

Abstract
The mechanisms by which oncolytic vaccinia virus induces tumor cell death are poorly understood. We have evaluated cell death pathways following infection of ovarian cancer cells with both wild-type and thymidine kinase-deleted (dTK) Lister strain vaccinia. We show that death does not rely upon classical apoptosis despite the appearances of some limited apoptotic features, including phosphatidylserine externalization and appearance of sub-G1 DNA populations. Vaccinia infection induces marked lipidation of LC3 proteins, but there is no general activation of the autophagic process and cell death does not rely upon autophagy induction. We show that vaccinia induces necrotic morphology on transmission electron microscopy, accompanied by marked by reductions in intracellular adenosine triphosphate, altered mitochondrial metabolism, and release of high mobility group box 1 (HMGB1) protein. This necrotic cell death appears regulated, as infection induces formation of a receptor interacting protein (RIP1)/caspase-8 complex. In addition, pharmacological inhibition of both RIP1 and substrates downstream of RIP1, including MLKL, significantly attenuate cell death. Blockade of TNF-α, however, does not alter virus efficacy, suggesting that necrosis does not result from autocrine cytokine release. Overall, these results show that, in ovarian cancer cells, vaccinia virus causes necrotic cell death that is mediated through a programmed series of events.
Introduction
Vaccinia is an ideal oncolytic virus candidate owing to its ability to infect a broad range of cells, rapid replication cycle, and production of extracellular enveloped virions that evade the immune response1,2 and that may allow spread to distant metastases following local delivery.3 Systemic delivery of the oncolytic vaccinia JX-594 demonstrated safe and effective infection of tumor tissue,4 while randomized data indicate a survival advantage for patients with advanced hepatocellular carcinoma treated with high dose (109 plaque-forming units (pfu)) intratumoral JX-594 compared with low dose (108 pfu).5
The mechanism by which tumor cell death is induced by OVs remains poorly understood. Classical apoptosis, autophagy, and necrosis have all been implicated in vaccinia infection to varying degrees; cell lysis is a common endpoint of infection, apoptosis has been observed in some cancer cell lines6 and immune cells,7 and autophagy is disrupted in fibroblasts following infection.8 Programmed necrosis is also reported to have a role in the fate of vaccinia-infected T cells,9 while two previous studies indicated that tumor necrosis factor (TNF)-α treatment of vaccinia-infected mouse fibroblasts10 and Jurkat cells11 induced necrosis, which was dependent upon the viral caspase inhibitor B13R and receptor interacting protein (RIP)1, respectively.
Evasion of cell death is a hallmark of cancer, and little of the previous work attempting to characterize vaccinia-induced cell death has been performed in malignant cells. We have investigated cell death pathways in models of ovarian cancer following infection with Lister-dTK, an oncolytic Lister strain vaccinia virus bearing a deletion of the thymidine kinase gene. Our data show that classical apoptosis is not the primary mode of cell death execution. Vaccinia interferes with the autophagic process but does not increase autophagic flux and does not rely upon autophagy to induce death. Lister-dTK infection leads to both morphological and metabolic features of necrosis. We show that RIP1 and caspase-8 associate during vaccinia infection of ovarian cancer cells, while pharmacological inhibition of key necrosis proteins, including RIP1 and mixed lineage kinase domain-like protein (MLKL),12 significantly attenuates vaccinia-induced cell death. Inhibition of TNF-α signaling, by contrast, has no effect on viral efficacy. Along with visible necrosis in infected tumors observed in vivo, these data strongly suggest that vaccinia induces necrotic death in ovarian cancer.
Results
Activity of oncolytic vaccinia in ovarian cancer
We constructed a Lister strain vaccinia mutant deleted in thymidine kinase and expressing red fluorescent protein (RFP) in the TK locus under the control of the endogenous I1L promoter (Lister-dTK) and assessed its activity in human and murine ovarian cancer cells (Figure 1a). Infection with Lister-dTK induced similar levels of cytotoxicity in six human ovarian cancer cell lines (Figure 1a). Unlike adenovirus,13 vaccinia virus can kill murine tumor cells, including MOSEC/ID8 and MOVCAR7 (Figure 1a). In the experimental conditions used below (72 hour following infection with multiplicity of infection (MOI) 1 or 10), the large majority of cells are infected (Supplementary Figure S1) but ~40–80% cells remain viable (Figure 1b).
Figure 1.

Activity of Lister-dTK vaccinia in ovarian cancer. (a) Deletion of the TK gene in Lister-dTK was confirmed by PCR (left). Cytotoxicity of vaccinia in human (A2780, A2780CP, IGROV1, SKOV3ip1, OVCAR-4, and TOV21G) and murine (MOSEC, MOVCAR7) ovarian cell lines, as well as hTERT-immortalized human ovarian surface epithelial cells (IOSE25) as determined by MTS assay 72 hours postinfection. Mean ± SD IC50 is shown (left). All experiments were performed in triplicate at least three times. (b) Survival of five human ovarian cancer cells 72 hours following infection with Lister-WT and Lister-dTK at MOI 1 (A2780, A2780CP, IGROV1, and SKOV3ip1) and 10 (TOV21G). (c) Ratio of IC50 (mean ± SEM) for Lister-dTK compared with Lister-wt. All experiments were performed in triplicate at least twice. (d) Replication of vaccinia in ovarian cancer cell lines. Cells were infected with Lister-dTK (MOI 1 pfu/cell) and titer determined by TCID50 assay on CV1 cells as described in “Materials and Methods.” Points represent mean titer ± SD, n = 3. (e) Infection and replication of vaccinia virus in nude mice bearing advanced intraperitoneal SKOV3ip1 tumors following a single ip dose of 108 pfu Lister-dTK. Sequential 5 µm sections were stained with hematoxylin & eosin or for vaccinia virus proteins. L, liver, T, tumor, N, necrotic area. Bars represent 500 µm (image i) and 200 µm (images ii, iii and iv). Panels i and ii show an intraperitoneal deposit growing on the surface of the small bowel serosa adjacent to the omentum. hTERT, human telomerase reverse transcriptase.
Lister-dTK was more effective than wild-type Lister (Lister-wt) in all malignant cell lines but was attenuated in IOSE25 immortalized ovarian surface epithelial cells (Figure 1c). Replication of Lister-dTK in vitro was generally more rapid than Lister-wt. 102–103 pfu/cell were generated within the first 24 hours of infection, with maximum yields of ~104 pfu/cell (Figure 1d). In vivo replication and tumor specificity was further confirmed in mice bearing advanced SKOV3ip1 xenografts: following a single intraperitoneal dose of 108 pfu Lister-dTK, vaccinia virus proteins were not expressed in normal liver but were expressed in tumor tissue, with visible necrotic areas within and adjacent to the zones of vaccinia infection (Figure 1e, Supplementary Figure S2).
