

Abstract
To understand the molecular basis of sugarcane-smut interaction, it is important to identify sugarcane genes that respond to the pathogen attack. High-throughput tag-sequencing (tag-seq) analysis by Solexa technology was performed on sugarcane infected with Sporisorium scitaminea, which should have massively increased the amount of data available for transcriptome profile analysis. After mapping to sugarcane EST databases in NCBI, we obtained 2015 differentially expressed genes, of which 1125 were upregulated and 890 downregulated by infection. Gene ontology (GO) analysis revealed that the differentially expressed genes involve in many cellular processes. Pathway analysis revealed that metabolic pathways and ribosome function are significantly affected, where upregulation of expression dominates over downregulation. Differential expression of three candidate genes involved in MAP kinase signaling pathway, ScBAK1 (GenBank Accession number: KC857629), ScMapkk (GenBank Accession number: KC857627), and ScGloI (GenBank Accession number: KC857628), was confirmed by reverse transcription polymerase chain reaction (RT-PCR). Real-time quantitative PCR (qRT-PCR) analysis concluded that the expression of these genes were all up-regulated after the infection of S. scitaminea and may play a role in pathogen response in sugarcane. The present study provides insights into the molecular mechanism of sugarcane defense to S. scitaminea infection, leading to a more comprehensive understanding of sugarcane-smut interaction.
1. Introduction
Sugarcane is the most important sugar crop and accounts for more than 90 percent of total sugar production in China [1]. Sugarcane smut, caused by Sporisorium scitaminea, is an important sugarcane fungal disease worldwide and is also one of the most important sugarcane diseases in China [2]. It was firstly reported in 1887 from Natal, South Africa [1]. The loss of stalk yield for smut susceptible varieties is serious [3, 4]. Nowadays, sugarcane smut is prevalent in sugarcane producing areas all over the world and has caused the elimination of several sugarcane varieties with high yield and high sugar [5]. Because of this, smut resistance is a major objective of sugarcane variety breeding [6, 7].
Cultivation of resistant varieties is the most effective disease-control measure [6, 8]. In order to understand the sugarcane smut defense mechanism, research has been conducted on sugarcane bud morphology, cytology, and physiology with smut resistance [8–10]. Padmanaban et al. [11] studied the relationship between bud structure and the smut resistance of sugarcane varieties. The results revealed that the presence of resistance was associated with certain bud morphologies. Identification of differentially expressed genes under various stresses can give clues as to defence mechanisms and biochemical pathways regulated under each stress [12]. At present, research has focused on the molecular interaction between plant and pathogen using various techniques, including microarrays [13], representational difference analysis (RDA) [14], suppression subtractive hybridization (SSH) [15], cDNA-amplified fragment length polymorphism (cDNA-AFLP) [16], and serial analysis of gene expression (SAGE) [17]. As for the molecular mechanism of sugarcane-smut interaction, there have been several previous reports [18–24]. In the research of Thokoane and Rutherford [18], a cDNA-AFLP technique was applied to detect differential gene expression in smut resistant and susceptible sugarcane genotypes. Sequence homology analysis indicated that a Pto ser/threo protein kinase and an active gypsy-type LTR retro-transposon played a role in the response of sugarcane to the infection of S. scitaminea. Heinze et al. [19] conducted another study using suppression subtractive hybridization (SSH) method, which revealed sugarcane genes encoding proteins homologous to chitinases, as well as transcripts related to flavonoids, were involved in the sugarcane resistance after 7 days of S. scitamineum infection. Borrás-Hidalgo et al. [20] obtained 62 differentially expressed genes before and after the infection of S. scitaminea, among which 52 were upregulated and 10 were downregulated. LaO et al. [21] investigated differential expression by cDNA-AFLP method in the susceptible and the resistant genotypes. A total of 64 genes were proved to be differentially expressed, among which 67.2% were upregulated in the resistant cultivar. This result indicated that sugarcane response involved genes of the oxidative burst, defense response, and ethylene and auxin pathways. Que et al. [22] obtained 7 differentially expressed genes before and after inoculation of cultivars NCo376 and F134 with smut by DDRT-PCR. In Que et al. [23], 136 transcript-derived fragments (TDFs) were found to be differentially expressed in response to challenge by S. scitaminea. Que et al. [24] applied 2-DE and MALDI-TOF-TOF/MS to reveal the protein expression profile of sugarcane after inoculating with S. scitamineum. This is the first report of proteomic investigation of sugarcane exposed to S. scitamineum.
As discussed above, the mechanism of sugarcane smut defense has been studied using various techniques. However, sugarcane is a highly heterozygous crop and there is no genomic resource for this genus, so more attention is needed to establish the molecular interactions between sugarcane and pathogen. Differentially expressed genes can be investigated using a multitude of methods, such as cDNA microarray, cDNA-AFLP, RDA, SAGE, and SSH [13–17]. However, these techniques have some inherent limitations, such as an inability to detect low expression levels, and cross-hybridization problems [25–27]. The Solexa sequencing technology enables many applications, including whole genome resequencing, transcriptome sequencing, gene expression profiling, and epigenomic sequencing. The technique uses sequencing-by-synthesis on an eight-channel flow-cell to produce more than 10 million reads per channel with read lengths up to 100 bp [27–34]. Digital gene expression profiling (DGE) in the Illumina Solexa sequencing platform is capable of simultaneously sequencing a large number of DNA molecules and has become a powerful tool to detect the changes in gene expression [25, 28]. DGE can yield millions of short reads (32–40 nt) and is more suitable for tag-based transcriptome sequencing [25, 28].
The application of Solexa technology for the identification of differentially expressed genes associated with the molecular mechanism of interaction between the sugarcane-smut was therefore explored. In the present study, the Illumina Solexa sequencing technology was used to reveal the response of sugarcane to infection by S. scitaminea. Yacheng 05–179, a highly resistant sugarcane genotype, was used as plant material. RNA expression profile sequencing analysis of two samples, before and after sugarcane smut fungus inoculation, was then conducted by Solexa sequencing. This study aims to discover the pathogenesis-related differentially expressed genes and further understand the interaction mechanisms between sugarcane and S. scitaminea at the molecular level.
