controversy by showing that HbAS frequency does not differ between children diagnosed with eBL and healthy children. The study also examined cellular Epstein-Barr Virus (EBV) titers, which in contrast to plasma EBV levels may serve as an informative eBL biomarker.
Keywords: endemic Burkitt lymphoma, pediatric cancer, EBV, malaria, sickle cell trait, viral loads, Plasmodium falciparum, Kenya, Luo, Luhya, biomarker
Endemic Burkitt lymphoma (eBL) is the most common pediatric cancer in Equatorial Africa with a polymicrobial etiology involving two ubiquitous pathogens, Epstein–Barr virus (EBV) and Plasmodium falciparum.1,2 The syndemic relationship between malaria and EBV coinfections has been associated with immune deficiencies in EBV-specific interferon-gamma (IFN-γ) memory recall responses, significantly higher peripheral blood EBV loads and perturbations in the B-cell compartment.3–6 Malarial parasites through cysteine-rich interdomain region 1α (CIDR1-α) have been shown to induce indiscriminant polyclonal B-cell proliferation accompanied by EBV lytic reactivation and an increase in the number of latently infected B cells.7 However, eBL is a rare event (annual incidence 2 in 100,000 children1) relative to the prevalence of malaria and EBV infections within these pediatric populations, suggesting that additional factors are involved in its etiology.
Epidemiologic studies of eBL have not revealed significant family clusters, suggesting that risk does not appear to be driven by highly penetrant mutations.8 This likely implies that the main step in pathogenesis (e.g., 8; 14 translocation) is a somatic event rather than a heritable trait. However, this does not exclude the possibility that less penetrant common variants are modifiers of the overall risk of eBL providing a more permissive or susceptible background. For instance, genome-wide studies of childhood acute lymphoblastic leukemia have demonstrated a number of common genetic variants that appear to alter risk.9 An obvious choice of common variants that might impact eBL susceptibility is the numerous mutations providing resistance to malaria. This was first suggested in the 1960s as “the protective hypothesis” soon after the genetic polymorphism responsible for sickle cell trait was discovered.10
Malaria appears to have been one of the strongest selective forces to recently impact the human genome witnessed by the numerous human gene mutations that confer a protective advantage against falciparum malaria.11 One of the most notable, the first disease to have a proven molecularly defined mutation, is the point mutation in the beta-hemoglobin gene creating sickle hemoglobin that polymerizes in deoxygenated states.12 Homozygous individuals (HbSS) suffer from sickle cell disease, whereas heterozygosity (HbAS, sickle cell trait) confers protection against clinical malaria with ∼60% protection against high-density parasitemia13,14 and 70–90% protection against severe disease.15,16 If malaria disease severity and level of parasitemia play a causal role in eBL etiology, it has been postulated that the incidence of eBL would be lower in those with sickle cell trait.10,17,18 Yet, the historical studies examining the relative frequency of HbAS in eBL patients compared to healthy children have yielded inconsistent findings. The first published study from Nigeria in 1966 reported that 95 eBL children had a significantly lower frequency of HbAS (17.9%) compared to 320 hospital-based controls (29.7%, p-value = 0.03).17 However, HbAS was lower (25.6%) in village-matched Yoruba controls rendering the difference with eBL patients insignificant, p-value = 0.18.17 A second smaller study from Uganda in 1970 supported the “protective HbAS genotype hypothesis” but found that the HbAS frequency between 36 eBL patients and 50 “neighbor controls” was comparable (16.7 and 28.0%, respectively).10 The choice of controls in the Nigerian study was questioned as biased given the older age of the controls compared to the eBL patients (HbAS frequency in the population may increase as susceptible HbAA children succumb to malaria), and the assumption that HbAS prevalence lacked substructure within the Yoruban population despite varying levels of malaria endemicity. A third study from Ghana in 1976 selected more definitive control groups and compared HbAS frequency in 112 eBL patients to 78 age-matched neighbors (12.8 and 10.3%, respectively) and to 84 siblings (14.3 and 16.7%, respectively). They concluded that HbAS frequency in eBL patients did not differ from healthy controls.18
It has been 45 years since the “protective hypothesis” was proposed suggesting that human genetic mutations that confer protection against severe malaria would also be associated with decreased eBL risk; yet this question has remained unresolved within the scientific community. With the advent of advanced molecular techniques for quantifying and standardizing EBV load19,20 and renewed interest in directly studying eBL patients in Africa,2 our study set forth to reinitiate investigations into the association between human genetic variations, EBV control and eBL risk starting with the most likely candidate, sickle cell trait. We leveraged three groups of Kenyan children: those diagnosed with eBL, healthy children residing in the same malaria holoendemic area where the incidence of eBL is high and children from a hypoendemic region where eBL incidence is low.21 Our first objective was to examine the HbAS frequency in eBL patients compared to geographically matched holoendemic malaria-exposed controls. Because of the ethnic heterogeneity within the eBL patients, which has been previously described in Kenya22 but was not described in previous studies of eBL, we also matched our cases and controls on ethnicity. Furthermore, we investigated the relationship between HbAS/AA genotype and EBV loads, as a functional measure of viral control and as a potential precursor biomarker for eBL risk.
