

Abstract
Recent evidence suggests that rare genetic variants within the TREM2 gene are associated with increased risk for Alzheimer’s disease. TREM2 mutations are the genetic basis for a condition characterized by polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) and an early-onset dementia syndrome. TREM2 is important in the phagocytosis of apoptotic neuronal cells by microglia in the brain. Loss of function might lead to an impaired clearance and accumulation of necrotic debris and subsequent neurodegeneration. In this study, we investigated a consanguineous family segregating autosomal recessive behavioral variant FTLD from Antioquia, Colombia. Exome sequencing identified a nonsense mutation in TREM2 (p.Trp198X) segregating with disease. Next, using a cohort of clinically characterized and neuropathologically verified sporadic AD cases and controls we report replication of the AD risk association at rs75932628 within TREM2. These data suggest that mutational burden in TREM2 may serve as a risk factor for neurodegenerative disease in general and that potentially this class of TREM2 variant carriers with dementia should be considered a molecularly distinct form of neurodegenerative disease.
1. Introduction
TREM2, or triggering receptor expressed on myeloid cells 2, is an immunoreceptor expressed on activated macrophages, osteoclast, immature dendritic cells and microglia (Colonna, 2003). It is a 26kDa transmembrane glycoprotein that consists of a single extracellular immunoglobulin-like domain, a transmembrane region with a charged lysine residue and a short cytoplasmic tail lacking any signaling motifs (Colonna, 2003). TREM2 forms a receptor signaling complex with TYROBP (Paloneva et al., 2002). The charged lysine in the transmembrane domain of TREM2 is needed for its association with TYROBP (Bouchon et al., 2000; Bouchon et al., 2001) and as TREM2 lacks an intracellular signaling tail, it is completely dependent on the presence of the adaptor protein TYROBP (Colonna, 2003). The TREM2/TYROBP complex regulates key signaling events involved in immune responses, differentiation of dendritic cells and osteoclasts and phagocytic activity in microglia (Bouchon et al., 2001; Hsieh et al., 2009; Otero et al., 2012).
Following neuronal injury, microglia initiate repair by phagocytizing dead neurons without eliciting inflammation. TREM2 has been shown to play a role in the phagocytosis of apoptotic neuronal cells by microglia and resolution of inflammation (Hsieh et al., 2009). TREM2 can directly bind to neuronal cells, with increased binding to apoptotic neuronal cells. When neuronal cells undergo apoptosis, they increase the expression of TREM2-ligands, which mediate signal transduction by TREM2 on microglia and promote phagocytosis (Hsieh et al., 2009). In osteoclasts, TREM2 has been shown to regulate bone mass by regulating the rate of osteoclast generation (Otero et al., 2012).
Genetic mutations in either TREM2 or TYROBP cause a similar clinical phenotype, the Nasu-Hakola syndrome (or polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy; PLOSL) (Figure 1), which is characterized with cystic-like lesions of the bone and brain demyelination that lead to fractures and presenile dementia (Paloneva et al., 2002). The disease is characterized by different stages. The first symptoms present with an osseous stage at the third decade of life with pathological bone fractures. This is followed by the early neuropsychiatric stage in the fourth decade, presenting a frontal lobe syndrome, and the late neuropsychiatric stage, with profound dementia and usually death by the age of 50 (Bianchin et al., 2004; Numasawa et al., 2011). Neuropathological findings include loss of myelin and axons in the brain, with reactive astrocytosis and microglial activation (Klunemann et al., 2005). Mutations in TREM2 have also been described in pure early-onset dementia without bone cysts, and frontotemporal dementia (FTD)-like syndrome (Chouery et al., 2008). Recently, a variant in TREM2 (rs75932628) has also been implicated as a risk factor for both early-onset and late-onset Alzheimer’s disease (R. Guerreiro et al., 2012; Jonsson et al., 2012; Pottier et al., 2013).
Figure 1.
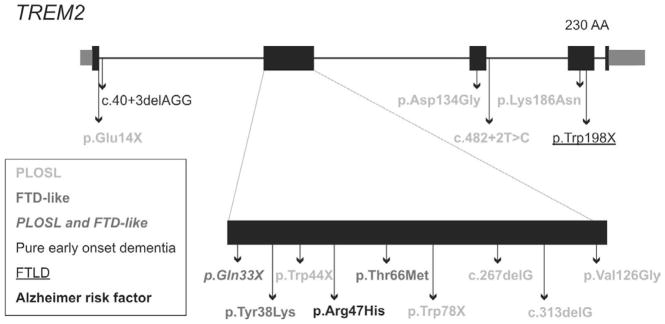
Overview of the mutations found in TREM2. ENST00000373113.3; ENSP00000362205.3.