2. Materials and Methods
2.1. Plant Materials and Treatment
The sugarcane genotypes, Yacheng 05–179 and Liucheng 03–182, were chosen as smut-resistant and smut-susceptible plant material, respectively. These two sugarcane genotypes were provided by the Key Lab of Sugarcane Biology and Genetic Breeding, Ministry of Agriculture (Fuzhou, China). Sugarcane smut fungus (S. scitaminea) was collected from Liucheng 03–182 plants and stored at 4°C.
A smut spore suspension of 5 × 106 spores/mL was needle-inoculated on to sugarcane buds as the treatment group, while sterile water inoculated buds were the mock group [9, 10]. These sugarcane buds were then cultured in an incubator at 28°C ± 0.5°C, and phenotypically normal bud tissue was then collected at time points of 12, 24, 36, 48, and 72 hours, respectively. All samples were immediately frozen in liquid nitrogen and stored in a refrigerator at −80°C until RNA extraction.
2.2. RNA Isolation
Total RNA was isolated using the TRIzol reagent according to the manufacturer's instructions (Invitrogen). Dried RNA samples were dissolved in diethylpyrocarbonate-treated H2O, and RNA quality was assessed on 1.0% denaturing agarose gels. RNA quality and quantity were verified using a NanoDrop 1000 spectrophotometer and an Agilent 2100 Bioanalyzer prior to Solexa sequencing at BGI. Total RNA samples of Yacheng 05–179 before and after pathogen inoculation at the time point of 48 h were subjected to digitized expression profiling sequencing at Beijing Genomics Institute (BGI; Shenzhen, China). Both RNA samples of Yacheng 05–179 and Liucheng 03–182 were used for Real-time quantitative PCR at time point 12, 24, 36, 48, and 72 hours.
2.3. Solexa Sequencing
The gene expression libraries were prepared using Illumina Gene Expression Sample Prep Kit according to the manufacturer's instructions. Specified Experimental Process, use magnetic oligo (dT) beads adsorption to purify mRNA from 6 μg total RNA, and then the first and second strand cDNA was synthesized. The bead-bound cDNA was subsequently digested with restriction enzyme Nla III, which recognized and cut off the CATG sites. The 3′ cDNA fragments attached to the oligo (dT) beads were washed away, and then 5′ cDNA fragments were ligated to the Illumina Adaptor 1, which contained a recognition site for the endonuclease Mme I for cutting 17 bp downstream of the recognition site (CATG), producing tags with adaptor 1. After removing 3′ fragments with magnetic beads precipitation, Illumina adaptor 2 is ligated to the 3′ ends of tags, acquiring tags with different adaptors of both ends to form a tag library. After 15 cycles of linear PCR amplification, 95 bp fragments are purified by 6% TBE PAGE Gel electrophoresis. The two constructed tag libraries were fixed onto the Illumina Sequencing Chip (flowcell) for cluster generation through situ amplification and were deep-sequenced using Illumina Genome Analyzer. After image analysis, base calling, and quality calibration, the raw data was produced [25, 26].
2.4. RT-PCR Confirmation and qRT-PCR Analysis of Three Candidate Genes
The expressions of three candidate genes in MAP kinase signaling pathway were determined by RT-PCR and Real-time quantitative PCR (qRT-PCR). Gene-specific primers were designed according to the gene sequences using the Primer Premier 5.0 software and were synthesized commercially (Shanghai Sangon, China).
The first-strand cDNA was synthesized from total RNA using PrimeScript 1st strand cDNA synthesis kit (Takara). The primers for RT-PCR confirmation are listed in Table 1. The 25 μL reaction mix contained 2.5 μL 10 × PCR Buffer (plus Mg2+), 2.5 μL deoxynucleotide triphosphates (dNTPs) (2.5 mM), 1.0 μL first-strand cDNA, 1.0 μL each of forward and reverse primers (10 μM), and 0.125 μL Ex-Taq enzyme (5 U/μL). The ddH2O was added as supplement. The PCR amplification program consisted of predenaturation for 4 min at 94°C; denaturation for 1 min at 94°C, annealing for 1 min at annealing temperature, and extension for 1.5 min at 72°C for 35 cycles; and a final extension for 10 min at 72°C. Then, 5 μL of the product was electrophoresed on 1.0% agarose gel and viewed under UV light.
Table 1.
Primers used for RT-PCR analysis.
| Genes | Forward primer (5′-3′) | Reverse primer (5′-3′) | Product size (bp) |
|---|---|---|---|
| ScBAK1 | TTTGAGTGGTCCAATCCC | CGAGTCATCCGTCAGGTC | 1,291 |
| ScMAPKK | CCTTCTTGGGTTCTTCCTCC | ATCCCTTCTCATAGTCTCATCTAG | 1,302 |
| ScGloI | AGCCAGAAGAAAGGGAGC | GTTCATCAAGGCGGAAAC | 1,091 |
The 25S rRNA (BQ536525) gene was chosen as the internal control in the qRT-PCR analysis [35]. The first-strand cDNA for qRT-PCR was synthesized using PrimeScript RT reagent kit (Takara). The primers for qRT-PCR analysis are listed in Table 2. In qRT-PCR analysis, 20 μL samples were run on the ABI PRISM7500 real time PCR System using SYBR Green PCR Master Mix (Applied Biosystems). The qRT-PCR reaction conditions were held at 50°C for 2 min, predenatured at 95°C for 10 min, and then kept at 94°C for 15 s and at 60°C for 60 s for 40 cycles. Three replicas were set for each sample. When the reaction was completed and the melting curve was analyzed. The 2−△△CT method was adopted to analyze the qRT-PCR results [36].
Table 2.
Primers used for qRT-PCR analysis.
| Genes | Forward primer (5′-3′) | Reverse primer (5′-3′) | Product size (bp) |
|---|---|---|---|
| 25S rRNA | GCAGCCAAGCGTTCATAGC | CCTATTGGTGGGTGAACAATCC | 109 |
| ScBAK1 | ACCTATGCCAATGTCTTACGG | GATGAAGCCAGTTGTAGCACC | 168 |
| ScMAPKK | TGAACTGCGGCTCAATCAAAG | TGCCTCACTAGCTGGACAACA | 180 |
| ScGloI | TGGACCGCACAATCAAATACTACAC | CAAAGCCCGTTCCAATGTCATAC | 108 |
3. Results and Analysis
3.1. Solexa Sequencing of Infected Sugarcane
After filtering 3′ adapter sequence, empty reads, low-complexity reads, and low-quality reads, a total of 4,847,568 and 4,883,691 21 bp length clean tags were obtained that corresponded to 446,284 and 423,464 distinct tags for Yacheng 05–179 before and after S. scitaminea inoculation, respectively (Table 3).
