

Abstract
Neuroinflammatory diseases, such as multiple sclerosis (MS), result from aberrant leucocyte traffic into the central nervous system (CNS). To breach the specialized blood-brain barrier, activated leucocytes interact with CNS endothelial cells (EC) and activate a CD54-mediated signaling pathway controlling the Rho GTPase. To function correctly Rho requires post-translational prenylation, and this can be inhibited by depleting the supply of isoprenoids through inhibition of the cholesterol synthesis pathway with HMG-CoA reductase inhibitors (statins). Here we show that treatment of brain EC in vitro with lovastatin inhibits Rho-mediated transendothelial T cell migration. This effect can be reversed by supplementation with mevalonolactone, the downstream product of HMG-CoA reductase, or by ectopic expression of myristoylated Rho, which remains active in the absence of prenylation. In a relapsing-remitting mouse model of MS, lovastatin treatment inhibited leucocyte migration into the CNS and significantly attenuated the development of both acute and relapsing clinical disease. These studies demonstrate that the indirect pharmacological inhibition of Rho proteins in brain EC by statins can inhibit a key stage in the pathogenesis of neuroinflammation, namely leucocyte migration across the blood-brain barrier. Theses studies demonstrate a novel effect of statins in modulating the immune response in neuroinflammtory diseases and may provide additional rationale for their use in the treatment of MS.
Keywords: Statins, Blood-brain barrier, Leucocyte recruitment, Rho, Multiple Sclerosis
INTRODUCTION
A key stage in the development of neuroinflammatory diseases, such as multiple sclerosis (MS), is the infiltration of leucocytes from the vasculature to the neural paranchyma. Transendothelial lymphocyte migration in the central nervous system (CNS) has been shown to be dependent on both lymphocyte activation (1) and an ability of leucocytes to effectively elicit signaling responses in endothelial cells (EC) (2-5). Although CNS endothelia are connected together by tight junctions, forming the blood-brain barrier (BBB), under normal conditions a low level of lymphocyte traffic into the CNS occurs which is independent of antigen-specificity of the invading lymphocyes (6). However, during the onset of immune-mediated diseases lymphocyte infiltration is markedly up regulated. It has previously been demonstrated that migration of lymphocytes across the CNS vasculature requires the interaction between intercellular adhesion molecule-1 (ICAM-1; CD54) expressed on brain EC cells and the T-cell integrin αLβ2 (LFA-1, CD11a/CD18) (7-9). Recent studies have further demonstrated that ICAM-1 on brain EC not only serves as a leucocyte adhesion molecule, but upon engagement results in EC intracellular signaling responses, leading to facilitation of lymphocyte transendothelial migration (2-5).
The efficient transduction of ICAM-1 mediated signaling responses in brain EC, and consequently transendothelial migration of T-lymphocytes, is critically dependent on functional EC Rho GTPase (2,3,10). To be functionally active, Rho proteins must undergo post-translational modification through addition of isoprenyl groups from isoprenoid pyrophosphate substrates (11,12). Inhibition of Rho prenylation with prenyl transferase inhibitors prevents membrane association and subsequent lack of Rho function. We have previously reported that treating brain EC with prenyl transferase inhibitors block lymphocyte migration across brain EC in vitro and attenuate clinical disease in an animal model of MS (10), further demonstrating the critical importance of Rho in lymphocyte recruitment through the BBB. As isoprenoid pyrophosphates are the downstream products of mevalonic acid metabolism, which is itself produced through the activity of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), there is compelling grounds for evaluating the effect of HMG-CoA reductase inhibitors (statins) on Rho prenylation and lymphocyte migration. This approach is appealing as statins are used clinically as hypolipidemic agents to reduce plasma cholesterol. As brain EC require functional Rho to support lymphocyte migration we therefore investigated whether a reduction in the supply of isoprenoids through inhibition of HMG-CoA reductase, would lead to reduced Rho prenylation and subsequent inhibition of transendothelial lymphocyte migration and neuroinflammation.
MATERIALS AND METHODS
Lymphocyte adhesion and migration assays
The extensively characterised immortalised Lewis rat brain EC line GP8/3.9, which retains phenotypic characteristics of primary cultures, was maintained as previously described (13). For migration assays encephalitogenic CD4+ myelin basic protein (MBP) T-cell lines were established from guinea pig MBP-primed Lewis rat lymph nodes and maintained as previously reported (14,15). For adhesion assays cervical and mesenteric peripheral lymph nodes were harvested from Lewis rats and activated overnight with 5μg/ml conconavalin A. Lymphocyte adhesion and transendothelial migration assays were carried out with GP8/3.9 EC as described in detail elsewhere (1,3). The adhesion assay was conducted over 90 min and the migration assay over a 4 h co-culture period.
Brain EC monolayers were treated with vehicle (PBS:DMSO; 1:1) or lovastatin (1-100 μM) for 24 h prior to and during the migration and adhesion assays. In addition, some cultures were supplemented with excess mevalonic acid (mevalonolactone, 5mM) for 24h prior to and during the migration and adhesion assays. EC were also pre-exposed to 10 μg/ml of the Clostridium botulinum toxin C3 transferase for 16 h (3).
