

Abstract
Enzymatic reactions occur in a crowded and confined environment in vivo, containing proteins, RNA and DNA. Previous reports have shown that interactions between macromolecules, and reactions rates differ significantly between crowded environments and dilute buffers. However, the direct effect of crowding on the level of high-resolution structures of macromolecules has not been extensively analyzed and is not well understood. Here we analyze the effect of macromolecular crowding on structure and function of the human translation initiation factors eIF4A, a two-domain DEAD-Box helicase, the HEAT-1 domain of eIF4G, and their complex. We find that crowding enhances the ATPase activity of eIF4A, which correlates with a shift to a more compact structure as revealed with small angle X-ray scattering. However, the individual domains of eIF4A, or the eIF4G-HEAT-1 domain alone show little structural changes due to crowding except for flexible regions. Thus, the effect of macromolecular crowding on activity and structure need to be taken into account when evaluating enzyme activities and structures of multi-domain proteins, proteins with flexible regions, or protein complexes obtained by X-ray crystallography, NMR or other structural methods.
Keywords: eIF4G, eIF4A, macromolecular crowding, enhancement, NMR, SAXS
INTRODUCTION
Cellular environments are crowded. Concentrations of macromolecules (DNA, RNA and proteins) vary widely between cell types and growth phase. For Escherichia coli concentrations are in the range of 300-400 g/L. These macromolecules occupy a large fraction of the cellular volume (20-30%)2. In the past, biophysical properties of macromolecules have been traditionally measured in dilute solutions. However, the complex environment inside cells changes these properties3 and the biochemical parameters such as equilibrium constants, association and dissociation rates. Thus, in-vitro structural parameters as obtained from solution of isolated components, are significantly different from those in a crowded compartment3. Recent progress has been made in studying the structure and function of proteins in cellular environments, for example by in-cell NMR6. However, such an endeavor is often not feasible for detailed structural and functional studies. However, cellular conditions can be mimicked using inert crowding agents, such as polyethylene glycol (PEG), Dextran, or Ficoll. Other proteins like bovine serum albumin are used as crowding agents as well7. However, the most widely used crowding agent is PEG. Numerous studies have shown the influence of PEG on enzymatic reaction rates, thermal denaturation, protein folding8-10 and renaturation. Several studies show the effect of crowding on protein structure using simulations11,12. We have shown recently the impact of macromolecular crowding on the structure and the function of the replisome of bacteriophage T714. To our knowledge, there are no experimental studies examining the influence of PEG on the atomic structure of folded proteins.
Here we investigate the effect of crowding on factors involved in human translation initiation. The control of translation (mRNA encoded protein synthesis) is crucial for cell proliferation and differentiation. More than ten eukaryotic translation initiation factors are known to be involved in the assembly of the 80S ribosome-RNA complex enabling placement of the initiator Met-tRNA at the correct start codon of the mRNA. The dominant mechanism used by cellular mRNAs, named cap dependent translation, requires the recognition of the 5′ m7G cap structure of the mRNA by the cap binding complex eIF4F. The eIF4F complex is composed of the cap binding subunit eIF4E, the RNA helicase eIF4A, and the scaffolding protein eIF4G15-17. The eukaryotic translation factor 4A (eIF4A) is the prototypic member of the DEAD-box family of RNA helicases18, a subfamily of superfamily 2 (SF2) of RNA helicases19. The ATP-dependent RNA helicase eIF4A unwinds secondary structure in the 5′ untranslated region (UTR) of mRNAs to facilitate 40S ribosome binding and scanning for the start codon. eIF4A by itself is a poor ATPase and helicase (kcat = 3/min)20. However, these activities are stimulated when eIF4A is part of a multiprotein complex consisting of eIF4G, eIF4E, eIF4B and/or eIF4H 21-25. eIF4G, a scaffolding protein that binds several initiation factors, contains three HEAT repeat domains in the C-terminal two thirds of its sequence. The first two, HEAT1 and HEAT2 bind to eIF4A. Binding of HEAT1 increases kcat by about 4-fold26. Binding of HEAT2 is thought to have only a modulatory role15,16,26. There are now high-resolution structures available for full-length human eIF4A, eIF4G-HEAT1 or their complex. However the domain orientation of eIF4A differs significantly between the various structures. Nevertheless, the structural information on the domains of this system allows now mechanistic analysis of crowding with structural methods, such as NMR spectroscopy or small angle X-ray scattering (SAXS).
Thus far, NMR studies of crowding effects on proteins under physiological conditions have focused primarily on measuring relaxation parameters and protein stability27 and targeted mainly disordered proteins. Studies of globular proteins and protein complexes, and experiments that could provide more mechanistic insights are hampered by increased line broadening and unfavorable relaxation properties. Nevertheless, recently the structure of a 66 amino acid protein, TTHA17178 from Thermus thermophilus, was determined in cells by NMR28. However, the samples used were highly concentrated (3-4 mM) and an attempt to determine the structure of a larger protein (17 kDa) was unsuccessful. Additionally, the RMSD of the final structures seems significantly higher than the RMSD of the structure of the same protein determined in solution. Thus, differences due to cellular crowding are difficult to assess.
In the present study we have used biochemical assays, SAXS and NMR to characterize the effect of macromolecular crowding on the structure and activity of eIF4A and eIF4G-HEAT1. The combination of SAXS and NMR overcomes some of the problems of studying proteins in a crowded medium by NMR only. There is no size limitation for SAXS, and SAXS also allows investigating proteins that are difficult to measure by NMR, such as eIF4A. Indeed, we find a clear correlation between structural and functional properties of the translation initiation complex. Addition of crowding agent, which mimics cellular conditions, shifts the conformational equilibrium of eIF4A from an inactive open state towards the closed active conformation. On the other hand, the crowding effect is rather minor for the folded part of isolated HEAT1 domain of eIF4G but significantly alters mobile regions.
MATERIALS AND METHODS
Protein expression and purification
Human eIF4GI (747-994, 4GH1) was expressed in E. coli Bl21(DE3) with N-terminal GB1 and His-tags and purified via metal immobilized affinity chromatography using TALON resin (Clontech, USA) followed by overnight TEV cleavage at 4°C. The cleaved product was purified by size exclusion chromatography. Human eIF4A was expressed in E. coli Bl21(DE3) with an N-terminal His-tag and purified via metal immobilized affinity chromatography using TALON resin (Clontech, USA) followed by Q-Sepharose and size exclusion chromatography. 15N, D labeled 4GH1 was expressed in M9 media dissolved in D2O with 15NH4Cl as the sole nitrogen source. Chemical shift assignments of eIF4G-HEAT1 were published previously29.
