

Abstract
Amyotrophic Lateral Sclerosis (ALS) is an adult-onset and fast progression neurodegenerative disease that leads to the loss of motor neurons. Mechanisms of selective motor neuron loss in ALS are unknown. The early events occurring in the spinal cord that may contribute to motor neuron death are not described, neither astrocytes participation in the pre-symptomatic phases of the disease. In order to identify ALS early events, we performed a microarray analysis employing a whole mouse genome platform to evaluate the gene expression pattern of lumbar spinal cords of transgenic SOD1G93A mice and their littermate controls at pre-symptomatic ages of 40 and 80 days. Differentially expressed genes were identified by means of the Bioconductor packages Agi4×44Preprocess and limma. FunNet web based tool was used for analysis of over-represented pathways. Furthermore, immunolabeled astrocytes from 40 and 80 days old mice were submitted to laser microdissection and RNA was extracted for evaluation of a selected gene by qPCR. Statistical analysis has pointed to 492 differentially expressed genes (155 up and 337 down regulated) in 40 days and 1105 (433 up and 672 down) in 80 days old ALS mice. KEGG analysis demonstrated the over-represented pathways tight junction, antigen processing and presentation, oxidative phosphorylation, endocytosis, chemokine signaling pathway, ubiquitin mediated proteolysis and glutamatergic synapse at both pre-symptomatic ages. Ube2i gene expression was evaluated in astrocytes from both transgenic ages, being up regulated in 40 and 80 days astrocytes enriched samples. Our data points to important early molecular events occurring in pre-symptomatic phases of ALS in mouse model. Early SUMOylation process linked to astrocytes might account to non-autonomous cell toxicity in ALS. Further studies on the signaling pathways presented here may provide new insights to better understand the events triggering motor neuron death in this devastating disorder.
Keywords: ALS, SOD1G93A, pre-symptomatic, spinal cord, microarray, laser microdissection, astrocytes
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fast disabling neurodegenerative disease characterized by upper and lower motor neuron loss of motor cortex, brainstem, and spinal cord leading to respiratory insufficiency and death (Turner et al., 2013). The incidence of ALS ranges from 1.7 to 2.3 cases per 100,000 population per year worldwide (Beghi et al., 2006). The mechanisms underlying neurodegeneration in ALS are multifactorial, and seem to involve neurons and non-neuronal cells (Boillee et al., 2006a,b; Yamanaka et al., 2008; Wang et al., 2011a) as well as several molecular pathways (Boillee et al., 2006a; Ferraiuolo et al., 2011b; Kiernan et al., 2011; Usuki et al., 2012). Approximately 5% of ALS cases are familial, and 20% of these have been linked to mutations in Cu/Zn superoxide dismutase 1 (SOD1) (Rosen et al., 1993; Andersen and Al-Chalabi, 2011). The first symptoms define the beginning of the clinical phase of the diagnosed cases of the more prevalent sporadic forms, consisting in muscle atrophy, weakness, fasciculations, and spasticity (Brooks et al., 2000). There is a lack of pathological studies on post mortem spinal cord from ALS patients that could add information about the triggering, initial time of motor neuron death and mechanisms of the disease. In fact, Fischer et al. (2004) reported the post-mortem evaluation in a patient with a short history of ALS, whose electromyography showed signs of acute and chronic denervation, coming out with an unexpected die without peripheral and central motor neuron death together with autolytic changes and a little axonal degeneration. Histological evaluations at the neuromuscular junctions and also electrophysiological analysis at the peripheral nerves in ALS patients have allowed authors to claim that motor neuron death correlates to the begging of clinical classical symptoms (Veugelers et al., 1996; Liu et al., 2013).
As the majority of familial ALS cases are linked to the mutations in SOD1 gene (Dion et al., 2009), transgenic mice expressing human mutant SOD1 (mSOD1) developing age-dependent clinical and pathological features of human ALS are current largely employed in the physiopathological studies of the disorder (Turner and Talbot, 2008). Using this mouse model, we previously described early behavior and electrophysiological alterations, prior the classical neurological symptoms and the beginning of motor neuron death (Alves et al., 2011). In fact, several early events demonstrated in animal models seemed to precede the neuronal death, remarkably the activation of glial cells (microglia and astrocytes) close to motor neurons (Graber et al., 2010; Wang et al., 2011b; Gerber et al., 2012), retraction of motor neuron fibers and neuromuscular junction displacement (Fischer et al., 2004; De Winter et al., 2006; Narai et al., 2009). It is still unknown whether the most claimed pathogenic processes for ALS, for instance oxidative stress, mitochondrial and neurofilament dysfunction, excitotoxicity, inflammation, non-autonomous cell toxicity, protein misfolding and abnormal RNA processing (Rothstein et al., 1992; Bergeron et al., 1994; Boillee et al., 2006a; Lemmens et al., 2010; Bendotti et al., 2012; Richardson et al., 2013) are taking place at the pre-symptomatic period of the disease.
The central question in understanding ALS facing therapeutic target development involves a further knowledge about the toxic mechanisms that trigger motor neuron death (Boillee et al., 2006a). Until scientific technology approaches do not overstep ethical limitations of clinical studies, the mutant SOD1-expressing mouse model may offer opportunity for a detailed analysis of intra and intercellular signaling-related to motor neuron toxicity.
The profiling of gene expression using different platforms have been largely employed in the ALS model in several stages of the disease course (Olsen et al., 2001; Dangond et al., 2004; Malaspina and De Belleroche, 2004; Jiang et al., 2005; Perrin et al., 2005; Ferraiuolo et al., 2007, 2011a; Yamamoto et al., 2007; Offen et al., 2009; Brockington et al., 2010; D'arrigo et al., 2010; Guipponi et al., 2010; Saris et al., 2013b; Yu et al., 2013), including the early symptomatic phase (Olsen et al., 2001; Yoshihara et al., 2002; Ferraiuolo et al., 2007; Yu et al., 2013), however, there is a lack of information on differential gene expression taking place before classical clinical symptoms (Olsen et al., 2001; Yoshihara et al., 2002; Ferraiuolo et al., 2007; Guipponi et al., 2010). Olsen et al. (2001) inaugurated that issue by looking at patterns of gene expression from SOD1G93A spinal cord by means of a murine restricted platform of oligonucleotide microarray and by describing negligible changes in the transcript profile at the pre-symptomatic phases. Other authors that have examined gene profiling in pre-symptomatic phases of ALS disease employed restricted platforms of cDNA arrays, used animals with an uncommon symptom onset (Yoshihara et al., 2002; Guipponi et al., 2010) or evaluated gene profiling in specific spinal cord cells (Ferraiuolo et al., 2007, 2011a). Authors have encountered gene expressions related to inflammation, apoptosis, oxidative stress, ATP biosynthesis, myelination, axonal transport as candidates of biological processes taking place in the pre-symptomatic periods of ALS.
By means of a high-density oligonucleotide microarrays linked to specific tools capable to identify enriched pathways, the aim of this work was to identify early molecular changes in the pre-symptomatic stage in the spinal cord of the SOD1G93A mouse model. The data showed important alterations at early 40 days pre-symptomatic period of disease and in 80 days old pre-symptomatic mice.
Materials and methods
Samples
Specific pathogen-free male SOD1G93A mice of preclinical 40 and 80 days old mice and their age-paired non-transgenic wild-type controls, 20–25 g body weight, from University of São Paulo Medical School (São Paulo, Brazil) were used in the experiments. A total of 5 animals were used in each group in microarray experiments, while in the verification experiments by quantitative polymerase chain reaction (qPCR) and laser microdissection, each group was comprised for 6 and 3 different animals, respectively. Animals were kept under standardized lighting conditions (lights on at 7:00 h and off at 19:00 h), at a constant temperature of 23°C and with free access to food pellets and tap water. The colony was derived from Jackson Laboratories (Bar Harbor, ME, USA) from G93A mutant mice with 25 ± 1.5 copies of the human SOD1 transgene (Gurney, 1994). Mouse identification (SODG93A or WT) in our colony was performed by genotyping (Scorisa et al., 2010). Animals were killed by decapitation and their lumbar spinal cords were collected for molecular analysis. The study was conducted according protocols approved by the Animal Care and Use of Ethic Committee at the University of São Paulo and in accordance with the Guide for Care and Use of Laboratory Animals adopted by the National Institutes of Health.
RNA extraction
Total RNA was isolated using the MiniSpin kit for RNA extraction (GE Healthcare, USA) according to the manufacturer's instructions. RNA quantity and integrity were assessed by spectrophotometry (Nanodrop, Thermo Scientific, USA) and microfluidics—based electrophoresis (Agilent 2100 Bioanalyzer, Agilent Technologies, USA), respectively. RNA samples with OD 260/280 of approximately 2.0 and RIN > 7.0 were used for microarray experiments and qPCR. A pool of RNAs from neonatal organs (heart, kidney, liver) was employed as reference sample. A representative eletropherogram from Bioanalyzer evaluation of RNA integrity is shown in supplementary material (Figure S1).
Microarray experiments
For samples and reference, respectively, 250 and 500 ng of RNA were reverse transcribed by the Low-input RNA Linear Amplification Kit (Agilent Technologies) and then transcribed to Cy3-labeled (samples) or Cy5-labeled (reference) cRNA according to the manufacturer. The labeled cRNA was purified (Minispin kit, GE Life Sciences), and the dye content and concentration of cRNA were measured by a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). A total of 850 ng of Cy3-labeled cRNA was hybridized together with the same amount of Cy5-labeled reference to Whole Mouse Genome Oligo 4 × 44 K microarrays overnight at 65°C, and then the slides were washed and treated with Stabilizing and Drying Solution (Agilent Technologies) and scanned by Agilent Microarray Scanner. All steps were performed according to the manufacturer (Agilent Technologies).
The raw data from hybridizations and experimental conditions are available on the Gene Expression Omnibus website under accession number GSE50642.
Data analyses
The Feature Extraction Software v9.1.3.1 (Agilent Technologies) was used to extract and analyze the assay signals and subsequently determine the signal-to-noise ratios from the microarray images. Microarrays without enough quality were taken out from further analysis. The analysis proceeded with 4 samples for each group. Microarray raw data (.txt files) were imported into R v. 3.0.1 (Team RDC, 2012) and analyzed with the Bioconductor (Gentleman et al., 2004) packages Agi4×44PreProcess and limma (Smyth, 2005). Briefly, after quality check, the microarray probes were filtered and their median foreground intensity was normalized within and between arrays according to Agi4×44Preprocess and limma user guides, respectively. Finally, the probes were tested for differential expression using a linear model followed by Bayes moderated t-test (Smyth, 2005) for the comparisons of interest. Genes with nominal p < 0.05 were accepted to be differentially expressed and further considered in the analysis.
FunNet analysis
In order to further identify over-represented pathways and biological process, the lists with differentially expressed genes for both 40 and 80 days old mice were split into lists of up and down regulated genes and submitted to FunNet web based tool (Functional Analysis of Transcriptional Networks), using Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) annotations (Prifti et al., 2008).
Laser microdissection of astrocytes
The lumbar spinal cord of mice were rapidly removed and immediately frozen in ice cold isopentane at −45°C and stored at −80°C until use. The labeling procedure was performed as described previously (De Oliveira et al., 2009) and modified according to our experience. Frozen sections (5μm) were rapidly defrosted for 30 s and fixed with ice cold acetone, for 3 min. Sections were then incubated during 3 min in phosphate buffered saline (PBS) containing 3% Triton X-100 and then incubated with primary antibody, a polyclonal rabbit anti-glial fibrillary acidic protein (GFAP; Dako Cytomation; 1:100) diluted in 0.3% Triton X-100 containing 1% BSA for 5 min. Sections were then washed in PBS for 3 times of 15 s and then incubated with texas red-conjugated goat-anti-rabbit secondary antibody, in the same diluent than primary antibody, in a final concentration of 1:50 during 5 min in the dark and at room temperature. Sections were rinsed carefully three times with PBS for 15 s and immediately submitted to laser microdissection.
Around 200 astrocytes were isolated from each 40 and 80 days old mice lumbar spinal cords using P.A.L.M. Microlaser Technologies (Zeiss). RNA was extracted using PicoPure RNA isolation kit (Arcturus) and linear amplification of RNA was performed following Eberwine's procedure (Van Gelder et al., 1990) using the RiboampHSplus kit (Arcturus) according to the manufacturer's protocol. The quantity (NanoDrop 1000 Spectrophotometer) and quality (Agilent 2100 bioanalyser, RNA 6000 Pico LabChip) of amplified RNA was analyzed as described above. Also, the astrocytes enriched samples were submitted to PCRs in order to access contamination from other cell types. Protocol and results of astrocyte samples enrichment are presented in the supplementary material (Figure S2).