All migration and adhesion data were obtained from a minimum of three independent experiments using a minimum of 10 wells per assay. Data are expressed as mean ± SEM percent of control migration. Statistical significance between groups was determined by Student’s t test.
Myristoylated RhoB expressing brain endothelial cell lines
pcDNA3 RhoB containing an N-terminal 16 amino acid sequence from the v-src oncoprotein which encodes the myristoylation site of v-src (16) was restricted with HindIII and XhoI and a fragment containing the v-src/RhoB chimera blunted with klenow fragment of DNA polymerase and cloned into SnaB1 digested pBabepuro. GP8/3.9 cells were transfected with pBabepuro-myr-RhoB or empty vector using Fugene (Boehringer) according to manufacturer’s instructions, selected in 20μg/ml puromycin, removed from plates using cloning rings and expanded. One myristoylated RhoB clone and one pBabepuro control clone were used in all subsequent assays. The myristoylated RhoB and pBabepuro clones were treated with vehicle or lovastatin (10μM) for 24 h prior to and during the migration and adhesion assays or 10μg/ml C3 transferase for 16 h prior to the assays.
Preparation of cellular membrane fractions and ADP-ribosylation of endothelial Rho
Cells transfected with pBabepuro vector or cells expressing myristoylated RhoB were lysed in 10mM Tris-HCl pH 7.4, 1 mM EDTA, 2mM MgCl2 and 1mM AEBSF. Nuclear material was removed by centrifugation at 800g for 5 min at 4°C. A cellular membrane fraction was isolated following centrifugation of supernatants at 75,000g for 90 min at 4°C. Protein concentrations of resuspended membrane fractions were determined using BCA reagent (Pierce UK). 10μg of membrane protein was then added to reactions containing 50mM Tris-HCl, pH 7.4, 1mM EDTA, 2mM MgCl2, 0.5mM ATP, 0.3mM GTP, 5μCi [α-32P]NAD and 250ng/ml recombinant C3 transferase and incubated at 37°C for 1 h. Reactions were stopped following the addition of 2 volumes of 30%w/v trichloroacetic acid on ice for 30 min. Precipitated proteins were pelleted at 14,000g for 10 min and pellets washed three times with ethanol at −20°C. Pellets were dried, solubilised in 10 mM Tris-HCl, pH6.8, 4% SDS, 20% glycerol, 0.4% dithiothreitol and 0.1% bromophenol blue and proteins resolved on 15% SDS-PAGE. Gels were dried and exposed to autoradiographic film for 48 h.
Induction and treatment of EAE in Biozzi ABH mice
Relapsing-remitting EAE was induced in 6-8 week old Biozzi ABH mice as previously described (17) by subcutaneous injection in the flank with 1mg of syngeneic spinal cord homogenate in complete Freund’s adjuvant on day 0 and 7. Animals were monitored daily and clinical signs ranked as follows: normal = 0, limp tail = 1, impaired righting reflex = 2, partial hind limb paralysis = 3 and complete hind limb paralysis = 4 (18). Histological assessment of the brain and spinal cord was carried out on vehicle (n=3) and lovastatin-treated (n=3) mice 18 days post induction on haematoxylin and eosin stained parafin wax-embedded tissue.
Lovastatin (0.1mg-20mg/kg) was administered intraperitoneally in DMSO:PBS (1:1) mixed immediately prior to injection. Treatment efficacy was evaluated following daily intraperitoneal injection with, in some instances, co-administration of 2mg/kg i.p. mevalonolactone. Results represent the mean maximal score of all animals in the group (group score). Differences between groups were assessed using Mann Whitney U statistics.
Delayed hypersensitivity assays
In vivo lymphocyte proliferation in Biozzi ABH mice was initiated following treatment of ABH mice with lovastatin (10 or 20mg/kg/day) for 2 days prior to topical application of 25μl of 2.5% oxazolone in acetone:olive oil (4:1) to the ear. This was followed by a further 3 days treatment with lovastatin. Animals were killed and draining auricular lymph node lymphocytes isolated. Peripheral lymph node cells were washed in RPMI 1640 and 200μl of cells (2.5 × 106/ml) in RPMI 1640 plus 10% FCS placed in microtitre plates. Lymphocyte proliferation was evaluated in a cell proliferation assay (Aqueous One™, Promega) or by pulsing with 1μCi [3H]thymidine for 8 h prior to cell harvesting and measurement of [3H]thymidine incorporation by β–scintillation spectrometry.
Delayed type hypersensitivity effector function was examined following sensitisation of ABH mice with 50μl of the contact sensitizer oxazolone (2.5%), applied to the flank and skin-tested three weeks later with 0.25% oxazolone to the ear following 6 days treatment with vehicle or 20mg/kg lovastatin. 24 h following the re-challenge, ear thickness was measured using a micrometer gauge and the percentage increase over baseline measurement recorded. Significant differences between groups were determined by Student’s t test.