Small/Wide Angle Scattering (SAXS/WAXS)
SAXS/WAXS data were collected at beamline X9 of the National Synchrotron Light Source at Brookhaven National Laboratory in New York as described previously30 on a MarCCD SAXS detector and Photonic Science WAXS detector at 14.5 keV, respectively. The X-ray wavelength was 0.855 Å. All samples were measured at a temperature of 15°C. SAXS data was also measured at the Cornell High Energy Synchroton Source (CHESS, beamline G1) at an X-ray energy of 9.467 keV (λ = 1.3096 Å) using a CCD detector.
Samples were dialyzed overnight against SAXS buffer (eIF4G: 20 mM HEPES at pH 6.5, 2 mM DTT, 300 mM KCl, 0.01% NaN3 , 10% glycerol, eIF4A: 20 mM HEPES, pH 7.5, 130 mM KCl, 0.01% NaN3, 10% glycerol; eIF4A/eIF4G: 20 mM HEPES, pH 7, 130 mM KCl, 0.01% NaN3, 10% glycerol) using Slide-A-Lyzer cassettes (pierce, 10kDa MWCO). 5 mM ATP and 3 mM MgCl2 were added to the buffer for samples containing ATP. Dialysis buffers were kept as reference samples for buffer subtraction. Samples for measuring concentration series were adjusted to 1 - 10 mg/mL. Samples for measuring the effect of PEG were adjusted to 5 mg/mL. Polyethylene glycol 1k was added to 8% to the sample and the corresponding buffer sample. Samples were exposed for 60s and 120s. Scattering of the buffer alone was measured with 60s and 120s exposure as well and subsequently subtracted from the sample scattering. The magnitude of the scattering vector (q) is defined as
| (equation 1) |
where 2θ is the scattering angle and λ is the wave length.
Scattering curves were analyzed using PRIMUS11, GNOM12 and in-house scripts31. No radiation damage or aggregation was observable. SAXS and WAXS scattering curves were merged using in-house scripts31. Values for the radius of gyration were obtained from the linear region of the Guinier plot, using data from the low angle region with qRg < 1.3, the Guinier approximation
| (equation 2) |
and the program GNOM, which was used to fit the experimental data. DAMMIN1 was used to model the structural envelope based on the GNOM fit. 10 models per sample were calculated and averaged using DAMMAVER14.
NMR Spectroscopy
All NMR measurements were conducted at 298 K on Bruker Avance 750 MHz and Avance 600 spectrometers equipped with TXI cryoprobes with Z gradient. Data were processed and analyzed using NMRPipe32 and NMRView33. 2D [1H,15N] TROSY HSQC experiments were acquired at 750 MHz using 256 t1 increments with a sweep width of 2659 Hz and 2048 t2 points with a sweep width of 9014 Hz. Samples containing 0.5 mM 15N,D-eIF4G-HEAT1 in 10 mM phosphate buffer pH 6.5, 300 mM KCl, 2 mM DTT, 0.5 mM EDTA, 0.01% NaN3, 5% D2O. PEG 400 was titrated to concentrations of 1%, 2%, 5%, 8%, and 16%. eIF4A-CTD was titrated to 0.36 mM 15N,D-eIF4G-HEAT1 at ratios of 0:1, 0.25:1, 0.5:1, 0.75:1 and 2:1. Binding of full length eIF4A was evaluated by comparing the spectra of 15N,D-eIF4G-HEAT1 and 15N,D-eIF4G-HEAT1 with eIF4A at a 1:1 ratio. Chemical shift changes were calculated based on the formula
| (equation 3) |
Linewidth analysis was performed using NMRView34. Measurements of R1 and R2 were performed at 600 MHz using previously reported sequences35. R1 measurements were based on the acquisition of 7 timepoints with delays of 20 −1500 ms. R2 measurements were based on 6 timepoints with delays of 16.8 −100.8 ms. Peak intensities were determined using NMRViewJ peak picking procedures. R1 and R2 values were obtained by non-linear least square fitting of the data to exponential curves I(t) = I0exp(–Rjt) with j=1 or 2 using NMRViewJ and in-house Matlab scripts. Relaxation data was analyzed using NMRView, Matlab and in-house scripts. Tumbling times τc were determined using the software TENSOR36.
Modeling the structures of eIF4A and eIF4G-HEAT1
eIF4G-HEAT1 was modeled using SWISS-MODEL5 based on the structures of isoform II of eIF4G4 (PDB code 1HU3) and the structure of yeast eIF4G-HEAT1 37 (PDB code 2VSO), the open and closed form of eIF4A and the complex between eIF4G-HEAT1 and eIF4A were modeled based on the structures of the corresponding yeast proteins 37,38 (PDB codes 2VSO and 1FUU).
TLC assay
ATP hydrolysis by eIF4A was determined using thin-layer chromatography (TLC). Reaction mixtures contained 3 μM eIF4A, 3 μM poly(U), 5 mM ATP, 0.1 mCi of [α-32P] ATP, and polyethylene glycol (PEG) of varying molecular weights and concentrations (0-8%) as indicated in the corresponding figures. After 1-hr incubation at 37 °C, 0.4 μL of the reaction mixture was spotted onto a ESI cellulose TLC plate and developed using 0.5 M LiCl, 0.5 M Na-formate. The amount of [32P]-Pi formed was analyzed using a Fuji BAS 1000 Bioimaging analyzer. TLC spots were further analyzed using Melanie 7 (GeneBio, Geneva, Switzerland).
Norit assay
The standard assay for ATPase measures the formation of Norit-adsorbable nucleoside arising from the hydrolysis of [γ-32P]-ATP. The reaction mixture (20 μl) contained 10 mM Na-phosphate buffer (pH 7), 80 mM KCl, 3 mM MgCI2, 50 mM EDTA, 2 mM DTT, 50 μM [γ-32P]-ATP (30 cpm/pmol), and 3 μM eIF4A. Incubation was at 37 °C for 60 minutes. The reaction was stopped by the addition of 200 μL of a solution consisting of 0.6 ml of 1 M HCl, 0.1 M Na4P2O7, 0.02 M KH2PO4, 0.2 ml of a Norit A suspension (20% packed volume), and 0.1 ml of bovine serum albumin (5 mg/ml). The solutions were mixed and allowed to sit for 5 min at 0 °C. After centrifugation, 4 μL of the supernatant was spotted on a filter paper and the radioactivity was determined.