Quantitative PCR
A proportion of genes identified as differentially expressed were selected for verification by qPCR, on the basis of robust microarray data confirming differential gene expression. The genes were chosen for verification based on their possible involvement in ALS related mechanisms. Verification addresses the possibility of false positive microarray signals, due to cross-hybridization with related genes, concern about the accuracy of array probe sets, and uncertainty about the hybridization kinetics of multiple reactions occurring on the miniature scale of an array chip. The qPCR verification of microarray results were performed on independent sample, as described above. cDNA was synthesized from 1 μg of total RNA treated with DNAse by a reverse transcription reagent kit (Applied Biosystems Life Technologies) according to manufacturer. qPCR reactions were carried out in duplicate with 40 ng cDNA, the DyNAmo ColorFlash SYBR Green qPCR kit (Thermo Scientific, USA) and 400 nM of each primer in a final volume reaction of 20 μl, by using the PikoReal Real-Time PCR System (Thermo Scientific). The information for SYBR primers can be found in Table 1. For astrocytes enriched samples, 1 μg of amplified RNA was reverse transcribed to cDNA by a reverse transcription reagent kit (Applied Biosystems Life Technologies) modified from original protocol in order to improve efficiency. Briefly, Oligo(dT)16 primer was added to samples and incubated at 70°C during 5 min, then the other required reagents, such as reaction buffer, MgCl2, dNTPs, RNAse inhibitor, in the same concentrations than manufacturer protocol, and 156,25 U of Reverse transcriptase (Multiscribe), were added to reaction and incubated at 37°C for 60 min followed by 95°C for 5 min. qPCR reactions were carried out in duplicate using Taqman master mix and the following assays were used: Ube2i (Mm04243971_g1) and Gapdh (Mm99999915_g1).
Table 1.
Information for primers used in SYBR qPCR experiments of 40 and 80 days old pre-symptomatic SOD1G93A and wild-type mice.
| Gene ID | Primer sequences (5′-3′) | Amplicon (bp) |
|---|---|---|
| Glg1 | F: GAGTGAGATTGCAGCCAGAG | 143 |
| R:CAGGATGTAGTTCTTTGAGGGAG | ||
| Aqp4 | F: GCTCGATCTTTTGGACCCG | 112 |
| R: AGACATACTCATAAAGGGCACC | ||
| Calca | F: TGCAGATGAAAGCCAGGG | 149 |
| R: CTTCACCACACCTCCTGATC | ||
| Eef2 | F: CATGTTTGTGGTCAAGGCATAC | 141 |
| R:TTGTCAAAAGGATCCCCAGG | ||
| Nsg1 | F: AAGTGTACAAGTATGACCGCG | 128 |
| R: GACAGTGTAAAATTTCTCCCGG | ||
| Syt10 | F: AGACCATTGGAACGAGATGC | 148 |
| R: TGGAGGCTTTTATGGTGTGG | ||
| NORMALYZER | ||
| Gapdh | F: GAGTAAGAAACCCTGGACCAC | 109 |
| R: TCTGGGATGGAAATTGTGAGG | ||
The Glg1, Aqp4, Calca genes and Eef2, Nsg1, Syt10 genes were verified in 40 and 80 days mice, respectively, according to microarray results.
For SYBR reactions the cycling was composed by an initial denaturation at 95°C for 10 min, templates were amplified by 40 cycles of 95°C for 15 s and 60°C for 30 s. A dissociation curve was then generated to ensure amplification of a single product, and absence of primer dimers. For each primer pair, a standard curve was generated to determine the efficiency of the PCR reaction over a range of template concentrations from 0.032 ng/μ l to 20 ng/μ l, using cDNA synthesized from mouse reference RNA. The efficiency for each set of primers was 100 ± 5%. For Taqman reactions, cycling was composed by an initial step of 50°C for 2 min, followed by denaturation at 95°C for 10 min, templates were amplified by 40 cycles of 95°C for 15 s and 60°C for 1 min. Gene expressions, normalized to Gapdh, could be determined using the ΔΔCt mathematical model (ABI PRISM 7700 Sequence Detection System protocol; Applied Biosystems). One-tailed unpaired t-test was used to determine the statistical significance of any differences in gene expression [GraphPad (San Diego, CA) Prism 5]. Gapdh was chosen as a housekeeping gene to normalize the qPCR values because the microarray analysis showed that its expression was stable across samples.
Results
General features of differential gene expression between SOD1G93A and wild-type mice
Statistical analysis has pointed to 492 differentially expressed genes at the lumbar region of 40 days SOD1G93A, compared to the age matched wild-type mice, being 155 up and 337 down regulated genes, respectively, while 1105 genes were found differentially expressed by 80 days old ALS mice compared to age matched controls, being 433 up and 672 down regulated genes, respectively. The whole list with differentially expressed gene for both age mice can be found in Tables S1 and S2 in the Supplementary material. Of interest, among differentially expressed genes, 66 are common to both ages; they are presented in the Table 2.
Table 2.
Differentially expressed genes common to both gene lists of 40 and 80 days old pre-symptomatic SOD1G93A and wild-type mice.
| Gene symbol | Fold change 40 days | Fold change 80 days |
|---|---|---|
| Ocel1 | −1.68 | −1.92 |
| Fam32a | −1.45 | −1.61 |
| Trim37 | 1.22 | −1.34 |
| Lsm6 | −1.3 | −1.29 |
| Map1a | 1.2 | −1.15 and −1.28 |
| Bmpr2 | 1.21 | −1.26 |
| Eif3j2 | 1.29 | −1.26 |
| Foxn3 | 1.28 | −1.25 |
| Plekha5 | 1.17 | −1.25 |
| Rfxank | −1.12 | −1.22 |
| Malat1 | 1.1 | −1.21 |
| Ddx6 | 1.3 | −1.19 |
| Huwe1 | 1.12 | −1.19 |
| Snx27 | 1.19 | −1.19 |
| Srrm3 | 1.18 | −1.19 |
| Dzip1 | 1.14 | −1.17 |
| Hook3 | 1.14 | −1.17 |
| Nemf | 1.15 | −1.16 |
| Pdlim5 | 1.09 | −1.16 |
| Plvap | −1.14 | −1.16 |
| Thrap3 | 1.15 | −1.16 |
| Srsf11 | −1.1 | −1.15 |
| Azin1 | 1.12 | −1.14 |
| Eif5b | 1.19 | −1.14 |
| Hspa4 | 1.1 | −1.14 |
| Kras | 1.19 | −1.14 |
| Sp4 | 1.12 | −1.14 |
| Tusc3 | −1.1 | −1.14 |
| Vegfa | 1.12 | −1.14 |
| Fam133b | 1.09 | −1.13 |
| Fam81a | 1.12 | −1.13 |
| Prkrir | −1.1 | −1.13 |
| Ptrf | −1.3 | −1.13 |
| 6330411E07Rik | 1.09 | −1.12 |
| Gria4 | 1.18 | −1.12 |
| Zfp866 | 1.07 | −1.12 |
| Marc-2 | −1.07 | −1.11 |
| Hadh | 1.12 | −1.11 |
| Mtf2 | 1.1 | −1.11 |
| Dhps | −1.09 | −1.1 |
| Nsd1 | 1.12 | −1.1 |
| Mier1 | 1.13 | −1.09 |
| U2surp | 1.13 | −1.09 |
| 2610507B11Rik | 1.07 | 1.1 |
| Ncam1 | 1.17 | 1.12 |
| Rtn1 | −1.17 | 1.12 |
| Chd5 | 1.12 | 1.13 |
| Dhcr7 | −1.08 | 1.15 |
| Strbp | 1.15 | 1.15 |
| Synm | −1.12 | 1.15 |
| Map7d1 | 1.14 | 1.16 |
| Specc1 | 1.1 | 1.17 |
| Maea | 1.1 | 1.19 |
| Ncl | 1.1 and −1.1 | 1.19 |
| Glg1 | 1.29 | 1.2 |
| Dnajc27 | 1.17 | 1.21 |
| Ncdn | 1.14 | 1.21 |
| Lpcat2 | 1.16 | 1.22 |
| D17Wsu92e | 1.11 | 1.24 |
| Nisch | 1.08 | 1.24 |
| Tkt | −1.1 | 1.25 |
| Tmem59l | 1.29 | 1.26 |
| Mast3 | 1.24 | 1.28 |
| Plac9a | −1.45 | 1.35 |
| Nsg1 | −1.17 | 1.22 and 1.35 |
| Trappc3 | −1.2 | 1.32 and 1.41 |
Map1a, Ncl, Nsg1, and Trappc3 genes were represented by an additional probe.
Verification of microarray results by qPCR
The results of qPCR verification for the six representative genes are shown in Table S3 (Supplementary material). The up and down regulations of the verified genes in the 40 days and 80 days old SOD1G93A mice by means of qPCR were coincident and supported the microarray findings of correspondent animal ages (Table S3).
FunNet analysis
KEGG terms which were significantly enriched (at level p < 0.05) amongst differentially expressed genes between SOD1G93A and wild-type mice were identified for both 40 days and 80 days old pre-symptomatic ALS mice. Over-represented KEGG pathways and respective genes taking part of them are given in Tables 3, 4. Of importance, differentially expressed genes from 40 and 80 days old mice allowed to recognize 7 pathways common among both periods (Figure 1). Those were glutamatergic synapse, ubiquitin mediated proteolysis, chemokine signaling pathway, endocytosis, oxidative phosphorylation, antigen processing and presentation and tight junction. The number of transcripts in each pathway is also shown in Figure 1. Moreover, other interesting pathways could also be identified to appear only in 40 days (Table 3) or 80 days old SOD1G93A mice (Table 4). Furthermore, among pathways common to both ages, ubiquitin mediated proteolysis, chemokine signaling pathway and endocytosis were over-represented by up regulated genes and oxidative phosphorylation was pointed by down regulated genes (Figure 1). Furthermore, glutamatergic synapse and tight junction were pointed by the genes that were up regulated in 40 days and also up or down regulated in 80 days gene expression lists (Figure 1). Finally, antigen processing and presentation was pointed for down regulated genes in 40 days and up regulated genes in 80 days lists (Figure 1).
Table 3.
KEGG pathways enriched amongst differentially expressed up or down regulated genes at 40 days old mice.