In this study, we identified a nonsense mutation in TREM2 in a consanguineous Colombian family segregating autosomal recessive FTLD. Frontotemporal lobar degeneration (FTLD) is the second most common cause of early-onset dementia and fourth most common cause of late-onset dementia, and is characterized by atrophy of the prefrontal and anterior temporal lobes. FTLD is a clinically and genetically heterogeneous degenerative disorder. Patients usually show prominent behavioral and/or language deficits, which evolve gradually into cognitive impairment and dementia (McKhann et al., 2001; Neary et al., 1998). The most common clinical manifestation of FTLD consists of patients who present behavioral or personality changes (behavioral variant frontotemporal dementia or bvFTD) (Neary et al., 1998). Two other prototypic clinical phenotypes that occur in FTLD are language impairment disorders: semantic dementia (SD) and progressive nonfluent aphasia (PNFA) (Neary et al., 1998).
In this manuscript, we report that a nonsense mutation in TREM2 is the cause of FTLD in a Colombian family from the province Antioquia. Additionally, we provide replicative evidence demonstrating a role for the rs75932628 TREM2 variant in Alzheimer’s disease thereby suggesting TREM2 mutations as a risk factor for neurodegenerative disease in general.
2. Material and methods
2.1 Clinical diagnosis
A large consanguineous Colombian family segregating autosomal recessive FTLD was collected through the Grupo Neurosciencias, University of Antioquia, Colombia (Figure 2). Three patients and five unaffected relatives from the family were included. Our patients met published criteria for behavioral variant FTD (Rascovsky et al., 2011). The index case was a female offspring of first cousins who first showed symptoms of sexual disinhibition at age 47. She was excessively familiar with strangers and had abandoned all her responsibilities in the home. A paternal uncle and a brother had similar symptoms prior to age 60.
Figure 2.
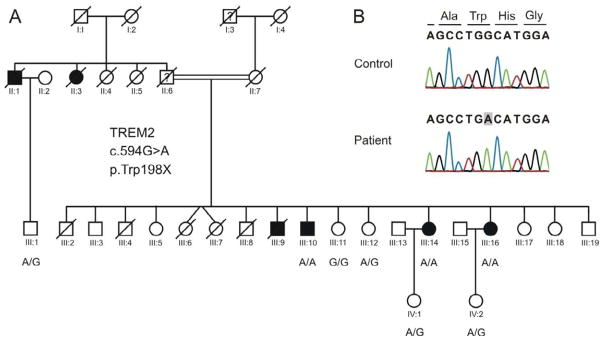
A. Inheritance of the nonsense mutation in TREM2 in the Colombian family. B. Sanger sequencing results. hg19/GRC37:chr6:41126693T>C; ENST00000373113.3:c.594G>A; ENSP00000362205.3:p.Trp198X.
Description of the clinical tests
All patients and controls underwent a standard medical and neurological history, physical examination, and office-based clinical cognitive assessment. Patient III:16 (index patient) and her brother (patient III:10) additionally underwent more detailed neuropsychological assessment that included:
General: mental status examinations (maximum score 30 points)
Memory: 10 item, 3 learning trials word-list learning with delayed free recall and recognition, and the Rey-Osterrieth Complex Figure Test (copy and recall, 36 points maximum for each)
Attention/Psychomotor Speed: Trail Making Test (seconds to complete), Wechsler Adult Intelligence Scale-Revised Digit Symbol Substitution Test (raw total score)
Language: FAS letter fluency (numbers of words generated in one minute for each letter), Category Fluency (Animals named in one minute), and sentence writing
Executive functions: Wisconsin Card Sorting Test (Categories and perseverative responses)
This study was conducted according to the guidelines of the ethical committee at the University of Antioquia. Informed consent was obtained from all individuals or from family members in cases of impaired cognition.
2.2 Exome sequencing
Exome sequencing was performed on two affected individuals and two unaffected individuals from the family (identify in the family tree who they are). First, 1.2ug of DNA was fragmented to 300–400bp fragments with a Covaris E210 instrument (Covaris Inc., Woburn, US). Next, libraries were prepared with Illumina’s Truseq DNA Sample Preparation Kit v2 (Illumina inc, San Diego, USA), following the manufacturer’s protocol. All four samples were labeled using four different indices. Final PCR enriched fragments were validated on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, US). In addition, samples were quantified with PicoGreen (Molecular Probes, Eugene, Oregon, USA), and 500ng of each sample was pooled before entering Truseq 62Mb Exome Enrichment Kit (Illumina inc, San Diego, USA). The final library was then validated on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, US), quantified using qPCR (7900 HT, Applied Biosystems, Foster City, CA; Kapa Biosystems, Woburn, MA) and sequenced by 100bp paired-end sequencing on a HiSeq2000 instrument (Illumina inc, San Diego, USA).