Normal Mode Analysis (NMA)
The force constant matrix based on atomic potentials with respect to Cartesian coordinates can be used to describe the motion of a molecule39,40. The eigenvectors of this matrix are called normal modes. The vibrational frequency is determined by the eigenvalues. NMA can be used to examine vibrational and thermal properties of molecular structures at the atomic level. We calculated the normal modes of eIF4G-HEAT1 based on the model using the NOMAD-REF server13.
RESULTS
Macromolecular crowding alters the rate of enzymatic reactions. Using polyethylene glycol (PEG) as an inert crowding agent, we examined the effect of macromolecular crowding on the ATPase activity of the eukaryotic translation initiation factor 4A (eIF4A) and its dependence on the eukaryotic translation initiation factor 4G (eIF4G). We also examined the effect of PEG on the structures of these proteins.
PEG is the most widely used crowding agent. It is a straight chain polymer with repeating subunits of the formula HO-CH2-[CH2-O-CH2]n-CH2-OH. It was shown that PEG and other crowding agents increase the rate of enzymatic reactions41, alter reaction products42, protect macromolecules from thermal denaturation11, accelerate protein folding43 and facilitate nucleic acid renaturation44. At higher concentrations (up to about 30%) PEG is used to precipitate proteins to aid in protein purification45.
The ATPase activity of eIF4A is enhanced by PEG
eIF4A is an RNA helicase that utilizes ATP as a source of energy to unwind RNA46. By itself the rate of ATP hydrolysis eIF4A is extremely poor (3 min−1)47. We used thin layer chromatography (TLC) and a Norit assay to determine the effect of PEG on the ATPase activity of eIF4A. The effect of PEG depends on the size of PEG as shown in Figure 1a. The effect is due to the increase of the excluded volume with increasing PEG size. At very large PEG sizes (35k PEG) eIF4A tends to precipitate. To determine if PEG changes the ATPase activity by affecting the eIF4A:RNA or the eIF4A:ATP interaction, we performed the same experiment with and without 3 μM RNA (poly(U)). The trend of the curve is the same with and without RNA, indicating that PEG effects the eIF4A:ATP interaction.
Figure 1.

The effect of PEG on the ATPase activity of eIF4A. a) The influence of the size of PEG on the ATPase activity. eIF4A at 3 μM was incubated for 1h with 3 μM poly(U) (black) and without RNA (grey), 5 mM ATP with 0.1 mCi of [α-32P] ATP and 4% PEG of varying sizes. The amount of released Pi was determined using the Norit assay. b) Ratio of Pi:ATP after incubation of 3 μM eIF4A at varying concentrations of PEG 8k. Amounts of Pi and ATP in the samples (same content as described in a) containing varying concentrations of PEG 8k were determined using a TLC assay. c) Dependence of the ATPase activity for increasing amounts of ATP with and without 8% PEG 8k. Sample content was the same as described in A) with the exception of varying ATP concentrations. All data points are averages of duplicates.
The effect of PEG 8k on the ATPase activity of eIF4A was determined using TLC. Figure 1b shows the ratio [32P]-Pi versus [32P]-ATP on the TLC plate with increasing amounts of PEG 8k. As seen in the figure, PEG enhances the ratio of Pi to ATP, which shows an increase of the ATP hydrolysis, up to a factor of 6 at 10% PEG 8k. Figure 1c documents the effect of PEG on the ATPase activity of eIF4A with increasing amounts of ATP and constant concentration of eIF4A and RNA.
The activity of eIF4A without PEG increases only slightly with increasing amounts of ATP (red circles). After addition of PEG the activity increases significantly with increasing amounts of PEG (black circles).
Several other crowding agents are frequently used. We compared the effect on the eIF4A activity of several agents besides PEG, namely Dextran (6k, 40k and 70k), Ficoll (70k) and PVP (10k and 40k) over a concentration range of 0-4%. The results are shown in Figure S1. All these crowding agents show an increase in activity with increasing concentration of the crowding agent. PEG 8k leads to the largest increase followed by PVP. Ficoll and Dextran show a smaller increase. These crowding agents are most often used at much higher concentrations, so we would expect a larger effect at higher concentrations.
Enhancement of eIF4A ATPase activity by eIf4G-HEAT1 and PEG
eIF4A is a poor, non-processive helicase when acting alone20. It’s activity is enhanced in the heterotrimeric translation initiation complex with eIF4G and eIF4E, and additionally by eIF4B and/or eIF4H48,49. eIF4G contains two eIF4A binding sites, one in the first and the other in the second HEAT domain50. A construct containing the first HEAT domain increases RNA affinity and the catalytic step but does not affect ATP affinity. However, a construct containing the second HEAT domain decreases affinity to ATP and RNA with no effect on the catalytic step16,26. The enhancement of the ATPase activity of eIF4A by the first HEAT domain of eIF4G (eIF4G-HEAT1) with and without PEG as a crowding agent is shown in Figure 2. First, we tested the effect of the eIF4A:eIF4G-HEAT1 ratio on the ATPase activity. As can be seen in Figure 2a, addition of eIF4G-HEAT1 enhances the activity by a factor of 1.6 for a 1:1 ratio of eIF4A:eIF4G-HEAT1, and up to a factor of 3.6 at a 1:10 ratio of eIF4A:eIF4G-HEAT1. Figure 2b shows the effect of increasing PEG concentrations at constant eIf4A:eIF4G-HEAT ratios of 1:0, 1:1 and 1:10. Increasing amounts of PEG increase the ATPase activity of eIF4A alone (red diamonds) and for the complex (blue circles: 1:1 ratio, black squares: 1:10 ratio).
Figure 2.