Material and Methods
Study populations
Between 2003 and 2012, children (aged 2–14 years, median age 7 years, with a ratio of 1.5 males to females as previously reported22) with a confirmed diagnosis of eBL (n = 636) were prospectively enrolled after informed consent from Nyanza General Provincial Hospital (renamed the Jaramogi Oginga Odinga Teaching and Referral Hospital in 2012), which is the regional referral hospital for cancer treatment in western Kenya. The catchment area for this hospital includes Provinces formerly known as Nyanza, Rift Valley and Western Provinces. Kenya was subdivided into counties in 2009 but we will retain the older names that encompass broader geographic areas. Histopathologic diagnosis was confirmed by two independent pathologists from fine needle aspirations stained with May-Grunewald Giemsa. An admission blood sample was drawn before chemotherapy induction. The sample was used for hemoglobin levels measured on-site by portable β-hemoglobin photometer (Hemocue AB Angelholm, Sweden) and the remainder was stored at −80°C for DNA extraction. HIV testing for all pediatric cancer patients is standard of care at hospitals in Kenya. Patients enrolled in this study with a diagnosis of eBL had a 1.3% HIV prevalence (unpublished finding). These eight children were excluded from the analysis. On the basis of prior studies that indicate the probability of sickle cell trait among controls is 0.2623 and the assumption that sickle cell trait protects cases from cancer by a factor of 50% [odds ratio (OR) for disease in HbAS relative to HbAA individuals is 0.5], we would be able to reject the null hypothesis that the OR equals 1 with a power of 0.976 for Fisher's exact test or corrected chi-square test using a sample size of 337 cases and 537 controls.
Between 2005 and 2009, healthy controls (aged 2–14 years, median 6.7 years with a roughly equal number of males as females) were enrolled from two rural areas of western Kenya, Nyanza (n = 547) and Rift Valley (n = 486) Provinces, which have divergent malaria transmission patterns and accordingly different incidences of eBL.21 Study sites have been described in more detail elsewhere.3,21,24 In brief, Nyanza residents experience stable and perennial P. falciparum transmission patterns (holoendemic malaria) and higher eBL incidence, whereas residents from Rift Valley located at high altitudes exceeding 3,000 m above sea level experience low and sporadic P. falciparum transmission (hypoendemic malaria) and lower eBL incidence.21 Healthy controls were frequency matched on age range and malaria exposure and enrolled by voluntary convenience sampling within each geographically defined study site. Inclusion criteria consisted of children having a normal full blood count and being EBV seropositive. Hematological indices were compared across study groups and values were within normal range for children residing in Kenya.25 All children were EBV seropositive, consistent with previous studies showing that primary infection occurs before the age of 3 years.3,24,26 Ethnicity of the child was reported by the parent as part of the demographic data collection survey.
Ethical approval
Ethical review and approval for this study was obtained from the Institutional Review Board at The University of Massachusetts Medical School and the Ethical Review Committee at the Kenya Medical Research Institute, Kenya. Parents of minor study participants provided individual, written informed consent.
Microscopic examination of P. falciparum
Whole blood was used for malaria parasite density determination. Giemsa-stained thick and thin blood smears were examined at 1,000× under oil immersion. Malaria parasitemia was quantified by counting the number of parasites per 200 leukocytes and multiplying by 40, assuming an average white blood cell count of 8,000 cells per microliter of blood, as per the World Health Organization standard criteria.27 Any child testing positive for blood-stage malaria parasites was treated free of charge following the Kenya National Ministry of Health Guidelines. All healthy control children were asymptomatic at time of enrollment; therefore, any malaria infections were classified as uncomplicated and treated accordingly.
PCR for sickle cell trait (HbAS)
HbAS trait was characterized as previously published.28 Briefly, a 772-bp PCR product within the human beta-globin gene was amplified from DNA extracted from whole blood using the following primers: HbB1 (5′-TCCTAAGCCAGTGCCAGAAG-3′) and HbB2 (5′-GAATTCGTCTGTTTCCCATTCTAAAC-3′). The PCR amplicon was subsequently subjected to restriction enzyme digestion using Bsu361 with resulting fragment sizes visualized by horizontal gel electrophoresis. A 430-bp DNA fragment was found to be associated with the mutant locus, whereas 228- and 202-bp DNA fragments were generated from the normal locus.
Quantitative PCR for cellular and plasma EBV
Peripheral whole blood was collected by venipuncture under sterile conditions. The plasma fraction was separated from peripheral blood cells by centrifugation before freezing. The cell pellet was then washed twice in sterile isotonic phosphate-buffered saline and resuspended volume/volume then frozen until batch DNA extraction was performed. DNA for cellular EBV load was extracted from ethylenediaminetetraacetic acid anticoagulated whole blood cell pellets, whereas DNA for EBV viremia was measured from the plasma using QIAamp DNA Mini Kit (Qiagen, Valencia, CA). Because of laboratory constraints DNA extraction could not be performed on plasma samples before freezing. However, in an attempt to distinguish free EBV DNA from lytic virions, plasma samples were analyzed before and after DNase I treatment.29 An aliquot of plasma was digested with 5 U DNase I (Promega, Madison, WI) at 37°C for 1 hr; then stop solution was added and incubated at 56°C bath for 10 min. Viral quantification was then performed on DNA extracted from the peripheral blood cell pellet and plasma before and after DNase I digestion as previously described for EBV BALF51.19,24 The quantitative (q)-PCR cycle was 3 min at 95°C, 10 sec at 95°C and 30 sec at 62.5°C repeated for 40 cycles and read by a Bio-Rad CFX96™ Real-Time System C1000™ Thermal Cycler base (BioRad Laboratories, Hercules, CA). IQ Supermix (BioRad Laboratories) was used for all reactions. Standard curves were generated using EBV-positive Namalwa (American Type Cell Culture Collection, USA, CRL-1432) and EBV-negative BL41 cell lines (gift from Dr. Rosemary Rochford). Cellular viral loads were normalized based on EBV genome copies per microgram of human beta-actin DNA multiplexed23 in the qPCR assay, whereas plasma viremia was normalized to EBV copies per microliter of plasma. The lowest level of detection was calculated to be two EBV copies per microliter whole blood.