In vivo lymphocyte migration assay
Antigen-activated lymphocytes were prepared following sensitising ABH mice with 2.5% oxazolone (25μl) into the ear, fore and hind limbs. After three days, the draining auricular, axcillary and inguinal lymph nodes were removed, lymphocytes separated and labelled with the cell-tracker dye CMFDA (5-chloromethylfluorescein diacetate; 5μg per 5×107 cells. Molecular Probes, OR, USA) for 30 min at 37°C in the dark. After washing (×2) and filtering through a 25μm nylon mesh, 5×107 cells were injected via the tail vein into recipient mice. Recipient ABH mice were previously treated with vehicle (n=3) or 4 daily doses of lovastatin (n=3, 20mg/kg) prior to injection of cells. After 14 h the animals were anaesthetised and blood flushed from the vasculature by transcardial perfusion with PBS. This was followed by 5 ml of 0.5mg/ml DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; Molecular Probes) to label the vasculature. Finally, the perfusate was switched to 10ml of 4% paraformaldehyde in PBS. Brain tissue was removed and 12 × 100 μm coronal sections cut at the level of the optic chiasm, mounted and viewed using confocal laser scanning microscopy (Zeiss LSM510, Herts, UK). Cell counts were made of labeled leucocytes and expressed as number of cells per section.
RESULTS
Lovastatin inhibits lymphocyte migration through brain EC monolayers
Treatment of both brain EC and lymphocytes with lovastatin during a 4 h co-culture had no effect on transendothelial lymphocyte migration (Fig1a) whilst pre-treatment of brain EC for 24 h resulted in a significant dose-dependent inhibition of lymphocyte migration (Fig 1b). This is consistent with the reported half-lives of Rho (19). The degree of inhibition was similar to that achieved following treatment of EC with C3 transferase, a toxin that specifically ribosylates and inactivates Rho. Furthermore, addition of mevalonolactone was able to reverse lovastatin-induced inhibition of migration (Fig 1c) demonstrating that the effect of lovastatin was due to inhibition of HMG-CoA reductase.
Figure 1.
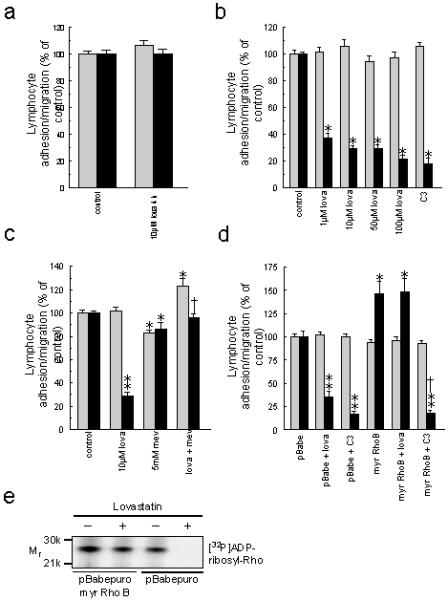
(a) Lovastatin treatment during the 4h EC/lymphocyte co-culture does not affect lymphocyte adhesion (shaded bars) or transendothelial migration (solid bars). (b) Pre-treatment of brain EC for 24 h with lovastatin or C3 transferase inhibits transendothelial lymphocyte migration without affecting adhesion. *p<0.0001 cf. control. (c) Mevalonolactone reverses lovastatin-induced inhibition of lymphocyte migration. *p<0.02, **p<0.001 cf. control. †p<0.0001 cf. lovastatin treated cells. (d) Over expression of myristolated Rho B confers resistance to the inhibitory effect of lovastatin but not C3 transferase. *p<0.02, **p<0.0001 cf. control migration through pBabepuro cell line. †p<0.0001 cf. EC expressing myristoylated RhoB. (e) Myristolated Rho, but not endogenous Rho, associates with EC membranes following treatment with lovastatin.
Brain EC expressing myristoylated Rho support lymphocyte migration in the presence of lovastatin
To determine whether lovastatin-induced inhibition of lymphocyte migration operates through depletion of isoprenoids, and thus prenylation of EC Rho, a brain EC line was generated expressing myristoylated Rho. This modification enables Rho to associate with cellular membranes within the correct cellular compartment and hence function in the absence of prenylation (16). T cell migration through EC expressing myristoylated Rho was enhanced by 50% over control vector-transfected EC (Fig 1d), which is consistent with endothelial Rho being necessary for lymphocyte migration through the BBB (2-4). Crucially, however, when EC expressing myristoylated Rho were pre-treated with lovastatin there was no alteration in lymphocyte transmigration (Fig 1d). Treatment of the myristoylated Rho expressing EC with C3 transferase was, as predicted, still able to effectively inhibit lymphocyte migration as Rho inhibition is mediated through a different mechanism, namely ADP ribosylation. These observations correlate with the finding that endogenous EC Rho did not associate with EC membranes following exposure to lovastatin whilst myristolated Rho was still able to associate with the membrane fraction under these conditions (Fig 1e). This demonstrates that a key mechanism of lovastatin-mediated inhibition of lymphocyte transendothelial migration is due to the prevention of Rho prenylation within EC.