| Gene ID | Gene symbol | Gene name |
|---|---|---|
| PATHWAYS POINTED BY UP REGULATED GENES | ||
| Fructose and manose metabolism | ||
| 170768 | Pfkfb3 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| 18640 | Pfkfb2 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 |
| 18642 | Pfkm | Phosphofructokinase, muscle |
| 230163 | Aldob | Aldolase B, fructose-bisphosphate |
| 54384 | Mtmr7 | Myotubularin related protein 7 |
| Tight junction | ||
| 14677 | Gnai1 | Guanine nucleotide binding protein (G protein), alpha inhibiting 1 |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 18176 | Nras | Neuroblastoma ras oncogene |
| 18417 | Cldn11 | Claudin 11 |
| 192195 | Ash1l | Ash1 (absent, small, or homeotic)-like (Drosophila) |
| Glutamatergic synapse | ||
| 140919 | Slc17a6 | Solute carrier family 17 (sodium-dependent inorganic phosphate cotransporter), member 6 |
| 14677 | Gnai1 | Guanine nucleotide binding protein (G protein), alpha inhibiting 1 |
| 14802 | Gria4 | Glutamate receptor, ionotropic, AMPA4 (alpha 4) |
| 14810 | Grin1 | Glutamate receptor, ionotropic, NMDA1 (zeta 1) |
| 20511 | Slc1a2 | Solute carrier family 1 (glial high affinity glutamate transporter), member 2 |
| Axon guidance | ||
| 12767 | Cxcr4 | Chemokine (C-X-C motif) receptor 4 |
| 14677 | Gnai1 | Guanine nucleotide binding protein (G protein), alpha inhibiting 1 |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 18176 | Nras | Neuroblastoma ras oncogene |
| 22253 | Unc5c | Unc-5 homolog C (C. elegans) |
| 56637 | Gsk3b | Glycogen synthase kinase 3 beta |
| Ubiquitin mediated proteolysis | ||
| 107568 | Wwp1 | WW domain containing E3 ubiquitin protein ligase 1 |
| 17999 | Nedd4 | Neural precursor cell expressed, developmentally down-regulated 4 |
| 22210 | Ube2b | Ubiquitin-conjugating enzyme E2B |
| 59026 | Huwe1 | HECT, UBA and WWE domain containing 1 |
| 68729 | Trim37 | Tripartite motif-containing 37 |
| 70790 | Ubr5 | Ubiquitin protein ligase E3 component n-recognin 5 |
| Chemokine signaling pathway | ||
| 12767 | Cxcr4 | Chemokine (C-X-C motif) receptor 4 |
| 14677 | Gnai1 | Guanine nucleotide binding protein (G protein), alpha inhibiting 1 |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 18176 | Nras | Neuroblastoma ras oncogene |
| 18708 | Pik3r1 | Phosphatidylinositol 3-kinase, regulatory subunit, polypeptide 1 (p85 alpha) |
| 56637 | Gsk3b | Glycogen synthase kinase 3 beta |
| 73178 | Wasl | Wiskott-Aldrich syndrome-like (human) |
| Endocytosis | ||
| 107568 | Wwp1 | WW domain containing E3 ubiquitin protein ligase 1 |
| 12767 | Cxcr4 | Chemokine (C-X-C motif) receptor 4 |
| 13854 | Epn1 | Epsin 1 |
| 17999 | Nedd4 | Neural precursor cell expressed, developmentally down-regulated 4 |
| 193740 | Hspa1a | Heat shock protein 1A |
| 26431 | Git2 | G protein-coupled receptor kinase-interactor 2 |
| 78618 | Acap2 | ArfGAP with coiled-coil, ankyrin repeat and PH domains 2 |
| PATHWAYS POINTED BY DOWN REGULATED GENES | ||
| Nucleotide excision repair | ||
| 19718 | Rfc2 | Replication factor C (activator 1) 2 |
| 66979 | Pole4 | Polymerase (DNA-directed), epsilon 4 (p12 subunit) |
| DNA replication | ||
| 19718 | Rfc2 | Replication factor C (activator 1) 2 |
| 66979 | Pole4 | Polymerase (DNA-directed), epsilon 4 (p12 subunit) |
| Fatty acid metabolism | ||
| 11363 | Acadl | Acyl-Coenzyme A dehydrogenase, long-chain |
| 74205 | Acsl3 | Acyl-CoA synthetase long-chain family member 3 |
| TGF-beta signaling pathway | ||
| 12167 | Bmpr1b | Bmpr1b bone morphogenetic protein receptor, type 1B |
| 15902 | Id2 | Inhibitor of DNA binding 2 |
| 19651 | Rbl2 | Retinoblastoma-like 2 |
| Antigen processing and presentation | ||
| 12010 | B2m | Beta-2 microglobulin |
| 12317 | Calr | Calreticulin |
| 19727 | Rfxank | Regulatory factor X-associated ankyrin-containing protein |
| ECM-receptor interaction | ||
| 11603 | Agrn | Agrin |
| 12814 | Col11a1 | Collagen, type XI, alpha 1 |
| 16773 | Lama2 | Laminin, alpha 2 |
| GnRH signaling pathway | ||
| 12314 | Calm2 | Calmodulin 2 |
| 16440 | Itpr3 | Inositol 1,4,5-triphosphate receptor 3 |
| 16476 | Jun | Jun oncogene |
| 17390 | Mmp2 | Matrix metallopeptidase 2 |
| Parkinson's disease | ||
| 104130 | Ndufb11 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 11 |
| 12857 | Cox4i1 | Cytochrome c oxidase subunit IV isoform 1 |
| 66576 | Uqcrh | Ubiquinol-cytochrome c reductase hinge protein |
| 67264 | Ndufb8 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8 |
| Oxidative phosphorylation | ||
| 104130 | Ndufb11 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 11 |
| 12857 | Cox4i1 | Cytochrome c oxidase subunit IV isoform 1 |
| 66576 | Uqcrh | Ubiquinol-cytochrome c reductase hinge protein |
| 67264 | Ndufb8 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8 |
| Alzheimer's disease | ||
| 104130 | Ndufb11 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 11 |
| 12314 | Calm2 | Calmodulin 2 |
| 12857 | Cox4i1 | Cytochrome c oxidase subunit IV isoform 1 |
| 16440 | Itpr3 | Inositol 1,4,5-triphosphate receptor 3 |
| 66576 | Uqcrh | Ubiquinol-cytochrome c reductase hinge protein |
| 67264 | Ndufb8 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8 |
Table 4.
KEGG pathways enriched amongst differentially expressed up or down regulated genes at 80 days old mice.
| Gene ID | Gene symbol | Gene name |
|---|---|---|
| PATHWAYS POINTED BY UP REGULATED GENES | ||
| Vascular smooth muscle contraction | ||
| 104111 | Adcy3 | Adenylate cyclase 3 |
| 12315 | Calm3 | Calmodulin 3 |
| 14673 | Gna12 | Guanine nucleotide binding protein, alpha 12 |
| 14674 | Gna13 | Guanine nucleotide binding protein, alpha 13 |
| 18751 | Prkcb | Protein kinase C, beta |
| 213498 | Arhgef11 | Rho guanine nucleotide exchange factor (GEF) 11 |
| 224129 | Adcy5 | Adenylate cyclase 5 |
| 26413 | Mapk1 | Mitogen-activated protein kinase 1 |
| Antigen processing and presentation | ||
| 14963 | H2-Bl | Histocompatibility 2, blastocyst |
| 14972 | H2-K1 | Histocompatibility 2, K1, K region |
| 15006 | H2-Q1 | Histocompatibility 2, Q region locus 1 |
| 15007 | H2-Q10 | Histocompatibility 2, Q region locus 10 |
| 15013 | H2-Q2 | Histocompatibility 2, Q region locus 2 |
| 15018 | H2-Q7 | Histocompatibility 2, Q region locus 7 |
| 15039 | H2-T22 | Histocompatibility 2, T region locus 22 |
| 15040 | H2-T23 | Histocompatibility 2, T region locus 23 |
| 15481 | Hspa8 | Heat shock protein 8 |
| 21355 | Tap2 | Transporter 2, ATP-binding cassette, sub-family B (MDR/TAP) |
| Tight junction | ||
| 11465 | Actg1 | Actin, gamma, cytoplasmic 1 |
| 13043 | Cttn | Cortactin |
| 13821 | Epb4.1l1 | Erythrocyte protein band 4.1-like 1 |
| 13822 | Epb4.1l2 | Erythrocyte protein band 4.1-like 2 |
| 14924 | Magi1 | Membrane associated guanylate kinase, WW and PDZ domain containing 1 |
| 16897 | Llgl1 | Lethal giant larvae homolog 1 |
| 17475 | Mpdz | Multiple PDZ domain protein |
| 18751 | Prkcb | Protein kinase C, beta |
| 67374 | Jam2 | Junction adhesion molecule 2 |
| 71960 | Myh14 | Myosin, heavy polypeptide 14 |
| Chemokine signaling pathway | ||
| 104111 | Adcy3 | Adenylate cyclase 3 |
| 14083 | Ptk2 | PTK2 protein tyrosine kinase 2 |
| 14688 | Gnb1 | Guanine nucleotide binding protein (G protein), beta 1 |
| 14693 | Gnb2 | Guanine nucleotide binding protein (G protein), beta 2 |
| 14697 | Gnb5 | Guanine nucleotide binding protein (G protein), beta 5 |
| 14701 | Gng12 | Guanine nucleotide binding protein (G protein), gamma 12 |
| 14708 | Gng7 | Guanine nucleotide binding protein (G protein), gamma 7 |
| 18751 | Prkcb | Protein kinase C, beta |
| 224129 | Adcy5 | Adenylate cyclase 5 |
| 26413 | Mapk1 | Mitogen-activated protein kinase 1 |
| 277360 | Prex1 | Phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 1 |
| Ubiquitin mediated proteolysis | ||
| 103583 | Fbxw11 | F-box and WD-40 domain protein 11 |
| 15204 | Herc2 | Hect (homologous to the E6-AP (UBE3A) carboxyl terminus) domain and RCC1 (CHC1)-like domain (RLD) 2 |
| 17237 | Mgrn1 | Mahogunin, ring finger 1 |
| 19823 | Rnf7 | Ring finger protein 7 |
| 217342 | Ube2o | Ubiquitin-conjugating enzyme E2O |
| 22192 | Ube2m | Ubiquitin-conjugating enzyme E2M |
| 22196 | Ube2i | Ubiquitin-conjugating enzyme E2I |
| 22213 | Ube2g2 | Ubiquitin-conjugating enzyme E2G 2 |
| 229615 | Pias3 | Protein inhibitor of activated STAT 3 |
| 50754 | Fbxw7 | F-box and WD-40 domain protein 7 |
| 63958 | Ube4b | Ubiquitination factor E4B, UFD2 homolog (S. cerevisiae) |
| Regulation of actin cytoskeleton | ||
| 11465 | Actg1 | Actin, gamma, cytoplasmic 1 |
| 14083 | Ptk2 | PTK2 protein tyrosine kinase 2 |
| 14673 | Gna12 | Guanine nucleotide binding protein, alpha 12 |
| 14674 | Gna13 | Guanine nucleotide binding protein, alpha 13 |
| 14701 | Gng12 | Guanine nucleotide binding protein (G protein), gamma 12 |
| 18717 | Pip5k1c | Phosphatidylinositol-4-phosphate 5-kinase, type 1 gamma |
| 192897 | Itgb4 | Integrin beta 4 |
| 226970 | Arhgef4 | Rho guanine nucleotide exchange factor (GEF) 4 |
| 227753 | Gsn | Gelsolin |
| 26413 | Mapk1 | Mitogen-activated protein kinase 1 |
| 67771 | Arpc5 | Actin related protein 2/3 complex, subunit 5 |
| 71960 | Myh14 | Myosin, heavy polypeptide 14 |
| Glutamatergic synapse | ||
| 104111 | Adcy3 | Adenylate cyclase 3 |
| 110637 | Grik4 | Glutamate receptor, ionotropic, kainate 4 |
| 14645 | Glul | Glutamate-ammonia ligase (glutamine synthetase) |
| 14688 | Gnb1 | Guanine nucleotide binding protein (G protein), beta 1 |
| 14693 | Gnb2 | Guanine nucleotide binding protein (G protein), beta 2 |
| 14697 | Gnb5 | Guanine nucleotide binding protein (G protein), beta 5 |
| 14701 | Gng12 | Guanine nucleotide binding protein (G protein), gamma 12 |
| 14708 | Gng7 | Guanine nucleotide binding protein (G protein), gamma 7 |
| 18751 | Prkcb | Protein kinase C, beta |
| 216456 | Gls2 | Glutaminase 2 (liver, mitochondrial) |
| 224129 | Adcy5 | Adenylate cyclase 5 |
| 26413 | Mapk1 | Mitogen-activated protein kinase 1 |
| Phagosome | ||
| 11465 | Actg1 | Actin, gamma, cytoplasmic 1 |
| 14963 | H2-Bl | Histocompatibility 2, blastocyst |
| 14972 | H2-K1 | Histocompatibility 2, K1, K region |
| 15006 | H2-Q1 | Histocompatibility 2, Q region locus 1 |
| 15007 | H2-Q10 | Histocompatibility 2, Q region locus 10 |
| 15013 | H2-Q2 | Histocompatibility 2, Q region locus 2 |