Statistical analysis
Chi-square tests and ORs with 95% confidence intervals (CIs) were used to test for differences in HbAS genotype frequencies between cases and controls. Comparison of log-transformed EBV load used a Kruskal–Wallis test, whereas post hoc Dunn's test was used to evaluate differences between all pairs of viral loads between study populations. All tests were two-tailed with p-values less than 0.05 considered statistically significant. Receiver operating characteristic (ROC) curves and putative thresholds for EBV loads and other statistical analyses were performed using Graphpad Prism version 5 (Graphpad Software, La Jolla, CA).
Results
Study population comparisons
As expected (Table1), children from malaria holoendemic areas were more likely to have a positive P. falciparum blood smear with higher density infections compared to children from a hypoendemic area (42 and 1%, respectively, p-value <0.001). Only 19% of the eBL children were parasitemic by blood smear on admission even though the vast majority resided in malaria holoendemic areas.21 However, 28% of parents reported their child having received antimalarial treatment within the 2 weeks before referral to the cancer ward. Of those with blood-stage parasitemia, geometric mean parasite densities were significantly lower for eBL patients compared to asymptomatic malaria-infected children from holoendemic areas (Table1: 764 versus 1,098 parasites per microliter of blood, p-value = 0.02, respectively).
Table 1.
Study population comparisons for malaria endemicity metrics, ethnicity and HbAS frequency
| Study group (defined by malaria endemicity) | Malaria exposure | Sample size (n) | P. falciparum parasitemic (%) | Geometric mean parasite density (per microliter blood) | HbAS frequency (%) |
|---|---|---|---|---|---|
| All eBL patients | Holoendemic | 413 | 19 | 7641 | 21.3 |
| Luo eBL patients | 306 | 23.8 | |||
| Luhya eBL patients | 81 | 15.9 | |||
| Nyanza Province controls | Holoendemic | 547 | 42 | 1,0981 | 26.5 |
| Luo controls | 537 | 27.0 | |||
| Rift Valley Province controls | Hypoendemic | 486 | 1 | 80 | 3.9 |
Difference in P. falciparum malaria parasite density between eBL patients and controls from holoendemic area, p-value = 0.02.
Sickle cell trait does not protect against eBL for children residing in malaria holoendemic areas after controlling for ethnicity
Of the children with confirmed eBL enrolled over a 10-year period, 413 DNA samples collected during the same time period as controls were screened for this study. Of those eBL patients, 21.3% carried the HbAS genotype compared to 26.5% of the Nyanza malaria holoendemic controls (Table1). The odds of developing eBL hinted at being protective for HbAS compared to HbAA when matched with geographically defined holoendemic malaria exposed children (OR = 0.75; 95% CI 0.55–1.02; p = 0.07) (Table2). However, a more in-depth investigation into the ethnicity of eBL patients revealed a predominance of Luo (74.1%) yet with a significant number of Luhya (19.6%) and 6.3% from other self-reported ethnicities relative to our controls who were predominantly Luo. Therefore, Luo eBL patients (n = 306) were compared to Luo controls (n = 537) rendering the OR for HbAS versus HbAA not significant [23.8 and 27.0% HbAS for Luo eBL patients compared to Luo controls, respectively (Table1); OR = 0.85; 95% CI 0.61–1.17; p-value = 0.33 (Table2)]. We further restricted our analysis to a smaller geographic area (nearest neighbors). Matching both by district and ethnicity completely abrogated any apparent association between HbAS frequency and eBL (Table2). Sufficient controls identified as Luhya were not available within our control group. However, using historical data, HbAS frequency among Luhya has been reported at 12.5%,30 similar to 15.9% HbAS frequency we observed for Luhya eBL patients (Table1).
Table 2.
Odds ratios for developing eBL (HbAS against HbAA)
| Group comparisons | Odds ratio | 95% CI | p-Value | |
|---|---|---|---|---|
| a) | Holoendemic malaria controls and all eBL patients | 0.75 | 0.55–1.02 | 0.07 |
| b) | Luo-matched controls and only Luo eBL patients | 0.85 | 0.61–1.17 | 0.33 |
| c) | Luo- and district-matched controls and eBL patients | 1.00 | 0.49–2.24 | 1.00 |
For the children from Nyanza Province, HbAS genotype was associated with lower point-prevalence malaria parasite density infections (geometric mean parasite density for HbAS versus HbAA, 1,788 vs. 914 parasites per microliter of blood, respectively) and was similar to other studies.31 This was not assessed for the Rift Valley Province children owing to low prevalence of infection and HbAS genotype or for eBL patients who may have received antimalarial treatment before enrollment. HbAS was infrequent in Rift Valley controls at 3.9% where malaria transmission is historically sporadic and less intense.