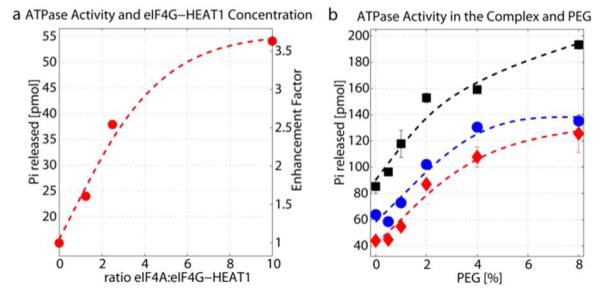
Enhancement of the eIF4A ATPase activity by eIF4G-HEAT1. a) Effect of the eIF4G-HEAT1:eIF4A ratio on the ATPase activity of eIF4A determined using the Norit assay. b) Influence of PEG on the ATPase activity of eIF4A alone and at two selected ratios of eIF4G-HEAT1:eIF4A (1:1 and 1:10). All data points are averages of duplicates.
Structural changes induced by PEG as measured by SAXS
We measured the SAXS curves of eIF4A, eIF4G-HEAT1 and their complex in the absence and presence of 8% PEG 1000. The Scattering curves of the three samples without PEG are shown in Figure 3a. Figure 3b-d depicts the Guinier plot of the small angle region for samples with (grey) and without (black) PEG as crowding agent. The solid lines represent the regression to the linear part of the Guinier plot. The slope of this regression is proportional to the square of the radius of gyration. Results for the Rg values extracted from the Guinier plot are shown in Table 1.
Figure 3.
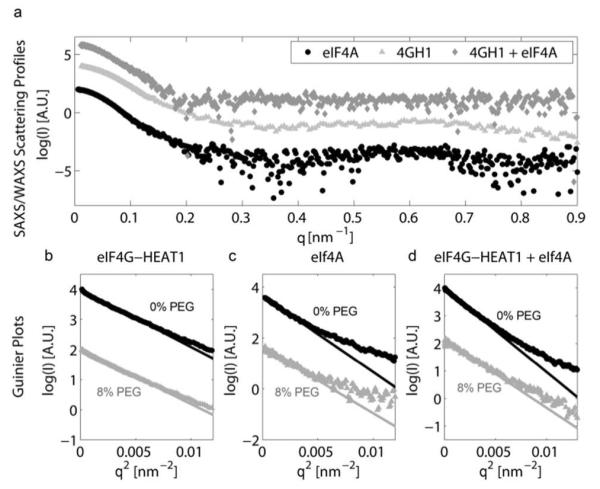
a) SAXS/WAXS scattering profiles and Guinier plots for eIF4A, eIF4G-HEAT and their complex. SAXS/WAXS Scattering profiles of eIF4A (circles), eIF4G-HEAT1 (triangles) and their complex (diamonds). b-d) Guinier plots and linear fit for the scattering profiles of eIF4G-HEAT1, eIF4A and their complex for samples with (grey) and without (black) the addition of PEG 1k. Note that all curves were translated vertically to avoid overlap and gain better visibility.
Table 1.
Rg and Dmax Values Derived From SAXS Data
| Sample | Rg [Å] (Guinier)a |
Rg [Å] (GNOM)b |
Dmax [Å]b |
|
|---|---|---|---|---|
| 0% PEG 1k | eIf4G-HEAT1 | 23.6 ± 0.04 | 23.41 ± 0.02 |
68 |
| eIF4G-HEAT1/ eIF4A/ATP |
30.6 ±0.2 | 30.62 ±0.05 | 95 | |
| eIF4A/ATP | 29.7 ±0.3 | 29.8 ±0.1 | 85 | |
| 8% PEG 1k | eIf4G-HEAT1 | 23.3 ±0.1 | 23.32 ±0.02 | 71 |
| eIF4G-HEAT1/ eIF4A/ATP |
26.4 ±0.4 | 27.4 ±0.1 | 82 | |
| eIF4A/ATP | 27.6 ±0.3 | 25.2 ±0.1 | 68 |
We also determined Rg and Dmax (the longest distance within the molecule) using the fitting software GNOM51. Results are summarized in Table 1. It is evident from the data, that the crowding agent has a large effect on the Rg and Dmax of eIF4A and its complex with eIF4G-HEAT1, but only a small effect on the size of eIF4G-HEAT1 alone. GNOM fits using the small angle regions and resulting distance distribution functions are shown in Figure S2. Prior to measuring the samples with and without PEG we measured a concentration series of eIF4G-HEAT and eIF4A to evaluate the effect of protein concentration on the Rg value. These concentration curves are shown in Figure S3. No significant effect of the concentration on Rg was found for eIF4A. A very slight increase in Rg of <3% was found for eIF4G-HEAT1, however, Rg values for 5 mg/mL protein (as used for further experiments) and 1 mg protein (~ infinite dilution) are still within errors.
Using the software DAMMIN we determined the structural envelopes from the SAXS curves of the three samples with and without PEG. We compared these envelopes with structural models of eIF4A, eIF4A in complex with eIF4G-HEAT1 and eIF4G-HEAT1 derived from crystal structures of isoform II of eIF4G-HEAT14, the structure of eIF4G-HEAT1 from yeast37, the open form of eIF4A from yeast and the eIF4G-HEAT1/eIF4A complex from yeast37,38 (Figure 4). Figure 4a and 4b show the filtered average structures of eIF4A without (a, red) and with (b, blue) PEG.
Figure 4.
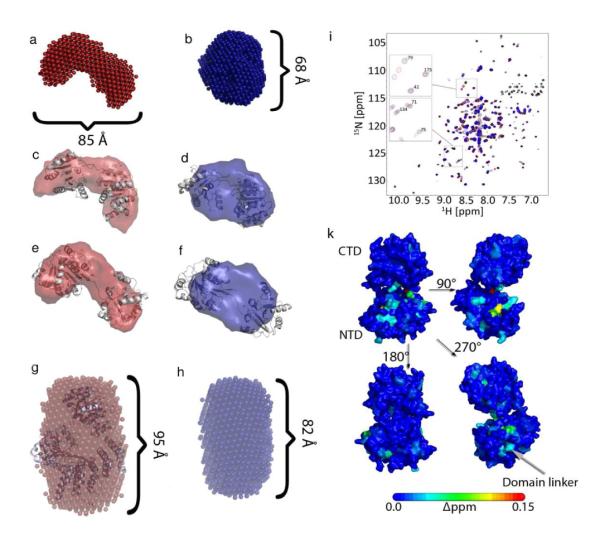
Structural envelopes determined based on SAXS data of eIF4A and the complex between eIF4A and eIF4G-hEAT1. The structural envelopes were calculated using DAMMIN1. a) and b) show the structure calculated for eIF4A without (a, red) and with 8% PEG 1k (b, blue). C) and d) show the overlay of the SAXS envelope for EIF4A without (c, red) and with 8% PEG 1k (d, blue) with the model of the closed conformation of eIF4A. E) and f) show the overlay of the same SAXS envelopes with the open conformation of eIF4A. G) and h) show the structural envelopes for the eIF4A/eIF4G-HEAT1 complex without (g, red) and with 10% PEG 1000 (h, blue). i) [15N,H]-TROSY HSQC spectra of the titration of PEG 400 to 0.3 mM eIF4A NTD (black: 0% PEG, red: 4% PEG, blue: 8% PEG). Selected peaks are shown in the excerpts. k) Mapping of chemical shift changes induced by PEG onto the structures of eIF4A-CTD and eIF4A-NTD.