| 15018 | H2-Q7 | Histocompatibility 2, Q region locus 7 |
| 15039 | H2-T22 | Histocompatibility 2, T region locus 22 |
| 15040 | H2-T23 | Histocompatibility 2, T region locus 23 |
| 15239 | Hgs | HGF-regulated tyrosine kinase substrate |
| 17113 | M6pr | Mannose-6-phosphate receptor, cation dependent |
| 21355 | Tap2 | Transporter 2, ATP-binding cassette, sub-family B (MDR/TAP) |
| 22142 | Tuba1a | Tubulin, alpha 1A |
| 22151 | Tubb2a | Tubulin, beta 2A class IIA |
| Protein processing in endoplasmic reticulum | ||
| 100037258 | Dnajc3 | DnaJ (Hsp40) homolog, subfamily C, member 3 |
| 108687 | Edem2 | ER degradation enhancer, mannosidase alpha-like 2 |
| 12955 | Cryab | Crystallin, alpha B |
| 15481 | Hspa8 | Heat shock protein 8 |
| 20014 | Rpn2 | Ribophorin II |
| 20338 | Sel1l | Sel-1 suppressor of lin-12-like (C. elegans) |
| 216440 | Os9 | Amplified in osteosarcoma |
| 22213 | Ube2g2 | Ubiquitin-conjugating enzyme E2G 2 |
| 269523 | Vcp | Valosin containing protein |
| 50907 | Preb | Prolactin regulatory element binding |
| 54197 | Rnf5 | Ring finger protein 5 |
| 56453 | Mbtps1 | Membrane-bound transcription factor peptidase, site 1 |
| 56812 | Dnajb2 | DnaJ (Hsp40) homolog, subfamily B, member 2 |
| 63958 | Ube4b | Ubiquitination factor E4B, UFD2 homolog (S. cerevisiae) |
| Cell adhesion molecules (CAMS) | ||
| 14963 | H2-Bl | Histocompatibility 2, blastocyst |
| 14972 | H2-K1 | Histocompatibility 2, K1, K region |
| 15006 | H2-Q1 | Histocompatibility 2, Q region locus 1 |
| 15007 | H2-Q10 | Histocompatibility 2, Q region locus 10 |
| 15013 | H2-Q2 | Histocompatibility 2, Q region locus 2 |
| 15018 | H2-Q7 | Histocompatibility 2, Q region locus 7 |
| 15039 | H2-T22 | Histocompatibility 2, T region locus 22 |
| 15040 | H2-T23 | Histocompatibility 2, T region locus 23 |
| 17967 | Ncam1 | Neural cell adhesion molecule 1 |
| 18007 | Neo1 | Neogenin |
| 19274 | Ptprm | Protein tyrosine phosphatase, receptor type, M |
| 20340 | Glg1 | Golgi apparatus protein 1 |
| 20970 | Sdc3 | Syndecan 3 |
| 58235 | Pvrl1 | Poliovirus receptor-related 1 |
| 67374 | Jam2 | Junction adhesion molecule 2 |
| Endocytosis | ||
| 11771 | Ap2a1 | Adaptor-related protein complex 2, alpha 1 subunit |
| 12757 | Clta | Clathrin, light polypeptide (Lca) |
| 13196 | Asap1 | ArfGAP with SH3 domain, ankyrin repeat and PH domain1 |
| 13429 | Dnm1 | Dynamin 1 |
| 14963 | H2-Bl | Histocompatibility 2, blastocyst |
| 14972 | H2-K1 | Histocompatibility 2, K1, K region |
| 15006 | H2-Q1 | Histocompatibility 2, Q region locus 1 |
| 15007 | H2-Q10 | Histocompatibility 2, Q region locus 10 |
| 15013 | H2-Q2 | Histocompatibility 2, Q region locus 2 |
| 15018 | H2-Q7 | Histocompatibility 2, Q region locus 7 |
| 15039 | H2-T22 | Histocompatibility 2, T region locus 22 |
| 15040 | H2-T23 | Histocompatibility 2, T region locus 23 |
| 15239 | Hgs | HGF-regulated tyrosine kinase substrate |
| 15481 | Hspa8 | Heat shock protein 8 |
| 16835 | Ldlr | Low density lipoprotein receptor |
| 18717 | Pip5k1c | Phosphatidylinositol-4-phosphate 5-kinase, type 1 gamma |
| 234852 | Chmp1a | Charged multivesicular body protein 1A |
| 243621 | Iqsec3 | IQ motif and Sec7 domain 3 |
| 67588 | Rnf41 | Ring finger protein 41 |
| 98366 | Smap1 | Stromal membrane-associated protein 1 |
| PATHWAYS POINTED BY DOWN REGULATED GENES | ||
| VEGF signaling pathway | ||
| 11651 | Akt1 | Thymoma viral proto-oncogene 1 |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 19056 | Ppp3cb | Protein phosphatase 3, catalytic subunit, beta isoform |
| 22339 | Vegfa | Vascular endothelial growth factor A |
| Long-term depression | ||
| 14678 | Gnai2 | Guanine nucleotide binding protein (G protein), alpha inhibiting 2 |
| 14683 | Gnas | GNAS (guanine nucleotide binding protein, alpha stimulating) complex locus |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 18795 | Plcb1 | Phospholipase C, beta 1 |
| 60596 | Gucy1a3 | Guanylate cyclase 1, soluble, alpha 3 |
| Gap junction | ||
| 14678 | Gnai2 | Guanine nucleotide binding protein (G protein), alpha inhibiting 2 |
| 14683 | Gnas | GNAS (guanine nucleotide binding protein, alpha stimulating) complex locus |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 18795 | Plcb1 | Phospholipase C, beta 1 |
| 60596 | Gucy1a3 | Guanylate cyclase 1, soluble, alpha 3 |
| RNA degradation | ||
| 104625 | Cnot6 | CCR4-NOT transcription complex, subunit 6 |
| 13209 | Ddx6 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 6 |
| 66373 | Lsm5 | LSM5 homolog, U6 small nuclear RNA associated (S. cerevisiae) |
| 72662 | Dis3 | DIS3 mitotic control homolog (S. cerevisiae) |
| 78651 | Lsm6 | LSM6 homolog, U6 small nuclear RNA associated (S. cerevisiae) |
| Parkinson's disease | ||
| 12866 | Cox7a2 | Cytochrome c oxidase subunit VIIa 2 |
| 333182 | Cox6b2 | Cytochrome c oxidase subunit VIb polypeptide 2 |
| 66142 | Cox7b | Cytochrome c oxidase subunit VIIb |
| 66495 | Ndufb3 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3 |
| 66916 | Ndufb7 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7 |
| 68202 | Ndufa5 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 |
| Oxidative phosphorylation | ||
| 12866 | Cox7a2 | Cytochrome c oxidase subunit VIIa 2 |
| 333182 | Cox6b2 | Cytochrome c oxidase subunit VIb polypeptide 2 |
| 66142 | Cox7b | Cytochrome c oxidase subunit VIIb |
| 66495 | Ndufb3 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3 |
| 66916 | Ndufb7 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7 |
| 68202 | Ndufa5 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 |
| Tight junction | ||
| 11651 | Akt1 | Thymoma viral proto-oncogene 1 |
| 14678 | Gnai2 | Guanine nucleotide binding protein (G protein), alpha inhibiting 2 |
| 16653 | Kras | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| 17888 | Myh6 | Myosin, heavy polypeptide 6, cardiac muscle, alpha |
| 30960 | Vapa | Vesicle-associated membrane protein, associated protein A |
| 58187 | Cldn10 | Claudin 10 |
| Glutamatergic synapse | ||
| 14678 | Gnai2 | Guanine nucleotide binding protein (G protein), alpha inhibiting 2 |
| 14683 | Gnas | GNAS (guanine nucleotide binding protein, alpha stimulating) complex locus |
| 14702 | Gng2 | Guanine nucleotide binding protein (G protein), gamma 2 |
| 14802 | Gria4 | Glutamate receptor, ionotropic, AMPA4 (alpha 4) |
| 14805 | Grik1 | Glutamate receptor, ionotropic, kainate 1 |
| 18795 | Plcb1 | Phospholipase C, beta 1 |
| 19056 | Ppp3cb | Protein phosphatase 3, catalytic subunit, beta isoform |
| 216227 | Slc17a8 | Solute carrier family 17 (sodium-dependent inorganic phosphate cotransporter), member 8 |
| Huntington's disease | ||
| 12866 | Cox7a2 | Cytochrome c oxidase subunit VIIa 2 |
| 18795 | Plcb1 | Phospholipase C, beta 1 |
| 333182 | Cox6b2 | Cytochrome c oxidase subunit VIb polypeptide 2 |
| 66142 | Cox7b | Cytochrome c oxidase subunit VIIb |
| 66495 | Ndufb3 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3 |
| 66916 | Ndufb7 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7 |
| 68202 | Ndufa5 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 |
| 69920 | Polr2i | Polymerase (RNA) II (DNA directed) polypeptide I |
| Alzheimer's disease | ||
| 11820 | App | Amyloid beta (A4) precursor protein |
| 12866 | Cox7a2 | Cytochrome c oxidase subunit VIIa 2 |
| 18795 | Plcb1 | Phospholipase C, beta 1 |
| 19056 | Ppp3cb | Protein phosphatase 3, catalytic subunit, beta isoform |
| 333182 | Cox6b2 | Cytochrome c oxidase subunit VIb polypeptide 2 |
| 66142 | Cox7b | Cytochrome c oxidase subunit VIIb |
| 66495 | Ndufb3 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3 |
| 66916 | Ndufb7 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7 |
| 68202 | Ndufa5 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 |
| Ribosome | ||
| 19951 | Rpl32 | Ribosomal protein L32 |
| 19981 | Rpl37a | Ribosomal protein L37a |
| 19982 | Rpl36a | Ribosomal protein L36A |
| 20068 | Rps17 | Ribosomal protein S17 |
| 20085 | Rps19 | Ribosomal protein S19 |
| 22186 | Uba52 | Ubiquitin A-52 residue ribosomal protein fusion product 1 |
| 57294 | Rps27 | Ribosomal protein S27 |
| 66489 | Rpl35 | Ribosomal protein L35 |
| 67945 | Rpl41 | Ribosomal protein L41 |
| 68028 | Rpl22l1 | Ribosomal protein L22 like 1 |
| 75617 | Rps25 | Ribosomal protein S25 |
Figure 1.

KEGG pathways classification showing the number of transcripts up regulated and down regulated per category in 40 and 80 days pre-symptomatic SOD1G93A mice in relation to age matched wild-types. Bars on the left indicate the number of down regulated genes, and bars on the right indicate the number of up regulated genes, for each category.
Some pathways pointed by FunNet were omitted from table because they were composed by genes already presented in other pathways and also genes apparently not related to ALS. They were melanoma, measles, hepatitis C, melanogenesis, pathways in cancer and prostate cancer at 40 days and viral myocarditis and melanogenesis in 80 days results.
The results for GO enriched terms can be found in Tables S4 and S5 in Supplementary material.
Laser microdissection of astrocytes and qPCR experiment
The profile for GFAP immunofluorescence for specific identification of astrocytes can be found in Figure 2. Our protocol allowed easily identifying the astrocytic profiles (Figure 2A) to be microdissected (Figure 2B). The procedure allowed a complete microdissection of the desired cell type (Figure 2C), the astrocytes in our case. The results of qPCR for Ube2i, using the two cycle amplified RNA, from 40 and 80 days mouse laser microdissected astrocytes have shown increased gene expressions in transgenic mice of both pre-symptomatic ages (Figure 3). The Ube2i expression was increased by 5.53-fold in the astrocytes from 40 days old SOD1G92A mice and by 1.77-fold change in astrocytes from 80 days old SOD1G92A mice compared to respective age matched wild-type samples.
Figure 2.
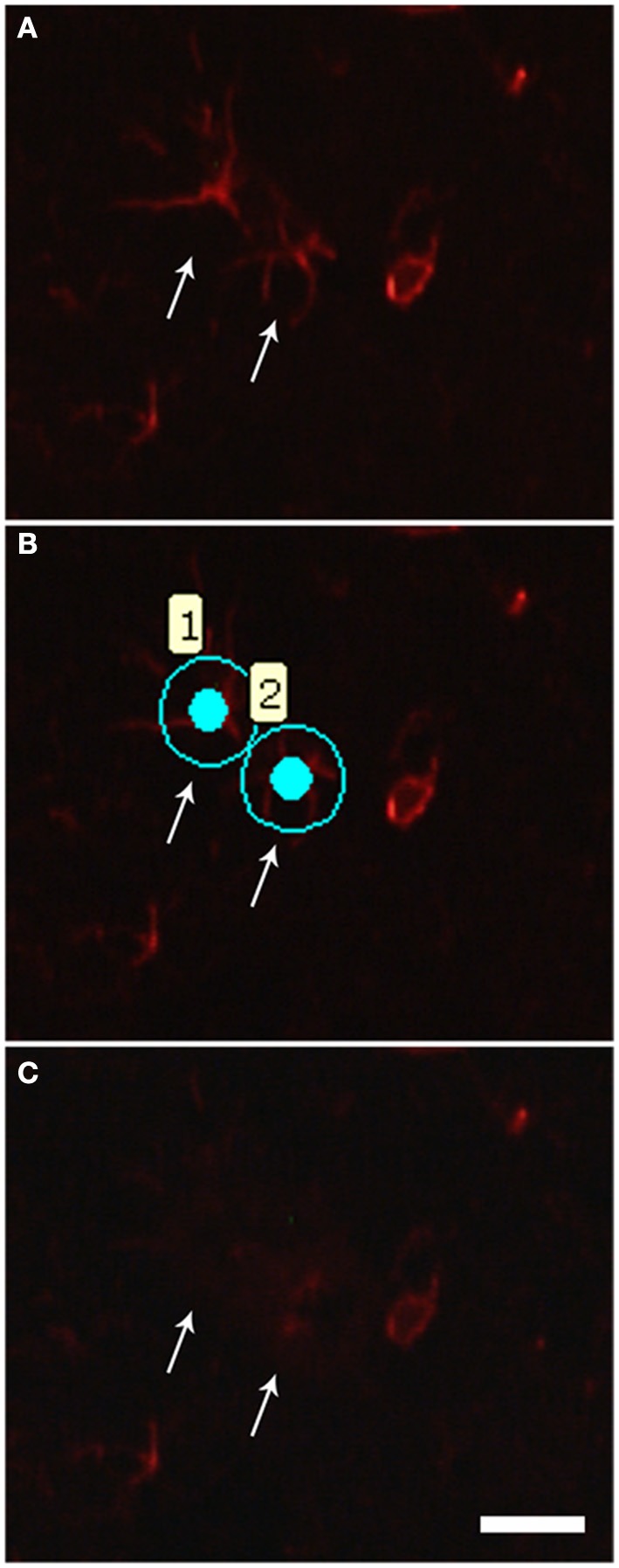
Photomicrographs illustrating astrocyte laser microdissection process. (A) The quick GFAP immunofluorescence allows recognizing the astrocytic profiles (arrows). (B) Astrocytes (1 and 2) were then selected for microdissection. (C) After laser firing and microdissection, selected cells (arrows) can no longer be visualized in the tissue. Scale bars of 20 μm.
Figure 3.