EBV loads vary across study populations but are not associated with HbAA/AS genotype
There is no empirical evidence indicating if measurements of EBV infection dynamics are informative within the context of eBL risk, although it has been shown that the age of primary EBV infection occurs significantly earlier in children residing in malaria holoendemic areas compared to areas with lower malaria incidence3,24,26 and circulating plasma EBV has been associated with acute malaria infections.32 Additionally, holoendemic malaria exposure has been associated with lack of viral control.4,33–38 Therefore, we measured EBV from fractionated peripheral blood samples and compared virus isolated from the plasma (with and without DNase I digestion) and cellular virus as an indicator of the number of latently infected B cells.39 Within our previous studies of children with eBL and those experiencing their primary EBV infection within the first few years of life,3,24 we measured EBV in DNA isolated from whole bloods. Consistent with our previous studies, children with eBL had significantly higher median cellular viral loads compared to healthy control children from Nyanza (14,810 versus 35.7 EBV copies per microgram of human DNA, respectively, p-value < 0.0001) and significant differences existed between children from Nyanza and Rift Valley (35.7 and 0.0 EBV copies per microgram of human DNA, respectively, p-value = 0.0003).3 When we compared EBV levels in the plasma, we found significantly higher viremia in eBL children (median 11,117 EBV copies per microliter of plasma before DNase I digestion) compared to healthy malaria holoendemic children (p-value < 0.0001). However, median EBV plasma levels did not significantly differ between children from Nyanza and Rift Valley (5.4 and 4.1 EBV copies per microliter of plasma, respectively). When we compared the DNase I-treated plasma, viremia across study groups demonstrated the same trend but was fivefold lower in concentration, suggesting a mixture of free viral DNA and lytic virions as previously found in other EBV-associated conditions.29 Plasma EBV levels before and after DNAse I treatment in the healthy children were highly correlated (r = 0.59, p-value < 0.001) and were not associated with point-prevalence malaria parasitemia.
Next, we sought to determine whether EBV loads differed by beta-hemoglobin genotype (HbAA/AS). As EBV levels were significantly higher in eBL patients, we examined groups independently and then stratified by HbAA/AS genotypes. We found no significant differences in cellular EBV levels based on beta-hemoglobin genotype for eBL patients (Fig. 1a) or healthy children from Nyanza (Fig. 1b) or Rift Valley (Fig. 1c). HbAS was also not associated with differences in plasma levels of EBV within any group (data not shown).
Figure 1.
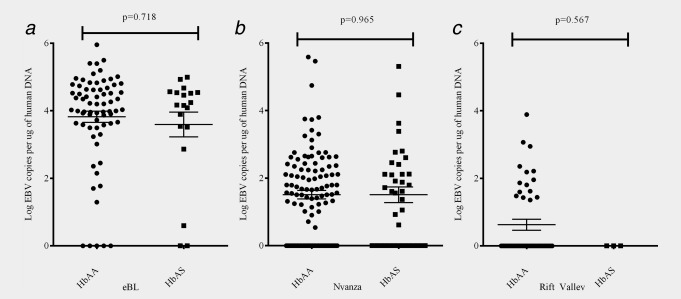
Cellular EBV load stratified by HbAA/AS genotype. Cellular EBV levels were compared for (a) children diagnosed with eBL (n = 89), (b) Nyanza (n = 149) and (c) Rift Valley (n = 64) children after stratification by HbAA/AS genotype. There were no significant differences in cellular viral load associated with genotype.
Correlation between cellular and plasma EBV levels
Past studies measuring peripheral EBV levels have quantified virus from whole blood or plasma levels, yet few have ascertained if viral compartment is informative for eBL risk.39 Therefore, we sought to assess the correlation between cellular and plasma EBV levels to determine if measuring virus from one compartment was equally informative as the other (Fig. 2). We found minimal correlation between viral levels measured from cellular compared to plasma sources for children who had been diagnosed with eBL (r = 0.243, p-value = 0.05), similar to other published studies.39 For healthy children from malaria holoendemic areas, there was a significant albeit minimal correlation between cellular and plasma virus (r = 0.177, p-value = 0.01), suggesting that although latently infected B cells and lytic reactivation may be elevated within the same children, their relative magnitude is not commensurate. The lack of correlation for the children drawn from Rift Valley was due to the significantly lower levels of cellular virus detected. Our observations are consistent with a cross-sectional study measuring plasma and whole-blood viral loads for 12 eBL patients from Malawi.39 We next assessed ROC curves comparing eBL cases to malaria holoendemic controls to determine if EBV load could be used as a predictive biomarker. We found the area under the ROC to be 0.812 for cellular and 0.962 for plasma levels (before DNase I treatment) both with a p-value < 0.0001. A cellular level of 1,000 EBV copies per microgram of human DNA (3 log-fold) would provide 90% specificity and 83% sensitivity, whereas plasma levels greater than 120 EBV copies per microliter of plasma (2 log-fold) would provide 90% specificity and 90% sensitivity. Comparing children from Nyanza and Rift Valley, the area under the ROC curve was 0.713 for cellular EBV (p-value < 0.0001) but for EBV measured in the plasma the area under the ROC curve was 0.553, p-value = 0.086, suggesting that cellular viral loads might be more informative for assessing eBL risk than free viral DNA or lytic virions in the plasma. However, a level of 1,000 EBV copiers per microgram of human DNA would provide 95% specificity with only 10% sensitivity. Figure 2 presents data on viral loads for both healthy Nyanza children and eBL patients to visualize relative differences in cellular and plasma EBV levels (before DNase I treatment) and suggests potential thresholds based on the ROC analysis. Cellular EBV loads might serve as a more sensitive biomarker of eBL risk than virus in the plasma, but other factors would have to be included in the development of a “BL risk score” to provide a reasonable false-positive rate given the rarity of eBL.