Features of classical apoptosis
We first investigated the role of apoptosis in vaccinia virus cytotoxicity using biochemical assays. Following Lister-dTK infection, there was an increase in the percentage of apoptotic (annexin V+/DAPI−) cells in all tested lines at 72 hours pi (Figure 2a). This was also observed following infection with Lister-wt, although to a lesser degree (not shown). Similarly, there was a significant increase in hypodiploid DNA 96-hour postinfection with Lister-dTK (Figure 2b) but not at 48 hours (Supplementary Figure S3). However, Bcl2 overexpression had no effect on vaccinia-induced cytotoxicity, but significantly reduced sensitivity to cisplatin as described previously (Figure 2c).14 Vaccinia induced minimal cleavage of caspase-3, although there was evidence of poly (ADP ribose) polymerase (PARP) cleavage in all cell lines (Figure 2d). Treatment with pan-caspase inhibitor zVAD-fmk significantly reduced cisplatin-induced cell death but did not inhibit vaccinia cytotoxicity (Figure 2e)—indeed, cell death increased marginally in IGROV1 cells. Overall, classical apoptosis did not appear to be a dominant mode of cell death following vaccinia infection.
Figure 2.

Vaccinia does not kill ovarian cancer cells using caspase-dependent apoptosis. (a) Externalization of phosphatidylserine on the cell membrane 72 hours after infection with Lister-dTK (MOI 1). Cells were stained with DAPI and annexin V. The percentage viable cells staining positive for annexin V in the lower right quadrant (Q4) are presented. Bar graph shows mean ± SD of three experiments in duplicate. *P = 0.04, **P = 0.005, ***P ≤ 0.0001 for all. (b) DNA fragmentation 96 hours after infection with Lister-dTK (MOI 1). Cells were fixed and stained with propidium iodide. Representative plots from a single sample show percentage gated cells with sub-G1 DNA. Bar graph shows mean ± SD sub-G1 DNA from 2 to 4 experiments, in duplicate. *P = 0.014 (A2780CP) and 0.01 (IGROV1), **P = 0.0055 (IGROV1) and 0.002 (SKOV3ip1), ***P < 0.001 (both). (c) Sensitivity of OVCAR-4 (O4) and Bcl2 over-expressing cells (O4-Bcl2) to Lister-dTK. Cell survival was measured by MTS assay. Dose-response curve shown from one experiment, mean IC50 ± SD is from three triplicate experiments. Bcl2 overexpression, confirmed by western blot, significantly inhibits cell death after exposure to 10 µmol/l cisplatin for 72 hours. Mean ± SD from three experiments, each in triplicate. ***P < 0.0001. (d) Cleavage of PARP and caspase-3 in untreated cells (U) and cells infected with Lister-wt (WT) or Lister-dTK (dTK; both MOI 1) for 72 hours. A2780 cells treated with 2 µmol/l staurosporine overnight were used as a positive control for apoptosis (+). Blots are representative of three independent experiments. (e) Caspase inhibition with zVAD-fmk (10 µmol/l added daily) in Lister-dTK (MOI 1) infection. Cell survival was measured by MTT assay 96 hours postinfection. In combination with 10 µmol/l cisplatin, a single dose of 10 µmol/l zVAD-fmk was used and cell survival determined by MTT assay after 48 hours. Mean ± SD from three experiments, each in triplicate. **P = 0.0055 (A2780CP) and 0.0011 (IGROV1).
Autophagy is dysregulated in vaccinia infection but does not contribute to cytotoxicity
There are few data on the role of autophagy in vaccinia-induced cell death, particularly in malignant cells, which may have aberrant autophagy pathways.15 During autophagy, light chain 3 (LC3) B-II, which is generated by the lipidation of LC3B-I, forms in the outer and inner membrane of the autophagosome and can also be detected as a faster migrating band on sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE). Coinfection of IGROV1 and A2780CP cells with Ad-LC3-GFP and either Lister-dTK (Figure 3a) or Lister-wt (Supplementary Figure S4) produced some punctate LC3 staining within infected cells, while infection with both Lister-wt and Lister-dTK induced abundant LC3B-II by immunoblot (Figure 3b).
Figure 3.

Vaccinia does not require autophagy for cytotoxicity. (a) LC3B localization in IGROV1 (i–iii) and A2780CP (iv) cells. Cells were co-infected with Ad GFP-LC3 (MOI 30) and Lister-dTK (MOI 1) and imaged 72 hours postinfection. (iii) is a magnified image from (ii). (b) Appearance of LC3B-II following infection with Lister-wt (WT) or Lister-dTK (dTK; both MOI 1). Protein was extracted 72 hours postinfection and subjected to SDS-electrophoresis. Representative blot of three experiments is shown. Quantification (below) of three independent experiments shows the mean ± SEM LC3B-II:LC3B-I relative expression. (c) Upper blot: accumulation of LC3B-II in A2780CP cells following 24 hours of serum starvation (lane 2, HBSS treatment) or treatment with the rapamycin. Lower blot: accumulation of LC3B-II in A2780 cells following 24 hours treatment with both rapamycin and chloroquine. (d) Lysosomal turnover of LC3B-II. Cells were infected with Lister-dTK (MOI 1) in the presence or absence of lysosomal protease inhibitors (LPI) pepstatin A (10 µg/ml) and E64d (10 µg/ml) for 72 hours. Protein was subjected to SDS-electrophoresis. Bar graph (right) shows quantification of LC3B-II:LC3B-I relative expression. (e) IC50 of Lister-dTK in the presence of the autophagy inhibitor 3-methyladenine relative to the IC50 of Lister-dTK alone. Cell survival was determined by MTS assay and represents the mean ± SD relative IC50. Lines present linear regression of the four IC50 values—the line slope does not differ significantly from 0 in any of the analyses. HBSS, Hanks buffered salt solution.
LC3B-II levels increase during autophagy, while it is degraded upon lysosome fusion. Therefore, an increase in LC3B-II levels can represent induction of autophagy or inhibition of the final step of the autophagic process (see ref. 16). Induction of autophagy by either rapamycin or serum starvation led to an increase in LC3B-II in A2780CP cells (Figure 3c), while inhibition of autophagosome–lysosome fusion using chloroquine also results in dose-dependent accumulation of LC3B-II by immunoblotting (Figure 3c).
We sought to determine whether the increase in LC3B-II levels following vaccinia infection resulted from autophagy induction or inhibition of the final fusion step, using the cysteine protease inhibitor E64d and the aspartic acid protease inhibitor Pepstatin A. These proteases inhibit lysosomal turnover of LC3B-II by blocking the activity of lysosome hydrolases, including cathepsins B and L. Thus, lysosomal protease inhibitors (LPI) alone increased levels of LC3B-II (Figure 3d). In three out of the four cells, these levels did not increase further in the presence of both LPI and Lister-dTK, whilst in the fourth, A2780, the extra increase was modest. This suggests that vaccinia does not induce autophagy but instead alters the rate of lysosomal degradation of LC3B-II. Confirming this, 3-methyladenine (3-MA), which blocks early autophagy signaling, did not alter the cytotoxicity of Lister-dTK in five different lines (Figure 3e). Chloroquine did not produce any consistent effect on vaccinia efficacy, with small but significant increases in IC50 in A2780 and A2780CP cells, no effect in SKOV3ip1 and TOV21G cells, and a significant decrease in IGROV1 cells (Supplementary Figure S5).