Table 3.
Categorization and abundance of clean tags.
| Summary | Inoculation | Control |
|---|---|---|
| Clean tags | ||
| Total number | 4,847,568 | 4,883,691 |
| Distinct tag number | 446,284 | 423,464 |
| All tag mapping to genes | ||
| Total number | 2,974,532 | 2,826,151 |
| Total % of clean tag | 61.36% | 57.88% |
| Distinct number | 95,633 | 82,429 |
| Distinct tag % of clean tag | 21.43% | 18.81% |
| One tag mapping to unique genes | ||
| Total number | 639,019 | 610,306 |
| Distinct number | 72,812 | 64,815 |
| One tag mapping to multiple genes | ||
| Total number | 1,996,930 | 1,902,489 |
| Distinct number | 85,308 | 75,512 |
| Unknown tags | ||
| Total number | 1,376,922 | 1,532,237 |
| Total % of clean tag | 28.40% | 31.37% |
| Distinct number | 288,186 | 283,137 |
| Distinct tag % of clean tag | 64.57% | 66.86% |
Matching the tags to genes is an important step to annotate sequences and can reveal the molecular events behind the gene expression [26]. In this study, all clean tags were aligned to the reference sugarcane EST database in NCBI (http://www.ncbi.nlm.nih.gov/). After comparing with sugarcane EST database, 61.36% and 57.88% of clean tags, and 21.43% and 18.81% of distinct clean tags could be matched exactly with the reference sequences in two samples, respectively. Then, there are 639,019 and 610,306 clean tags for inoculation and mock libraries are uniquely mapped to database, while the number of distinct clean tags are 72812 and 64,815 (Table 3; see Supplementary Material available online at http://dx.doi.org/10.1155/2013/298920), respectively. However, because of incomplete sequences, there are a large proportion total clean tags (about 30%) and distinct tags (about 65%) which could not be aligned to the reference sequences (Supplementary Material).
Heterogeneity and redundancy are two significant characteristics of mRNA expression [26]. We analyzed the distribution of clean tag copy number in the two libraries and found that the copy number of total clean tags and distinct clean tags showed very similar tendencies for two samples, respectively (Supplementary Material). In regard to the detection of low abundance expressed genes, the proportion of tags greater than 100-fold was over 54% and the proportion of distinct clean tags which was greater than 100-fold was very small (1.82% and 1.92%), but there are more than 80% distinct clean tags with a ratio within 5-fold (Figure 1 and Supplementary Material).
Figure 1.
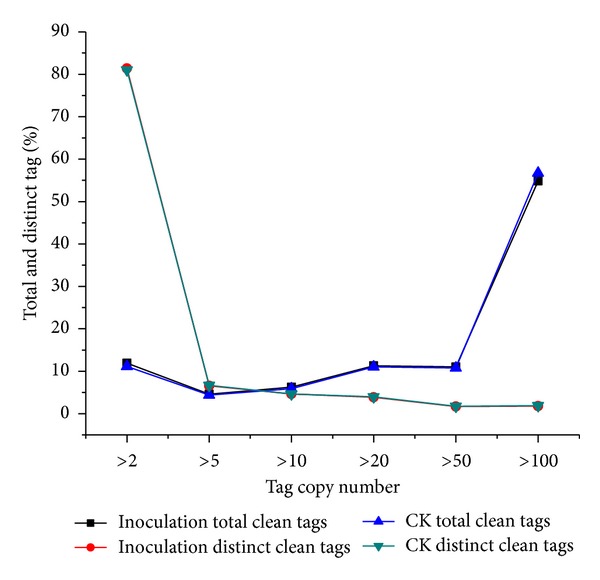
Distribution of total clean tag and distinct clean tag counts.
3.2. Differentially Expressed Genes in Two Libraries
By using blast against the reference sugarcane EST database in NCBI and putative differentially expressed genes were selected based on the following two criteria: (1) if the average fold change of gene expression before and after S. scitaminea inoculation was more than or equal to twofold (|log2 Ratio| ≥ 1) and (2) if the false discovery rate value of the single sample was less than 0.001 (FDR ≤ 0.001) [25]. By using this approach we obtained 2015 differentially expressed genes, and the expression of 1125 genes were identified as upregulated in the Yacheng 05–179 after inoculation as compared with that in Yacheng 05–179 before inoculation (Supplementary Material). The expression of 890 genes was decreased by more than twofold in Yacheng 05–179 before inoculation. We then screened 48 candidate genes for further study (31 upexpressed and 17 downexpressed) using the conditions of FDR ≤ 0.001 and |log2 Ratio| ≥ 5, and 3 upexpressed genes in the MAP kinase signaling pathway (Table 4).
Table 4.
Some selected differentially expressed genes identified using Solexa sequencing in sugarcane.