Lovastatin treatment attenuates acute phase EAE in Biozzi mice
Our observation that lovastatin is a potent anti-migratory compound in vitro, suggested that it may possess therapeutic potential in controlling CNS inflammation. Vehicle treated-animals displayed characteristic paralysis with onset at around day 15 post-inoculation (Fig 2a). Lovastatin administered daily from day 8 onwards caused a significant dose-dependent reduction in the severity of clinical EAE (Table 1). Although some treated animals developed clinical disease, even at the lowest dose tested (0.1mg/kg), the onset of disease was delayed and the maximal severity of disease was significantly reduced (Table 1). As predicted from the proposed mechanism, when lovastatin treatment was discontinued, EAE developed rapidly suggesting that lovastatin had not restricted initial T cell priming (Fig 2a). Consistent with our in vitro data, the therapeutic effects of lovastatin were also, at least, partially reversed in mice following co-administration of mevalonolactone (Table 1).
Figure 2.
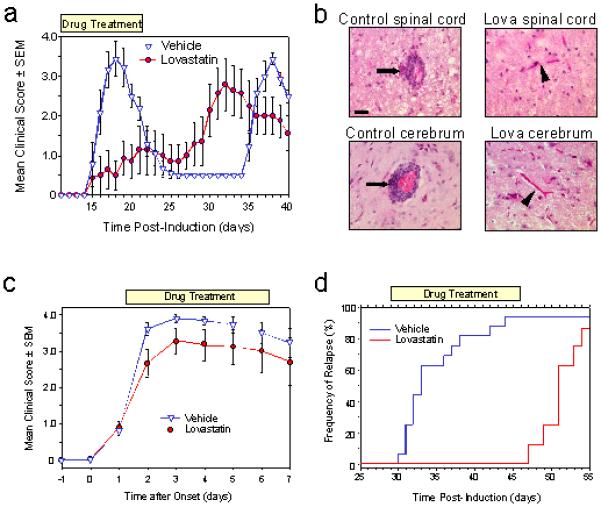
(a) Lovastatin treatment (10mg/kg; days 8-22) inhibits the development of disease until cessation of treatment. (b) Spinal cord and cerebrum from control and lovastatin treated (10mg/kg) ABH mice. EAE in untreated mice showed marked perivascular leucocyte cuffing (arrows) that was absent in treated mice (arrow heads). Scale bar = 20μm. (c) Lovastatin treatment (10mg/kg) of acute phase EAE after onset does not attenuate disease progression whereas (h) treatment during remission (20mg/kg) prevents the onset of disease relapse.
Table 1.
Lovastatin ameliorates the development of both acute phase and relapse EAE. Results represent the mean maximal score of all animals in the group (group score).
| Treatment | No. EAE/ Total |
Group Score ± SEM |
Onset Day ± SD |
|---|---|---|---|
| Acute disease (Treatment days 8-21) | |||
| Vehicle | 14/14 | 4.0 ± 0.1 | 14.7 ± 1.1 |
| Lovastatin 0.1 mg/kg | 10/10 | 3.0 ± 0.5* | 16.1 ± 1.1* |
| Lovastatin 1mg/kg | 9/10 | 2.7 ± 0.5** | 16.0 ± 1.7 |
| Lovastatin 10mg/kg | 7/12 | 1.7 ± 0.5*** | 17.3 ± 1.5* |
| Lovastatin 20mg/kg | 4/10* | 1.0 ± 0.5*** | 16.0 ± 0.0** |
| Vehicle | 8/9 | 3.8 ± 0.4 | 14.9 ± 0.4 |
| Lovastatin 20mg/kg | 2/8* | 0.3 ± 0.2*** | 19.5 ± 3.5 |
| Mevalonolactone 2mg/kg | 7/7 | 3.8 ± 0.2 | 16.1 ± 1.2 |
| Lovastatin + Mevalonolactone |
6/9† | 2.0 ± 0.6**† | 17.0 ± 2.5* |
| Relapse disease‡ (Treatment days 29-40) | |||
| Vehicle | 13/14 | 3.7 ± 0.2 | 33.0 ± 2.5 |
| Lovastatin (20mg/kg) | 0/8*** | 0.5 ± 0.0*** | n/a |
p<0.05
p<0.01
p<0.001 significantly different from vehicle treated control group.
p<0.05 compared to lovastatin treated group.
n/a = not applicable
The severity of clinical EAE in ABH mice has been shown to correlate closely with the degree of CNS leucocyte infiltration (20). Histological examination of the CNS of vehicle-treated control animals at the peak of clinical disease (day 18) revealed characteristic lesions consisting of perivascular mononuclear cell cuffing and parenchymal invasion within the spinal cord and brain (Fig 2b) which was absent in normal-appearing, lovastatin-treated animals (10 mg/kg from day 8) (Fig 2b).
To evaluate whether treatment prior to disease onset was necessary to elicit a therapeutic effect, lovastatin was administered at disease onset when animals exhibited a limp tail (clinical score 1). Under these conditions, lovastatin had little effect on the development of acute disease (Fig 2c) presumably as leucocytic infiltration into the CNS is already occurring at this stage (18). Moreover, this is also consonant with the half-lives of prenylated Rho (16,19,21) such that 24 h of exposure to lovastatin would be required to ensure adequate EC depletion of isoprenoids and subsequent inactivation of Rho. This was further demonstrated in an additional experimental group (n=8) where lovastatin treatment was shifted from day 8 to day 12 resulting in a less effective therapeutic effect (group score 3.0 ± 0.4).