Figure 2.
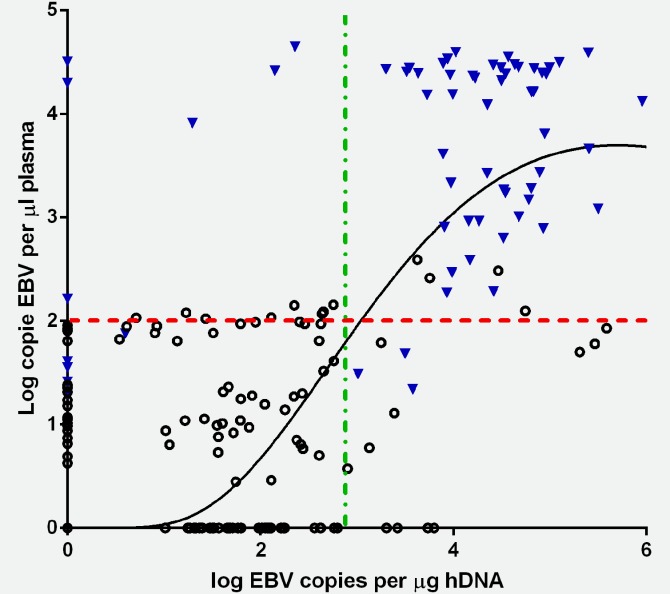
Correlation between cellular and plasma EBV levels for healthy and eBL children. There was a significant correlation between cellular and plasma EBV levels in healthy Kenyan children from Nyanza (triangle) r = 0.177, p-value = 0.010. Children diagnosed with eBL had a nonsignificant correlation coefficient of r = 0.243, p-value = 0.05 (open circles). Based on the area under the ROC curve to assess sensitivity and specificity, dashed lines at 2-log EBV copies per microliter of plasma and 3-log EBV copies per microgram of human DNA were selected to visualize viral loads in healthy Kenyan children (triangle) and eBL patients (open circles) as a proposed threshold if either were used as a biomarker. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
The primary objective of our study was to apply a sufficiently powered sample set to examine the long-standing question of whether sickle cell trait, which is associated with protection from severe malaria and hyperparasitemia, decreases eBL risk—referred to as the “protective hypothesis.”10,17 Genotyping an unprecedented number of children with confirmed diagnosis of eBL (n = 413), we found a frequency of 21.3% HbAS. However, when compared to healthy children who were matched for malaria exposure and ethnicity, the odds of developing eBL were not significantly associated with HbAA/AS genotype (OR = 0.85; 95% CI 0.61–1.17, p-value = 0.33) and this slight effect all but disappeared when eBL cases were matched by district (neighborhood controls). This suggests that eBL risk is not significantly predicated on HbAA/AS genotype to the same degree as the protective effect it engenders for malaria in terms of severe disease and hyperparasitemia.40 Although it is certainly possible that HbAS exerts a small effect given the range of our CIs, in light of its lack of association with EBV load it is more likely that eBL pathogenesis is due to malaria-induced immunological dysregulation, which is not impacted by this structural mutation in hemoglobin beta chain.
Sickle cell trait significantly protects children from the clinically severe manifestations of malaria that can result in death before the age of 5 years41,42 but does not protect a child from all malaria infections. Children with sickle cell trait are able to harbor asymptomatic parasitemias, which fits with our current model of eBL pathogenesis. Furthermore, as the incidence of eBL in Kenya peaks between 5 and 9 years of age, we propose that acute uncomplicated and/or chronic asymptomatic malaria infections have a more profound impact on EBV persistence and subsequent risk of eBL than severe malaria (reviewed in Ref.43). A syndemic model for EBV and malaria related to the progression of eBL pathogenesis over time is corroborated by our clinical data collected prospectively on more than 600 children diagnosed with eBL in western Kenya, whose parents reported their child's first severe illness was presenting to hospital with cancer—not severe malaria (Moormann, unpublished data).
Our secondary aim was to measure peripheral EBV load as an indicator of viral control and associate levels with HbAA/AS genotype. Within this study, we confirmed that significantly higher median cellular EBV levels existed in malaria-exposed compared to nonexposed children3 but these levels were not associated with HbAS genotype. Differences in peripheral EBV level were apparent for cellular virus but not plasma viremia (i.e., free viral DNA or lytic virions), suggesting that measuring the frequency of latently infected B cells could be exploited as a minimally invasive biomarker for eBL risk.
The annual incidence of eBL in western Kenya is 2–5 per 100,000 children22 making it orders of magnitude less frequent than the ubiquitous coinfections implicated in its etiology. Therefore, an easily measured biomarker would be of use to identify children at risk for eBL. EBV loads have the potential to be used as predictive and prognostic biomarkers once a child has been diagnosed with eBL. Studies of EBV-associated malignancies such as non-Hodgkin's lymphoma, post-transplant lymphoproliferative disorders and nasopharyngeal carcinoma have measured EBV levels,44–48 providing a clinically useful metric for monitoring disease regression or as a harbinger of relapse. To date, no such studies had been carried out to evaluate EBV levels as prognostic indicators for eBL.