| Gene | log2 Ratio | P Value | FDR | Annotation |
|---|---|---|---|---|
| gi|35266134 | 10.6 | 4.44E −16 | 2.90E −14 | Zea mays STIP1 and U box protein 1 |
| gi|35111854 | 9.2 | 1.67E −09 | 4.70E −08 | Sorghum bicolor hypothetical protein |
| gi|34977253 | 9.0 | 1.35E −08 | 3.42E −07 | Saccharum SP-80-3280 chloroplast |
| gi|35208604 | 8.8 | 1.09E −07 | 2.43E −06 | S. bicolor hypothetical protein |
| gi|35293529 | 8.8 | 1.09E −07 | 2.43E −06 | S. bicolor hypothetical protein |
| gi|26879857 | 8.8 | 2.19E −07 | 4.62E −06 | Mus musculus chromosome 1 |
| gi|34921564 | 8.6 | 8.82E −07 | 1.69E −05 | S. bicolor hypothetical protein |
| gi|34942866 | 8.6 | 8.82E −07 | 1.69E −05 | S. bicolor ATP synthase complex subunit 9 |
| gi|34967006 | 8.5 | 3.55E −06 | 5.99E −05 | Z. ays transcription factor GT-3b |
| gi|36001135 | 8.5 | 3.55E −06 | 5.99E −05 | Z. mays omega-3 fatty acid desaturase |
| gi|35094785 | 8.4 | 7.14E −06 | 0.000113 | S. bicolor hypothetical protein |
| gi|35229762 | 8.4 | 7.14E −06 | 0.000113 | Z. mays nucleotide adenylyltransferase 1 |
| gi|35294230 | 8.4 | 7.14E −06 | 0.000113 | Z. mays clone 294529 ATEB1A mRNA |
| gi|35981936 | 8.3 | 1.43E −05 | 0.000214 | Z. mays dihydrolipoamide S-acetyltransferase 1 |
| gi|34965850 | 8.3 | 1.43E −05 | 0.000214 | Z. mays h/ACA ribonucleoprotein mRNA |
| gi|34942070 | 8.3 | 1.43E −05 | 0.000214 | S. bicolor mitochondrion |
| gi|35052194 | 8.3 | 1.43E −05 | 0.000214 | S. officinarum receptor kinase 1 (BAK1) |
| gi|35275212 | 8.3 | 1.43E −05 | 0.000214 | S. bicolor hypothetical protein |
| gi|34917382 | 8.2 | 2.88E −05 | 0.000401 | S. bicolor hypothetical protein |
| gi|34973370 | 8.2 | 2.88E −05 | 0.000401 | Z. mays myb-like protein mRNA |
| gi|34942843 | 8.2 | 2.88E −05 | 0.000402 | S. bicolor hypothetical protein |
| gi|35074510 | 8.1 | 5.77E −05 | 0.000741 | No match |
| gi|35090069 | 8.1 | 5.77E −05 | 0.000741 | Saccharum NCo 310 chloroplast DNA |
| gi|34977027 | 8.1 | 5.77E −05 | 0.000741 | S. bicolor mitochondrion |
| gi|35203085 | 8.1 | 5.77E −05 | 0.000741 | S. bicolor hypothetical protein |
| gi|35942526 | 8.1 | 5.77E −05 | 0.000741 | S. bicolor hypothetical protein |
| gi|35278085 | 8.1 | 5.77E −05 | 0.000741 | S. bicolor hypothetical protein |
| gi|35006270 | 8.1 | 5.77E −05 | 0.000739 | Z. mays GAPDH mRNA, partial cds |
| gi|35265314 | 8.1 | 5.77E −05 | 0.000739 | No match |
| gi|34957782 | 8.1 | 5.77E −05 | 0.000739 | S. bicolor hypothetical protein |
| gi|34964700 | 5.8 | 1.65E −05 | 0.000243 | S. bicolor hypothetical protein |
| gi|35245148 | 1.7 | 7.75E −07 | 1.50E −05 | NSP-interacting kinase 1 |
| gi|36032703 | 3.3 | 1.21E −08 | 3.11E −07 | MKK6-putative MAPKK mRNA |
| gi|33461608 | 2.2 | 2.22E −06 | 3.89E −05 | No match |
| gi|36001222 | −16.2 | 0 | 0 | Kladothrips maslini 16S ribosomal RNA |
| gi|35990824 | −11.2 | 5.91E −37 | 6.64E −35 | Manduca sexta actin mRNA, complete cds |
| gi|33461608 | −10.3 | 2.74E −19 | 2.03E −17 | No match |
| gi|36014330 | −10.2 | 2.17E −18 | 1.54E −16 | Z. mays subtilisin-chymotrypsin inhibitor, |
| gi|34922345 | −9.5 | 1.67E −11 | 6.06E −10 | Z. perennis isolate per7a MPI gene |
| gi|34971519 | −9.3 | 2.63E −10 | 8.38E −09 | S. bicolor hypothetical protein |
| gi|35975174 | −8.5 | 4.09E −06 | 6.81E −05 | Z. mays subtilisin-chymotrypsin inhibitor |
| gi|35056509 | −8.3 | 1.63E −05 | 0.000240497 | S. bicolor hypothetical protein |
| gi|35008679 | −8.1 | 6.45E −05 | 0.000819694 | Z. mays clone 261727 mRNA sequence |
| gi|35998230 | −8.1 | 2.16E −85 | 4.06E −83 | Z. mays subtilisin-chymotrypsin inhibitor |
| gi|36040747 | −7.3 | 3.31E −48 | 4.57E −46 | No match |
| gi|31072203 | −6.5 | 6.96E −52 | 1.02E −49 | No match |
| gi|35989329 | −5.7 | 1.29E −58 | 2.02E −56 | Z. mays adhesive/proline-rich protein |
| gi|35013208 | −5.4 | 5.53E −37 | 6.23E −35 | S. bicolor hypothetical protein |
| gi|36034301 | −5.2 | 7.59E −14 | 1.65E −141 | Z. mays xylanase inhibitor protein |
| gi|36008557 | −5.1 | 1.67E −10 | 5.41E −09 | S. officinarum clone SCQGLR1019F04 |
| gi|34929144 | −5.0 | 5.60E −52 | 8.24E −50 | U-box domain-containing protein 21 |
3.3. Gene Annotation and Functional Classification
Gene ontology (GO) analysis was performed by mapping each differentially expressed gene on to the reference sugarcane EST database in NCBI (http://www.ncbi.nlm.nih.gov/). As shown in Figure 2, the proportions and comparisons of these differentially expressed genes were summarized in three main functional categories, cellular component with ten categories of genes, biological process with thirteen categories of genes, and molecular function with nine categories of genes. What should be stressed is that among these ten types of genes involved in molecular function, catalytic activity and binding are major molecular function (Figure 2 and Supplementary Material). This suggests that biological metabolism and catalytic activity are the major responsive classes; biological metabolic processes are enhanced and catalytic activity increases after the inoculation with the pathogen. In addition, the products of different genes usually cooperate with each other to exercise their biological function, so pathway-based analysis helps to further understand genes biological functions. We obtained 303 differentially expressed genes with pathway annotation involving in 79 biological pathways (Supplementary Material), among which metabolic pathways and ribosome were major pathways, and the number of upexpressed genes was significantly higher than that of those downexpressed (Figure 3). As indicated in Figure 4, it is interesting that, when sugarcane was infected by smut pathogen, the expression of three key enzymes, ScBAK1, ScMapkk, and ScGloI, which belongs to MAP kinase signaling pathway, were upregulated (Figure 4).