Lovastatin treatment prevents the onset of disease relapse
To investigate whether lovastatin could attenuate disease relapse, the drug was administered during remission from the acute (grade 4) paralytic phase. This resulted in complete abrogation of disease relapse (Fig 2d) during treatment (grade 0.5 ± 0.0 n=8) compared to untreated mice where 13/14 animals relapsed with a maximal group score of 3.7 ± 0.1 and a mean day of onset of 33.0 ± 2.5 days (Table 1). However, upon cessation of statin treatment on day 46 post-inoculation, most animals rapidly (within 5 days) developed paralytic relapse (Fig 2d).
Lovastatin induces a modest inhibition of T cell proliferation in the Biozzi mouse
There are a number of reports demonstrating that statins have anti-proliferative activity in vitro (22,23). Such potential immunosuppression could account for the ameliorative effects of lovastatin, particularly when statins are administered during the sensitisation period, as occurred previously in MBP-induced hyperacute EAE (24,25). The anti-proliferative effect of statins cannot be readily tested in spinal cord-induced EAE in the ABH mouse as it is not possible to routinely induce an in vitro T cell recall responses to the dominant encephalitogen (proteolipid protein, residues 56-70) for both acute and relapsing spinal cord induced disease (26). However to address whether the lovastatin was anti-proliferative in vivo we investigated the primary proliferative response to the contact sensitizer oxazolone following epicutaneous application to the ear. Lymphocytes isolated from the lymph nodes of both lovastatin and vehicle-treated animals both exhibited significant proliferation over background with a modest but statistically significant reduction in the lovastatin treated animals (Fig 3a).
Figure 3.
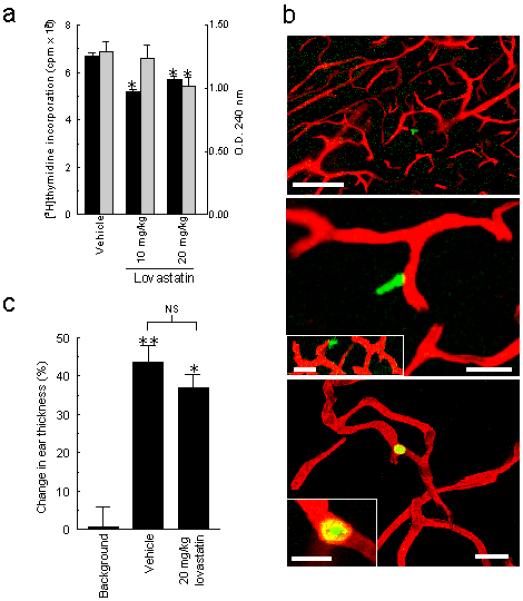
(a) Proliferation of auricular lymph node cells evaluated by [3H]thymidine incorporation (left abscissa, solid bars) or by a colorimetric cell proliferation assay (right abscissa, shaded bars) from vehicle and lovastatin treated ABH mice. *p<0.05 cf. vehicle-treated animals. (b) Representative confocal fluorescence projected images of cerebral vasculature (red) and labelled lymphocytes (green). Top panel (bar = 100 μm) shows extravasated lymphocyte and middle panel (bar = 20 μm), a lymphocyte in the cerebral parenchyma adjacent to a microvessel from vehicle treated animals. Insert represents projection of middle panel Z stack. Lower panel (bar = 20 μm) shows a lymphocyte confined to the intravascular compartment from a lovastatin pre-treated mouse (20 mg/kg/day for 4days). Insert shows higher power X-Y section detail of lower panel (bar = 10 μm). (c) Delayed type hypersensitivity function in vehicle or lovastatin treated (20mg/kg) mice expressed as the percentage increase in ear thickness over baseline measurements. **p<0.001, *p<0.01 cf. non-oxazolone primed animals.
Migration of donor lymphocytes into the CNS of lovastatin-treated recipient mice is severely inhibited
To further test the premise that a critical therapeutic effect of lovastatin is inhibition of migration across the BBB and is independent of its potential immunosuppressive action on T cells, ABH mice were pre-treated with vehicle or 20mg/kg lovastatin prior to intravenous delivery of fluorescently labelled antigen-activated (oxazolone) lymphocytes that have been shown previously to migrate across the BBB irrespective of their antigen-specificity (1,6). After 14 h, labelled lymphocytes were observed in the cerebral parenchyma of those animals treated with vehicle alone (Fig 3b) but were rarely seen in animals treated with lovastatin. The average number of labelled lymphocytes per 100 μm coronal section was significantly greater for vehicle treated animals (14.2 ± 3.0 Mean ± SEM) than for the lovastatin treated animals (0.75 ± 0.3, p <0.001). Leucocytes observed in statin treated animals were principally restricted to the vascular lumen of small blood vessels (Fig 3b).
Lovastatin has a limited effect on the DTH response
When we evaluated effector delayed type hypersensitivity function in these mice following skin-testing with oxazolone, lovastatin failed to attenuate the response (Fig 3c), indicating that leucocyte extravasation into the skin was largely unaffected. These results imply some selectivity for the anti-migratory effects of lovastatin at the level of the CNS EC and corroborate other observed differences between brain and non-brain endothelium in how they support lymphocyte migration (5).