Necrotic morphology accompanies ovarian cancer cell death
To determine if vaccinia induces necrosis, we first examined the cellular morphology using transmission electron microscopy (TEM; Figure 4a). Immature and mature virions were seen within the cytoplasm of infected cells, and cytoplasmic vacuoles and condensed mitochondria were both observed. Additionally, ruptured nuclear and cell membranes were seen with release of cellular content and virions. Together, these features suggest a necrosis-like phenotype. Release of the nuclear protein HMGB1 (high mobility group box 1 protein) is a feature of necrosis as cell membranes disintegrate; we detected HMGB1 in the supernatant of Lister-dTK infected cells 48 and 72 hours postinfection (Figure 4b) suggesting passive release from necrotic cells.
Figure 4.

Vaccinia infection induces necrotic features in ovarian cancer cells. (a) Electron microscopy. A2780 cells were mock infected (i–ii) or infected with Lister-dTK (MOI 10) for 72 hours (iii–viii). Infected cells with cytoplasmic vacuoles (V) flank an already dead cell (iii and iv). Nuclear and cytoplasmic membrane rupture (v). Dense mitochondria (M) in an infected cell (vi). A cell full of infectious virions (electron dense structures) displays a swollen nucleus and ruptured cell membrane (arrowed) (vii). Cytoplasmic vacuoles and mitochondria in a cell harboring both mature and immature virions (viii). (b) Release of HMGB1 from infected cells. Supernatant and cell lysates were collected 72 hours following infection with Lister-dTK (MOI 10). (c) Intracellular ATP levels following infection with Lister-dTK (MOI 10). ATP concentration was quantified using a luciferase based ATP assay. Data show mean ± SD, n = 6. ***P < 0.001 for all analyses. (d) The relative rate of glycolysis and oxidative metabolism in A2780CP cells following Lister-dTK infection (MOI 1). Extracellular acidification rate (ECAR) was used to determine glycolysis and oxygen consumption rate (OCR) to quantify mitochondrial respiration. Rates are displayed relative to control (uninfected) cells. Cells were treated with TNF-α (10 ng/ml) and zVAD-fmk (25 µmol/l) for 4 hours as positive control. Bars represent mean ± SEM, n = 7. *P = 0.049 (OCR, Lister-dTK) and 0.04 (ECAR, TNF/zVAD), ***P < 0.0001 (ECAR, Lister-dTK) and 0.0002 (OCR, TNF/zVAD).
Biomarkers of necrosis
There are few definitive biochemical markers that indicate necrosis. However, a decrease in intracellular adenosine triphosphate (ATP) levels is frequently seen.17 ATP levels were steady over time in uninfected cells but significantly reduced in all cell lines infected with Lister-dTK (Figure 4c). Another feature of necrosis induction is an initial increase in mitochondrial oxidative metabolism with generation of reactive oxygen species (ROS).18 Using an extracellular flux analyzer, we measured extracellular acidification rate (ECAR, a measure of glycolysis) and oxygen consumption rate (OCR, a measure of mitochondrial respiration) following vaccinia infection. For both the parameters, there was a significant increase after 24 hours of infection (especially ECAR), followed by a steep decline below baseline levels (Figure 4d). Four hours of exposure to TNF-α/zVAD-fmk acted as positive necrosis control.
Necrosis mediators in vaccinia virus cytotoxicity
At least two necrotic death-inducing platforms have been described. The necrosome (or necroptosome) contains caspase-8, TRADD, FADD, RIP1, and RIP3 and classically forms in response to TNF-α/TNFR1 binding. It can lead to either apoptotic or necrotic death depending on the activity of caspase-8. The recently described “ripoptosome”, which contains RIP1, FADD, and caspase-8,19 forms independently of TNFR1 or death receptor activation but can be induced by genotoxic stress. In addition, the recruitment of caspase 10 and caspase inhibitor cFLIP isoforms to this complex has also been described.20 Ripoptosome activity is regulated by cFLIP, cIAP1, cIAP2, and XIAP, which inactivate components of the complex or target them for ubiquitylation. Poxviruses encode potential regulators of these complexes, including viral FLIP21 and caspase-8 inhibitors,22 so we investigated whether these cell death–inducing complexes were evident in vaccinia-infected ovarian cancer cells. In these experiments, we also investigated the cell line TOV21G, which expresses high levels of RIP3. In addition, the production of inflammatory cytokines, including TNF-α and IL-6, has been extensively modeled in both IGROV1 and TOV21G cells.23,24
Infection with Lister-dTK led to the formation of a RIP1/caspase-8 complex in A2780 and TOV21G cells (Figure 5a), as did etoposide and TNF-α, which initiate ripoptosome19 and necrosome formation,9 respectively. In TOV21G cells, which produce extremely high levels of endogenous TNF-α, addition of extra TNF-α did not lead to a RIP1/caspase-8 complex. RIP3 could be coprecipitated with caspase-8 in absence of infection in TOV21G and, although RIP3 levels reduced following infection in these cells, RIP3 was still detected coprecipitated with caspase-8 (Figure 5a). Levels of RIP1, RIP3, and caspase-8 generally decreased 72 hours postinfection (Figure 5b) and MG132 treatment suggested that proteasomal degradation was not responsible for these changes in protein expression, at least at this time point.
Figure 5.
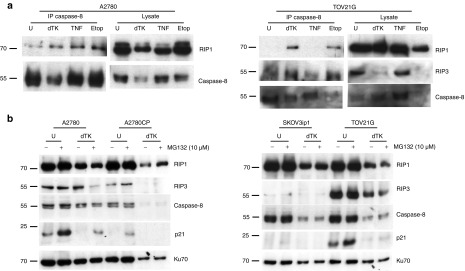
Necrosis mediators following vaccinia infection. (a) Formation of a RIP1/RIP3/caspase 8 complex following infection with Lister-dTK (MOI 10) for 96 hours. Caspase-8 was immunoprecipitated and the presence of RIP1, RIP3, and caspase-8 detected by immunoblotting. Cells were treated with 100 µmol/l etoposide or 10 ng/ml TNF-α in the presence of zVAD-fmk (25 µmol/l) as positive controls for necrosis. (b) Expression of RIP1, RIP3, and caspase-8 was assessed following infection with Lister-dTK (MOI 10) for 72 hours. The proteasome inhibitor MG132 (10 µmol/l) was added 5 hours before cell lysates were collected, and p21 expression assessed as positive control for MG132 activity.