Figure 2.
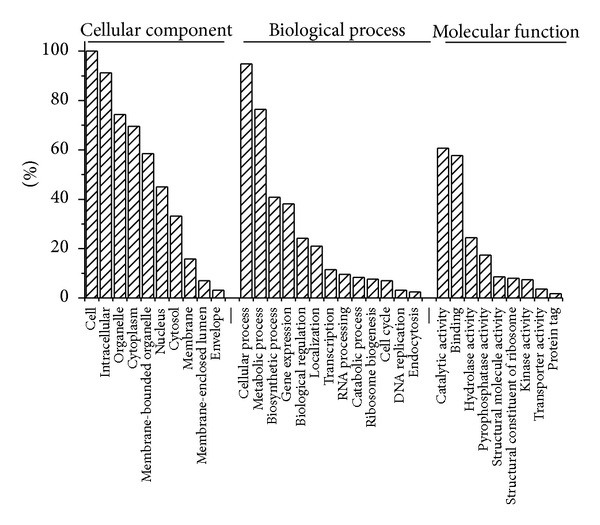
Gene ontology analysis for differentially expressed genes obtained using Solexa sequencing.
Figure 3.
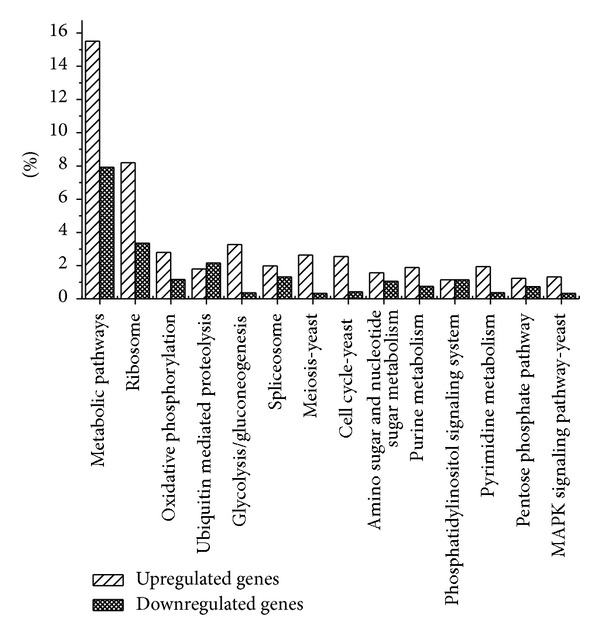
Pathway classifications for upregulated and downregulated genes.
Figure 4.
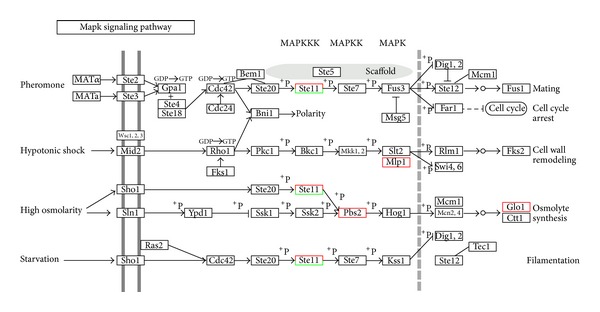
MAP kinase signaling pathway of sugarcane infected by S. scitaminea.
3.4. RT-PCR Confirmation and qRT-PCR Analysis of Three Candidate Genes
Three differentially expressed genes screened in Solexa sequencing were confirmed by RT-PCR. Full-length cDNA sequence of three upregulated genes in MAP kinase signaling pathway designated as ScBAK1 (GenBank Accession number: KC857629), ScMapkk (GenBank Accession number: KC857627), and ScGloI (GenBank Accession number: KC857628) were obtained from sugarcane based on the bud full-length cDNA library, of which the length is 1,291 bp, 1,302 bp, and 1,091 bp, with ORF length of 1,004 bp, 1,068 bp, and 885 bp, respectively (Figure 5).
Figure 5.
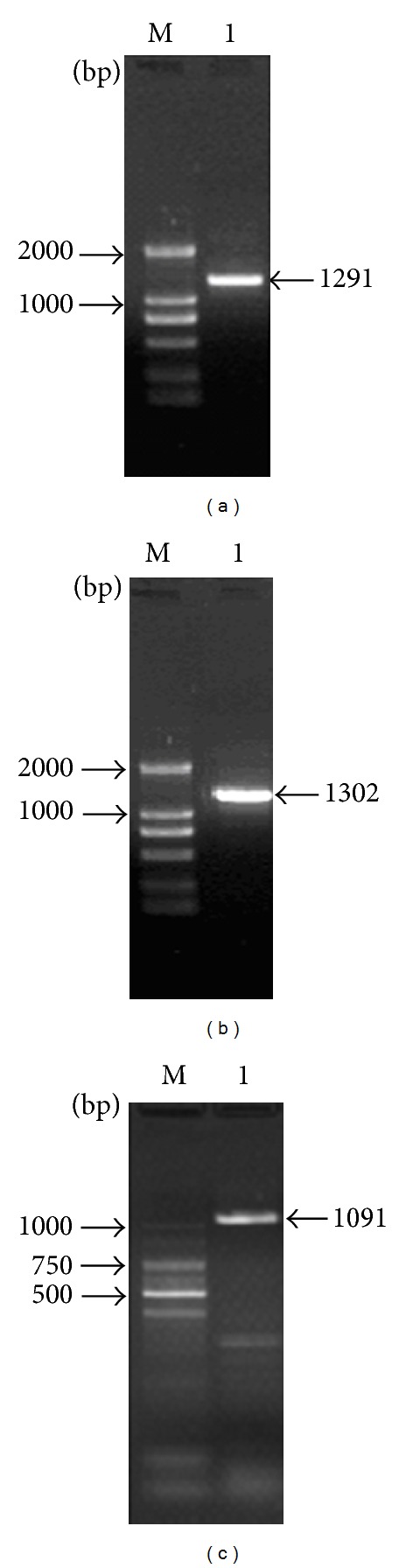
RT-PCR products for amplification of three upregulated genes in MAP kinase signaling pathway. (a) M, DL2000; Lane 1, PCR product of ScBAK1 gene; (b) M, DL2000; Lane 1, PCR product of ScMapkk gene; (c) M, 100 bp Ladder; Lane 1, PCR product of ScGloI gene.