Our lack of association with HbAS does not preclude other malaria-associated host genetic variants having protective effects for eBL or viral control. Rather, we would suggest that traits associated with eBL pathogenesis, such as immune modulation of malaria that allows this infection to remain asymptomatic (and therefore chronic), will be more likely to influence risk than protective variants due to structural alterations of the red cell that inhibit parasite growth. Additionally, other immune-modulating human genetic variants unrelated to malaria may be important for viral control and eBL risk. In addition, eBL risk may be associated with genomic instability given the requisite c-myc-Ig chromosomal translocation. This is supported by examples of common genetic variants being associated with acute lymphoblastic leukemia that has no known infectious risk factor.9 Therefore, a combinatorial model for eBL etiology emerges that incorporates human genetic variation as well as factors that influence immunity to EBV during malaria coinfections.
This study although having a large enough sample size had two limitations. The case–control study design was unable to capture the longitudinal changes in EBV infection dynamics as a consequence of repeated malaria infections. However, this limitation may be addressed by the fact that the study populations resided in areas of western Kenya with well-defined malaria endemicity, thus allowing us to make conclusions about the effect of cumulative malaria exposure on EBV persistence. Furthermore, our longitudinal studies recently reported that primary EBV infections occur at a significantly younger age in children residing in a malaria holoendemic area compared to a hypoendemic area, 7.28 compared to 8.39 months of age, respectively, and that 35% of children resident in Kisumu experience primary EBV infection before 6 months of age.24 The cumulative-negative impact of chronic malaria on EBV control is supported by a recent study from The Gambia, where malaria control programs have been very successful, showing that residents were no longer impaired as measured by classical EBV regression assay49—in contrast to their earlier studies conducted in 1984 when malaria transmission was high.34 The second limitation is the appropriate choice of controls. We selected geographically and ethnicity matched controls; however, malaria-exposure history, selective pressure and thus allele frequencies can vary within microenvironments.21 Therefore, future studies should strive to include sibling and parent–child matches necessary for definitive genetic comparisons as well as the development of individual measures of malaria exposure and resulting immunity.
In summary, this study concludes that HbAS genotype does not confer protection against eBL. This is consistent with the lack of an effect for HbAS genotype on peripheral blood EBV levels within the eBL-at-risk malaria holoendemic controls. In addition, plasma EBV levels (i.e., free viral DNA or lytic virions) do not have predictive power to serve as an eBL biomarker. However, high levels of cellular EBV (i.e., latently infected B cells) may have utility as a precursor for eBL in combination with other risk factors. This study does not eliminate a role for human genetics/genomics in eBL pathogenesis but supports a more encompassing syndemic model that includes modulations of the host response to EBV during malaria coinfections.
Acknowledgments
This work was supported by grants from the NIH RO1 CA134051 (A.M.M.) and the Thrasher Research Fund (A.M.M.) and NIH 1KL2RR031981 (J.A.B.). The authors thank the parents and guardians for enrolling their children in this study. They are also indebted to the medical staff at Nyanza Provincial General Hospital (renamed Jaramogi Oginga Odinga Teaching and Referral Hospital in 2012) and field teams in Nyanza and Rift Valley Provinces for assisting in both recruitment and sample blood collection. This manuscript has been approved by the Director of the Kenya Medical Research Institute.
Glossary
- CI
confidence interval
- DNA
deoxyribonucleic acid
- eBL
endemic Burkitt lymphoma
- EBV
Epstein–Barr virus
- HbAA
normal hemoglobin
- HbAS
hemoglobin AS, sickle cell trait
- HbSS
sickle cell disease
- HIV
human immunodeficiency virus
- IFN-γ
interferon-gamma
- CIDR1-α
cysteine-rich interdomain region 1α
- NIH
National Institutes of Health
- OR
odds ratio
- q-PCR
quantitative polymerase chain reaction
- ROC
receiver operating characteristic
- WHO
World Health Organization
References
- Morrow RH., Jr Epidemiological evidence for the role of falciparum malaria in the pathogenesis of Burkitt's lymphoma. IARC Sci Publ. 1985:177–86. [PubMed] [Google Scholar]
- Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt's lymphoma: a polymicrobial disease? Nat Rev Microbiol. 2005;3:182–7. doi: 10.1038/nrmicro1089. [DOI] [PubMed] [Google Scholar]
- Moormann AM, Chelimo K, Sumba OP, et al. Exposure to holoendemic malaria results in elevated Epstein-Barr virus loads in children. J Infect Dis. 2005;191:1233–8. doi: 10.1086/428910. [DOI] [PubMed] [Google Scholar]