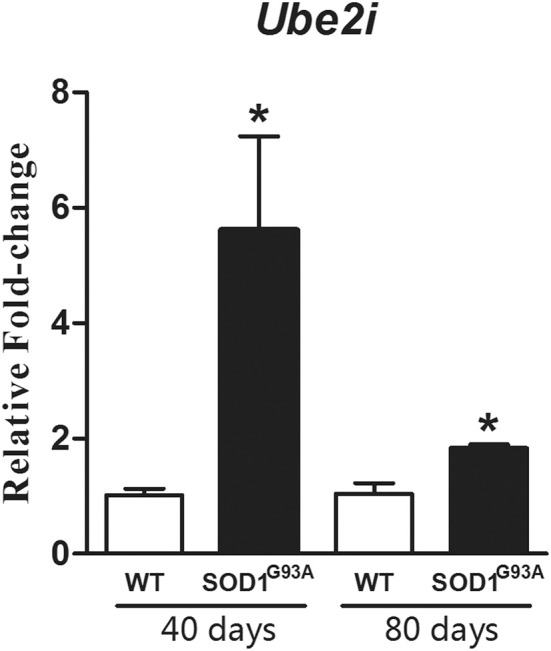
Graph shows relative fold change values for Ube2i in microdissected astrocytes from 40 and 80 days old SOD1G93A mice compared to the age matched wild-type controls (WT). Significant increases are seen in both transgenic astrocytes enriched samples. Results are presented as means ± s.e.m. from 3 samples used for each group. *p-value < 0.05, according to unpaired t-test.
Discussion
Gene-expression profiling studies have been conducted in the search of molecular pathways related to motor neuron death in ALS by employing animal models in different phases of the disease and human post mortem material at the very end stage of motor neuron degeneration (Olsen et al., 2001; Dangond et al., 2004; Malaspina and De Belleroche, 2004; Jiang et al., 2005; Perrin et al., 2005; Ferraiuolo et al., 2007, 2011a; Yamamoto et al., 2007; Offen et al., 2009; Brockington et al., 2010; D'arrigo et al., 2010; Guipponi et al., 2010; Saris et al., 2013b).
The analysis of the mechanisms that trigger motor neuron death in the ALS may include evaluation of the altered molecular pathways that are taking place in compromised regions before the occurrence of cell death. Previous works have attempted to describe gene profiling in the pre-symptomatic phases of ALS animal model by employing distinct methodologies (Ferraiuolo et al., 2007; D'arrigo et al., 2010; Guipponi et al., 2010). This is the first work to analyze gene expression profile in the whole lumbar spinal cord of early 40 and 80 days old pre-symptomatic SOD1G93A mouse in a whole genome array platform, which allowed depicting enriched pathways related to possible mechanisms of neuronal toxicity in ALS. Our analysis has pointed to up to 1105 differentially expressed genes in pre-symptomatic periods of SOD1G93A mouse model, a larger number of than described elsewhere (Perrin et al., 2005, 2006). It should be pointed that the average of fold change described in previous publications is about 3, which is higher than that found in our microarray analysis. However, it must be emphasized that subtle changes in gene expression are exactly those that occur in initial stages of disease before the onset of clinical symptoms (Druyan et al., 2008). Moreover, some authors have argued that even small differences can be biologically relevant (Pedotti et al., 2008). Indeed, our qPCR verification analysis revealed higher fold changes than in the microarray, reaching values higher than 2 in the 80 days pre-symptomatic phase, which is closer to the symptom onset. The use of qPCR analysis to qualitatively verify the microarray results is largely accepted in the literature. However, it is well recognized that both methods have quantitative differences (Chuaqui et al., 2002), which are thought to be related to the variation in the hybridization kinetics of the technologies, low fold changes or lack of concordance between transcripts accessed in each method. The number of genes employed in qPCR validation is comparable to that found by other studies (Dallas et al., 2005; Brockington et al., 2010).
The differentially expressed genes with a p-value lower than 0.05 were submitted to enrichment analyses based on GO and KEGG databases, which correlated genes to already described related pathways and processes. Modulated genes based on GO evidenced more general biological processes that might be implicated in the ALS mechanisms. Of interest, regulation of astrocyte differentiation, protein retention in endoplasmatic reticulum lumen, Golgi vesicle transport and fructose metabolism, among others, were pointed at the pre-symptomatic 40 days old transgenic mice. At later pre-symptomatic phase of 80 days, the pattern of gene expression identified the GO terms post-Golgi vesicle-mediated transport, tricarboxylic acid cycle (TCA) and mRNA processing, among others. GO database analyses have been largely employed in the ALS research in several phases of the disease (Ferraiuolo et al., 2007, 2011a; Brockington et al., 2010).
Authors have also used the KEGG database to identify overrepresented pathways based on differentially expressed genes obtained by the microarray technique (Mougeot et al., 2011; Kalathur et al., 2012). The KEGG database analysis in the present work pointed to pathways that might be related to ALS mechanism at the pre-symptomatic ages of SOD1G93A mice. Some pathways were found to be common to both pre-symptomatic periods, emphasizing the putative toxic triggering that may last before the onset of classical ALS symptoms with possible significance to mechanisms of initiation of motor neuron degeneration. Those pathways are going to be discussed below. It should be mentioned that alternative splicing have been recently implicated in ALS mechanisms (Lenzken et al., 2011; Singh and Cooper, 2012), however we could not access this biological event because the present analysis employed a platform designed to gene expression studies on 3′UTR that does not allow evaluation of alternative splicing variants.
Glutamatergic synapse
The microarray profiling study by means of KEEG enriched analysis pointed to the category of glutamatergic synapse pathway in the lumbar spinal cord of ALS SOD1G93A. The large number of up regulated genes at 40 and 80 days underlines the excitotoxicity estate mediated by glutamatergic synapse of motor neurons in the pre-symptomatic condition of ALS disease (Bendotti et al., 2001; Gibb et al., 2007; Zhao et al., 2008; Jiang et al., 2009; Sunico et al., 2011). The modulation of GluR4, by means of Gria4 findings in our microarray analysis, might reflect the dynamic state of the AMPA receptor subunit in the course of pre-symptomatic stages of ALS. At the early phases of the pre-symptomatic period, highly expressed Gria4 gene might contribute to the AMPA receptor-mediated motor neuron toxicity, being a very early mechanism of the disease. The down regulation of the Gria4 at the late pre-symptomatic stage could reflect a transient reactive mechanism to excitotoxic condition preceding motor neuron death. Reductions of GluR4 have been described at cellular level in the late disease stage of SOD1 mice, without alterations at the pre-symptomatic periods (Petri et al., 2005) thus, reflecting a disappearance of GluR4 containing neurons. In fact, imbalance of excitatory to inhibitory synaptic function precedes motor neuron degeneration as described in the spinal cord motor neurons in the late stage of pre-symptomatic phase of SOD1 ALS model by means of cellular analyses (Schutz, 2005). It should be mentioned that Ca2+ permeability of the AMPA receptor seems to occur mainly by the presence of the GluR2 subunit in the receptor complex. In fact, GluR2 deficiency clearly accelerated the motor neuron degeneration and shortened the life span of mutant SOD1G93A double transgenic mice (Tateno et al., 2004). Synaptic GluR1 increases/mRNA up regulation, and decreases of synaptic and total GluR2 were found at early ages prior to disease onset thus prompting motor neurons to a higher Ca2+-permeable AMPA receptors -induced excitotoxicity (Zhao et al., 2008). The variant C-terminus of GluR4 (GluR4c), an alternative splicing isoform, stabilizes and locates AMPA receptors in the cell membrane, and also seems to potentate actions of GluR2 (Kawahara et al., 2004), thus highlighting the pivotal role of GluR4 subunit in regulating channel properties and trafficking of AMPA receptors. It must be then further clarified the role of GluR4 in the ALS mechanisms and possible dynamic interaction with that subunit with other AMPA receptor subtypes, especially GluR2.
The regulation of Slc1a2 glial glutamate transporter (named EAAT2 or glial glutamate transporter GLT1) has not been evaluated in details. Excitotoxicity caused by a down-regulation of EAAT2 is thought to be a contributing factor to motor neuron death in ALS. Several mechanisms may account for impairment of EAAT2 function, for instance altered transcription/splicing, post-translational modifications, accelerated degradation, intracelular trafficking and inactivation by caspase-3 cleavage (Heath and Shaw, 2002; Boston-Howes et al., 2006) but not directly to gene regulation processes. It is possible that the impaired EAAT2 function could take place at the very early period of the pre-symptomatic stage, a matter that remains to be elucidated (Bendotti et al., 2001; Sasaki et al., 2001), thus, explaining the Slc1a2 expression possibly related to motor neuron protection at those ages. The absence of this genomic process in the late pre-symptomatic period might potentiate loss of function of GLT1 thus culminating with the motor neuron death in ALS. Furthermore, the vesicular glutamate transporter 2 (VGLUT2), codified by Slc17a6 gene, was found to be regulated and related to neuronal death in the pre-symptomatic stage of ALS model (Schutz, 2005; Sunico et al., 2011). The genetic reduction of VGLUT2 protein level in the ALS mouse model accounted for motor neuron rescue without modifying functional impairment (Wootz et al., 2010). It is possible that the up regulation of the Slc17a6 gene at the early pre-symptomatic stage of the 40 days old SOD1G93A mice potentiates the toxic state of motor neurons.
Ubiquitin mediated proteolysis and oxidative phosphorylation
A recent meta-analysis study of the reported gene lists has described the evidences for a shared dysfunction in protein turnover in the ubiquitin-proteasome system in ALS mouse models and ALS patients (Saris et al., 2013a). Moreover, constitutive proteasome was decreased in motor neurons at the pre-symptomatic stage of SOD1G93A (Cheroni et al., 2005), an alternative processes to decrease aggregate formation, thus an attempt to neuroprotect motor neurons of preclinical SOD1G93A mice before the onset of clinical symptoms (Bendotti et al., 2012). That should be the case of Nedd4 and Fbxw7 expressions described here, whose encoded molecules have been already correlated to neuroprotection in ALS (Nateri et al., 2004; Matsumoto et al., 2011; Kwak et al., 2012). Moreover, it should be taken into attention the elevation of Ubc12 in the spinal cord of SOD1G93A mice at the pre-symptomatic phase (Massignan et al., 2007). Ubc12 is an ubiquitin E2 ligase that adds NEDD-8 to substrates. Ubc12 elevation in pre-symptomatic ALS was correlated to a tentative response to protein aggregation (Massignan et al., 2007). Interestingly, Nedd8 gene was down regulated in our microarray analysis only in 80 days old mice, possibly representing a failure of the above described process close to the period of clinical onset.
The down regulation of genes over-representing the oxidative phosphorylation category at both pre-symptomatic ages of ALS mice seen in this work may be related to the progressive deteriorations of mitochondrial function and oxidative phosphorylation system described at pre-symptomatic ALS phases (Lin et al., 2009; Chen et al., 2010; Martin, 2010, 2011; Koopman et al., 2013), thus triggering reactive oxygen species (ROS) production (Manfredi and Xu, 2005) and motor neuron vulnerability before the onset of clinical symptoms. It is also interesting to notice that the TCA was seen as an over-represented GO term (Table S5, Supplementary material) in the up-regulated 80 days gene expression list. The TCA cycle is responsible to provide substrate to oxidative phosphorylation (Koopman et al., 2013) and its up regulation was previously seen in laser microdissected motor neurons from a VEGF model of ALS already in the pre-symptomatic period (Brockington et al., 2010). All in all, a possible mechanism of oxidative phosphorylation in the astrocyte-neuronal unit taking place in pre-symptomatic ALS might amplify motor neuron vulnerability to ROS damage.
Chemokine signaling pathway and tight junction
The up regulation of all genes in the category chemokine signaling pathway in the pathogenesis of ALS is in agreement to previous publications (Henkel et al., 2006; Zhang et al., 2006; Rentzos et al., 2007; Kuhle et al., 2009; Sargsyan et al., 2009; Tateishi et al., 2010; Gupta et al., 2012). The up regulation of Cxcr4 and Pik3r1 described in this work is an important finding because the genes might be involved in non-autonomous toxicity in the early phase of ALS (Shideman et al., 2006; Luo et al., 2007; Manzano et al., 2011). Furthermore, disruption of blood-brain barrier and blood-spinal cord barrier are described as early events in ALS, thus impairing neurovascular unit prior motor neuron degeneration (Garbuzova-Davis et al., 2011, 2012; Grammas et al., 2011; Miyazaki et al., 2011). Indeed, reduced levels of adhesion molecules and the tight junction proteins zona occludens-1, occludin and claudin-5 are shown in post mortem tissue from patients and in ALS animal models (Zhong et al., 2008; Arhart, 2010; Garbuzova-Davis et al., 2012).
Our KEGG enriched analysis also demonstrated the modulation of tight junction related genes. Of substantial interest, we might point out the up regulation of Cldn11 at 40 days and the down regulation of Cldn10 at 80 days pre-symptomatic ALS mice, in agreement to previous description on differential regulation of tight junction genes related to specific characteristics of ALS clinical evolution (Henkel et al., 2009).