Vaccinia infection can induce TNF-α production, and it has previously been shown that, in vaccinia-infected T cells and mouse embryonic fibroblasts, RIP3 knockout attenuates killing induced by CD3 and TNF-α, respectively.9 To investigate whether viral necrosis was a primary event or simply occurred secondary to induced TNF-α production, we infected ovarian cancer cells with Lister-dTK in the presence and absence of a blocking anti-TNF-α antibody (1 µg/ml)—there was no change in viral efficacy (Supplementary Figure S6). Similarly, stable shRNA-mediated TNF-α knockdown in IGROV1 cells25 did not alter Lister-dTK–mediated cytotoxicity significantly (Figure 6a). Moreover, knockdown of RIP3 alone had a minimal effect on Lister-dTK efficacy (Figure 6b). These results strongly suggest that vaccinia-induced cell death does not rely upon secondary necrosis induced by autocrine/paracrine TNF-α production.
Figure 6.

TNF-α blockade, necrostatin, and RNAi knockdown of RIP1 and RIP3. (a) IGROV1 cells with stable shRNA-mediated TNF-α knockdown (IGROV1-shTNF) and control cells encoding scrambled shRNA (IGROV1-shControl) were infected with Lister-dTK in triplicate. Cell survival was assessed 72 hours postinfection (left). Experiments were repeated twice—representative data are presented. TNF-α expression was quantified by quantitative RT-PCR, normalized to 18S. Bars represent mean ± SD, n = 3. *P < 0.05. (b) A2780, SKOV3ip1, and TOV21G were transfected with 10 nmol/l RIP3 siRNA (R3) or nontargeting siRNA (NT). Protein lysates were harvested 24 and 96 hours post-transfection and levels of RIP3 determined by immunoblotting. Cells were infected with Lister-dTK (MOI 10) at 24 hours after siRNA transfection and cell survival determined by MTT assay 72 hours postinfection. (c) Effect of inhibiting RIP1 kinase activity on Lister-dTK induced cell death. Cells were treated with necrostatin-1 (100 µmol/l) ± zVAD-fmk (25 µmol/l) immediately following infection with Lister-dTK and cell survival determined by MTS assay 72 hours postinfection. Representative dose response curves are shown. Mean IC50 ± SEM of a minimum of three experiments (right). (d) A2780 and SKOV3ip1 cells were transfected with 10 nmol/l RIP1 siRNA (R1) or nontargeting siRNA (NT) and treated as in (b). *P < 0.05, **P < 0.005.
Inhibitors of necrosis impair vaccinia cytotoxicity
We then investigated the effect of inhibition and knockdown of various steps of the necrotic pathway on cell death. Necrostatin-1 inhibits the kinase activity of RIP126 while necrosulfonamide targets MLKL, which has recently emerged as a key signaling protein that mediates binding to downstream targets.12,27 Necrostatin-1, either alone or in combination with the caspase inhibitor zVAD-fmk, markedly inhibited the sensitivity of cells to Lister-dTK, while zVAD-fmk alone again had no significant effect on virus activity (Figure 6c). RNAi-mediated knockdown of RIP1 also significantly attenuated viral cytotoxicity (Figure 6d), as did double knockdown of RIP1 and RIP3 in TOV21G cells (Supplementary Figure S7), in contrast to RIP3 knockdown alone (Figure 6b).
Finally, necrosulfonamide had a dose-dependent effect on the activity of Lister-dTK in all four ovarian cancer cells lines, and completely prevented virus-induced cell death even at viral doses of 100 pfu/cell (Figure 7a). Necrostatin-1 and necrosulfonamide were also able to inhibit vaccinia-induced cell death early (24 hours) after infection, suggesting that necrosis is not a secondary late phenomenon induced after multiple rounds of virus replication (Figure 7b). In addition, mitochondrial division inhibitor 1 (mdivi-1), which is reported to block the latter stages of necrosis by inhibiting Drp1, also partially protected cells from Lister-dTK cytotoxicity (Supplementary Figure S8). Necrostatin and necrosulfonamide did not reduce viral replication in the short-term (Supplementary Figure S9), suggesting that the effects on cytotoxicity resulted from blockade of direct virus-induced necrosis rather than an indirect effect via reduced virion production.
Figure 7.

Necrosulfonamide and virus replication after necrosis inhibition. (a) Effect of inhibiting MLKL on Lister-dTK induced cell death. Cells were treated with necrosulfonamide (MLKL inhibitor) following infection with virus, and cell survival determined by MTT assay 72 hours postinfection. (b) A2780CP and TOV21G cells were infected with Lister-dTK at MOI 100. Infectivity was assessed by RFP flow cytometry 12 hours postinfection (left). Cell survival was assessed 24 hours postinfection in the presence and absence of 100 µmol/l necrostatin-1 or 10 µmol/l necrosulfonamide.
Discussion
These results provide evidence that vaccinia virus induces necrosis in ovarian cancer cells. The data also support findings that vaccinia infection does not lead to apoptotic cell death. It is known that vaccinia encodes various inhibitors of apoptosis including F1L, N1L,28,29 and SPI-222 and that these combine to prevent premature cell death that might restrict virus replication and spread. Apoptosis has been observed following infection with the Western Reserve in various malignant cells,6,30,31 although this may be strain specific, as there was no apoptosis following Wyeth strain infection. Here, infection with Lister strain vaccinia virus led to phosphatidylserine externalization (Figure 2a), DNA fragmentation (Figure 2b), and some PARP cleavage (Figure 2d) and, superficially, these observations imply that vaccinia induces apoptosis. Although statistically significant, the proportion of cells expressing these markers was low. Moreover, caspase-3 cleavage was not apparent (Figure 2d), while inhibition of apoptosis by both Bcl2 overexpression (Figure 2c) and zVAD-fmk treatment completely failed to attenuate the cytotoxicity of Lister-dTK in vitro, despite inhibiting cisplatin-induced death (Figure 2e)—indeed, in IGROV1 cells, zVAD-fmk increased vaccinia cytotoxicity. It has been reported that necrotic cells can also demonstrate annexin V+/PI− staining before they become PI+.32 This highlights the need to use multiple assays to evaluate apoptosis, particularly as the release of apoptosis-inducing factor can also occur as a result of ATP depletion during programmed necrosis,33 leading to some features similar to apoptosis.34 Bax-mediated release of apoptosis-inducing factor was described as a critical step in programmed necrosis,35 and it may well be that the markers of apoptosis observed coexist with more striking necrotic traits, as has been acknowledged in the classification of cell death.36
Interestingly, HMGB1 release was also previously observed alongside features of apoptosis following Western Reserve infection in H460 and HT-29 cells,30 the cause of which is unclear, although it is entirely logical that cell type and virus strain play a role. While vaccinia virus encodes numerous inhibitors of apoptosis and immunogenic cell death (reviewed in ref. 37), these are not shared by all strains; SPI-1, for example, is expressed in Western Reserve, Lister, and Copenhagen, whereas SPI-2 is expressed in Western Reserve only.38 Furthermore, there are widespread differences in the expression of proteins involved in cell death between normal and malignant cells, as well as between different populations of cancer cells. For instance, malignant ovarian tumors demonstrate lower expression of the autophagy proteins Beclin1 and LC3, although there are few data on the involvement of autophagy in vaccinia-induced cell death, or indeed the vaccinia lifecycle.15 It was hypothesized that vaccinia might hijack autophagy machinery for formation of the virion envelope, but vaccinia replicates and matures in Atg 5−/− mouse embryonic fibroblasts and Beclin 1−/− embryonic stem cells to the same degree as in the wild-type counterparts.39 We found only limited markers of autophagy following infection with Lister-dTK: some limited evidence of autophagosome formation was seen by confocal microscopy, but this could not be confirmed by electron microscopy (Figure 3a); LC3B-II formation was observed (Figure 3b), but autophagic flux was not evident (Figure 3d); 3-methyladenine did not alter the cytotoxicity of Lister-dTK in any ovarian cancer cell line (Figure 3e). Consistent with our results, it was recently demonstrated that vaccinia initiates massive LC3 lipidation (conversion of LC3B-I to LC3B-II) in mouse fibroblast cells, and this too was not accompanied by an increase in autophagic flux, nor were autophagosomes evident on electron microscopy.8 Together, these data and the work presented here suggest that, while LC3B-I is readily converted to LC3B-II, a true increase in the autophagic process (i.e., autophagic flux and autophagosome formation) is not a predominant feature of vaccinia infection. The implications of modulation of LC3B by vaccinia are unclear, and it is not known what role this plays in infected cells if downstream autophagy does not take place.