The structure of eIF4A with PEG is much more compact indicating closure of the main cleft between the two RecA like domains. Figures 4c and 4d show the overlay of the representative structures of eIF4A without (4c) and with (4d) PEG with the model of the open form of eIF4A (non-ATP bound). Figures 4e and 4f show the overlay of the same representative structures with the model of the closed form of eIF4A. The open form overlays well with the representative structure of eIF4A without PEG, whereas the closed form overlays well with the envelope of eIF4A with PEG, suggesting that the conformation most prevalent in a crowded environment is more similar to the closed/active form than to an open, more flexible form.
We measured the TROSY HSQC spectra of the two domains of eIF4A with and without PEG. Whereas eIF4A-CTD does not show any significant chemical shift changes, residues of eIF4A-NTD facing eIF4A-CTD shift significantly. An overlay of the HSQC spectra for eIF4A-NTD is shown in Figure 4i. Additional peaks that shift originate from residues in the linker between the 2 domains as shown in Figure 4k. This data supports that the structures of the domains do not change significantly (except for some residues on eIF4A-NTD facing towards eIF4A-CTD) and the main conformational change originates from the domain-domain orientation.
Figures 4g and 4h depict the average structure of the eIF4A/eIF4G-HEAT1 complex without (g, red) and with (h, blue) PEG and the overlay with the model derived from the YEAST crystal structure. No major structural rearrangement can be observed, however, the structure of the complex is significantly more compact in the presence of crowding agent. This result corresponds well with the hypothesis that eIF4G confers the active conformation upon eIF4A when bound, so no additional rearrangement would be expected upon addition of PEG. The change of Dmax from 95 Å to 82 Å, and of Rg from 30.6 Å to 27.6 Å (Table 1 and Fig. 4) is consistent with a significant compaction of the structure.
Figure 5a is a homology model of eIF4G-HEAT1 obtained based on the crystal structure of isoform II of eIF4G-HEAT14. Figure 5b-c show the average SAXS structure of eIF4G-HEAT1 without (red) and with PEG (blue).
Figure 5.
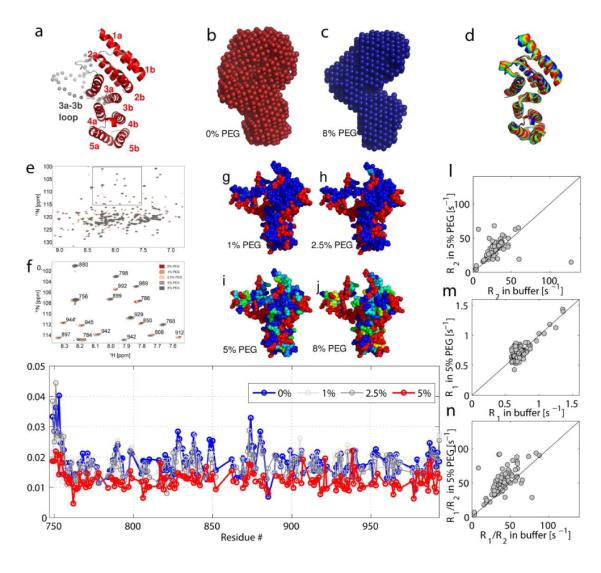
Effect of PEG on the structure and flexibility of eIF4G-HEAT1. a) Model of eIF4G-HEAT1 based on the structure of isoform II of eIF4G (PDB code 1HU34) determined using SWISS-MODEL5. The loop between residues 848-881 that was not visible in the electron density map is shown in gray spheres. All helices are labeled. b) and c) Structural models of eIF4G-HEAT1 without (b, red) and with PEG (c, blue) calculated by DAMMIN1 based on the SAXS experimental data. The location of the loop between 848 and 881 is indicated with an arrow. d) Overlay of snapshots of confirmations for motion of eIF4G-HEAT1 as determined by Normal Mode Analysis using the NOMAD-REF sever13. We only show the motion described by mode 14. The first 6 modes describe translational and rotational motions. The other modes are very similar. e) Overlay of the [15N,H]-TROSY HSQC spectra of the titration of PEG 400 to 0.5 mM 15N,D -eIF4G-HEAT1. f) shows an excerpt of e) with peak assignments. g)-j) Mapping of the changes in peak intensity with increasing concentrations of PEG (g: 1%; h: 2.5%; i: 5%; j: 8%). The degree of changes is indicated by the color coding as shown in the color bar on the bottom. k) Apparent T2 values for the samples described in e-j) plotted versus residue numbers. *T2 values were calculated according to linewidth=1/(πT2). l-n) 15N-R1 and 15N-R2 values measured for eIF4G-HEAT1. l) Plot of 15N-R2 with 5% PEG versus 15N-R2 in buffer. m) Plot of 15N-R1 with 5% PEG versus 15N-R1 in buffer. n) Plot of the R1/R2 ratio with 5% PEG versus the ratio in buffer.