- Moormann AM, Chelimo K, Sumba PO, et al. Exposure to holoendemic malaria results in suppression of Epstein-Barr virus-specific T cell immunosurveillance in Kenyan children. J Infect Dis. 2007;195:799–808. doi: 10.1086/511984. [DOI] [PubMed] [Google Scholar]
- Asito AS, Moormann AM, Kiprotich C, et al. Alterations on peripheral B cell subsets following an acute uncomplicated clinical malaria infection in children. Malar J. 2008;7:238. doi: 10.1186/1475-2875-7-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moormann AM, Heller KN, Chelimo K, et al. Children with endemic Burkitt lymphoma are deficient in EBNA1-specific IFN-gamma T cell responses. Int J Cancer. 2009;124:1721–6. doi: 10.1002/ijc.24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati D, Zhang LP, Chen Q, et al. Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infect Immun. 2004;72:5412–18. doi: 10.1128/IAI.72.9.5412-5418.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey JJ, Rochford R, Sumba PO, et al. Family environment is associated with endemic Burkitt lymphoma: a population-based case-control study. Am J Trop Med Hyg. 2008;78:338–43. [PubMed] [Google Scholar]
- Levine RL. Inherited susceptibility to pediatric acute lymphoblastic leukemia. Nat Genet. 2009;41:957–8. doi: 10.1038/ng0909-957. [DOI] [PubMed] [Google Scholar]
- Pike MC, Morrow RH, Kisuule A, et al. Burkitt's lymphoma and sickle cell trait. Br J Prev Soc Med. 1970;24:39–41. doi: 10.1136/jech.24.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verra F, Mangano VD, Modiano D. Genetics of susceptibility to Plasmodium falciparum: from classical malaria resistance genes towards genome-wide association studies. Parasite Immunol. 2009;31:234–53. doi: 10.1111/j.1365-3024.2009.01106.x. [DOI] [PubMed] [Google Scholar]
- Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326–8. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg. 1954;48:312–18. doi: 10.1016/0035-9203(54)90101-7. [DOI] [PubMed] [Google Scholar]
- Williams TN. Human red blood cell polymorphisms and malaria. Curr Opin Microbiol. 2006;9:388–94. doi: 10.1016/j.mib.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Aidoo M, Terlouw DJ, Kolczak MS, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002;359:1311–12. doi: 10.1016/S0140-6736(02)08273-9. [DOI] [PubMed] [Google Scholar]
- Gilles HM, Fletcher KA, Hendrickse RG, et al. Glucose-6-phosphate-dehydrogenase deficiency, sickling, and malaria in African children in South Western Nigeria. Lancet. 1967;1:138–40. doi: 10.1016/s0140-6736(67)91037-9. [DOI] [PubMed] [Google Scholar]
- Williams AO. Haemoglobin genotypes, ABO blood groups, and Burkitt's tumour. J Med Genet. 1966;3:177–9. doi: 10.1136/jmg.3.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkrumah FK, Perkins IV. Sickle cell trait, hemoglobin C trait, and Burkitt's lymphoma. Am J Trop Med Hyg. 1976;25:633–6. doi: 10.4269/ajtmh.1976.25.633. [DOI] [PubMed] [Google Scholar]
- Kimura H, Ito Y, Suzuki R, et al. Measuring Epstein-Barr virus (EBV) load: the significance and application for each EBV-associated disease. Rev Med Virol. 2008;18:305–19. doi: 10.1002/rmv.582. [DOI] [PubMed] [Google Scholar]
- Hayden RT, Hokanson KM, Pounds SB, et al. Multicenter comparison of different real-time PCR assays for quantitative detection of Epstein-Barr virus. J Clin Microbiol. 2008;46:157–63. doi: 10.1128/JCM.01252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey JJ, Mwanda WO, Wairiumu P, et al. Spatial distribution of Burkitt's lymphoma in Kenya and association with malaria risk. Trop Med Int Health. 2007;12:936–43. doi: 10.1111/j.1365-3156.2007.01875.x. [DOI] [PubMed] [Google Scholar]
- Mwanda OW, Rochford R, Moormann AM, et al. Burkitt's lymphoma in Kenya: geographical, age, gender and ethnic distribution. East Afr Med J. 2004:S68–S77. doi: 10.4314/eamj.v81i8.9210. [DOI] [PubMed] [Google Scholar]
- Moormann AM, Embury PE, Opondo J, et al. Frequencies of sickle cell trait and glucose-6-phosphate dehydrogenase deficiency differ in highland and nearby lowland malaria-endemic areas of Kenya. Trans R Soc Trop Med Hyg. 2003;97:513–14. doi: 10.1016/s0035-9203(03)80010-x. [DOI] [PubMed] [Google Scholar]
- Piriou E, Asito AS, Sumba PO, et al. Early age at time of primary Epstein-Barr virus infection results in poorly controlled viral infection in infants from Western kenya: clues to the etiology of endemic Burkitt lymphoma. J Infect Dis. 2012;205:906–13. doi: 10.1093/infdis/jir872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulstaert F, Hannet I, Deneys V, et al. Age-related changes in human blood lymphocyte subpopulations. II. Varying kinetics of percentage and absolute count measurements. Clin Immunol Immunopathol. 1994;70:152–8. doi: 10.1006/clin.1994.1023. [DOI] [PubMed] [Google Scholar]
- de-The G. Is Burkitt's lymphoma related to perinatal infection by Epstein-Barr virus? Lancet. 1977;1:335–8. doi: 10.1016/s0140-6736(77)91137-0. [DOI] [PubMed] [Google Scholar]
- Dowling MAC, Shute GT. A comparative study of thick and thin blood films in the diagnosis of scanty malaria parasitaemia. Bull World Health Organ. 1966;34:249–67. [PMC free article] [PubMed] [Google Scholar]