Lister-dTK infection resulted in morphological changes indicative of necrosis, including ruptured membranes, swollen nuclei, and cytoplasmic vacuolation. In addition, the release of nuclear HMGB1 from infected cells was demonstrated. Characteristic of necrosis, a significant increase in glycolysis and mitochondrial respiration, was observed 24 hours postinfection. However, the rate of glycolysis then fell below baseline levels, corresponding with a sharp and significant decline in intracellular ATP levels. These data, consistent with the presence of areas of necrosis observed in vivo, strongly suggest that vaccinia infection is associated with necrosis.
Key components of programmed necrosis, including RIP1 and caspase-8, associate during vaccinia infection of ovarian cancer cells. A RIP1/RIP3 complex was previously detected in the liver of wild-type mice infected with vaccinia and was proposed to mediate the inflammatory response to infection.9 In the same study, Cho et al. showed that vaccinia-infected mouse embryonic fibroblasts and T cells were sensitized to TNF-α- and CD3-induced necrosis respectively, and, while formation of the RIP1/RIP3 complex was associated with extensive necrosis and inflammation in the liver of infected mice, infection was controlled. In RIP3-deficient mice, fat-pad necrosis and liver inflammation were not apparent, but mice were unable to control virus replication and succumbed to infection, suggesting that TNF-α–induced RIP3-mediated necrosis is essential in the host response to vaccinia. Our results indicate that the necrosis in ovarian cancer cells does not depend on virus-induced autocrine TNF-α production as both TNF-α neutralizing antibodies and stable TNF-α knockdown had no effect on overall efficacy. In addition, although we were able to detect complexes containing caspase-8, RIP1, and RIP3, RIP3 knockdown alone has a minimal effect. We have previously shown that TNF-α promotes tumor growth in ovarian cancer and sits at the heart of a proinflammatory network:24,40 thus, the response to virus-induced TNF-α production in malignant epithelial cells is markedly different to that seen in mouse embryonic fibroblasts or T cells. Our data show that ovarian cancer cells can respond to TNF-α, with increased extracellular acidification and oxygen consumption (Figure 4d) as well as necrosome formation (Figure 5a). To underline the complexity of vaccinia infection, the Chan laboratory also found that the stable expression of the viral FLIP MC159 in transgenic mice reduced rather than increased vaccinia replication in the fat pad and liver, in marked contrast to RIP3 knockdown.41
There is now convincing evidence to support a role for MLKL as an important downstream effector of TNF-α–induced necrosis following recruitment to the necrosome by RIP3.27 An intriguing aspect of data presented here is that MLKL appears to have a role in vaccinia virus infection of ovarian cancer cells, but not RIP3, suggesting that vaccinia may activate MLKL independently of RIP3. Zhao et al.27 demonstrated some phosphorylation of MLKL in 293 cells, which lack RIP3—thus, other cellular or viral proteins may contribute to induction of necrosis. Unlike murine cytomegalovirus, which expresses the M45 protein that acts to modulate programmed necrosis through its interactions with RIP1 and RIP3,42,43 no similar protein encoded by vaccinia has yet been identified. How vaccinia triggers and signals necrosis must be an important focus of future work.
Together, our results demonstrate, for the first time to our knowledge, a defined pathway of programmed necrosis in malignant cells following infection with vaccinia virus. Ongoing work will dissect which vaccinia proteins are key to necrosis induction, how necrosis induction impacts upon immune responses to virus-infected cells in vivo, and how clinical resistance to vaccinia might arise via pertubations in necrosis execution in malignant cells.
Materials and Methods
Cell lines and virus construction. A2780, A2780CP, SKOV3ip1, IGROV1, TOV21G, and OVCAR-4 are all human ovarian carcinoma cell lines and were authenticated by 16 locus STR analysis (LGC Standards, Middlesex, UK). All were maintained in Dulbecco's Modified Eagle Medium supplemented with penicillin/streptomycin and 10% fetal calf serum, (PAA Laboratories, Austria). OVCAR-4 Bcl2-overexpressing cells14 were cultured in the presence of 1 mg/ml of G418. IGROV1 cells with stable shRNA-mediated TNF-α knockdown have been described previously.40 IOSE25 are human telomerase reverse transcriptase–immortalized human ovarian surface epithelial cells44 and were maintained in normal ovarian cancer surface epithelium-complete medium as described previously.45 MOSEC/ID846 and MOVCAR747 are murine ovarian cancer cells and were kindly donated by Drs Roby and Connolly, respectively.