qRT-PCR analysis was applied to validate the expressions of ScBAK1, ScMapkk, and ScGloI genes during the infection with S. scitaminea (Figure 6 and Supplementary Material). The smut-resistant genotype Yacheng 05–179 and smut-susceptible genotype LiuCheng 03–182 were selected as plant material. The results showed that during 0 h–12 h after pathogen infection, the expression of these three genes in both smut-resistant genotype and smut-susceptible genotype were up-regulated, but their expressions in smut-resistant genotype were much more significant than that in the smut-susceptible genotype especially for ScMapkk and ScGloI genes. Therefore, the expression of these genes may play a role in defence during S. scitaminea infection. However, the expression of ScGloI gene was up-regulated again after infection for 48 h in both genotypes of the resistant and susceptible one, and the expression of ScMapkk gene was up-regulated significantly again after 72 hpi in resistant Yacheng 05–179 but not in susceptible LiuCheng 03–182. This suggests that the smut-resistance function mechanism is different between these three genes, ScBAK1, ScMapkk, and ScGloI.
Figure 6.
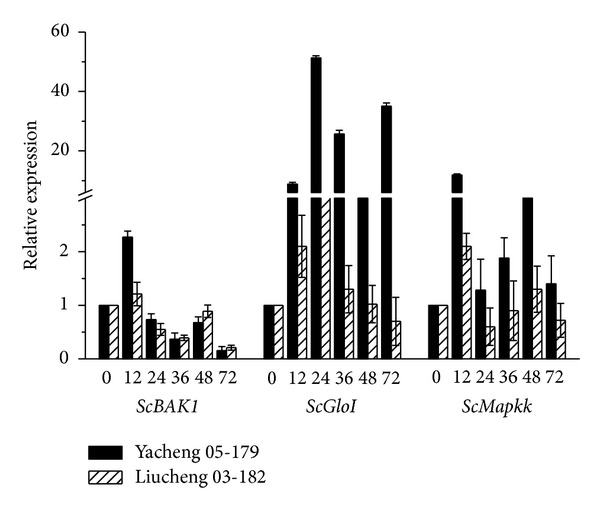
qRT-PCR expression profiles for ScBAK1, ScGloI, and ScMapkk gene under S. scitaminea stress in resistant and susceptible sugarcane genotypes.
4. Discussion
The transcriptome, which can vary with external environmental conditions, is the set of all RNA molecules, including mRNA, rRNA, tRNA, and noncoding RNAs produced in one or a population of cells [32]. Transcriptome analysis using high-throughput short-read sequencing technology, such as Solexa sequencing, is straightforward, and does not have to be restricted to the genome of model organisms [25, 28, 29]. This analysis can provide information on gene expression and gene regulation and thus is essential to interpret the functional elements of the genome and reveal molecular mechanisms [32]. The nonmodel species that lack a reference genome, where RNA-Seq analysis has been applied, include Eucalyptus grandis [29], Persea americana [30], Artemisia annua [31], Vitis vinifera L. [28], Sesamum indicum L. [32], Cicer arietinum [34], Medicago sativa L. [33], and Myrica rubra [27].
Solexa sequencing is a high-throughput, short-read, massively parallel sequencing platform, of which the read length is relatively short (21 bp), and bioinformatics analysis of the corresponding differentially expressed genes has to only rely on sugarcane EST databases. Therefore, in the absence of corresponding sequenced genome information as a reference, many of the differentially expressed genes cannot be functionally annotated. However, the genomes of sorghum, maize and rice can act as reference information. According to sugarcane UniGenes identified and annotated by RNA-Seq and sorghum, maize and rice reference genome, we hope to establish a platform for future genetic and functional genomic research in sugarcane. In the present study, a total of 4,847,568 and 4,883,691 21 bp length clean tags that corresponded to 446,284 and 423,464 distinct tags for Yacheng 05–179 before and after S. scitaminea inoculation were successfully obtained. The results showed that Solexa technology can quickly assess millions of short sequences of both high-abundance and low-abundance expressed genes in sugarcane, similar to previous research reports [27–34]. This study was restricted by the absence of sugarcane genome sequence, thus, yielded an incomplete picture, and the completion of the sugarcane genome project should significantly contribute to a more detailed picture of the gene expression profiles related to sugarcane-smut interaction mechanism.
Differential gene expression during sugarcane-smut interaction is likely to be induced following pathogen challenge, which leads to up or downregulation of gene expression [18, 20, 22–24]. The influence of environment, evolution of S. scitaminea and molecular changes of the host sugarcane variety makes the interactions between sugarcane, the environment, and S. scitaminea more complicated. Previous studies on sugarcane-smut interaction revealed that sugarcane genes encoding proteins homologous to chitinases as well as transcripts related to the pathways of both phenylpropanoids and flavonoids were shown to be involved in the sugarcane resistance after 7 days of S. scitamineum infection [19]. Sugarcane-smut interaction was also carried out using a somaclonal genotype showing stable resistance for longer than 10 years [20]. Two months after fungal inoculation, some differentially expressed TDFs were similar to those identified in this study, that is, genes encoding NBS-LRR-like proteins, protein kinases, and proteins related to both auxin and ethylene pathways. In our study, we used samples after sugarcane smut fungus inoculation to characterize the transcriptome using Solexa sequencing. This generated more than 4.8 million tags of two samples after sequencing. However, there are a vast majority of tags (Table 3 and Supplementary Material, about 30% of total clean tags and about 65% of distinct tags) which cannot be aligned to the reference sequences. The functional categorization shows a complex linkage between genes involved in cellular and metabolic processes, gene expression, biological regulation, and localization. As indicated in Figure 3, metabolic pathway and ribosome were major pathways. Theoretically, the identification of differentially expressed genes has great potential to help to understand the molecular mechanism of smut resistance.