2.3 Data-analysis
Reads were aligned to the Human genome (Hg19/GRC37) using the Burrows-Wheeler transform (BWA v.0.5.9) (Li and Durbin, 2009). PCR duplicates were removed using Picard v1.51(H. Li et al., 2009) and base quality recalibration, indel realignment and SNP and INDEL discovery was performed using the Genome analysis toolkit (GATK v1.1-31-gdc8398e) (McKenna et al., 2010). Data were filtered against dbSNP135, 1000 genomes, an in-house exome database, and then annotated with snpEff v2.0.2 against ensembl v63 to identify new damaging mutations (nonsense, splice site, frameshift, missense) (Cingolani, 2011). Only homozygous variants present in the two affected individuals and either heterozygous or not present in the controls were considered as candidates.
2.4 Sanger DNA sequencing
The c.594G>A mutation in TREM2 was confirmed with Sanger sequencing in the samples used for exome sequencing along with three additional controls and one additional case from the family. Primers surrounding the identified variants were designed using Primer3. Polymerase chain reaction (PCR) was carried out under standard conditions. Direct sequencing of the PCR product was performed on an ABI3130XL sequencer (Applied Biosystems Inc., Foster City, USA).
2.5 APOE genotyping
APOE genotyping was performed on all family members by the method of Crook et al (Crook et al., 1994).
2.6 Clinically Characterized and Neuropathologically Verified Subjects
Our US series was obtained from 21 National Alzheimer’s Coordinating Center (NACC) brain banks and from the Miami Brain Bank as previously described (Corneveaux et al., 2010; Myers et al., 2007; Webster et al., 2009). Additional cohorts from the United Kingdom and Europe were obtained in the same manner as the original US series. Our criteria for inclusion were as follows: self-defined ethnicity of European descent (in an attempt to control for the known allele frequency differences between ethnic groups), neuropathologically confirmed AD or no neuropathology present, and age of death greater than 65. Neuropathological diagnosis was defined by board-certified neuropathologists according to the standard NACC protocols (Beekly et al., 2004). Samples derived from subjects with a clinical history of stroke, cerebrovascular disease, Lewy bodies, or comorbidity with any other known neurological disease were excluded. AD or control neuropathology was confirmed by plaque and tangle assessment with 45% of the entire series undergoing Braak staging (Braak and Braak, 1995). Samples were de-identified before receipt, and the study met human studies institutional review board and HIPPA regulations. This work is declared not human-subjects research and is IRB exempt under regulation 45 CFR 46. See the Acknowledgements section for a list of individual sites that contributed samples to this effort.
2.7 Genome-wide SNP Genotyping
Genomic DNA samples were analyzed on the Genome-Wide Human SNP 6.0 Array (Affymetrix, Inc. Santa Clara, CA) according to the manufacturer’s protocols (Affymetrix Genome-Wide Human SNP Nsp/Sty 6.0 User Guide;Rev. 1 2007) and as described previously (Corneveaux et al., 2010).
2.8 SNP Imputation
Genotypes were imputed using MACH v.1.0.16 (Y. Li et al., 2009) as described previously (Corneveaux et al., 2010).
3. Results
3.1 Clinical syndrome
Initial symptoms began between ages 45 and 50 involving altered social behavior in all three, as well as an oropharyngeal tic in one that subsequently developed in the other two as well. Two of the three developed the new onset of substance use including alcohol and tobacco as well as complex partial seizures characterized by versive head and eye turning and speech arrest. Eventually, all three developed a severe pan-frontal syndrome with apathy, disinhibition and impulsivity, obsessive and perseverative behavior, impaired social behavior and cognitive decline that included psychomotor slowing and impaired problem solving with increased perseverative errors, as well as constructional apraxia, and memory loss (Table 1). Physical exam revealed frontal release signs but no evidence of motor neuron disease. MRI (index patient) showed asymmetric bifrontal atrophy (Figure 3).
Table 1.