The Lister wild-type vaccinia virus strain (Lister-wt) was obtained from ATCC. Lister-dTK was constructed by a process of recombination, plaque purification to select positive clones followed by virus expansion and purification. CV1 cells were infected with 0.05 pfu/cell of Lister-wt virus and then transfected 0.5 µg Lister-wt viral DNA (extracted using a QiagenQIAmp DNA Blood Mini Kit) and 0.3 µg of pDR-TK-I1L-RFP, a plasmid containing a mammalian codon-optimized RFP gene from the bubble tip anemone Entacmaea quadricolor under control of the I1L promoter, flanked by sequences homologous to the VV Lister thymidine kinase gene to allow homologous recombination with the Lister-wt virus (Supplementary Figure S10). Recombination was confirmed by the presence of RFP using a fluorescent microscope and cells harvested and freeze-thawed three times. A plaque assay was then performed to select a pure recombinant virus. CV1 cells were infected with serial dilutions of Lister-dTK for 2 hours, washed with PBS and then overlaid with 1% bacteriological agar/10% fetal calf serum-Dulbecco's Modified Eagle Medium. Positive plaques expressing RFP were picked 48 hours later, dissolved in Hanks buffered salt solution, and freeze-thawed three times. Viruses were subjected to further rounds of plaque purification until all plaques resulting from infection expressed RFP. Bulk virus was prepared by infecting CV1 cells in a multilayer CF-10 (Fisher Scientific, Loughborough, UK) flasks. These were harvested when evidence of infection was observed. Cells were collected, freeze-thawed three times (liquid N2/37 °C), centrifuged at 900 rpm, and the supernatant was collected. The pellet was resuspended in 10 mmol/l Tris pH9 and centrifuged again at 900 rpm before combining the supernatants in a total volume of 30 ml of 10 mmol/l Tris pH 9 after sonification. This was layered onto four SW27 ultracentrifuge tubes containing 36% sucrose in 10 mmol/l Tris pH 9 and centrifuged at 13,500 rpm for 80 minutes. The viral pellet was then resuspended in 1 mmol/l Tris and titer determined by TCID50 assay. Deletion of TK in Lister-dTK was confirmed by PCR using the following primers: forward: 5′-AGCTACCACCGCAATAGAT and reverse: 5′-CGAGTGCGGCTACTATAAC. Control primers for A52R were: forward: 5′-ATGATGCGGAAGAACAAT and reverse: 5′-TTGCGGTATATGTATGAGGTG.
Cell viability assays. Totally, 104 cells were infected with MOI 0.0001—1,000 pfu/cell in serum-free medium and re-fed with Dulbecco's Modified Eagle Medium containing 2% fetal calf serum 2 hours later. Inhibitors (zVAD-fmk (Calbiochem); 3-MA, chloroquine, necrostatin-1, and mdivi-1 (all from Sigma-Aldrich, Poole, UK); necrosulfonamide (Toronto Research Chemicals, Toronto, Ontario, Canada); and neutralizing anti-TNF-αAb (clone 1825; R and D Systems, catalogue MAB210)) were added at the time of re-feeding. Cell viability was assayed by MTT or MTS assay. Dose–response curves were generated using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA). All cell viability assays were performed a minimum of three times, with at least triplicate wells, unless stated.
TCID50 assays. A total of 2 × 105 cells were infected (MOI 1 pfu/cell) in serum-free medium. After 2 hours, cells were re-fed with Dulbecco's Modified Eagle Medium containing 2% fetal calf serum. Cells were harvested and subjected to three rounds of freeze/thawing (liquid N2/37 °C), after which they were centrifuged. The supernatant was titered by TCID50 assay on CV1 cells according to the following method: Log TCID50/ml = A − D (S − 0.5), where A = log of the highest dilution showing CPE in >50% of the wells, D = log of the dilution factor, and S = summation of the proportion of positive wells in each row. Titer (pfu/ml) was calculated according to the formula pfu/ml = 10n−0.7, where n = logTCID50/ml as originally described for vesicular stomatitis virus.48 Assays were performed twice, in triplicate.
Immunoblotting/coimmunoprecipitation. For immunoblots, cells were lysed in radio immunoprecipitation assay buffer (50 mmol/l Tris HCl, pH 8.0, 150 mmol/l NaCl, 1 mmol/l ethylenediaminetetraacetic acid, 1% NP-40, 0.1% SDS) containing one protease inhibitor tablet (Roche, Welwyn, UK) per 20 ml buffer. Protein lysates were electrophoresed on SDS-PAGE and transferred onto polyvinylidene diflouride membranes by semi-dry blotting. Antibody binding was visualized using enhanced chemiluminescence (GE Healthcare, Bucks, UK). Antibodies were as follows: HMGB1 (#ab18256; Abcam, Cambridge, UK), antivaccinia virus (#9503-2057; AbDSerotec, Kidlington, UK), caspase-8 (#51-80841N; BD Pharmingen, Oxford, UK), cleaved PARP (#9541), caspase-3 (#9662), LC3B (#2775), RIP1 (#3493; all from Cell Signaling, Beverly, MA), RIP3 (#IMG-5846A; Imgenex, San Diego, CA), Ku70 (#sc-1486), Bcl2 (#sc509), p21 (#sc-397), PGAM5L (#sc-161156; all Santa Cruz Biotechnology, Santa Cruz, CA), Caspase-8 Ab for ip, (#sc-6136, Santa Cruz).
For assessment of HMGB-1 release, 2 ml cell supernatant was concentrated approximately fourfold by centrifugation via Amicon 3K Ultra-4 centrifugal filter units (Millipore, UK) at 4,300 rpm for 30 minutes. Thirty microliters concentrate was electrophoresed on SDS-PAGE cells as above.
For co-immunoprecipitation, cells were lysed in 30 mmol/l Tris pH 7.4, 120 mmol/l NaCl, 2 mmol/l EDTA, 2 mmol/l KCl, 10% glycerol, 0.2% NP-40 buffer containing protease inhibitors as above. Lysates were incubated with polyclonal goat caspase-8 antibody (Santa Cruz Biotechnology) for 2 hours at 4 °C followed by addition of Protein G Sepharose 4 fast flow (GE Healthcare, Buckinghamshire, UK) overnight. Samples were then washed in 50 mmol/l Tris, resuspended in sample buffer (5% SDS, 20% buffer IIb (0.5 mol/l Tris, 0.2 mol/l NaH2PO4 pH 7.8), 50% glycerol, bromophenol blue, 20% distilled water)) containing 100 μmol/l of the reducing agent DTT. To avoid detection of the similar sized IgG heavy chain on SDS-PAGE, antimouse and antirabbit HRP conjugated secondary antibodies were specific for the light chain of IgG (Jackson Immuno Research Laboratories, Suffolk, UK).
Confocal microscopy. Cells were seeded in eight-well chamber slides (Fisher Scientific) and then co-infected with vaccinia virus (MOI 1) and Ad LC3-GFP (MOI 30), a replication-defective adenovirus expressing LC3B tagged to GFP, a kind gift of Dr Aviva Tolkovsky, University of Cambridge, UK, for 72 hours. The generation of Ad LC3-GFP has been described previously.49 Cells were fixed in 4% paraformaldehyde for 30 minutes at room temperature, washed in PBS. Slides were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen) and allowed to set for 30 minutes at room temperature before storage at 4 °C, protected from the light. Cells were viewed using a confocal laser scanning microscope (LSM 510 META, Carl Zeiss, Inc., Germany) with a 63×/1.4 NA Plan-Apochromat oil immersion objective. A Diode 405 nm laser, 488 nm Argon laser and Helium Neon 543 nm laser were used to excite DAPI, GFP, and RFP, respectively. High quality (12 bit, 1,024 pixels) images were captured using LSM 5 Software, version 3.2.
siRNA transfection. siRNA oligonucleotides targeted against RIP1 and RIP3 were obtained as mixed pools of four separate siRNAs (ON-TARGET plus SMART pool, Dharmacon). A nontargeting (NT) pool of siRNA was also used. All cell lines were transfected using DharmaFECT 1 transfection reagent (Dharmacon). Cells were transfected with 10 nmol/l siRNA and protein expression determined by immunoblot.