It is also important to highlight the particular modulation of the Kras gene, which has been up regulated at the age of 40 days and down regulated at the age of 80 days. The Kras gene is an oncogene that was located in the tight junction category by the KEGG analysis probably due its relation to topography of invading/proliferating cells in the scenario of neurodegenerative processes. Moreover, Kras proteins regulate cell activities such as proliferation, differentiation, apoptosis, and cell migration, those taking place in neurodegenerative processes-induced astroglial/microglial activation as well as expression of inflammatory and neurotrophic/neurotoxic mediators (Rotshenker, 2009). There is a marked proliferation/activation of both microglia and astrocytes at specific disease stages in ALS mouse models (Hall et al., 1998; Weydt et al., 2002) leading to the production of neuroprotective or pro-inflammatory molecules, which can decrease or increase the rate of primary motor neuron degeneration, respectively. Taken all together, up regulation of Kras gene at the early pre-symptomatic phase is in line with the early glial proliferative and reactivity events that will initiate the toxic triggering of non-autonomous cells and also the glial neuroprotective mechanisms to maintain temporarily the motor neurons. Later in that period, still before neuronal degeneration taking place, Kras gene down regulation might allow glial cells to drive toxic insult.
Endocytosis and antigen processing and presentation
Endocytosis was an additional over-represented pathway in the pre-symptomatic stage of ALS. Genes found before clinical onset pointing to endocytosis have been related to clathrin-dependent/independent endocytosis, autophagy and also neurotransmission (Massey et al., 2006; Luo et al., 2007; Kon and Cuervo, 2010; McMahon and Boucrot, 2011; Elmer and McAllister, 2012), thus, related to extracellular turnover, repair of molecular processes and neuroprotection (Le Roy and Wrana, 2005; Doherty and McMahon, 2009; McMahon and Boucrot, 2011; Polymenidou and Cleveland, 2011). Disruption of these processes has been implicated as a general feature in the pathogenesis of ALS (Otomo et al., 2012), whereas there is a lack of information on that issue in pre-symptomatic periods (Morimoto et al., 2007; Tian et al., 2011). Clathrin-mediated endocytosis has a range of different physiological functions, remarkably the regulation of surface proteins, nutrition, activation of signaling pathways, protein trafficking and degradation of membrane components, in fact, mechanisms that might occur at the pre-symptomatic phases of ALS.
It is likely that the regulation of the genes for the heat shock proteins Hspa1a and Hspa8 (also known as Hsp70-3 and Hsc70, respectively), described in our work is related to neuroprotective events before neurodegeneration, once treatment with recombinant human Hsp70 was able to both increase lifespan (Gifondorwa et al., 2007) and decrease neuromuscular junction denervation (Gifondorwa et al., 2012) in the SOD1G93A mouse model. This protective role of Hsp70 has been also supported by other authors (Bruening et al., 1999; Takeuchi et al., 2002; Kieran et al., 2004). Actually, the increase of Hsc70 in the spinal cord of transgenic mice at pre-symptomatic ages of disease (Basso et al., 2009) and the demonstration of ubiquitinated Hsc70-induced degradation of mutant SOD1 (Urushitani et al., 2004) emphasized the possible neuroprotective role of heat shock protein regulation described in our work.
Furthermore, antigen processing and presentation pathway was also pointed as enriched among down regulated genes in 40 days old and up regulated genes in 80 days old pre-symptomatic SOD1G93A mice. Genes presented in 40 and 80 days lists are mostly related to major histocompatibility complex (MHC) class I (H2-Bl, H2-K1, H2-Q1, H2-Q10, H2-Q2, H2-Q7, H2-T22, H2-T23—80 days ALS mice), molecules necessary for peptide loading (Tap2—80 days ALS mice) and to surface expression (B2m—40 days ALS mice) (Kimura and Griffin, 2000). B2m gene, possibly via cell surface MHC class I molecules, has been implicated in the synaptic plasticity at dendrites and axonal regeneration after peripheral nerve axotomy (Oliveira et al., 2004). It is possible that the down regulation of B2m in spinal cord from SOD1G93A at pre-symptomatic ages is related to axonal and dendritic retractions and displacement of neuromuscular junction described as one of the earliest events faced by motor neurons in ALS models (Fischer et al., 2004). Our findings are in line with a description of down regulation of B2m protein reported in cerebrospinal fluid of ALS patients (Brettschneider et al., 2008), thus emphasizing the importance of its regulation in ALS. Additionally, Rfxank was down regulated at 40 days in our analysis, which is in agreement to a loss of MHC-II neuronal expression concurrent with abundant MHCII-positive microglia surrounding motor neurons in the pre-symptomatic SOD1G93A mice (Casas et al., 2013), thus, interfering with the neuroimmunemodulation mediated by microglia (Graber et al., 2010; Sanagi et al., 2010). All in all, dysregulation of genes related to antigen processing and presentation might account for a number of intercellular mechanisms able to amplify the harmful non-autonomous cell toxicity at the pre-symptomatic stages of ALS.
Laser microdissection of astrocytes
We performed laser microdissection of GFAP positive astrocytes from lumbar spinal cord ventral horn of SOD1G93A transgenic and wild-type mice in the same pre-symptomatic ages of microarray analysis. The use of laser microdissection has been gained importance in recent years, once it allows specific cell enrichment from complex tissues, revealing to be a powerful tool in the study of neurodegenerative disorders in which individual cell types are known to be differentially involved in disease stages. The advantage of the methodology is the possibility to address molecular biology in the context of in vivo cellular analysis. The method is of substantial importance to evaluate changes in the astrocytes, the glial cell involved remarkably in toxic mechanisms of ALS. A previous study has employed laser microdissection of astrocytes to perform microarray experiments in ALS mouse model (Ferraiuolo et al., 2011a). The pattern of gene expression was first evaluated in the lumbar regions of the spinal cord in the present analysis, thus, taking into account all cell types from tissue. The depicted pathways represented the state of intercellular interaction in the pre-symptomatic studied periods of the ALS mouse model. The selected genes to be evaluated in type-specific cell, which is the case of Ube2i in the laser microdissected astrocytes described herein, would allow a closer analysis of astrocyte participation in the context of the neighbor cell toxicity. The Ube2i gene was then chosen for further evaluation in astrocytes by qPCR because astrocytes exert a non-autonomous cell toxicity to motor neurons and because SUMOylation pathway has gained importance in ALS mechanisms recently (for review, see Dangoumau et al., 2013). Increases of gene expression for Ube2i were found in enriched astrocytes samples from 40 and 80 days old pre-symptomatic mice, a regulation still not presented in the literature in that stage of disease, thus, entering in the context of ALS pathogenesis. In fact, conjugation of small ubiquitin-like modifier (SUMO) molecules involves a series of steps, being the ubiquitin conjugating enzyme E2, codified by Ube2i gene, responsible for the recognition of the target protein. SUMOylation is involved in the cellular response to oxidative stress, hypoxia, glutamate excitotoxicity and proteasome impairment, events that have been linked to motor neuron toxicity in ALS (Xu et al., 2011). Moreover, studies are required to determine the precise implication of the SUMO pathway in regulating the balance between cellular adaptive and neuroprotective response to stress (Fei et al., 2006; Dangoumau et al., 2013) with a special importance to motor neuron in the pre-symptomatic stage of ALS. Nevertheless, as discussed previously in this report, glutamate astroglial excitotoxicity faced by motor neurons in ALS is also hamfull by the cleavage of EAAT2 in the ventral horn of the spinal cord (Martin et al., 2007; Foran et al., 2011). The proteolytic fragments may be SUMOylated and accumulated in the nucleus of astrocytes (Boston-Howes et al., 2006; Foran et al., 2011) as described in SOD1G93A mice, worsening the gliotoxic effects of astrocytes to motor neurons (Foran et al., 2011). Taking together, SUMOylation process and expression of Ube2i might participate in complex events related to the astrocyte-neuron unit in ALS, and future works are required to address specific cellular events.
In conclusion, the present work gives further evidence about molecular events taking place in the spinal cord from ALS mouse model before the onset of classical symptoms. The gene expression changes reflect responses for both neuroprotection and toxicity at the spinal cord in the evaluated periods. Indeed, the study of Ube2i expression in astrocytes adds novel insights for the participation of this cell type on the early mechanisms in ALS.
Author contributions
Gabriela Pintar de Oliveira and Chrystian J. Alves performed the experiments. All authors designed the study, analyzed the results and wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Grant #2010/20457-7, São Paulo Research Foundation (FAPESP). I would like to thanks Dr. Jessica Ruivo Maximino for establishing the SOD1G93A mouse colony in the Animal Facility of FMUSP and advices on animal handling and tissue processing. We also thanks Drs. Dirce Maria Carraro and Alex Fiorini de Carvalho from Laboratory of Genomics and Molecular Biology, A.C. Camargo Hospital, São Paulo, Brazil, for expertise on microarray experiments and Dr. Chin Jia Lin, responsible for Laser Microdissection Microscope Facility at FMUSP, for his expertise on laser microdissection experiments. Indeed, we thank Dr. Pamela J Shaw, from Sheffield University, and her research team for the support in the microarray analysis.
Supplementary material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fncel.2013.00216/abstract
References
- Alves C. J., De Santana L. P., Dos Santos A. J., De Oliveira G. P., Duobles T., Scorisa J. M., et al. (2011). Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 1394, 90–104 10.1016/j.brainres.2011.02.060 [DOI] [PubMed] [Google Scholar]
- Andersen P. M., Al-Chalabi A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615 10.1038/nrneurol.2011.150 [DOI] [PubMed] [Google Scholar]
- Arhart R. W. (2010). A possible haemodynamic mechanism for amyotrophic lateral sclerosis. Med. Hypotheses 75, 341–346 10.1016/j.mehy.2010.03.017 [DOI] [PubMed] [Google Scholar]
- Basso M., Samengo G., Nardo G., Massignan T., D'alessandro G., Tartari S., et al. (2009). Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS ONE 4:e8130 10.1371/journal.pone.0008130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beghi E., Logroscino G., Chio A., Hardiman O., Mitchell D., Swingler R., et al. (2006). The epidemiology of ALS and the role of population-based registries. Biochim. Biophys. Acta 1762, 1150–1157 10.1016/j.bbadis.2006.09.008 [DOI] [PubMed] [Google Scholar]
- Bendotti C., Marino M., Cheroni C., Fontana E., Crippa V., Poletti A., et al. (2012). Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog. Neurobiol. 97, 101–126 10.1016/j.pneurobio.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Bendotti C., Tortarolo M., Suchak S. K., Calvaresi N., Carvelli L., Bastone A., et al. (2001). Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J. Neurochem. 79, 737–746 10.1046/j.1471-4159.2001.00572.x [DOI] [PubMed] [Google Scholar]
- Bergeron C., Beric-Maskarel K., Muntasser S., Weyer L., Somerville M. J., Percy M. E. (1994). Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J. Neuropathol. Exp. Neurol. 53, 221–230 10.1097/00005072-199405000-00002 [DOI] [PubMed] [Google Scholar]
- Boillee S., Vande Velde C., Cleveland D. W. (2006a). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59 10.1016/j.neuron.2006.09.018 [DOI] [PubMed] [Google Scholar]
- Boillee S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., et al. (2006b). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 10.1126/science.1123511 [DOI] [PubMed] [Google Scholar]
- Boston-Howes W., Gibb S. L., Williams E. O., Pasinelli P., Brown R. H., Jr., Trotti D. (2006). Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J. Biol. Chem. 281, 14076–14084 10.1074/jbc.M600653200 [DOI] [PubMed] [Google Scholar]
- Brettschneider J., Mogel H., Lehmensiek V., Ahlert T., Sussmuth S., Ludolph A. C., et al. (2008). Proteome analysis of cerebrospinal fluid in amyotrophic lateral sclerosis (ALS). Neurochem. Res. 33, 2358–2363 10.1007/s11064-008-9742-5 [DOI] [PubMed] [Google Scholar]