Neuropsychological Test Scores in two TREM2 carrier members of the Colombian kindred.
| Patient 1 | Patient 2 | |
|---|---|---|
| Age years | 48 | 55 |
| Gender | F | M |
| Education years | 10 | 14 |
| Handedness | Right | Right |
| Occupation | Homemaker | Unemployed |
| Minimental Status Exam | 24/30 | 23/30 |
| Trail Making Test-A (seconds) | 99 | 154 |
| Trail Making Test-B (seconds) | 278 | na |
| WAIS-R* Digit Symbol (raw total) | 23 | 11 |
| 10 word list | ||
| -3 learning trials | 2--3--3 | 2--3--3 |
| -spontaneous recall | 1 | 0 |
| -true positive recognition | 7 | 1 |
| -true negative recognition | 9 | 10 |
| Complex Figure Test-copy | 18.5 | 16.5 |
| -recall (%) | 5 (27%) | 0 (0%) |
| Animal fluency (1 minute) | 8 | 3 |
| Wisconsin Cart Sorting Test | ||
| -Categories | 1 | 0 |
| perseverative responses (%) | 28 (58.3%) | 47 (97.9%) |
| FAS letter fluency | 15 | 2 |
| Frontal Behavioral Inventory | 34/72 | 54/72 |
WAIS-R = Wechsler Adult Intelligence Scale-Revised
Figure 3.
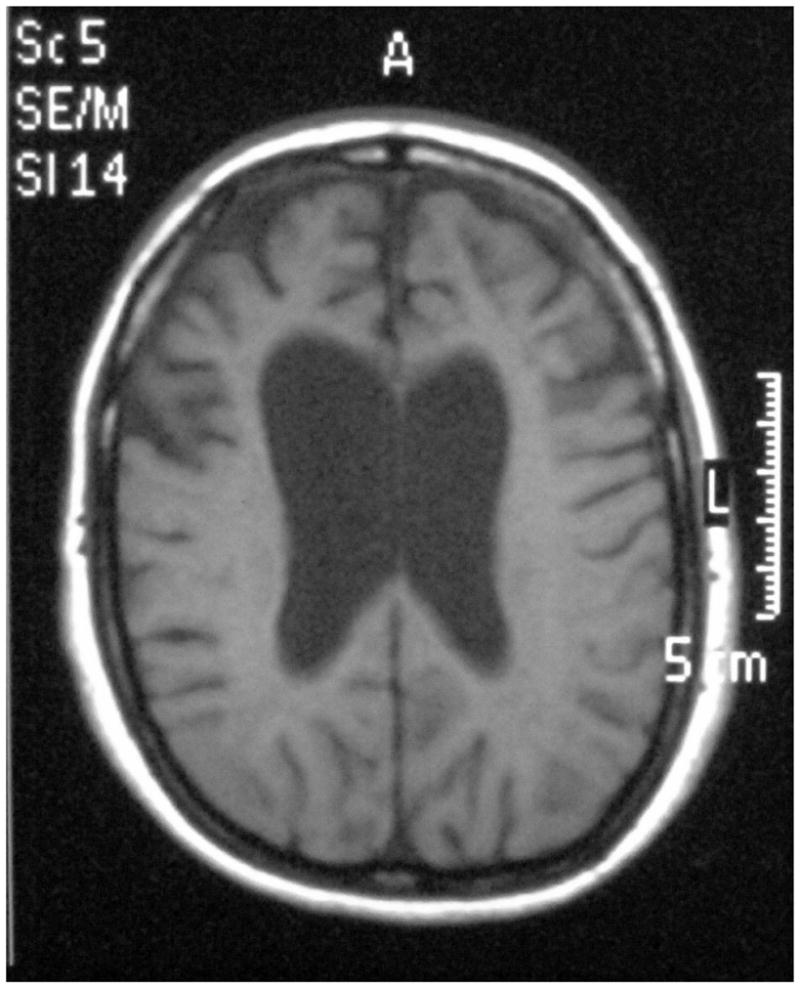
MRI of the index case patient demonstrating the frontal lobe atrophy.
Patient III:16 (Index patient) began to express noticeable behavioral and personality changes at age 47, starting with sexual and social dis-inhibition, mood changes and neglect of personal hygiene. In addition, her abstract reasoning, decision-making, planning and judgment decreased severely. Her brother (patient III:10), presented behavioral changes at the age of 50. He became apathetic, showed progressive memory loss and obsessive compulsive behaviors. Later, he suffered from insomnia, presented motor and phonetic tics, and seizures characterized by versive head movement, gaze deviation, and speech arrest. The older sister (patient III:14), started showing personality changes, dis-inhibition and progressive memory loss at the age of 45. She emits guttural speech sounds and presents similar seizures as her brother characterized by a versive head, eye motions and speech arrest. All three patients’ functionality has been reduced to only basic self-caring activities.