DISCUSSION
In this report we show, for the first time, that treatment of brain EC with the HMG-CoA reductase inhibitor, lovastatin, results in a substantial inhibition of transendothelial lymphocyte migration. Direct evidence is provided that this is mediated through the downstream restriction of Rho prenylation and hence loss of activity. Thus, brain EC over expressing myristoylated Rho are insensitive to lovastatin as myristoylation and subsequent activation of Rho in these cells is independent of the supply of isoprenoid precursors. The findings presented here are consistent with previous reports showing that Rho GTPases are an essential component of the ICAM-1 mediated signaling pathway necessary for the support of T cell migration through CNS EC monolayers (2,3,10). The ability of statins to inhibit lymphocyte migration now provides further evidence that brain EC Rho proteins are critically important in orchestrating the endothelial response to T-lymphocyte adhesion and subsequent transendothelial migration.
Whilst previous reports have indicated that HMG-CoA reductase inhibitors may interfere with adhesion molecule expression (27-29) and function (30), we have found that the expression of ICAM-1 on brain EC was unaffected by lovastatin (data not shown). It is also unlikely that in this setting lovastatin is significantly altering the LFA-1/ICAM-1 interaction, as exposure to lovastatin during a 4h lymphocyte co-culture period did not affect migration. Furthermore, the LFA-1/ICAM-1 dependent adhesion of lymphocytes to endothelium was not affected by lovastatin. Finally, supplementing brain EC cultures, and importantly animals, with mevalonolactone (a precursor of mevalonic acid) was able to ablate the inhibitory effects of lovastatin on transendothelial lymphocyte migration and attenuate the development of EAE. Together, this data supports the premise that lovastatin is acting largely through restriction of the downstream products of HMG-CoA reductase and not through staeric interference.
The novel finding that lovastatin treatment of brain EC in vitro results in inhibition of lymphocyte migration prompted us to evaluate its effect on lymphocyte migration in vivo. Firstly, the migration of donor lymphocytes across the BBB of lovastatin treated recipient mice was inhibited within a timeframe that is inconsistent with it being the result of a change in donor T cell function. Next, treatment of EAE induced Biozzi ABH mice with lovastatin dramatically attenuated the infiltration of leucocytes into the CNS and substantially alleviated clinical signs in both the acute, and importantly, the relapse phase of disease. This treatment regimen reduced both the number and severity of animals showing clinical signs of EAE and in most cases delayed disease onset. Although this data would suggest that a major action of lovastatin is via inhibition of leucocyte migration across the BBB, we recognise that due to the pleiotropic action of statins, other immunomodulatory effects are likely to contribute to the amelioration of disease in vivo. These include immunosuppression (24,25,31) and modulation of MHC class II expression (31-33). However, in our model we believe the evidence suggests that these are not the primary effector mechanisms and some of the described in vivo effects (24,25,33), may be an indirect consequence of reduced inflammation. Whilst it has been found that statins can inhibit an in vitro T cell proliferative response (31-33) we found little evidence, albeit with a different antigen (oxazolone), that lovastatin inhibited a primary proliferative, sensitising response in vivo. Furthermore, statin treatment was not initiated during the sensitising period and this suggests that the inhibition of disease was not due to the failure of lymphocytes to become sensitised. Moreover, in ABH mice we were unable to observe a dramatic attenuation of leucocyte infiltration into the skin during a DTH response. Indeed, it has been shown previously that potent peripherally-acting immunosuppressive agents have only a marginal effect on disease relapse in this model (18), and that significant immunosuppression has not been observed in statin-treated humans (23). It has also been proposed that alterations in matrix metalloprotease (MMP) secretion (34) may alter lymphocyte endothelial cell interactions. This may have some bearing on migration but it is important to note that MMP secretion is a key event in post-diapedesis parenchymal migration (35) and is not thought to be important in the crossing the vascular barrier per se.
Finally, it has recently been reported that atorvastatin promotes a Th2 bias resulting in reversal of clinical disease in three EAE models (34). This is an interesting concept but is unlikely to explain all of our in vivo observations, particularly where we tested the effect of lovastatin on trafficking and disease relapse. In this latter paradigm lovastatin treatment was started just prior to predicted relapse and, unlike the untreated group, animals remained disease-free during treatment. Importantly when drug was removed animals rapidly developed disease, some within 48 hours. This time frame is probably too quick for either the generation of a new population of encephalitogenic cells, which would be required if lovastatin had a major anti-proliferative effect preventing the generation of effector cells, or the loss of protective Th2 cells and a reversion to a Th1 phenotype generated from Th0 cells. Furthermore in contrast to that observed here with lovastatin, disease did not reappear in atorvastatin treated animals (34) and may suggest some differences in statin activity. Such differences have been observed previously (30,31), especially in their inhibition of CIITA and MHC class II expression (32). Nevertheless, even though some of these discrepancies may be explained by differences in the model used, inhibition of T cell responses, the generation of a Th2 bias, and inhibition of migration may all contribute to the in vivo attenuation of clinical signs. Whatever multiple effects lovastatin may exert on the immune system, the data provided by our in vitro assay provides compelling evidence that it will also impinge on a critical stage of lesion formation, namely lymphocyte diapedesis at the BBB. Furthermore, the severely reduced migration of T cells transferred from control animals into the brain of animals pre-treated with lovastatin adds further weight to the premise that lovastatin is also affecting migration at the BBB in vivo.