Although the overall effect of crowding on the structure of eIF4G-HEAT1 is less pronounced than for eIF4A, there are several observable effects. First, the loop between residues 848 to 881 was not observed in the crystal structure of isoform II of eIF4G-HEAT1 and is shown with spheres in the model for isoform I in Figure 5a. Tihs loop is significantly more compact in the presence of PEG as indicated by a smaller occupied volume of the SAXS envelope in the region of the loop. The second difference is a larger Dmax in the presence of PEG (Table 1) and a slightly different angle between the upper and lower half of the molecule. In order to take the flexibility of the molecule into account, we calculated normal modes using the NOMAD-REF server13. Mode 14 shows a breathing type motion of eIF4G. Snapshots of the motion of mode 14 are shown in figure 5d. The two extrema of this motion show similar changes as the structures of eIF4G-HEAT with and without PEG. This would suggest that PEG influences the equilibrium between conformations shifting it towards a different conformation than is prevalent in buffer.
PEG reduces flexibility
To further explore the effect of PEG on the structure of eIF4G-HEAT1, we performed an NMR titration experiment. We measured the [15N,1H]-TROSY-HSQC spectra of 15N, D-eIF4G-HEAT1 with increasing amounts of PEG. Figure 5e shows the overlay of these spectra and Figure 5f show an excerpt of Figure 5e. Only very small changes in chemical shifts are observed upon addition of PEG indicating that PEG does not induce major structural changes and does not bind to EIF4G-HEAT1. Figure S4 shows the mapping of the chemical shift changes on the structure of eIF4G-HEAT1. The largest changes are observed for residues in the loop between residue 848-881 (3a-3b loop, see Figure S4a,c) and in helix 1a.
Additionally, we observed changes in the peak intensity upon addition of PEG. We therefore determined proton T2* values with increasing PEG 400 concentrations (0-5%) based on the line width of the HSQC peaks as shown in Figure 5g. This graph clearly shows an overall decrease in T2* values and high decrease in the regions containing the 3a-3b loop (residues 848-881), the 2b-3a loop (residues 820-833) as well as the N-terminus. Figure 5h-k shows the mapping of the changes in peak intensity on the structure of eIF4G-HEAT1. The changes in peak intensities/line width are not related to the charges or hydrogen bond network as was suggested for GB17 but are highest in flexible regions and especially the region between 848 and 881. The SAXS data suggests that the 3a-3b loop occupies less volume in the crowded medium, supporting the results from the proton T2* values. This region also shows the largest conformational changes as calculated by the normal mode analysis. Other regions that undergo large changes are on the face of the protein opposite to the flexible loop and the termini. Chemical shift changes are largest for the above described regions corroborating the notion of small local conformational changes..
Additionally, we measured the relaxation rates R1 and R2 for eIF4G-HEAT1 with and without 5% PEG 400. Plots of R1 and R2 as well as the ratio R1/R2 in PEG vs. buffer are shown in Figure X. Figure S5 shows T1 vs. T2 plots for the samples in buffer and PEG.
Tumbling rates τc were determined using the software TENSOR as 20.0 ns ± 0.1 ns for the sample without PEG and 20.1 ns ± 0.1 ns for the sample with PEG. Although not significant difference could be observed under these conditions, 15N-R2 values are slightly longer with PEG and the spread of 15N-R1 values is larger as well. The addition of PEG to eIF4G-HEAT1 causes very small chemical shift changes spreading through the entire molecule. As these changes are very small and effect residues on the surface as well as buried residues with no preference for residue type, it is highly unlikely that they are cause by binding of PEG. Major structural changes would result in large chemical shift changes, so we conclude that these minor changes might be caused by minor conformational changes.
Independently, we measured the chemical shift changes upon binding of eIF4A-CTD and eIF4A full length to EIF4G-HEAT1. eIF4A-CTD binds in the -terminal part of EIF4G-HEAT1. All residues in this binding site located on helix 1a, 1b, 2a and 2b and their connecting loops disappear in the TROSY HSQC spectrum upon binding. These residues are represented as grey spheres in Figure S4c. In addition to those residues, several other residues show minor chemical shift changes. Addition of eIF4A full length to eIF4G-HEAT leads to additional chemical shift changes.
Just like for the sample with PEG, these chemical shift changes spread over the entire molecule suggesting minor conformational changes such as a small shift in the conformational equilibrium.
Comparing the chemical shift changes upon binding of eIF4A-CTD/eIF4A and PEG shows that structural changes occur in similar positions throughout the molecule (see Figure S4). Therefore, although eIF4G-HEAT1 is more rigid, it does adopt a slightly different conformation in the bound form and that this conformation is sampled more frequently in the unbound form in a crowded environment.
DISCUSSION
We have studied the effect of macromolecular crowding on the structure and activity of the translation initiation factor eIF4A, the HEAT1 domain of eIF4G and their complex. Addition of high concentration of PEG has a significant effect on structure, dynamics and activity but the effect on eIF4A and its complex with eIF4G-HEAT-1 is much larger than that on HEAT-1 alone.
Conformation in a crowded environment
The scattering pattern of a protein in solution represents the average of all conformations present in the solution. The envelope obtained for a highly dynamic protein would therefore have a larger volume compared to the static protein. eIF4G-HEAT1 is a domain consisting of 10 helices. PEG affected the overall structure of this domain only slightly. We made two significant observations. First, the flexible linker between residues 111 and 144 is more confined in the crowded environment. Second, the overall flexibility of the molecule is affected and a slightly different conformation is preferred in the crowded medium. We used NMA to assess this difference in conformations. The range of motion determined by NMA explains the minor changes in the structures with and without PEG and suggests that the equilibrium between the conformations is shifted in the crowded environment.
In contrast to eIF4G-HEAT1, eIF4A is a two-domain protein with a flexible linker connecting the two domains. All DEAD-box helicases show a similar domain structure, however, the domain-domain orientations are different in all known structures of DEAD-box helicases due to the flexibility of the linker. The currently accepted model is that the apoenzyme (no ATP or ADP bound) adopts an open form with a large inter-domain angle. Binding of ATP to eIF4A leads to transformation to a closed, active conformation. eIF4A alone is a poor helicase whose activity is enhanced by binding to eIF4G. In this model eIF4G stabilizes the closed conformation of eIF4A and thereby enhances the activity.
We modeled the structure of eIF4A in the open and closed conformations based on the crystal structures of yeast eIF4A (PDB codes 1FUU and 2VSO). Comparing the SAXS envelope of eIF4A to the models (Figure 4) clearly shows that the structure of eIF4A in buffer is more similar to the open conformation and the structural envelope of eIF4A in the crowded medium is more similar to the closed conformation. Dmax decreases by 20% in the crowded vs. non-crowded environment. Current models suggest that eIF4A prefers the closed form upon binding to ATP and/or eIF4G. However, here we show, that the ATP bound form of eIF4A without a crowding agent is more similar to an open conformation. After addition of PEG as a crowding agent, the conformation becomes more compact and resembles the closed conformation of eIF4A.