3.2 Identification of a nonsense mutation in TREM2
Two patients (III:10 and III:14) and two controls (III:11 and III:12) from the Colombian family were sequenced using the Illumina’s 62Mb Truseq Exome Enrichment Kit. Variants identified by next generation sequencing were filtered by dbSNP135, the 1000 genomes project and an in-house list of exomes. Only variants homozygous in the two patients were further prioritized. This lead to the identification of a nonsense mutation in exon 4 of the gene TREM2, that leads to premature truncation of the protein (c.594G>A;p.Trp198X). We confirmed the presence of the mutation in the patients by Sanger sequencing, and subsequently checked the inheritance of the mutation in the family (Figure 2).
No possible disease-causing variants were detected in the known FTLD genes (GRN, MAPT, VCP, CHMP2B, TARDP or C9ORF72 and TMEM106B). In addition, all family members were APOE E3/E3 homozygotes.
3.3 TREM2 as a risk factor for Alzheimer disease
Recent studies show that the p.Arg47His variant (rs75932628) is associated with a significantly increased risk for Alzheimer’s disease (R. Guerreiro et al., 2012; Jonsson et al., 2012; Pottier et al., 2013). We saw an enrichment of this variant by a factor of 2.8 in our collection of clinically characterized and neuropathologically confirmed AD cases which was significant when combined with the data of Guerreiro et al, 2012 (Table 2) (R. Guerreiro et al., 2012).
Table 2.
Association of p.Arg47His in a cohort of clinically characterized and neuropathologically verified Alzheimer’s disease cases and controls (“TGenII”). AD Meta-Analysis: combination of TGenII Cohort results and Guerreiro et al., 2012.
| Group | MAF case | MAF control | Allele case | Allele control | Enrichment | p-value |
|---|---|---|---|---|---|---|
| TGen II AD | 0.0025 | 0.0009 | 5 | 1990 | 1 | 1158 | 2.8 | 0.4242 |
| AD meta-analysis | 0.0088 | 0.0027 | 27 | 3081 | 6 | 2263 | 3.3 | 0.0044 |
4. Discussion
TREM2 is a membrane protein that forms a receptor-signaling complex with TYROBP that triggers activation of the immune response, differentiation of dendritic cells and osteoclasts and phagocytosis in microglia. We identified a mutation in exon 4 of the TREM2 gene (p.Trp198X) in a consanguineous Colombian family segregating autosomal recessive FTLD (Figure 2). The clinical phenotype of our family seems very similar to PLOSL, except for the complete absence of a bone phenotype in our family. Chouery and colleagues also reported a mutation in TREM2 (c.40+3delAGG) in a Lebanese family segregating pure early-onset dementia without bone cysts (Figure 1) (Chouery et al., 2008). The affected individuals presented personality and behavioral changes at age of 30–35, culminating in severe dementia. Brain MRI showed either a diffuse brain atrophy or cortical atrophy in the temporal lobes (Chouery et al., 2008). Similar to our patients, a subsequent report in Turkish patients (R.J. Guerreiro et al., 2012), also described a frontotemporal dementia (FTD)- like syndrome without bone cysts. Neither prior report described the tics we found in our patients, but two of the three Turkish patients described had generalized seizures.
Also recently, a rare missense mutation in TREM2 (p.Arg47His; rs75932628), was found to significantly increase the risk of late-onset Alzheimer disease (R. Guerreiro et al., 2012; Jonsson et al., 2012) and early-onset Alzheimer disease (Pottier et al., 2013). The mutation may lead to a decreased affinity of TREM2 for its natural ligand or affect its signaling. Although this variant is less frequent than APOE E4, it confers a risk for Alzheimer’s disease with an effect similar to that of APOE E4 (Jonsson et al., 2012). The clinical phenotype in all TREM2 associated dementia cases reported to date have striking panfrontal syndromes quite distinct from the typical AD phenotype. How different genetic variants result in topographically altered patterns of pathology is not yet known, but it has been previously shown that presenilin 1 mutations, unlike sporadic AD or APOE e4 carrier cases, are associated with early striatal deposition of amyloid (Klunk et al., 2007).
We saw an enrichment of this variant in TREM2 (p.Arg47His) in our cohort of clinically characterized and neuropathologically verified Alzheimer’s disease cases and control (Table 2) (Corneveaux et al., 2010), which is in line with the data of Guerreiro et al. (R. Guerreiro et al., 2012). Because TREM2 has been shown to be involved in PLOSL, FTD-like disease, Alzheimer’s disease and now also FTLD, we hypothesize that TREM2 might be a risk factor for neurodegeneration in general, and other neurodegenerative disease cohorts should be examined for this TREM2 variant to better understand its role in the pathophysiology of neurodegeneration.