We conclude that leucocyte migration across the BBB is a pivotal stage in the development of inflammatory lesions and, as statins inhibit this process, their use in the treatment of early relapsing-remitting multiple sclerosis may prove to be of significant therapeutic potential. Moreover, these data demonstrate the principle of therapeutic targeting of BBB signaling pathways that are central to the facilitation of transvascular leucocyte migration.
Acknowledgements
The Multiple Sclerosis Society of Great Britain and Northern Ireland, The Wellcome Trust and the Association pour la Recherche sur la sclérose en plaques (France) supported this work. The authors would like to thank G. Prendergast, Wistar Institute, Philadelphia, USA for the myristoylated RhoB cDNA.
References
- 1.Pryce G, Male DK, Campbell I, Greenwood J. Factors controlling T-cell migration across rat cerebral endothelium in vitro. J. Neuroimmunol. 1997;75:84–94. doi: 10.1016/s0165-5728(97)00006-4. [DOI] [PubMed] [Google Scholar]
- 2.Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud P-O. Rho-dependent signaling pathways coupled to ICAM-1 in microvascular brain endothelial cells. J. Immunol. 1998;161:5755–5761. [PubMed] [Google Scholar]
- 3.Adamson P, Etienne S, Couraud P-O, Calder V, Greenwood J. T-lymphocyte migration through CNS endothelial cells involves signalling through endothelial ICAM-1 via a rho dependent pathway. J. Immunol. 1999;162:2964–2973. [PubMed] [Google Scholar]
- 4.Etienne-Manneville S, Adamson P, Manneville JB, Wilbourn B, Greenwood J, Couraud P-O. ICAM-1 coupled cytoskeletal rearrangements and lymphocyte migration across brain endothelium involves intracellular calcium signalling. J. Immunol. 2000;165:3375–3383. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- 5.Adamson P, Wilbourn B, Etienne-Manneville S, Calder V, Beraud ES, Milligan G, Couraud P-O, Greenwood J. Lymphocyte trafficking through the blodd-brain barrier is dependent on endothelial cell heterotrimeric G-protein signaling. FASEB J. 2002;16:1185–1194. doi: 10.1096/fj.02-0035com. [DOI] [PubMed] [Google Scholar]
- 6.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 7.Male D, Rahman J, Pryce G, Tamatani T, Miyasaka M. Lymphocyte migration into the CNS modelled in vitro: roles of LFA-1, ICAM-1 and VLA-4. Immunology. 1994;81:366–372. [PMC free article] [PubMed] [Google Scholar]
- 8.Greenwood J, Wang Y, Calder V. Lymphocyte adhesion and transendothelial migration in the CNS: the role of LFA-1, ICAM-1, VLA-4 and VCAM-1. Immunology. 1995;86:408–415. [PMC free article] [PubMed] [Google Scholar]
- 9.Reiss Y, Hoch G, Deutsch U, Engelhardt B. T cell interaction with ICAM-1-deficient endothelium in vitro: essential role for ICAM-1 and ICAM-2 in transendothelial migration of T cells. Eur. J. Immunol. 1998;28:3086–3099. doi: 10.1002/(SICI)1521-4141(199810)28:10<3086::AID-IMMU3086>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 10.Walters CE, Pryce G, Baker D, Sebti SM, Greenwood J, Adamson P. Inhibition of rho GTPases with protein prenyl transferase inhibitors prevent leukocyte recruitment to the CNS and attenuate clinical signs of disease in an animal model of multiple sclerosis. J. Immunol. 2002;168:4087–4094. doi: 10.4049/jimmunol.168.8.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adamson P, Paterson HF, Hall A. Intracellular localisation of the p21rho proteins. J. Cell. Biol. 1992;119:617–629. doi: 10.1083/jcb.119.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of the p21rho proteins. J. Biol. Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 13.Greenwood J, Pryce G, Devine L, Male DK, dos Santos WLC, Calder VL, Adamson P. SV40 large T immortalised cell lines of the rat blood-brain and blood-retinal barriers retain their phenotypic and immunological characteristics. J. Neuroimmunol. 1996;71:51–63. doi: 10.1016/s0165-5728(96)00130-0. [DOI] [PubMed] [Google Scholar]
- 14.Beraud E, Reshef T, Vandenbark AA, Offner H, Friz R, Chou CH, Bernard D, Cohen IR. J. Immunol. 1986;136:511–515. [PubMed] [Google Scholar]
- 15.Beraud E, Balzano C, Zamora AJ, Varriale S, Bernard D, Ben Nun A. J. Neuroimmunol. 1993;47:41–53. doi: 10.1016/0165-5728(93)90283-5. [DOI] [PubMed] [Google Scholar]