- Brockington A., Heath P. R., Holden H., Kasher P., Bender F. L., Claes F., et al. (2010). Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFdelta/delta mouse model of amyotrophic lateral sclerosis. BMC Genomics 11:203 10.1186/1471-2164-11-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R., Miller R. G., Swash M., Munsat T. L. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- Bruening W., Roy J., Giasson B., Figlewicz D. A., Mushynski W. E., Durham H. D. (1999). Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J. Neurochem. 72, 693–699 10.1046/j.1471-4159.1999.0720693.x [DOI] [PubMed] [Google Scholar]
- Casas C., Herrando-Grabulosa M., Manzano R., Mancuso R., Osta R., Navarro X. (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav 3, 145–158 10.1002/brb3.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Northington F. J., Martin L. J. (2010). Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 214, 219–234 10.1007/s00429-009-0226-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroni C., Peviani M., Cascio P., Debiasi S., Monti C., Bendotti C. (2005). Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol. Dis. 18, 509–522 10.1016/j.nbd.2004.12.007 [DOI] [PubMed] [Google Scholar]
- Chuaqui R. F., Bonner R. F., Best C. J., Gillespie J. W., Flaig M. J., Hewitt S. M., et al. (2002). Post-analysis follow-up and validation of microarray experiments. Nat. Genet. 32(Suppl.), 509–514 10.1038/ng1034 [DOI] [PubMed] [Google Scholar]
- Dallas P. B., Gottardo N. G., Firth M. J., Beesley A. H., Hoffmann K., Terry P. A., et al. (2005). Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR—how well do they correlate? BMC Genomics 6:59 10.1186/1471-2164-6-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangond F., Hwang D., Camelo S., Pasinelli P., Frosch M. P., Stephanopoulos G., et al. (2004). Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genomics 16, 229–239 10.1152/physiolgenomics.00087.2001 [DOI] [PubMed] [Google Scholar]
- Dangoumau A., Veyrat-Durebex C., Blasco H., Praline J., Corcia P., Andres C. R., et al. (2013). Protein SUMOylation, an emerging pathway in amyotrophic lateral sclerosis. Int. J. Neurosci. 123, 366–374 10.3109/00207454.2012.761984 [DOI] [PubMed] [Google Scholar]
- D'arrigo A., Colavito D., Pena-Altamira E., Fabris M., Dam M., Contestabile A., et al. (2010). Transcriptional profiling in the lumbar spinal cord of a mouse model of amyotrophic lateral sclerosis: a role for wild-type superoxide dismutase 1 in sporadic disease? J. Mol. Neurosci. 41, 404–415 10.1007/s12031-010-9332-2 [DOI] [PubMed] [Google Scholar]
- De Oliveira G. P., Maximino J. R., Lin C. J., Chadi G. (2009). A method to immunolabel rodent spinal cord neurons and glia for molecular study in specific laser microdissected cells involved in neurodegenerative disorders. J. Mol. Histol. 40, 217–225 10.1007/s10735-009-9233-2 [DOI] [PubMed] [Google Scholar]
- De Winter F., Vo T., Stam F. J., Wisman L. A., Bar P. R., Niclou S. P., et al. (2006). The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci. 32, 102–117 10.1016/j.mcn.2006.03.002 [DOI] [PubMed] [Google Scholar]
- Dion P. A., Daoud H., Rouleau G. A. (2009). Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat. Rev. Genet. 10, 769–782 10.1038/nrg2680 [DOI] [PubMed] [Google Scholar]
- Doherty G. J., McMahon H. T. (2009). Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 10.1146/annurev.biochem.78.081307.110540 [DOI] [PubMed] [Google Scholar]
- Druyan S., de Oliveira J. E., Ashwell C. M. (2008). Focused microarrays as a method to evaluate subtle changes in gene expression. Poult. Sci. 87, 2418–2429 10.3382/ps.2007-00513 [DOI] [PubMed] [Google Scholar]
- Elmer B. M., McAllister A. K. (2012). Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci. 35, 660–670 10.1016/j.tins.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei E., Jia N., Yan M., Ying Z., Sun Q., Wang H., et al. (2006). SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 347, 406–412 10.1016/j.bbrc.2006.06.092 [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L., Heath P. R., Holden H., Kasher P., Kirby J., Shaw P. J. (2007). Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 27, 9201–9219 10.1523/JNEUROSCI.1470-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L., Higginbottom A., Heath P. R., Barber S., Greenald D., Kirby J., et al. (2011a). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641 10.1093/brain/awr193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L., Kirby J., Grierson A. J., Sendtner M., Shaw P. J. (2011b). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630 10.1038/nrneurol.2011.152 [DOI] [PubMed] [Google Scholar]
- Fischer L. R., Culver D. G., Tennant P., Davis A. A., Wang M., Castellano-Sanchez A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240 10.1016/j.expneurol.2003.10.004 [DOI] [PubMed] [Google Scholar]
- Foran E., Bogush A., Goffredo M., Roncaglia P., Gustincich S., Pasinelli P., et al. (2011). Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia 59, 1719–1731 10.1002/glia.21218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S., Hernandez-Ontiveros D. G., Rodrigues M. C., Haller E., Frisina-Deyo A., Mirtyl S., et al. (2012). Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 1469, 114–128 10.1016/j.brainres.2012.05.056 [DOI] [PubMed] [Google Scholar]
- Garbuzova-Davis S., Rodrigues M. C., Hernandez-Ontiveros D. G., Louis M. K., Willing A. E., Borlongan C. V., et al. (2011). Amyotrophic lateral sclerosis: a neurovascular disease. Brain Res. 1398, 113–125 10.1016/j.brainres.2011.04.049 [DOI] [PubMed] [Google Scholar]
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber Y. N., Sabourin J. C., Rabano M., Vivanco M., Perrin F. E. (2012). Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 7:e36000 10.1371/journal.pone.0036000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb S. L., Boston-Howes W., Lavina Z. S., Gustincich S., Brown R. H., Jr., Pasinelli P., et al. (2007). A caspase-3-cleaved fragment of the glial glutamate transporter EAAT2 is sumoylated and targeted to promyelocytic leukemia nuclear bodies in mutant SOD1-linked amyotrophic lateral sclerosis. J. Biol. Chem. 282, 32480–32490 10.1074/jbc.M704314200 [DOI] [PubMed] [Google Scholar]
- Gifondorwa D. J., Jimenz-Moreno R., Hayes C. D., Rouhani H., Robinson M. B., Strupe J. L., et al. (2012). Administration of recombinant heat shock protein 70 delays peripheral muscle denervation in the SOD1(G93A) mouse model of Amyotrophic Lateral Sclerosis. Neurol. Res. Int. 2012, 170426 10.1155/2012/170426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifondorwa D. J., Robinson M. B., Hayes C. D., Taylor A. R., Prevette D. M., Oppenheim R. W., et al. (2007). Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 27, 13173–13180 10.1523/JNEUROSCI.4057-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber D. J., Hickey W. F., Harris B. T. (2010). Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J. Neuroinflammation 7, 8 10.1186/1742-2094-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammas P., Martinez J., Miller B. (2011). Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev. Mol. Med. 13, e19 10.1017/S1462399411001918 [DOI] [PubMed] [Google Scholar]
- Guipponi M., Li Q. X., Hyde L., Beissbarth T., Smyth G. K., Masters C. L., et al. (2010). SAGE analysis of genes differentially expressed in presymptomatic TgSOD1G93A transgenic mice identified cellular processes involved in early stage of ALS pathology. J. Mol. Neurosci. 41, 172–182 10.1007/s12031-009-9317-1 [DOI] [PubMed] [Google Scholar]
- Gupta P. K., Prabhakar S., Sharma N. K., Anand A. (2012). Possible association between expression of chemokine receptor-2 (CCR2) and amyotrophic lateral sclerosis (ALS) patients of North India. PLoS ONE 7:e38382 10.1371/journal.pone.0038382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney M. E. (1994). Transgenic-mouse model of amyotrophic lateral sclerosis. N. Engl. J. Med. 331, 1721–1722 10.1056/NEJM199412223312516 [DOI] [PubMed] [Google Scholar]
- Hall E. D., Oostveen J. A., Gurney M. E. (1998). Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23, 249–256 10.1002/(SICI)1098-1136(199807)23:3 [DOI] [PubMed] [Google Scholar]
- Heath P. R., Shaw P. J. (2002). Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26, 438–458 10.1002/mus.10186 [DOI] [PubMed] [Google Scholar]
- Henkel J. S., Beers D. R., Siklos L., Appel S. H. (2006). The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol. Cell. Neurosci. 31, 427–437 10.1016/j.mcn.2005.10.016 [DOI] [PubMed] [Google Scholar]
- Henkel J. S., Beers D. R., Wen S., Bowser R., Appel S. H. (2009). Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology 72, 1614–1616 10.1212/WNL.0b013e3181a41228 [DOI] [PubMed] [Google Scholar]
- Jiang M., Schuster J. E., Fu R., Siddique T., Heckman C. J. (2009). Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 15031–15038 10.1523/JNEUROSCI.0574-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. M., Yamamoto M., Kobayashi Y., Yoshihara T., Liang Y., Terao S., et al. (2005). Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251 10.1002/ana.20379 [DOI] [PubMed] [Google Scholar]
- Kalathur R. K., Hernandez-Prieto M. A., Futschik M. E. (2012). Huntington's disease and its therapeutic target genes: a global functional profile based on the HD Research Crossroads database. BMC Neurol. 12:47 10.1186/1471-2377-12-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y., Ito K., Sun H., Ito M., Kanazawa I., Kwak S. (2004). Regulation of glutamate receptor RNA editing and ADAR mRNA expression in developing human normal and Down's syndrome brains. Brain Res. Dev. Brain Res. 148, 151–155 10.1016/j.devbrainres.2003.11.008 [DOI] [PubMed] [Google Scholar]
- Kieran D., Kalmar B., Dick J. R., Riddoch-Contreras J., Burnstock G., Greensmith L. (2004). Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 10, 402–405 10.1038/nm1021 [DOI] [PubMed] [Google Scholar]
- Kiernan M. C., Vucic S., Cheah B. C., Turner M. R., Eisen A., Hardiman O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955 10.1016/S0140-6736(10)61156-7 [DOI] [PubMed] [Google Scholar]
- Kimura T., Griffin D. E. (2000). The role of CD8(+) T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J. Virol. 74, 6117–6125 10.1128/JVI.74.13.6117-6125.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon M., Cuervo A. M. (2010). Chaperone-mediated autophagy in health and disease. FEBS Lett. 584, 1399–1404 10.1016/j.febslet.2009.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman W. J., Distelmaier F., Smeitink J. A., Willems P. H. (2013). OXPHOS mutations and neurodegeneration. EMBO J. 32, 9–29 10.1038/emboj.2012.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhle J., Lindberg R. L., Regeniter A., Mehling M., Steck A. J., Kappos L., et al. (2009). Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 16, 771–774 10.1111/j.1468-1331.2009.02560.x [DOI] [PubMed] [Google Scholar]
- Kwak Y. D., Wang B., Li J. J., Wang R., Deng Q., Diao S., et al. (2012). Upregulation of the E3 ligase NEDD4-1 by oxidative stress degrades IGF-1 receptor protein in neurodegeneration. J. Neurosci. 32, 10971–10981 10.1523/JNEUROSCI.1836-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens R., Moore M. J., Al-Chalabi A., Brown R. H., Jr., Robberecht W. (2010). RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 33, 249–258 10.1016/j.tins.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Lenzken S. C., Romeo V., Zolezzi F., Cordero F., Lamorte G., Bonanno D., et al. (2011). Mutant SOD1 and mitochondrial damage alter expression and splicing of genes controlling neuritogenesis in models of neurodegeneration. Hum. Mutat. 32, 168–182 10.1002/humu.21394 [DOI] [PubMed] [Google Scholar]
- Le Roy C., Wrana J. L. (2005). Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 6, 112–126 10.1038/nrm1571 [DOI] [PubMed] [Google Scholar]
- Lin J., Diamanduros A., Chowdhury S. A., Scelsa S., Latov N., Sadiq S. A. (2009). Specific electron transport chain abnormalities in amyotrophic lateral sclerosis. J. Neurol. 256, 774–782 10.1007/s00415-009-5015-8 [DOI] [PubMed] [Google Scholar]
- Liu J. X., Brannstrom T., Andersen P. M., Pedrosa-Domellof F. (2013). Distinct changes in synaptic protein composition at neuromuscular junctions of extraocular muscles versus limb muscles of ALS donors. PLoS ONE 8:e57473 10.1371/journal.pone.0057473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Xue H., Pardo A. C., Mattson M. P., Rao M. S., Maragakis N. J. (2007). Impaired SDF1/CXCR4 signaling in glial progenitors derived from SOD1(G93A) mice. J. Neurosci. Res. 85, 2422–2432 10.1002/jnr.21398 [DOI] [PubMed] [Google Scholar]
- Malaspina A., De Belleroche J. (2004). Spinal cord molecular profiling provides a better understanding of amyotrophic lateral sclerosis pathogenesis. Brain Res. Brain Res. Rev. 45, 213–229 10.1016/j.brainresrev.2004.04.002 [DOI] [PubMed] [Google Scholar]
- Manfredi G., Xu Z. (2005). Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 5, 77–87 10.1016/j.mito.2005.01.002 [DOI] [PubMed] [Google Scholar]
- Manzano R., Toivonen J. M., Olivan S., Calvo A. C., Moreno-Igoa M., Munoz M. J., et al. (2011). Altered expression of myogenic regulatory factors in the mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 8, 386–396 10.1159/000324159 [DOI] [PubMed] [Google Scholar]