It is also important to note that the rarity of the p.Arg47His variant results in most carriers existing in the heterozygous state in the general population. This is important as they likely express a wild type version of TREM2 in addition to the mutant version. Under the assumption that the TREM2 mutant doesn’t function as a dominant negative molecule this may represent a therapeutic avenue for treatment or prevention of the neurodegenerative disease in the p.Arg47His heterozygotes. Chemical or biological agonists of TREM2 could be explored for use in these individuals.
Mutations in TREM2 leading to PLOSL are mainly loss of function mutations caused by premature truncation of the protein, lacking the complete transmembrane and cytoplasmic domains (Figure 1) (Klunemann et al., 2005). Although the effect of some of the non-synonymous mutations might be unclear, the p.Lys186Asn mutation changes the positively charged lysine in the transmembrane part of the protein to an asparagine, which disrupts the association with and expression of TYROBP, also resulting in a loss-of-function (Bouchon et al., 2000; Klunemann et al., 2005; Paloneva et al., 2002). The c.40+3delAGG mutation, which leads to pure early-onset dementia without bone cysts, weakens the splice site at the 5′donor site of intron 1. Quantitative RT-PCR showed an over two-fold down regulation of wild-type TREM2 transcripts because of this mutation (Chouery et al., 2008). The presence of functional wild-type protein in these patients, although reduced, might explain why these patients only present a neurodegenerative phenotype and lack a bone phenotype.
A genotype-phenotype correlation is much less clear in our family. The p.Trp198X mutation truncates only the last 33 AA of the protein (Figure 1), leaving the extracellular and transmembrane part of the protein almost intact, but missing the short cytoplasmic domain (Bouchon et al., 2000). Since the transmembrane region mediates the formation of a complex with TYROBP, the protein might therefore still be expressed and be able to form a partly functional complex with TRYOBP on the cell surface, leading to a less severe phenotype in our family. However a genotype-phenotype correlation is less clear for the p.Gln33X mutation, which has been described to cause both FTD-like syndrome and PLOSL. This suggests that environmental factors of modifier genes also influence the phenotype associated with TREM2 mutations. In addition, heterozygote carriers of p.Gln33X were reported to show memory deficits or and might have a higher risk for developing late-onset Alzheimer disease (R. Guerreiro et al., 2012).
The Colombian family in this study originates from Antioquia, which is a young admixture that was founded in the 16–17th century by only a few hundred American natives and immigrants mainly from Spain (Carvajal-Carmona et al., 2003). The people in the region have remained relatively isolated since its origin (Carvajal-Carmona et al., 2003). Young genetic isolates confer an advantage to gene-mapping, and are usually enriched for specific Mendelian traits that often occur in the population because of a founder effect (Sheffield et al., 1998). In addition to the founder effect, small effective population size and bottlenecks increase consanguinity, and therefore increase the opportunity to map recessive diseases. Rare Mendelian disease in isolated populations is almost always attributable to a single mutation that is shared identical-by-descent among affected individuals in the population. In the Antioquia population, founder mutations have been identified previously, for example, the c.G736A mutation in the PARK2 gene that causes early onset Parkinson’s Disease (Pineda-Trujillo et al., 2006). It is unclear however when the p.Trp198X TREM2 mutation arose in Antioquia. If the mutation originated before or close to the founding of the population, the carrier frequency could be very high in Antioquia. However, an analysis of the 1,000 Genomes Project dataset revealed no carriers of this variant in 60 individuals from Medellin, Colombia (referred to as “CLM”) suggesting a frequency of less than 1 in 120 chromosomes.
In conclusion, we identified a mutation in TREM2 that leads to FTLD in a consanguineous family from Colombia. The mechanisms of TREM2 mutations leading to FTLD and neurodegeneration in PLOSL and are unknown, but we can hypothesize that lack of TREM2 impairs the clearance of apoptotic neurons in microglia, leading to the accumulation of necrotic debris. We also replicate the association of the TREM2 p.Arg47His variant and Alzheimer’s disease risk. The involvement of TREM2 genetic variants in PLOSL, FTD-like syndrome, Alzheimer’s disease and now also FTLD, suggests that the TREM2 locus may be associated with neurodegenerative disease in general and raises the possibility that this variant in the context of other genetic variants or environmental factors are the determinants of the form of neurodegenerative disease that a carrier patient presents with.
Acknowledgments
This work was funded in part by COLCIENCIAS: proyect 111554531651, award to FL. IS is a postdoctoral fellow of the FWO-vlaanderen. This work was supported by Kronos Science; The National Institute of Neurological Disorders and Stroke [R01 NS059873 to MJH]; The National Institute on Aging [R01 AG031581 to EMR, P30 AG19610 to EMR, AG034504 to AJM]; The Banner Alzheimer’s Foundation; The Johnnie B. Byrd Sr. Alzheimer’s Disease Institute; The Medical Research Council; The Intramural Research Program of the National Institutes of Health; and the state of Arizona.