- 16.Lebowitz PF, Davide JP, Prendegast GC. Evidence that farnesyltransferase inhibitors suppress ras transformation by interfering with Rho activity. Mol. Cell. Biol. 1995;15:6613–6622. doi: 10.1128/mcb.15.12.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker D, O’Neill JK, Wilcox C, Gschmeissner S, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J. Neuroimmunol. 1990;28:261–271. doi: 10.1016/0165-5728(90)90019-j. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill JK, Baker D, Davison AN, Maggon KK, Jaffee BD, Turk JL. Therapy of chronic relapsing experimental allergic encephalomyelitis and the role of the blood-brain barrier: Elucidication by the action of brequinar sodium. J. Neuroimmunol. 1992;38:53–62. doi: 10.1016/0165-5728(92)90090-8. [DOI] [PubMed] [Google Scholar]
- 19.Backlund PS. Post-translational processing of RhoA. J. Biol. Chem. 1997;272:33175–33180. doi: 10.1074/jbc.272.52.33175. [DOI] [PubMed] [Google Scholar]
- 20.Allen SJ, Baker D, O’Neill JK, Davison AN, Turk JL. Kinetics of cellular infiltration of the central nervous system during chronic relapsing experimental allergic encephalomyelitis in the Biozzi AB/H mouse. Cell. Immunol. 1993;146:335–350. doi: 10.1006/cimm.1993.1031. [DOI] [PubMed] [Google Scholar]
- 21.Jahner D, Hunter T. The ras related gene rhoB is an immediate early gene inducible by v-fps, epidermal growth factor and platelet derived growth factor in rat fibroblasts. Mol. Cell. Biol. 1991;11:3682–3690. doi: 10.1128/mcb.11.7.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakrabarti R, Engleman EG. Interrelationships between mevalonate metabolism and the mitogenic signaling pathway in T lymphocyte proliferation. J. Biol. Chem. 1991;266:12216–12222. [PubMed] [Google Scholar]
- 23.McPherson R, Tsoukas C, Baines MG, Vost A, Melino MR, Zupkis RV, Pross HF. Effects of lovastatin on natural killer cell function and other immunological parameters in man. J. Clin. Immunol. 1993;13:439–444. doi: 10.1007/BF00920019. [DOI] [PubMed] [Google Scholar]
- 24.Stanislaus R, Pahan K, Singh AK, Singh I. Amelioration of experimental allergic encephalomyelitis in Lewis rats by lovastatin. Neurosci. Lett. 1999;269:71–74. doi: 10.1016/s0304-3940(99)00414-0. [DOI] [PubMed] [Google Scholar]
- 25.Stanislaus R, Singh AK, Singh I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J. Neurosci. Res. 2001;66:155–162. doi: 10.1002/jnr.1207. [DOI] [PubMed] [Google Scholar]
- 26.Amor S, O’Neill JK, Morris MM, Smith RM, Wraith DC, Groome N, Travers PJ, Baker D. Encephalitogenic epitopes of myelin basic protein, proteolipid protein, myelin oligodendrocyte glycoprotein for experimental allergic encephalomyelitis induction in Biozzi ABH (H-2Ag7) mice share an amino acid motif. J. Immunol. 1996;156:3000–3008. [PubMed] [Google Scholar]
- 27.Weber C, Erl W, Weber KSC, Weber PC. HMG-CoA reductase inhibitors decrease CD11b expression and CD11b-dependent adhesion of monocytes to endothelium and reduce increased adhesiveness of monocytes isolated from patients with hypercholesterolemia. J. Am. Coll. Cardiol. 1997;30:1212–1217. doi: 10.1016/s0735-1097(97)00324-0. [DOI] [PubMed] [Google Scholar]
- 28.Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–184. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida M, Sawada T, Ishii H, Gerszten RE, Rosenweig A, Gimbrone MA, Jnr., Yasukochi Y, Numano F. HMG-CoA reductase inhibitor modulates monocyte-endothelial cell interaction under physiological flow conditions in vitro. Arterioscler. Thromb. Vasc. Biol. 2001;21:1165–1171. doi: 10.1161/hq0701.092143. [DOI] [PubMed] [Google Scholar]
- 30.Weitz-Schmidt G, Welzebbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, Cottens S, Takada Y, Hommel U. Statins selectvely inhibit leukocyte function antigen-1 by binding to a novel integrin site. Nature Med. 2001;7:687–692. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- 31.Neuhaus O, Strasser-Fuchs S, Fazekas F, Kieseier BC, Niederwieser G, Hartung HP, Archelos JJ. Statins as immunomodulators. Comparison with interferon-ß1b in MS. Neurology. 2002;59:990–997. doi: 10.1212/wnl.59.7.990. [DOI] [PubMed] [Google Scholar]
- 32.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognised type of immunomodulator. Nature Med. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 33.Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 34.Wong B, Lumma WC, Smith AM, Sisko JT, Wright SD, Cai TQ. Statins suppress THP-1 cell migration and secretion of matrix metalloproteinase 9 by inhibiting geranylgeranylation. J. Leukoc. Biol. 2001;69:959–962. [PubMed] [Google Scholar]
- 35.Steinman L. Multiple sclerosis: a two-stage disease. Nature Immunol. 2001;2:762–764. doi: 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]