- Husain SM, Kalavathi P, Anandaraj MP. Analysis of sickle cell gene using polymerase chain reaction & restriction enzyme Bsu 361. Indian J Med Res. 1995;101:273–6. [PubMed] [Google Scholar]
- Ryan JL, Fan H, Swinnen LJ, et al. Epstein-Barr Virus (EBV) DNA in plasma is not encapsidated in patients with EBV-related malignancies. Diagn Mol Pathol. 2004;13:61–8. doi: 10.1097/00019606-200406000-00001. [DOI] [PubMed] [Google Scholar]
- Foy H, Kondi A, Timms GL, et al. The variability of sickle-cell rates in the tribes of Kenya and the Southern Sudan. Br Med J. 1954;1:294–7. doi: 10.1136/bmj.1.4857.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Maiteki-Sebuguzi C, Rosenthal PJ, et al. Evidence for both innate and acquired mechanisms of protection from Plasmodium falciparum in children with sickle cell trait. Blood. 2012;119:3808–14. doi: 10.1182/blood-2011-08-371062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati D, Espmark E, Kironde F, et al. Clearance of circulating Epstein-Barr virus DNA in children with acute malaria after antimalaria treatment. J Infect Dis. 2006;193:971–7. doi: 10.1086/500839. [DOI] [PubMed] [Google Scholar]
- Whittle HC, Brown J, Marsh K, et al. T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature. 1984;312:449–50. doi: 10.1038/312449a0. [DOI] [PubMed] [Google Scholar]
- Moss DJ, Burrows SR, Castelino DJ, et al. A comparison of Epstein-Barr virus-specific T-cell immunity in malaria-endemic and -nonendemic regions of Papua New Guinea. Int J Cancer. 1983;31:727–32. doi: 10.1002/ijc.2910310609. [DOI] [PubMed] [Google Scholar]
- Whittle HC, Brown J, Marsh K, et al. The effects of Plasmodium falciparum malaria on immune control of B lymphocytes in Gambian children. Clin Exp Immunol. 1990;80:213–18. doi: 10.1111/j.1365-2249.1990.tb05236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KM, Syed N, Whittle H, et al. Circulating Epstein-Barr virus-carrying B cells in acute malaria. Lancet. 1991;337:876–8. doi: 10.1016/0140-6736(91)90203-2. [DOI] [PubMed] [Google Scholar]
- Njie R, Bell AI, Jia H, et al. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J Infect Dis. 2009;199:31–8. doi: 10.1086/594373. [DOI] [PubMed] [Google Scholar]
- Snider CJ, Cole SR, Chelimo K, et al. Recurrent Plasmodium falciparum malaria infections in Kenyan children diminish T-cell immunity to Epstein Barr virus lytic but not latent antigens. PLoS One. 2012;7:e31753. doi: 10.1371/journal.pone.0031753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SJ, Pronk I, Middeldorp J. Toward standardization of Epstein-Barr virus DNA load monitoring: unfractionated whole blood as preferred clinical specimen. J Clin Microbiol. 2001;39:1211–16. doi: 10.1128/JCM.39.4.1211-1216.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TN, Mwangi TW, Wambua S, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis. 2005;192:178–86. doi: 10.1086/430744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arese P. How genetics and biology helped humanity to survive falciparum malaria. Parassitologia. 2006;48:553–9. [PubMed] [Google Scholar]
- Kreuels B, Kreuzberg C, Kobbe R, et al. Differing effects of HbS and HbC traits on uncomplicated falciparum malaria, anemia, and child growth. Blood. 2010;115:4551–8. doi: 10.1182/blood-2009-09-241844. [DOI] [PubMed] [Google Scholar]
- Moormann AM, Snider CJ, Chelimo K. The company malaria keeps: how co-infection with Epstein-Barr virus leads to endemic Burkitt lymphoma. Curr Opin Infect Dis. 2011;24:435–41. doi: 10.1097/QCO.0b013e328349ac4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher A, Armstrong AA, MacKenzie J, et al. Detection of Epstein-Barr virus (EBV) genomes in the serum of patients with EBV-associated Hodgkin's disease. Int J Cancer. 1999;84:442–8. doi: 10.1002/(sici)1097-0215(19990820)84:4<442::aid-ijc20>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Fan H, Gulley ML. Epstein-Barr viral load measurement as a marker of EBV-related disease. Mol Diagn. 2001;6:279–89. doi: 10.1054/modi.2001.29161. [DOI] [PubMed] [Google Scholar]
- Keegan THM, Glaser SL, Clarke CA, et al. Epstein-Barr virus as a marker of survival after Hodgkin's lymphoma: a population-based study. J Clin Oncol. 2005;23:7604–13. doi: 10.1200/JCO.2005.02.6310. [DOI] [PubMed] [Google Scholar]
- Jarrett RF. Risk factors for Hodgkin's lymphoma by EBV status and significance of detection of EBV genomes in serum of patients with EBV-associated Hodgkin's lymphoma. Leuk Lymphoma. 2003;44(Suppl 3):S27–S32. doi: 10.1080/10428190310001623801. [DOI] [PubMed] [Google Scholar]
- Gandhi MK, Lambley E, Burrows J, et al. Plasma Epstein-Barr virus (EBV) DNA is a biomarker for EBV-positive Hodgkin's lymphoma. Clin Cancer Res. 2006;12:460–4. doi: 10.1158/1078-0432.CCR-05-2008. [DOI] [PubMed] [Google Scholar]
- Jayasooriya S, Hislop A, Peng Y, et al. Revisiting the effect of acute P. falciparum malaria on Epstein-Barr virus: host balance in the setting of reduced malaria endemicity. PLoS One. 2012;7:e31142. doi: 10.1371/journal.pone.0031142. [DOI] [PMC free article] [PubMed] [Google Scholar]