- Martin L. J. (2010). Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel) 3, 839–915 10.3390/ph3040839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Wilkinson K. A., Nishimune A., Henley J. M. (2007). Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat. Rev. Neurosci. 8, 948–959 10.1038/nrn2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L. J. (2011). Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43, 569–579 10.1007/s10863-011-9395-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey A. C., Zhang C., Cuervo A. M. (2006). Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 73, 205–235 10.1016/S0070-2153(05)73007-6 [DOI] [PubMed] [Google Scholar]
- Massignan T., Casoni F., Basso M., Stefanazzi P., Biasini E., Tortarolo M., et al. (2007). Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem. Biophys. Res. Commun. 353, 719–725 10.1016/j.bbrc.2006.12.075 [DOI] [PubMed] [Google Scholar]
- Matsumoto A., Tateishi Y., Onoyama I., Okita Y., Nakayama K., Nakayama K. I. (2011). Fbxw7beta resides in the endoplasmic reticulum membrane and protects cells from oxidative stress. Cancer Sci. 102, 749–755 10.1111/j.1349-7006.2011.01851.x [DOI] [PubMed] [Google Scholar]
- McMahon H. T., Boucrot E. (2011). Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12, 517–533 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- Miyazaki K., Ohta Y., Nagai M., Morimoto N., Kurata T., Takehisa Y., et al. (2011). Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 89, 718–728 10.1002/jnr.22594 [DOI] [PubMed] [Google Scholar]
- Morimoto N., Nagai M., Ohta Y., Miyazaki K., Kurata T., Morimoto M., et al. (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1167, 112–117 10.1016/j.brainres.2007.06.045 [DOI] [PubMed] [Google Scholar]
- Mougeot J. L., Li Z., Price A. E., Wright F. A., Brooks B. R. (2011). Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med. Genomics 4:74 10.1186/1755-8794-4-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narai H., Manabe Y., Nagai M., Nagano I., Ohta Y., Murakami T., et al. (2009). Early detachment of neuromuscular junction proteins in ALS mice with SODG93A mutation. Neurol. Int. 1:e16 10.4081/ni.2009.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateri A. S., Riera-Sans L., Da Costa C., Behrens A. (2004). The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science 303, 1374–1378 10.1126/science.1092880 [DOI] [PubMed] [Google Scholar]
- Offen D., Barhum Y., Melamed E., Embacher N., Schindler C., Ransmayr G. (2009). Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 38, 85–93 10.1007/s12031-007-9004-z [DOI] [PubMed] [Google Scholar]
- Oliveira A. L., Thams S., Lidman O., Piehl F., Hokfelt T., Karre K., et al. (2004). A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. U.S.A. 101, 17843–17848 10.1073/pnas.0408154101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen M. K., Roberds S. L., Ellerbrock B. R., Fleck T. J., McKinley D. K., Gurney M. E. (2001). Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann. Neurol. 50, 730–740 10.1002/ana.1252 [DOI] [PubMed] [Google Scholar]
- Otomo A., Pan L., Hadano S. (2012). Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012, 498428 10.1155/2012/498428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedotti P., 't Hoen P. A., Vreugdenhil E., Schenk G. J., Vossen R. H., Ariyurek Y., et al. (2008). Can subtle changes in gene expression be consistently detected with different microarray platforms? BMC Genomics 9:124 10.1186/1471-2164-9-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin F. E., Boisset G., Docquier M., Schaad O., Descombes P., Kato A. C. (2005). No widespread induction of cell death genes occurs in pure motoneurons in an amyotrophic lateral sclerosis mouse model. Hum. Mol. Genet. 14, 3309–3320 10.1093/hmg/ddi357 [DOI] [PubMed] [Google Scholar]
- Perrin F. E., Boisset G., Lathuiliere A., Kato A. C. (2006). Cell death pathways differ in several mouse models with motoneurone disease: analysis of pure motoneurone populations at a presymptomatic age. J. Neurochem. 98, 1959–1972 10.1111/j.1471-4159.2006.04024.x [DOI] [PubMed] [Google Scholar]
- Petri S., Schmalbach S., Grosskreutz J., Krampfl K., Grothe C., Dengler R., et al. (2005). The cellular mRNA expression of GABA and glutamate receptors in spinal motor neurons of SOD1 mice. J. Neurol. Sci. 238, 25–30 10.1016/j.jns.2005.06.005 [DOI] [PubMed] [Google Scholar]
- Polymenidou M., Cleveland D. W. (2011). The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147, 498–508 10.1016/j.cell.2011.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prifti E., Zucker J. D., Clement K., Henegar C. (2008). FunNet: an integrative tool for exploring transcriptional interactions. Bioinformatics 24, 2636–2638 10.1093/bioinformatics/btn492 [DOI] [PubMed] [Google Scholar]
- Rentzos M., Nikolaou C., Rombos A., Boufidou F., Zoga M., Dimitrakopoulos A., et al. (2007). RANTES levels are elevated in serum and cerebrospinal fluid in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 8, 283–287 10.1080/17482960701419232 [DOI] [PubMed] [Google Scholar]
- Richardson K., Allen S. P., Mortiboys H., Grierson A. J., Wharton S. B., Ince P. G., et al. (2013). The effect of SOD1 mutation on cellular bioenergetic profile and viability in response to oxidative stress and influence of mutation-type. PLoS ONE 8:e68256 10.1371/journal.pone.0068256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- Rothstein J. D., Martin L. J., Kuncl R. W. (1992). Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468 10.1056/NEJM199205283262204 [DOI] [PubMed] [Google Scholar]
- Rotshenker S. (2009). The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J. Mol. Neurosci. 39, 99–103 10.1007/s12031-009-9186-7 [DOI] [PubMed] [Google Scholar]
- Sanagi T., Yuasa S., Nakamura Y., Suzuki E., Aoki M., Warita H., et al. (2010). Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J. Neurosci. Res. 88, 2736–2746 10.1002/jnr.22424 [DOI] [PubMed] [Google Scholar]
- Sargsyan S. A., Blackburn D. J., Barber S. C., Monk P. N., Shaw P. J. (2009). Mutant SOD1 G93A microglia have an inflammatory phenotype and elevated production of MCP-1. Neuroreport 20, 1450–1455 10.1097/WNR.0b013e328331e8fa [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saris C. G., Groen E. J., Koekkoek J. A., Veldink J. H., Van Den Berg L. H. (2013a). Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: a comparison between transgenic mouse models and human patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 177–189 10.3109/21678421.2012.729842 [DOI] [PubMed] [Google Scholar]
- Saris C. G., Groen E. J., Van Vught P. W., Van Es M. A., Blauw H. M., Veldink J. H., et al. (2013b). Gene expression profile of SOD1-G93A mouse spinal cord, blood and muscle. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 190–198 10.3109/21678421.2012.749914 [DOI] [PubMed] [Google Scholar]
- Sasaki S., Warita H., Abe K., Komori T., Iwata M. (2001). EAAT1 and EAAT2 immunoreactivity in transgenic mice with a G93A mutant SOD1 gene. Neuroreport 12, 1359–1362 10.1097/00001756-200105250-00014 [DOI] [PubMed] [Google Scholar]
- Schutz B. (2005). Imbalanced excitatory to inhibitory synaptic input precedes motor neuron degeneration in an animal model of amyotrophic lateral sclerosis. Neurobiol. Dis. 20, 131–140 10.1016/j.nbd.2005.02.006 [DOI] [PubMed] [Google Scholar]
- Scorisa J. M., Duobles T., Oliveira G. P., Maximino J. R., Chadi G. (2010). The review of the methods to obtain non-neuronal cells to study glial influence on Amyotrophic Lateral Sclerosis pathophysiology at molecular level in vitro. Acta Cir. Bras. 25, 281–289 10.1590/S0102-86502010000300011 [DOI] [PubMed] [Google Scholar]
- Shideman C. R., Hu S., Peterson P. K., Thayer S. A. (2006). CCL5 evokes calcium signals in microglia through a kinase-, phosphoinositide-, and nucleotide-dependent mechanism. J. Neurosci. Res. 83, 1471–1484 10.1002/jnr.20839 [DOI] [PubMed] [Google Scholar]
- Singh R. K., Cooper T. A. (2012). Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 18, 472–82 10.1016/j.molmed.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. (2005). Limma: linear models for microarray data, in Bioinformatics and Computational Biology Solutions using R and Bioconductor, eds Gentleman V. C. R., Dudoit S., Irizarry R., Huber W. (New York, NY: Springer; ), 397–420 [Google Scholar]
- Sunico C. R., Dominguez G., Garcia-Verdugo J. M., Osta R., Montero F., Moreno-Lopez B. (2011). Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol. 21, 1–15 10.1111/j.1750-3639.2010.00417.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H., Kobayashi Y., Yoshihara T., Niwa J., Doyu M., Ohtsuka K., et al. (2002). Hsp70 and Hsp40 improve neurite outgrowth and suppress intracytoplasmic aggregate formation in cultured neuronal cells expressing mutant SOD1. Brain Res. 949, 11–22 10.1016/S0006-8993(02)02568-4 [DOI] [PubMed] [Google Scholar]
- Tateishi T., Yamasaki R., Tanaka M., Matsushita T., Kikuchi H., Isobe N., et al. (2010). CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J. Neuroimmunol. 222, 76–81 10.1016/j.jneuroim.2010.03.004 [DOI] [PubMed] [Google Scholar]
- Tateno M., Sadakata H., Tanaka M., Itohara S., Shin R. M., Miura M., et al. (2004). Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum. Mol. Genet. 13, 2183–2196 10.1093/hmg/ddh246 [DOI] [PubMed] [Google Scholar]
- Tian F., Morimoto N., Liu W., Ohta Y., Deguchi K., Miyazaki K., et al. (2011). In vivo optical imaging of motor neuron autophagy in a mouse model of amyotrophic lateral sclerosis. Autophagy 7, 985–992 10.4161/auto.7.9.16012 [DOI] [PubMed] [Google Scholar]
- Turner B. J., Talbot K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134 10.1016/j.pneurobio.2008.01.001 [DOI] [PubMed] [Google Scholar]
- Turner M. R., Hardiman O., Benatar M., Brooks B. R., Chio A., De Carvalho M., et al. (2013). Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 10.1016/S1474-4422(13)70036-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani M., Kurisu J., Tateno M., Hatakeyama S., Nakayama K., Kato S., et al. (2004). CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 90, 231–244 10.1111/j.1471-4159.2004.02486.x [DOI] [PubMed] [Google Scholar]
- Usuki S., Kamitani T., Matsuo Y., Yu R. K. (2012). Pathobiochemical effect of acylated steryl-beta-glucoside on aggregation and cytotoxicity of alpha-synuclein. Neurochem. Res. 37, 1261–1266 10.1007/s11064-011-0662-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gelder R. N., Von Zastrow M. E., Yool A., Dement W. C., Barchas J. D., Eberwine J. H. (1990). Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. U.S.A. 87, 1663–1667 10.1073/pnas.87.5.1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veugelers B., Theys P., Lammens M., Van Hees J., Robberecht W. (1996). Pathological findings in a patient with amyotrophic lateral sclerosis and multifocal motor neuropathy with conduction block. J. Neurol. Sci. 136, 64–70 10.1016/0022-510X(95)00295-D [DOI] [PubMed] [Google Scholar]
- Wang L., Gutmann D. H., Roos R. P. (2011a). Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 20, 286–293 10.1093/hmg/ddq463 [DOI] [PubMed] [Google Scholar]
- Wang R., Yang B., Zhang D. (2011b). Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59, 946–958 10.1002/glia.21167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P., Weiss M. D., Moller T., Carter G. T. (2002). Neuro-inflammation as a therapeutic target in amyotrophic lateral sclerosis. Curr. Opin. Investig. Drugs 3, 1720–1724 10.1016/0022-510X(95)00295-D [DOI] [PubMed] [Google Scholar]
- Wootz H., Enjin A., Wallen-Mackenzie A., Lindholm D., Kullander K. (2010). Reduced VGLUT2 expression increases motor neuron viability in Sod1(G93A) mice. Neurobiol. Dis. 37, 58–66 10.1016/j.nbd.2009.09.006 [DOI] [PubMed] [Google Scholar]
- Xu R., Wu C., Zhang X., Zhang Q., Yang Y., Yi J., et al. (2011). Linking hypoxic and oxidative insults to cell death mechanisms in models of ALS. Brain Res. 1372, 133–144 10.1016/j.brainres.2010.11.056 [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Tanaka F., Sobue G. (2007). [Gene expression profile of spinal ventral horn in ALS]. Brain Nerve 59, 1129–1139 [PubMed] [Google Scholar]
- Yamanaka K., Chun S. J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 10.1038/nn2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara T., Ishigaki S., Yamamoto M., Liang Y., Niwa J., Takeuchi H., et al. (2002). Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 80, 158–167 10.1046/j.0022-3042.2001.00683.x [DOI] [PubMed] [Google Scholar]
- Yu L., Guan Y., Wu X., Chen Y., Liu Z., Du H., et al. (2013). Wnt signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem. Res. 38, 1904–1913 10.1007/s11064-013-1096-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Gascon R., Miller R. G., Gelinas D. F., Mass J., Lancero M., et al. (2006). MCP-1 chemokine receptor CCR2 is decreased on circulating monocytes in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 179, 87–93 10.1016/j.jneuroim.2006.06.008 [DOI] [PubMed] [Google Scholar]
- Zhao P., Ignacio S., Beattie E. C., Abood M. E. (2008). Altered presymptomatic AMPA and cannabinoid receptor trafficking in motor neurons of ALS model mice: implications for excitotoxicity. Eur. J. Neurosci. 27, 572–579 10.1111/j.1460-9568.2008.06041.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Deane R., Ali Z., Parisi M., Shapovalov Y., O'banion M. K., et al. (2008). ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 11, 420–422 10.1038/nn2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.