Many data and biomaterials were collected from several National Institute on Aging (NIA) and National Alzheimer’s Coordinating Center (NACC, grant #U01 AG016976) funded sites. Amanda J. Myers, PhD (University of Miami, Department of Psychiatry) and John A. Hardy, PhD (Reta Lila Weston Institute, University College London) collected and prepared the series. Marcelle Morrison-Bogorad, PhD., Tony Phelps, PhD and Walter Kukull PhD are thanked for helping to co-ordinate this collection. The directors, pathologist and technicians involved include: National Institute on Aging: Ruth Seemann, John Hopkins Alzheimer’s Disease Research Center (NIA grant # AG05146): Juan C. Troncoso, MD, Dr. Olga Pletnikova, University of California, Los Angeles (NIA grant # P50 AG16570):Harry Vinters, MD, Justine Pomakian, The Kathleen Price Bryan Brain Bank, Duke University Medical Center (NIA grant #AG05128, NINDS grant # NS39764, NIMH MH60451 also funded by Glaxo Smith Kline): Christine Hulette, MD, Director, John F. Ervin, Stanford University: Dikran Horoupian, MD, Ahmad Salehi, MD, PhD, New York Brain Bank, Taub Institute, Columbia University (NYBB): Jean Paul Vonsattel, MD, Katerina Mancevska, Massachusetts Alzheimer’s Disease Research Center (P50 AG005134): E. Tessa Hedley-Whyte, MD, MP Frosch, MD, Karlotta Fitch, University of Michigan (NIH grant P50-AG08671): Dr. Roger Albin, Lisa Bain, Eszter Gombosi, University of Kentucky (NIH #AG05144): William Markesbery, MD, Sonya Anderson, Mayo Clinic, Jacksonville: Dennis W. Dickson, MD, Natalie Thomas, University Southern California: Caroll A. Miller, MD, Jenny Tang, M.S., Dimitri Diaz, Washington University, St Louis Alzheimer’s Disease Research Center (NIH #P50AG05681): Dan McKeel, MD, John C. Morris, MD, Eugene Johnson, Jr., PhD, Virginia Buckles, PhD, Deborah Carter, University of Washington, Seattle (NIH #P50 AG05136):Thomas Montine, MD, PhD, Aimee Schantz, MEd., University of Pennsylvania School of Medicine, Alzheimer’s Disease Research Center: John Q Trojanowski, MD, Virginia M Lee, MD, Vivianna Van Deerlin, MD, Terry Schuck, Boston University Alzheimer’s Disease Research Center (NIH grant P30-AG13846): Ann C. McKee, Carol Kubilus Sun Health Research Institute Brain Donation Program of Sun City, Arizona (NIA #P30 AG19610; Arizona Alzheimer’s Disease Core Center, Arizona Department of Health Services, contract 211002, Arizona Alzheimer’s Research Center; Arizona Biomedical Research Commission, contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium; Michael J. Fox Foundation for Parkinson’s Research): Joseph Rogers, PhD, Thomas G. Beach, MD, PhD, Lucia I. Sue Emory University: Bruce H. Wainer, MD, PhD, Marla Gearing, PhD, University of Texas, Southwestern Medical School: Charles L. White, III, M.D., Roger Rosenberg, Marilyn Howell, Joan Reisch, University of California, Davis: William Ellis, MD, Mary Ann Jarvis, Rush University Medical Center, Rush Alzheimer’s Disease Center (NIH #AG10161): David A. Bennett, M.D. Julie A. Schneider, MD, MS, Karen Skish, MS, PA (ASCP)MT, Wayne T Longman, University of Miami/NPF Brain Endowment Bank: Deborah C. Mash, MD, Margaret J Basile, Mitsuko Tanaka Oregon Health & Science University: Randy Wotljer, PhD. Additional tissues include samples from the following sites: Newcastle Brain Tissue Resource (funding via the Medical Research Council, local NHS trusts and Newcastle University): C.M. Morris, MD, Ian G McKeith, Robert H Perry MRC London Brain Bank for Neurodegenerative Diseases (funding via the Medical Research Council): Simon Lovestone, Md PhD, Safa Al-Sarraj. MD, Claire Troakes, South West Dementia Brain Bank (funding via numerous sources including the Higher Education Funding Council for England (HEFCE), Alzheimer’s Research Trust (ART), BRACE as well as North Bristol NHS Trust Research and Innovation Department and DeNDRoN): Seth Love, MD, Patrick Kehoe, PhD, Laura Palmer, The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stiching Alzheimer Onderzoek): Inge Huitinga, MD, Marleen Rademaker, Michiel Kooreman, Institut de Neuropatologia, Servei Anatomia Patologica, Universitat de Barcelona: Isidre Ferrer Abizanda, MD, PhD, Susana Casas Boluda.