Structural envelopes obtained using SAXS data are just like NMR data a representation of the structure of a protein in solution. And although, the crystal structure represent static pictures of an open and closed conformation, they correspond well with the structural envelopes representing a snapshot of the structure over time. We therefore conclude that the equilibrium between the open and closed conformation is shifted towards the open conformation in buffer and towards the closed conformation a crowded environment.
The overall shape of the eIF4A/eIF4G-HEAT1 complex doesn’t change significantly after addition of PEG, however, the complex is more compact under crowding conditions. This result confirms that eIF4G-HEAT1 confers the closed/active confirmation of eIF4A without the presence of the crowding agent.
Effect of PEG and eIF4G-HEAT1 on the ATPase activity of eIF4A
PEG has a significant effect on the structure of eIF4A pushing the equilibrium between open and closed form towards the closed conformation. We also show that PEG increases the ATPase activity of eIF4A by a factor of 6. Thus, we conclude that the prevalent conformation of eIF4A in a crowded environment, such as inside the cell, is compact and is the active conformation. This suggests that eIF4A is more active in a cellular environment than when measured in vitro. Just like in vitro in the absence of crowding agent, eIF4G-HEAT1 enhances eIF4A activity in the crowded environment as well. The accepted mechanism for this enhancement was that eIF4G-HEAT1 confers the closed/active conformation upon eIF4A. However, since the conformation equilibrium in the crowded medium is already shifted towards the closed confirmation, we conclude, that eIF4G must play an additional role in the enhancement of ATPase activity.
Phosphate release is the rate-limiting step in the catalytic cycle for DEAD box RNA helicases52. The independence of the ATPase activity of eIF4A on ATP concentration in dilute buffer as shown in Figure 1c complies with this observation. However, in the presence of 8% PEG 1000, the ATPase activity increases with increasing ATP concentration, showing that phosphate release and ATP hydrolysis are not rate limiting under these conditions.
CONCLUDING REMARKS
Macromolecular crowding has different effects depending on the system concerned. Diffusion controlled reactions might be slowed, enzymatic reaction rates accelerated, protein folding hindered or enhanced, and it is difficult to predict the effect accurately. Therefore, experimental data are crucial to properly characterize the system in question. We showed here that macromolecular crowding has a significant effect on structure and activity. The ATPase activity of eIF4A is significantly enhanced and it seems that phosphate release, the rate limiting step in dilute buffer, is not rate limiting in the crowded medium. The increase in activity is associated with structural changes. The structural effect is more pronounced for multi-domain proteins with flexible linkers but can clearly be observed for single domain proteins and flexible loops as well. We have shown that the conformational equilibria of eIF4A as well as eIF4G-HEAT1 structure are shifted towards the active forms in the crowded environment. This shift towards the active eIF4A state also explains the enhanced ATPase activity.
To our knowledge, this study shows the first high-resolution structural data on the effect of macromolecular crowding. This structural insight provides important information that needs to be considered for any biological macromolecules, in particular those containing multiple domains or protein complexes.
Supplementary Material
ACKNOWLEDGMENT
We thank Scott A. Robson for helpful discussions about 15N relaxation.
Funding Sources Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-0936384, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award GM-103485 from the National Institute of General Medical Sciences, National Institutes of Health. This research was supported by NIH grant CA068262 and the Agilent Foundation. Purchase and maintenance of equipment used was supported by NIH grants GM047467, S0RR026417 and EB002026
ABBREVIATIONS
- eIF4A
eukaryotic translation initiation factor 4A
- eIF4G
eukaryotic translation initiation factor 4G
- eIF4G-HEAT1
first HEAT domain of eIF4G
- PEG
polyethylene glycol
- TLC
thin layer chromatography
- NMA
normal mode analysis
- SAXS
small angle X-ray scattering.
Footnotes
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Additional figures: ATP hydrolysis of eIF4A with different crowding agents, GNOM Fit and Distance Distribution Functions for eIF4G-HEAT1, concentration dependence of the radius of gyration, chemical shift changes on eIF4G-HEAT1 with PEG and eIF4A-CTD, and 15N-T1/15N-T2 plots for eIF4G-HEAT1 with and without PEG. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Svergun DI. Biophys. J. 1999;76:2879. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ellis RJ. Curr. Opin. Struct. Biol. 2001;11:114. doi: 10.1016/s0959-440x(00)00172-x. [DOI] [PubMed] [Google Scholar]
- (3).Zhou H-X, Rivas G, Minton AP. Annu. Rev. Biophys. 2008;37:375. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Marcotrigiano J, Lomakin IB, Sonenberg N, Pestova TV, Hellen CU, Burley SK. Mol. Cell. 2001;7:193. doi: 10.1016/s1097-2765(01)00167-8. [DOI] [PubMed] [Google Scholar]