The study provides the potential to develop a molecular marker for smut resistance selection if the up-regulation of expression of ScMapkk and ScGloI genes could be verified in further smut-resistant and smut-susceptible genotypes with different genetic backgrounds. This is important for sugarcane smut resistance breeding because the interaction of sugarcane, smut pathogen and environment leads to the instability of the resistance phenotype tested by field-inoculation. In addition, only 48 candidate genes were screened in this study, and there are still many differentially expressed genes to be tested.
Plants possess integrated signaling networks that mediate the perception and responses to biotic and abiotic stresses which govern plant growth and development. MAP kinase signaling pathway is a conserved signaling pathway common in plants. The present results permit us to hypothesize about the role of the MAP kinase pathway on the sugarcane early response against the S. scitaminea infection. When subjected to biotic and abiotic stress, the MAP kinase signaling pathway is activated [37–40] and plays an important role in plant resistance mechanisms [41, 42]. The results of qRT-PCR analysis revealed that the expression of these three candidate genes, ScBAK1, ScMapkk, and ScGloI were up-regulated in both smut-resistant genotype and smut-susceptible genotype, but the expression in smut-resistant genotype was greater than that in the smut-susceptible genotype especially for ScMapkk and ScGloI genes. The expression of these three genes may play a role in defending against infection by pathogens in sugarcane. When sugarcane was infected by the smut pathogen, the expression of key enzymes were up-regulated, leading to the activation of MAP kinase signaling pathway and the triggering of the genes involved with plant defence (Figure 4), which was similar to the previous reports [37–40].
5. Conclusions
In conclusion, the usefulness of the Solexa sequencing in identifying genes related to sugarcane smut defense has been successfully demonstrated in this study. However, most of the molecular mechanisms of sugarcane-smut interaction are as yet unknown. More genes related to sugarcane defense and their expression profiles in response to smut infection should be analyzed further. The present study provides a Solexa sequencing platform for gene expression research on this crop and also a reference for studying the molecular mechanism in nonmodel organisms.
Supplementary Material
Additional File 1: Statistics of clean tag alignment.pdf
Additional File 2: Distribution of clean tag copy number.pdf
Additional File 3: 2015 differentially expressed genes identified in sugarcane.xls
Additional File 4: Gene ontology analysis for differentially expressed genes.xls
Additional File 5: Pathway annotation of 305 differently expressed genes in sugarcane.xls
Additional File 6: Raw data of qRT-PCR for ScBAK1, ScMapkk and ScGloI genes.xls
Conflict of Interests
The authors declare that they have no conflict of interest.
Acknowledgments
This work was funded by National Natural Science Foundation of China (31101196), the earmarked fund for the Modern Agriculture Technology of China (CARS-20) and Research Funds for Distinguished Young Scientists in Fujian Agriculture and Forestry University (xjq201202). We appreciate all ideas and constructive criticism from the reviewers. We especially thank Andrew C Allan in The New Zealand Institute for Plant & Food Research Ltd, (Plant and Food Research), Mt Albert Research Centre, Private Bag 92169, Auckland, New Zealand, for his critical revision and valuable comments on this manuscript.
References
- 1.Chen RK, Xu LP, Lin YQ, et al. Modern Sugarcane Genetic Breeding. Beijing, China: China Agriculture Press; 2011. [Google Scholar]
- 2.Que YX, Xu LP, Lin JW, Chen RK, Grisham MP. Molecular variation of Sporisorium scitamineum in mainland China revealed by RAPD and SRAP markers. Plant Disease. 2012;96(10):1519–1525. doi: 10.1094/PDIS-08-11-0663-RE. [DOI] [PubMed] [Google Scholar]
- 3.Hoy JW, Hollier CA, Fontenot DB, Grelen LB. Incidence of sugarcane smut in Louisiana and its effects on yield. Plant Disease. 1986;70(1):59–60. [Google Scholar]
- 4.Padmanaban P, Alexander KC. Effect of smut on growth and yield parameters of sugarcane. Indian Phytopathology. 1988;41(3):367–369. [Google Scholar]
- 5.Xu LP, Lin YQ, Fu HY. Evaluation of smut resistance in sugarcane and identification of resistance in sugarcane varieties. Journal of Fujian Agricultural Science. 2000;29(3):292–295. [Google Scholar]
- 6.Hsieh WH, Lee CS. Compatability and pathogenicity of two races of Ustilago scitaminea Syd in Taiwan. Taiwan Sugar. 1978;25:46–48. [Google Scholar]
- 7.Legendre BL, Burner DM. New opportunities in the development of sugarcane cultivars. Sugar Cane. 1997;5:4–9. [Google Scholar]
- 8.Xu LP, Chen RK. Current status and prospects of smut and smut resistance breeding in sugarcane. Journal of Fujian Agricultural Science. 2000;15(2):26–31. [Google Scholar]
- 9.Que YX, Xu LP, Lin JW, Chen TS, Chen RK, Li YL. Establishment of evaluation system of smut resistance for sugarcane varieties. Journal of Plant Genetics and Resources. 2006;7(1):18–23. [Google Scholar]
- 10.Gong DM, Chen RK, Lin YQ. Relation between phenylproanoid metabolism in sugarcane and resistance to smut. Journal of Fujian Agricultural Science. 1995;24(4):394–398. [Google Scholar]
- 11.Padmanaban P, Alexander KC, Shanmugam N. Studies on certain characters associated with smut resistance. Indian Phytopathology. 1988;41(4):594–598. [Google Scholar]
- 12.Sävenstrand H, Brosché M, Ängehagen M, Strid Å. Molecular markers for ozone stress isolated by suppression subtractive hybridization: specificity of gene expression and identification of a novel stress-regulated gene. Plant, Cell and Environment. 2001;23(7):689–700. [Google Scholar]
- 13.Aharoni A, Vorst O. DNA microarrays for functional plant genomics. Plant Molecular Biology. 2002;48(1-2):99–118. doi: 10.1023/a:1013734019946. [DOI] [PubMed] [Google Scholar]
- 14.Hubank M, Schatz DG. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Research. 1994;22(25):5640–5648. doi: 10.1093/nar/22.25.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diatchenko L, Lau YF, Campbell AP, et al. Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(12):6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachem CWB, Oomen RJFJ, Visser RGF. Transcript imaging with cDNA-AFLP: a step-by-step protocol. Plant Molecular Biology Reporter. 1998;16(2):157–173. [Google Scholar]