Footnotes
Disclosure statement
The authors declare no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, Kukull WA. The National Alzheimer’s Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270–277. [PubMed] [Google Scholar]
- Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva JP, Junior, Neto Evaldo S, Poffo MA, Walz R, Carlotti CG, Junior, Sakamoto AC. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy--PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004;24:1–24. doi: 10.1023/B:CEMN.0000012721.08168.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–4995. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–284. [DOI] [PubMed] [Google Scholar]
- Carvajal-Carmona LG, Ophoff R, Service S, Hartiala J, Molina J, Leon P, Ospina J, Bedoya G, Freimer N, Ruiz-Linares A. Genetic demography of Antioquia (Colombia) and the Central Valley of Costa Rica. Hum Genet. 2003;112:534–541. doi: 10.1007/s00439-002-0899-8. [DOI] [PubMed] [Google Scholar]
- Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL, Megarbane A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum Mutat. 2008;29:E194–204. doi: 10.1002/humu.20836. [DOI] [PubMed] [Google Scholar]
- Cingolani P. snpEff: Variant effect prediction. 2011 http://snpeff.sourceforge.net.
- Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3:445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, Nalls MA, Chen K, Lee W, Chewning K, Villa SE, Meechoovet HB, Gerber JD, Frost D, Benson HL, O’Reilly S, Chibnik LB, Shulman JM, Singleton AB, Craig DW, Van Keuren-Jensen KR, Dunckley T, Bennett DA, De Jager PL, Heward C, Hardy J, Reiman EM, Huentelman MJ. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–3301. doi: 10.1093/hmg/ddq221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook R, Hardy J, Duff K. Single-day apolipoprotein E genotyping. J Neurosci Methods. 1994;53:125–127. doi: 10.1016/0165-0270(94)90168-6. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J. TREM2 Variants in Alzheimer’s Disease. N Engl J Med. 2012;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H, Emre M, Singleton A, Hardy J. Using Exome Sequencing to Reveal Mutations in TREM2 Presenting as a Frontotemporal Dementia-like Syndrome Without Bone Involvement. Arch Neurol. 2012:1–7. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Ph DS, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N Engl J Med. 2012;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005;64:1502–1507. doi: 10.1212/01.WNL.0000160304.00003.CA. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27:6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, Zismann VL, Joshipura K, Huentelman MJ, Hu-Lince D, Coon KD, Craig DW, Pearson JV, Holmans P, Heward CB, Reiman EM, Stephan D, Hardy J. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, Satoh JI. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. 2011;18:1179–1183. doi: 10.1111/j.1468-1331.2010.03311.x. [DOI] [PubMed] [Google Scholar]
- Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, Faccio R, Ross FP, Teitelbaum SL, Takayanagi H, Colonna M. TREM2 and beta-Catenin Regulate Bone Homeostasis by Controlling the Rate of Osteoclastogenesis. J Immunol. 2012;188:2612–2621. doi: 10.4049/jimmunol.1102836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Trujillo N, Apergi M, Moreno S, Arias W, Lesage S, Franco A, Sepulveda-Falla D, Cano D, Buritica O, Pineda D, Uribe CS, de Yebenes JG, Lees AJ, Brice A, Bedoya G, Lopera F, Ruiz-Linares A. A genetic cluster of early onset Parkinson’s disease in a Colombian population. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:885–889. doi: 10.1002/ajmg.b.30375. [DOI] [PubMed] [Google Scholar]
- Pottier C, Wallon D, Rousseau S, Rovelet-Lecrux A, Richard AC, Rollin-Sillaire A, Frebourg T, Campion D, Hannequin D. TREM2 R47H Variant as a Risk Factor for Early-Onset Alzheimer’s Disease. J Alzheimers Dis. 2013 doi: 10.3233/JAD-122311. [DOI] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield VC, Stone EM, Carmi R. Use of isolated inbred human populations for identification of disease genes. Trends Genet. 1998;14:391–396. doi: 10.1016/s0168-9525(98)01556-x. [DOI] [PubMed] [Google Scholar]
- Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, Rohrer K, Zhao A, Marlowe L, Kaleem M, McCorquodale DS, 3rd, Cuello C, Leung D, Bryden L, Nath P, Zismann VL, Joshipura K, Huentelman MJ, Hu-Lince D, Coon KD, Craig DW, Pearson JV, Heward CB, Reiman EM, Stephan D, Hardy J, Myers AJ. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]