Flow cytometry. For annexin V staining, cells were infected with vaccinia (MOI 1) for 72 hours, resuspended in 100 μl annexin V-binding buffer (10 mmol/l HEPES, 140 mmol/l NaCl and 2.5 mmol/l CaCl2, pH 7.4). 2.5 μl annexin V FITC-conjugate (Invitrogen) was added for 20 minutes at room temperature, followed by the addition of 2 μmol/l final concentration 4-6-diamidino-2-phenylindole (DAPI) (Invitrogen). Flow cytometry was performed on a Fortessa cytometer using FACS Diva software and cells were initially gated based on forward scatter vs. side scatter. Ten thousand events from this population were then collected and apoptotic cells quantified using a quadrant gating system. Apoptotic cells were defined as annexin V+/DAPI−.
For analysis of hypodiploid DNA (sub-G1), cells were infected with vaccinia (MOI 1) for 96 hours. Cells were pelleted by centrifugation and resuspended in 1 ml ice-cold 70% ethanol while vortexing before fixing for a minimum of 24 hours at 4 °C. The DNA content of cells was analyzed by washing cells with PBS and then incubating with 20 μg RNase A (Invitrogen) and 100 μg propidium iodide (Sigma) in a total volume of 300 μl of PBS for 30 minutes at 37 °C in the dark. Ten thousand events were acquired by flow cytometry on a FACScalibur cytometer (Becton Dickinson Immunocytometry Systems, Belgium) with CellQuest Pro Software version 4.0.2. A primary gate on forward scatter vs. side scatter was set to exclude cell debris, and a second gate to exclude doublets on a dot plot of pulse width (FL2-W) vs. pulse area (FL2-A). Propidium iodide fluorescence (FL2-H) was then plotted against cell counts on a linear scale to distinguish between the different phases of the cell cycle. A G1 marker was set around the first peak at 2N DNA and the cell population with <2N DNA quantified.
ATP and cellular metabolism assays. For ATP assays, protein extracts were suspended in standard reaction buffer containing luciferase and luciferin according to the manufacturer's instructions (ATP Determination Kit; Invitrogen), and luminescence read at 560 nm.
For cellular metabolism assays, an XF24 extracellular flux analyzer (Seahorse Bioscience, http://www.seahorsebio.com/company/about.php) was used to determine ECAR and OCR. A2780CP cells were seeded and infected with vaccinia (MOI 10) for 24, 48, or 72 h. As a positive control, cells were also pretreated with TNF-α (10 ng/ml) and zVAD-fmk (25 µmol/l) for 4 hours. Before measurement, cells were changed to unbuffered RPMI medium (pH 7.4 at 37 °C) for 60 minutes in a non-CO2 incubator. The cartridge was hydrated in unbuffered RPMI medium overnight. On the flux analyzer, the protocol included calibration and four readings to establish basal ECAR and OCR rates in both uninfected and infected cells. Cell number was then measured using a Coulter counter (Beckman, UK) and the rate of ECAR and OCR normalized to cell number and presented as a percentage of control (uninfected) cells.
Transmission electron microscopy. A2780 cells were seeded on glass coverslips and infected with Lister-dTK (MOI 1) for 72 hours. They were fixed in 3% glutaraldehyde, postfixed in 1% aqueous osmium tetroxide, washed in water and dehydrated in a graded ethanol series. The preparation was then cleared in propylene oxide and infiltrated with Araldite epoxy resin. The cells were embedded by inverting a “Beem” capsule filled with resin on top and curing for 48 hours at 60 °C. Ultrathin sections (60–90 nm) were cut and stained with uranyl acetate and lead citrate and viewed in a JEOL JEM1230 transmission electron microscope, with images being collected on an Olympus Morada digital camera.
Ethics statement and in vivo experiments. All experiments complied with the National Cancer Research Institute guidelines for the welfare and use of animals in cancer research50 and were conducted after specific UK government Home Office personal (reference 70/19481) and project (reference 70/7263) license approval. Experiments were approved locally by the ethics review committee of Queen Mary University of London Biological Services Unit (reference PAC60-3). CD1 nude/nude mice were injected with 5 × 106 SKOV3ip1 ovarian carcinoma cells intraperitoneally (ip). When ascites were clinically evident, mice were injected with a single dose of 108 pfu Lister-dTK ip in 400 μl PBS. Mice were killed 72 hours postinjection and tumor, liver, and spleen harvested. Tissue was fixed in formaldehyde after which it was transferred to 70% ethanol, processed, embedded in paraffin, and 5 µmol/l sections cut. Vaccinia virus proteins were detected using a polyclonal rabbit antivaccinia antibody (AbDSerotec, Oxford, UK). Analysis of tissue sections was performed using an Axiophot microscope connected to a Zeiss AxioCam camera and using Axiovision software (Zeiss).
Statistics. All dose–response curves and statistical analyses were generated using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). All comparisons are unpaired two-tailed t-tests.
Authors Contribution
Design of experiments: L.M.W., K.A., H.K., D.O., F.R.B., I.A.McN. Execution of experiments: L.M.W., K.A., D.O., I.A.McN. Analysis of data: L.M.W., K.A., I.A.McN. Manuscript preparation: L.M.W., I.A.McN.
SUPPLEMENTARY MATERIAL Figure S1. SKOV3ip1,A2780CP and A2780 cells were infected with Lister-dTK (MOI 1 and 10) for 12 or 72h. Expression of RFP was assessed by flow cytometry. Figure S2. Magnified image from Figure 1D (panel iv) demonstrating expression of vaccinia proteins in tumour regions with necrosis evident on H&E staining from a sequential 5 µm section. Figure S3. Externalisation of phosphatidylserine (left) and DNA fragmentation (right) 48hrs after infection with Lister-dTK and Lister-WT (both MOI 1). Figure S4. LC3B localisation in IGROV1 (left) and A2780CP (right) cells following Lister-WT infection. Figure S5. Cells were infected with Lister-dTK in the presence of chloroquine (1 – 20 µmol/l). Figure S6. Effects of TNF-α blockade. Figure S7. TOV21G cells were transfected with 10nM RIP1 siRNA (R1) 10nM RIP3 siRNA (R3), both (R1+3) or non-targeting siRNA (NT). Figure S8. Effect of inhibiting Drp1 on Lister-dTK induced cell death. Figure S9. Replication of Lister-dTK in A2780CP cells following treatment with necrosis inhibitors. Figure S10. pDR-TK-I1L-RFP, used to generate Lister dTK as described in Materials and Methods, contains a 2.5kb fragment with the following components: Lister Vaccinia virus (GenBank accession number AY678276)nucleotides 82375–83247 (left hand homology sequence,) and 83648–84441 (right hand homology sequence), a mammalian codon-optimised RFP gene from Entacmaea quadricolor under the control of Lister VV I1L promoter (nt 63306–63333).