- 17.Yamamoto M, Wakatsuki T, Hada A, Ryo A. Use of serial analysis of gene expression (SAGE) technology. Journal of Immunological Methods. 2001;250(1-2):45–66. doi: 10.1016/s0022-1759(01)00305-2. [DOI] [PubMed] [Google Scholar]
- 18.Thokoane LN, Rutherford RS. cDNA-AFLP differential display of sugarcane (Saccharum spp, hybrids) genes induced by challenge with the fungal pathogen Ustilago scitaminea (sugarcane smut) Proceedings of the South African Sugar Technologists' Association. 2001;75:104–107. [Google Scholar]
- 19.Heinze BS, Thokoane LN, Williams NJ, Barnes JM, Rutherford RS. The smut-sugarcane interaction as a model system for the integration of marker discover and gene isolation. Proceedings of the South African Sugar Technologists' Association. 2001;75:88–93. [Google Scholar]
- 20.Borrás-Hidalgo O, Thomma BPHJ, Carmona E, et al. Identification of sugarcane genes induced in disease-resistant somaclones upon inoculation with Ustilago scitaminea or Bipolaris sacchari . Plant Physiology and Biochemistry. 2005;43(12):1115–1121. doi: 10.1016/j.plaphy.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 21.LaO M, Arencibia AD, Carmona ER, et al. Differential expression analysis by cDNA-AFLP of Saccharum spp. after inoculation with the host pathogen Sporisorium scitamineum . Plant Cell Reports. 2008;27(6):1103–1111. doi: 10.1007/s00299-008-0524-y. [DOI] [PubMed] [Google Scholar]
- 22.Que YX, Yang ZX, Xu LP, Chen RK. Isolation and identification of differentially expressed genes in sugarcane infected by Ustilago scitaminea . Acta Agronomica Sinica. 2009;35(3):452–458. [Google Scholar]
- 23.Que YX, Lin JW, Song XX, Xu LP, Chen RK. Differential gene expression in sugarcane in response to challenge by fungal pathogen Ustilago scitaminea revealed by cDNA-AFLP. Journal of Biomedicine & Biotechnology. 2011;2011:p. 160934. doi: 10.1155/2011/160934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Que YX, Xu LP, Lin JW, Ruan MH, Zhang MQ, Chen RK. Differential protein expression in sugarcane during sugarcane: Sporisorium scitamineum interaction revealed by 2-DE and MALDI-TOF-TOF/MS. Comparative and Functional Genomics. 2011;2011:10 pages. doi: 10.1155/2011/989016.989016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu T, Qin Z, Zhou X, Feng Z, Du Y. Transcriptome profile analysis of floral sex determination in cucumber. Journal of Plant Physiology. 2010;167(11):905–913. doi: 10.1016/j.jplph.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Wang QQ, Liu F, Chen XS, Ma XJ, Zeng HQ, Yang ZM. Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics. 2010;96(6):369–376. doi: 10.1016/j.ygeno.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Feng C, Chen M, Xu CJ, et al. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genomics. 2012;13(1, article 19) doi: 10.1186/1471-2164-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zenoni S, Ferrarini A, Giacomelli E, et al. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiology. 2010;152(4):1787–1795. doi: 10.1104/pp.109.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novaes E, Drost DR, Farmerie WG, et al. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9, article 312 doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wall PK, Leebens-Mack J, Chanderbali AS, et al. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics. 2009;10, article 347 doi: 10.1186/1471-2164-10-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W, Wang Y, Zhang Q, Qi Y, Guo D. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genomics. 2009;10(1):p. 465. doi: 10.1186/1471-2164-10-465.1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei W, Qi X, Wang L, et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics. 2011;12, article 451 doi: 10.1186/1471-2164-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang SS, Tu ZJ, Cheung F, et al. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics. 2011;12(1, article 199) doi: 10.1186/1471-2164-12-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg R, Patel RK, Jhanwar S, et al. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiology. 2011;156(4):1661–1678. doi: 10.1104/pp.111.178616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iskandar HM, Simpson RS, Casu RE, Bonnett GD, Maclean DJ, Manners JM. Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Molecular Biology Reporter. 2004;22(4):325–337. [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Pedley KF, Martin GB. Role of mitogen-activated protein kinases in plant immunity. Current Opinion in Plant Biology. 2005;8(5):541–547. doi: 10.1016/j.pbi.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 38.Kiegerl S, Cardinale F, Siligan C, et al. SIMKK, a mitogen-activated protein kinase (MAPK) kinase, is a specific activator of the salt stress-induced MAPK, SIMK. Plant Cell. 2000;12(11):2247–2258. doi: 10.1105/tpc.12.11.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ichimura K, Shinozaki K, Tena G, et al. Mitogen-activated protein kinase cascades in plants: a new nomenclature. Trends in Plant Science. 2002;7(7):301–308. doi: 10.1016/s1360-1385(02)02302-6. [DOI] [PubMed] [Google Scholar]
- 40.Cuschieri J, Maier RV. Mitogen-activated protein kinase (MAPK) Critical Care Medicine. 2005;33(12):S417–S419. doi: 10.1097/01.ccm.0000191714.39495.a6. [DOI] [PubMed] [Google Scholar]
- 41.Yang HQ, Jie LY. The plant MAPK and its function in pathogen signaling cascades. Acta Phytopathological Sinica. 2003;33(1):8–13. [Google Scholar]
- 42.Zhang TG, Liu YB, Xia XH. Research advances about MAPK pathways in plants. Acta Botanica Boreali-Occidentalia Sinica. 2008;28(8):1704–1714. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional File 1: Statistics of clean tag alignment.pdf
Additional File 2: Distribution of clean tag copy number.pdf
Additional File 3: 2015 differentially expressed genes identified in sugarcane.xls
Additional File 4: Gene ontology analysis for differentially expressed genes.xls
Additional File 5: Pathway annotation of 305 differently expressed genes in sugarcane.xls
Additional File 6: Raw data of qRT-PCR for ScBAK1, ScMapkk and ScGloI genes.xls