- (5).Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- (6).Selenko P, Wagner G. J. Struct. Biol. 2007;158:244. doi: 10.1016/j.jsb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- (7).Selenko P, Serber Z, Gadea B, Ruderman J, Wagner G. Proc Natl. Acad. Sci. U.S.A. 2006;103:11904. doi: 10.1073/pnas.0604667103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tsao D, Minton AP, Dokholyan NV. Plos One. 2010;5:e11936. doi: 10.1371/journal.pone.0011936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tokuriki N, Kinjo M, Negi S, Hoshino M, Goto Y, Urabe I, Yomo T. Protein Sci. 2004;13:125. doi: 10.1110/ps.03288104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).van den Berg B, Ellis RJ, Dobson CM. EMBO J. 1999;18:6927. doi: 10.1093/emboj/18.24.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang Q, Liang KC, Czader A, Waxham MN, Cheung MS. PLoS Comput. Biol. 2011;7:e1002114. doi: 10.1371/journal.pcbi.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stagg L, Zhang SQ, Cheung MS, Wittung-Stafshede P. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18976. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lindahl E, Azuara C, Koehl P, Delarue M. Nucleic Acids Res. 2006;34:W52. doi: 10.1093/nar/gkl082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Akabayov B, Akabayov SR, Lee SJ, Wagner G, Richardson CC. Nat. Commun. 2013;4:1615. doi: 10.1038/ncomms2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marintchev A, Wagner GQ. Rev. Biophys. 2004;37:197. doi: 10.1017/S0033583505004026. [DOI] [PubMed] [Google Scholar]
- (16).Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, Herdy B, Sonenberg N, Wagner G. Cell. 2009;136:447. doi: 10.1016/j.cell.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, Sonenberg N. Nat. Rev. Mol. Cell Biol. 2011;12:235. doi: 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- (18).Linder P, Lasko PF, Ashburner M, Leroy P, Nielsen PJ, Nishi K, Schnier J, Slonimski PP. Nature. 1989;337:121. doi: 10.1038/337121a0. [DOI] [PubMed] [Google Scholar]
- (19).Gorbalenya AE, Koonin EV. Curr. Opin. Struct. Biol. 1993;3:419. [Google Scholar]
- (20).Lorsch JR, Herschlag D. Biochemistry. 1998;37:2180. doi: 10.1021/bi972430g. [DOI] [PubMed] [Google Scholar]
- (21).Pause A, Methot N, Sonenberg N. Mol. Cell. Biol. 1993;13:6789. doi: 10.1128/mcb.13.11.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Grifo JA, Tahara SM, Leis JP, Morgan MA, Shatkin AJ, Merrick WC. J. Biol. Chem. 1982;257:5246. [PubMed] [Google Scholar]
- (23).Grifo JA, Abramson RD, Satler CA, Merrick WC. J. Biol. Chem. 1984;259:8648. [PubMed] [Google Scholar]
- (24).Richter-Cook NJ, Dever TE, Hensold JO, Merrick WC. J. Biol. Chem. 1998;273:7579. doi: 10.1074/jbc.273.13.7579. [DOI] [PubMed] [Google Scholar]
- (25).Abramson RD, Dever TE, Lawson TG, Ray BK, Thach RE, Merrick WC. J. Biol. Chem. 1987;262:3826. [PubMed] [Google Scholar]
- (26).Korneeva NL, First EA, Benoit CA, Rhoads RE. J. Biol. Chem. 2005;280:1872. doi: 10.1074/jbc.M406168200. [DOI] [PubMed] [Google Scholar]
- (27).Miklos AC, Li C, Pielak GJ. Methods Enzymol. 2009;466:1. doi: 10.1016/S0076-6879(09)66001-8. [DOI] [PubMed] [Google Scholar]
- (28).Sakakibara D, Sasaki A, Ikeya T, Hamatsu J, Hanashima T, Mishima M, Yoshimasu M, Hayashi N, Mikawa T, Walchli M, Smith BO, Shirakawa M, Guntert P, Ito Y. Nature. 2009;458:102. doi: 10.1038/nature07814. [DOI] [PubMed] [Google Scholar]
- (29).Akabayov SR, Wagner G. Biomol. NMR Assign. 2013 doi: 10.1007/s12104-013-9459-5. Advance online publication doi: 10.1007/s12104-013-9459-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Allaire M, Yang L. J. Synchrotron Radiat. 2011;18:41. doi: 10.1107/S0909049510036022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Akabayov B, Akabayov SR, Lee SJ, Tabor S, Kulczyk AW, Richardson CC. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15033. doi: 10.1073/pnas.1010141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J. Biomol. NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- (33).Johnson BA, Blevins RA. J. Biomol. NMR. 1994;4:603. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- (34).Johnson BA. Methods Mol. Biol. 2004;278:313. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- (35).Zhu G, Xia Y, Nicholson LK, Sze KH. J. Magn. Reson. 2000;143:423. doi: 10.1006/jmre.2000.2022. [DOI] [PubMed] [Google Scholar]
- (36).Dosset P, Hus JC, Blackledge M, Marion D. J. Biomol. NMR. 2000;16:23. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- (37).Schutz P, Bumann M, Oberholzer AE, Bieniossek C, Trachsel H, Altmann M, Baumann U. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9564. doi: 10.1073/pnas.0800418105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Caruthers JM, Johnson ER, McKay DB. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13080. doi: 10.1073/pnas.97.24.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tama F, Sanejouand YH. Protein Eng. 2001;14:1. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- (40).Suhre K, Sanejouand YH. Nucleic Acids Res. 2004;32:W610. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Minton AP. J. Biol. Chem. 2001;276:10577. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- (42).Zimmerman SB, Minton AP. Annu. Rev. Biophys. Biomol. Struct. 1993;22:27. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- (43).van den Berg B, Wain R, Dobson CM, Ellis RJ. EMBO J. 2000;19:3870. doi: 10.1093/emboj/19.15.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sikorav JL, Church GM. J. Mol. Biol. 1991;222:1085. doi: 10.1016/0022-2836(91)90595-w. [DOI] [PubMed] [Google Scholar]
- (45).Ingham KC. Methods Enzymol. 1984;104:351. doi: 10.1016/s0076-6879(84)04101-x. [DOI] [PubMed] [Google Scholar]
- (46).Rogers GW, Komar AA, Merrick WC. In: Progress in Nucleic Acid Research and Molecular Biology. Moldave K, editor. Vol. 307. Academic Press; San Diego: 2002. [DOI] [PubMed] [Google Scholar]
- (47).Lorsch JR, Herschlag D. Biochemistry. 1998;37:2180. doi: 10.1021/bi972430g. [DOI] [PubMed] [Google Scholar]
- (48).Rogers GW, Richter NJ, Lima WF, Merrick WC. J. Biol. Chem. 2001;276:30914. doi: 10.1074/jbc.M100157200. [DOI] [PubMed] [Google Scholar]
- (49).Rogers GW, Richter NJ, Merrick WC. J. Biol. Chem. 1999;274:12236. doi: 10.1074/jbc.274.18.12236. [DOI] [PubMed] [Google Scholar]
- (50).Imataka H, Sonenberg N. Mol. Cell. Biol. 1997;17:6940. doi: 10.1128/mcb.17.12.6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Svergun DI. J. Appl. Crystallogr. 1992;25:495. [Google Scholar]
- (52).Henn A, Cao W, Hackney DD, De La Cruz EM. J. Mol. Biol. 2008;377:193. doi: 10.1016/j.jmb.2007.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.