Acknowledgments
We thank Graham McPhail, (Nanovision Centre, Queen Mary University of London) for his expertise and help with obtaining TEM images, and Keyur Trivedi and Mohammed Ikram (Molecular Pathology Department, Barts Cancer Institute) for immunohistochemistry. This work was supported by the Medical Research Council (grant reference G0601891) and Cancer Research UK.
Supplementary Material
References
- Vanderplasschen A, Hollinshead M, Smith GL. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J Gen Virol. 1997;78 (Pt 8):2041–2048. doi: 10.1099/0022-1317-78-8-2041. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci USA. 1998;95:7544–7549. doi: 10.1073/pnas.95.13.7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner S, Humrich JY, Thuman P, Sauter B, Schuler G, Jenne L. The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin Exp Immunol. 2006;146:344–353. doi: 10.1111/j.1365-2249.2006.03177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humlová Z, Vokurka M, Esteban M, Melková Z. Vaccinia virus induces apoptosis of infected macrophages. J Gen Virol. 2002;83 Pt 11:2821–2832. doi: 10.1099/0022-1317-83-11-2821. [DOI] [PubMed] [Google Scholar]
- Moloughney JG, Monken CE, Tao H, Zhang H, Thomas JD, Lattime EC, et al. Vaccinia virus leads to ATG12–ATG3 conjugation and deficiency in autophagosome formation. Autophagy. 2011;7:1434–1447. doi: 10.4161/auto.7.12.17793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Beg AA. Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol. 2000;74:7470–7477. doi: 10.1128/jvi.74.16.7470-7477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Young AM, Archibald KM, Tookman LA, Pool A, Dudek K, Jones C, et al. Failure of translation of human adenovirus mRNA in murine cancer cells can be partially overcome by L4-100K expression in vitro and in vivo. Mol Ther. 2012;26:1676–1688. doi: 10.1038/mt.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish IA, Bell S, McKay T, Tenev T, Marani M, Lemoine NR. Expression of Smac/DIABLO in ovarian carcinoma cells induces apoptosis via a caspase-9-mediated pathway. Exp Cell Res. 2003;286:186–198. doi: 10.1016/s0014-4827(03)00073-9. [DOI] [PubMed] [Google Scholar]
- Shen Y, Li DD, Wang LL, Deng R, Zhu XF. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy. 2008;4:1067–1068. doi: 10.4161/auto.6827. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Kettle S, Alcamí A, Khanna A, Ehret R, Jassoy C, Smith GL. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J Gen Virol. 1997;78 Pt 3:677–685. doi: 10.1099/0022-1317-78-3-677. [DOI] [PubMed] [Google Scholar]
- Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011;17:6083–6096. doi: 10.1158/1078-0432.CCR-11-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe H, Chakravarty P, Leinster DA, Charles KA, Kwong J, Thompson RG, et al. Australian Ovarian Cancer Study Group A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res. 2012;72:66–75. doi: 10.1158/0008-5472.CAN-11-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salako MA, Kulbe H, Ingemarsdotter CK, Pirlo KJ, Williams SL, Lockley M, et al. Inhibition of the inflammatory cytokine TNF-a increases adenovirus activity in ovarian cancer via modulation of cIAP1/2 expression. Mol Ther. 2011;19:490–499. doi: 10.1038/mt.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai D, Yu E, Jin C, Welsh K, Shiau CW, Chen L, et al. Vaccinia virus protein F1L is a caspase-9 inhibitor. J Biol Chem. 2010;285:5569–5580. doi: 10.1074/jbc.M109.078113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilenko ST, Banadyga L, Bond D, Barry M. The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J Virol. 2005;79:14031–14043. doi: 10.1128/JVI.79.22.14031-14043.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZS, Naik A, O'Malley ME, Popovic P, Demarco R, Hu Y, et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- Liskova J, Knitlova J, Honner R, Melkova Z. Apoptosis and necrosis in vaccinia virus-infected HeLa G and BSC-40 cells. Virus Res. 2011;160:40–50. doi: 10.1016/j.virusres.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Sawai H, Domae N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem Biophys Res Commun. 2011;411:569–573. doi: 10.1016/j.bbrc.2011.06.186. [DOI] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- Boujrad H, Gubkina O, Robert N, Krantic S, Susin SA. AIF-mediated programmed necrosis: a highly regulated way to die. Cell Cycle. 2007;6:2612–2619. doi: 10.4161/cc.6.21.4842. [DOI] [PubMed] [Google Scholar]
- Cabon L, Galán-Malo P, Bouharrour A, Delavallée L, Brunelle-Navas MN, Lorenzo HK, et al. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death Differ. 2012;19:245–256. doi: 10.1038/cdd.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Morselli E, Vitale I, Senovilla L, Pinti M, et al. Viral strategies for the evasion of immunogenic cell death. J Intern Med. 2010;267:526–542. doi: 10.1111/j.1365-2796.2010.02223.x. [DOI] [PubMed] [Google Scholar]
- Kettle S, Blake NW, Law KM, Smith GL. Vaccinia virus serpins B13R (SPI-2) and B22R (SPI-1) encode M® 38.5 and 40K, intracellular polypeptides that do not affect virus virulence in a murine intranasal model. Virology. 1995;206:136–147. doi: 10.1016/s0042-6822(95)80028-x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Monken CE, Zhang Y, Lenard J, Mizushima N, Lattime EC, et al. Cellular autophagy machinery is not required for vaccinia virus replication and maturation. Autophagy. 2006;2:91–95. doi: 10.4161/auto.2.2.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, et al. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007;67:585–592. doi: 10.1158/0008-5472.CAN-06-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa S, Woelfel M, Guildford M, Moquin D, Chan FK. Viral cell death inhibitor MC159 enhances innate immunity against vaccinia virus infection. J Virol. 2010;84:10467–10476. doi: 10.1128/JVI.00983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci USA. 2008;105:3094–3099. doi: 10.1073/pnas.0800168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li NF, Broad S, Lu YJ, Yang JS, Watson R, Hagemann T, et al. Human ovarian surface epithelial cells immortalized with hTERT maintain functional pRb and p53 expression. Cell Prolif. 2007;40:780–794. doi: 10.1111/j.1365-2184.2007.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li NF, Wilbanks G, Balkwill F, Jacobs IJ, Dafou D, Gayther SA. A modified medium that significantly improves the growth of human normal ovarian surface epithelial (OSE) cells in vitro. Lab Invest. 2004;84:923–931. doi: 10.1038/labinvest.3700093. [DOI] [PubMed] [Google Scholar]
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63:1389–1397. [PubMed] [Google Scholar]
- Hunt JM, Marcus PI. Mechanism of Sindbis virus-induced intrinsic interference with vesicular stomatitis virus replication. J Virol. 1974;14:99–109. doi: 10.1128/jvi.14.1.99-109.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Committee of the National Cancer Research Institute Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.